

Abstract
Parkinson's disease (PD) is an age-related neurodegenerative disorder characterized in part by the preferential loss of nigrostriatal dopaminergic neurons. Although the precise etiology of PD is unknown, accumulating evidence suggests that PD involves microglial activation that exerts neurotoxic effects through production of proinflammatory cytokines and increased oxidative and nitrosative stress. Thus, controlling microglial activation has been suggested as a therapeutic target for combating PD. Previously we demonstrated that pharmacological inhibition of a class of enzymes known as prolyl hydroxylases via 3,4-dihydroxybenzoate administration protected against MPTP-induced neurotoxicity, however the exact mechanisms involved were not elucidated. Here we show that this may be due to DHB's ability to inhibit microglial activation. DHB significantly attenuated LPS-mediated induction of nitric oxide synthase and pro-inflammatory cytokines in murine BV2 microglial cells in vitro in conjunction with reduced ROS production and activation of NFκB and MAPK pathways possibly due to up-regulation of HO-1 levels. HO-1 inhibition partially abrogates LPS-mediated NFκB activity and subsequent NO induction. In vivo, DHB pre-treatment suppresses microglial activation elicited by MPTP treatment. Our results suggest that DHB's neuroprotective properties could be due to its ability to dampen induction of microglial activation via induction of HO-1.
1. Introduction
Parkinson's disease (PD) is a slowly progressive age-related neurodegenerative disorder characterized by irreversible degeneration of the dopaminergic nigrostriatal pathway, resulting in marked impairments of motor control. Although PD has been heavily researched in the last two decades, the precise etiology of the disease is still unknown. However, research in recent years has provided substantial evidence supporting the hypothesis that oxidative stress and inflammation both likely play a major role in disease pathogenesis [1–4].
A growing body of both experimental and clinical studies suggests that inflammation may contribute to neurodegeneration associated with many neurological diseases including PD [5, 6]. The first evidence for a role for inflammation in PD came from a postmortem study by McGeer and colleagues who found activated microglia and T-lymphocytes in the SNpc of a PD patient [7]. Since then, there have been numerous reports supporting a role for neuroinflammatory processes in PD pathogenesis [8–10]. In addition to the presence of activated microglia, increased levels of proinflammatory cytokines including IL1β and IL-6 and enzymes associated with inflammation such as inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) have been observed in the Parkinsonian brain [11, 12].
Microglia are the resident immune-competent cells in the brain that act to amplify the effects of inflammation thereby mediating ongoing cellular degeneration [13, 14]. In the event of brain damage or infection, microglia cells become activated and secrete a variety of proinflammatory mediators and other potentially neurotoxic factors which can have deleterious effects on neighboring neurons. Suppression of microglia activation has been suggested as a possible therapeutic intervention that may alleviate the progression of various neurodegenerative diseases including PD [15].
Prolyl 4-hydroxylases (PHDs) are a family of enzymes that act to hydroxylate a variety of substrates, the most well studied of which is hypoxia-inducible factor (HIF). HIF is a transcription factor that plays a major role in the regulation of cellular and systemic oxygen homeostasis [16]. It is a heterodimer consisting of a constitutively expressed β subunit and one of two α subunits, HIF-1α or HIF-2α, which are mainly regulated by oxygen. During normoxia, HIFα is continuously synthesized and hydroxylated on a specific proline residue by specific PHD isoforms in a cell- and tissue-specific manner [17]. Hydroxylated HIFα is rapidly ubiquitylated and subsequently degraded by the proteasome. Under hypoxic conditions, PHDs are prevented from hydroxylating proline residues of the HIF-1α protein resulting in upregulation of HIFα isoforms. This results in accumulation of HIFα in the cytosol followed by its translocation to the nucleus where it binds HIF-1β. The heterodimer then binds to hypoxia response elements (HREs) found on a variety of genes including heme oxygenase-1 (HO-1), transferrin receptor (TfR), and tyrosine hydroxylase (TH), resulting in their transcriptional induction [18].
Previous studies have shown that broad pharmacological inhibition of the PHDs alleviates neurodegeneration associated with stroke and hypoxic-ischemic injuries [19, 20]. Recently, studies from our own laboratory found that PHD inhibition via the broad-acting inhibitor 3,4-dihydroxybenzoate (DHB) protects against MPTP-induced neurotoxicity [21]. The exact mechanisms involved, however, were not elucidated in this previous study. Here, we report that the neuroprotective effects of DHB could be due at least in part to antiinflammatory properties of the drug. Our results demonstrate that DHB prevents microglial activation that coincides with reduced neuronal cell loss in both in vitro and in vivo models of PD. These effects may be attributable to DHB's known ability to induce increases in HO-1 levels, in turn eliciting both anti-oxidant and anti-inflammatory effects.
2. Materials and Methods
2.1. Reagents
All cell culture reagents were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies against MAPKs, NFκB, iNOS, and actin were purchased form Cell Signaling Technology (Beverley, MA, USA). Reagents for qPCR were purchased from Promega (Madison, WI, USA) and Roche Applied Science (USA).
2.2. Cell Lines
The BV2 cell line was obtained from Dr. Luc Vallieres, Quebec City, Canada. Immortalization of the BV2 cell line via infection of murine primary microglial cell cultures with a v-raf/v-myc oncogene-carrying retrovirus (J2) has been described previously [22]. Phenotypically, BV2 cells tested positive for MAC1 and MAC2 antigens. BV2 cells were maintained in Dulbecco's modified essential medium supplemented with 10% heat-inactivated fetal bovine serum, streptomycin (10 mg/mL), and penicillin (10 U/mL) at 37°C. The dopaminergic neuronal cell line used in the in vitro neuronal viability studies, N27, is derived from embryonic rat dopaminergic mesencephalic neurons via SV40 large T-antigen immortalization. The cells were grown in RPMI medium 1640 containing 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 μg/mL) [23]. To examine the effects of DHB, microglial BV2 cells were treated with DHB for 1 h before stimulation with LPS. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reduction assay as previously described [24].
2.3. Mice
10-week-old C57BL/6 male mice (Jackson Laboratories, Bar Harbor, ME) were used for the described in vivo studies. Mice were housed according to standard animal care protocols, fed ad libitum, kept on a 12 h light/dark cycle, and maintained in a pathogen-free environment in the Buck Institute Vivarium. All experiments were approved by local IACUC review and conducted according to current NIH policies on the use of animals in research. For DHB studies (n = 10 per group), the drug was diluted to a final dose of 100 mg/kg (in 5% ethanol) and administered intraperitoneally to mice 6 h prior to 2 consecutive intraperitoneal injections of either saline vehicle or 20 mg/kg of MPTP given 12 h apart [21]. Age-matched controls (10 weeks of age) were also treated with 2 × 20 mg/kg MPTP or saline, 12 h apart. Seven days following the final MPTP or saline injection, mice were sacrificed for either tissue harvest for biochemical assays or brain fixation via intracardiac perfusion for immunohistochemistry.
2.4. RT-PCR
BV2 cells (7.5 × 105 cells on a 6 cm dish) were treated with LPS in the presence or absence of DHB and total RNA extracted with TRI reagent. Total RNA (1 μg) was then reverse-transcribed in a reaction mixture containing 1 U RNase inhibitor, 500 ng random primers, 3 mM MgCl2, 0.5 mM dNTP, and 10 U reverse transcriptase in RT buffer (Promega). The synthesized cDNA was used as a template for qPCR analysis using the universal probe library system from Roche. For in vivo studies, striatal tissue was dissected out from various treatment groups, total RNA extracted using the TRIZOL method, and reverse-transcribed to cDNA. qPCR analysis of GAPDH, TNF-α, IL-6, iNOS, and HO-1 was performed using the Roche universal probe library detection system. Relative quantification of gene expression was performed using the comparative threshold (CT) method. Changes in mRNA expression level were calculated following normalization to GAPDH. The ratios obtained after normalization are expressed as fold change over corresponding wild-type controls [25].
2.5. Measurement of IL-6 and Nitrate Levels in Conditioned Media (CM)
Microglial cells (1 × 105 cells per well in a 24-well plate) were pretreated with DHB or normal media for 1 h and then stimulated with LPS (100 ng/mL). CM was collected from the cultured microglia 24 h following LPS stimulation and the concentrations of IL-6 measured using the mouse IL-6 ELISA kit from BD Biosciences according to manufacturer's instructions [26]. Accumulated nitrite was measured in the CM using the Griess reagent (Sigma). For mouse IL-6 measurements, striatal tissue was dissected from saline or MPTP groups in the absence or presence of co-DHB treatment and IL-6 levels measured from tissue homogenates using the ELISA kit as described above. For HO-1 inhibition studies, BV2 were pretreated with ZnPPIX (10 μM) for 30 min and treated with DHB for another 1 h before LPS application. The NO levels were measured after 24 h following LPS stimulation.
2.6. Western Blot Analyses
Whole cell protein lysates from BV2 cells were prepared in lysis buffer, protein samples separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (Invitrogen). Membranes were blocked with 5% skim milk in 10 mM Tris-HCl containing 150 mM NaCl and 0.5% Tween 20 (TBST), then incubated with primary antibodies (1 : 1000) against iNOS, phosphorylated 65 subunit, phosphorylated p38, phosphorylated JNK, p38, JNK, HO-1, or actin [27]. After thoroughly washing with TBST, horseradish peroxidase-conjugated secondary antibodies (1 : 3000 dilution in TBST; Millipore, CA, USA) were applied and blots developed using an enhanced chemiluminescence detection kit (Pierce Biotechnology, Rockford, IL, USA).
2.7. Measurement of Intracellular Reactive Oxygen Species (ROS) Levels
Intracellular accumulation of ROS was measured using H2DCF-DA (Sigma) as previously described [28]. In brief, microglial cells were stimulated with LPS for 16 h in the absence or presence of DHB then stained with 20 μM H2DCF-DA in Hank's balanced salt solution buffer for 1 h at 37°C. DCF fluorescence intensity was measured on a fluorescence plate reader at 485 nm excitation and 535 nm emission (Molecular Devices, CA).
2.8. Transient Transfection of NFκB Reporter Construct and Assay by Luciferase
Transfection of the NFκB binding reporter gene into BV2 cells was performed using lipofectamine 2000 (Invitrogen, USA). The NFκB binding reporter plasmid contains three copies of the κB-binding sequence fused to the firefly luciferase gene (Clontech, Mountain View, CA, USA). BV2 cells (2 × 105 cells per well in a 12-well plate) were transfected with 1 μg of the reporter construct mixed with lipofectamine 2000. After 48 h, cells were harvested and luciferase activity assayed as previously described [28]. To determine the effect of DHB on reporter gene activity, cells were pretreated for 1 h with the agent before treating with LPS (0.1 μg/mL) for 4 h prior to cell harvest.
2.9. Immunocytofluorescent Staining to Assess Nuclear Translocation of NFκB
BV2 cells were seeded onto glass coverslips and stimulated with LPS (100 ng/mL) following pretreatment with DHB (100 μM) or media for 1 h. Then cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Trition X-100 for 30 min. After blocking with 5% nonfat milk in PBS buffer, cells were incubated with rabbit anti-p65 antibodies for 1 h at room temperature. After a brief wash, cells were incubated with Alexa fluor-conjugated secondary antibody (1: 500, Molecular Probes) [28]. Finally, the cells were washed again, mounted with vectashield hard mount with DAPI and visualized using a Zeiss LSM 510 confocal microscope.
2.10. Assay of Effects of CM from BV2 on Cell Viability in Dopaminergic N27 Cells
BV2 cells were stimulated with LPS (100 ng/mL) in the absence or presence or DHB (100 μM) for 24 h. CM from control, DHB treated, LPS-treated, or LPS + DHB-treated cells was added to dopaminergic N27 cells plated in 96 well plates. After 48 h, N27 cell viability was assessed via the MTT assay.
2.11. In Vivo Immunohistochemistry
Immunochemistry was performed on sections from the striata of fixed perfused brains from mice treated with MPTP in the absence or presence of DHB cotreatment versus saline-treated controls. Microglial activation was detected using primary antibodies against Iba1 [29]. Briefly, 7 μm SN sections from paraffin-embedded brains were cut and processed for staining. Sections were mounted onto slides and processed in 10 mM citrate buffer for enhancement of antigen retrieval. After blocking with 10% donkey serum for 1 h, primary antibody (1 : 500, anti-Iba1 antibody; Dako) was applied to the sections for overnight incubation at 4°C followed by biotinylated secondary antibody and 3,3′-diaminobenzidine processing.
2.12. Statistical Analysis
Unless otherwise stated, all experiments were performed in triplicate samples and repeated at least three times. The data are presented as the mean ± SE and statistical comparisons between groups were performed using one-way ANOVA followed by Student's t-test between two populations based on the assumption that both populations have normal distribution. A P value < 0.05 was considered significant.
3. Results
3.1. DHB Suppresses LPS-Mediated Upregulation of Both Proinflammatory Genes and Associated Gene Products in BV2 Microglial Cells
LPS stimulation is known to induce the expression of multiple proinflammatory genes that can in turn contribute to neuroinflammation and subsequent neurotoxicity. In our studies, LPS treatment of BV2 microglial cells was found to result in upregulation of mRNA levels for the proinflammatory cytokines TNF-α and IL-6 and for the nitric oxide (NO) producing enzyme iNOS (Figure 1(a)). Pretreatment with DHB not only significantly decreased proinflammatory gene expression but also iNOS protein levels and amounts of NO and IL-6 secreted into the CM in a concentration-dependent manner (Figures 1(b) and 1(c)). DHB at the concentrations used for these studies (10 to 100 μM) were not found to have any effects on cell viability as assessed by the MTT assay (data not shown).
Figure 1.
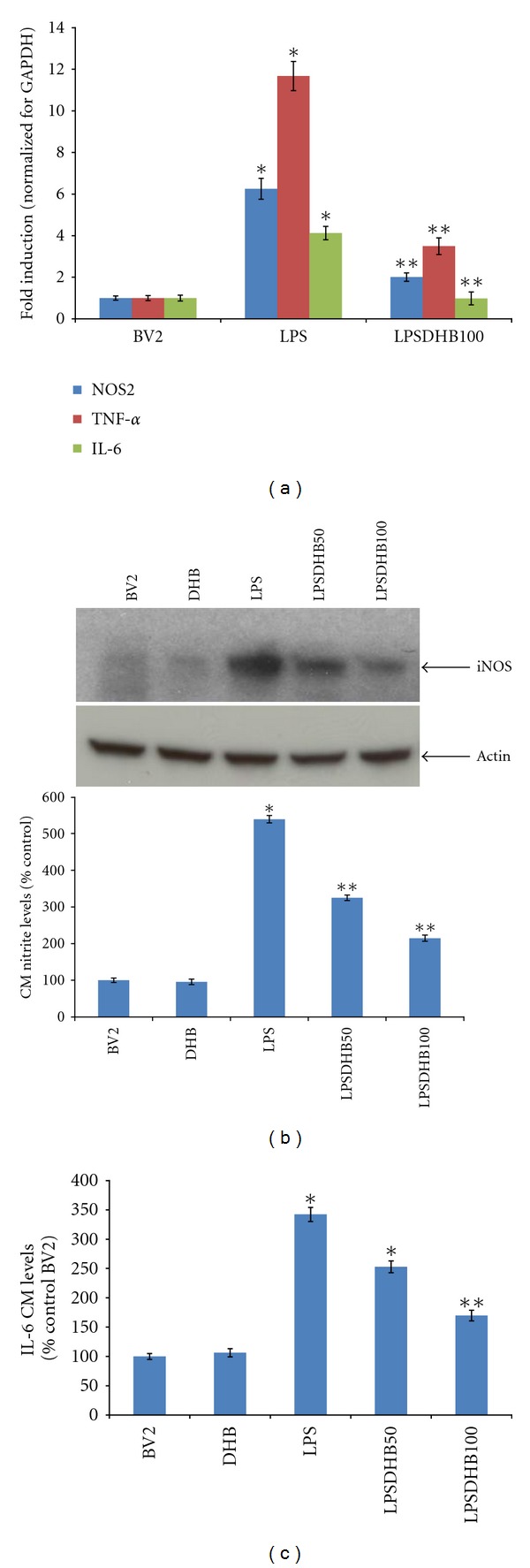
(a) BV2 microglial cells were pretreated with DHB (100 μM) for 1 h followed by cotreatment with LPS (100 ng/mL) for 6 h. Total RNA was isolated and real-time PCR analysis performed. Relative mRNA levels for iNOS, TNF-α, and IL-6 were normalized for GAPDH. Each bar represents means ± SEM. *P < 0.05 compared with the control group, **P < 0.05 compared with the LPS-treated group. (b) Protein was extracted from whole cell lysates and subjected to western blot analysis for iNOS protein levels; actin was used as a loading control. Conditioned media (CM) was collected after 24 h LPS (100 ng/mL) ± DHB treatment and nitrite levels determined using the Griess reagent. Data are presented as means ± SD of at least four independent experiments. *P < 0.05 compared with the control group, **P < 0.05 compared with the LPS treated group. (c) IL-6 levels in the CM were determined using an ELISA kit according to manufacturer's recommendations. Data are presented as means ± SD of at least four independent experiments. *P < 0.05 compared with the control group, **P < 0.01 compared with the LPS treated group.
3.2. DHB Attenuates LPS-Induced Intracellular ROS Production and Activation of NFκ B and MAP Kinase Pathways
LPS is known to induce the production of intracellular ROS that can in turn stimulate expression of proinflammatory genes via the activation of secondary messenger systems including NFκB and the MAP kinases [30]. LPS exposure was found to not only induce increased intracellular ROS production in microglial BV2 cells (Figure 2), but also phosphorylation of the p65 subunit of NFκB and the MAP kinases JNK and p38 necessary for their activation (Figure 3). Pretreatment with DHB was found to significantly block LPS-induced ROS production in a dose-dependent manner (Figure 2) and to modulate LPS-induced phosphorylation of these pathway components (Figure 3). Furthermore, it prevented nuclear translocation of p65 and subsequent NFκB-dependent transcriptional activity, the latter in a dosage-dependent fashion (Figure 4). These data suggest that DHB can prevent oxidative stress-mediated induction of these inflammatory pathways.
Figure 2.
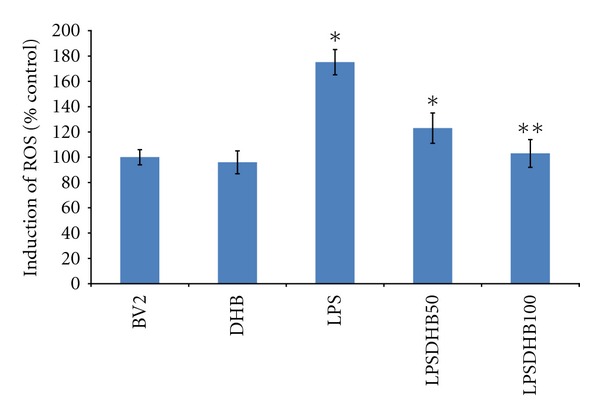
BV2 microglial cells were pretreated with DHB for 1 h followed by co-treatment with LPS (100 ng/mL) for 16 h. ROS levels were measured using the DCFDA method. *P < 0.05 compared with the control group, **P < 0.05 compared with the LPS-treated group.
Figure 3.
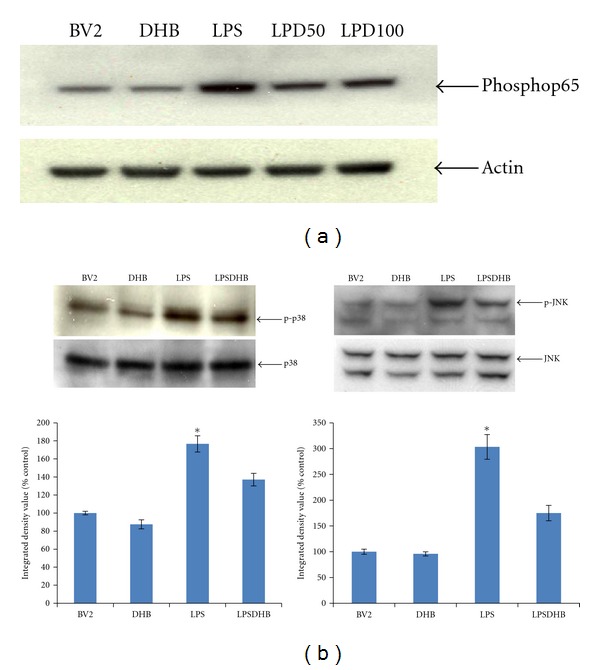
BV2 cells were pretreated with DHB for 1 h (50 & 100 μM) and then stimulated with LPS (100 ng/mL) for a 1 h incubation period. Cells were lysed, run on a SDS-PAGE gel, transferred to PVDF membranes, and blotted with specific antibodies to (a) phosphorylated p65 NFκB subunit or (b) phosphorylated or unphosphorylated p38 and JNK. Actin was used as a loading control. Band density (integrated density value) is expressed graphically as a percentage ratio of densitometric optical density of phosphorylated forms to that of nonphosphorylated p38 and JNK. Data (mean ± SD) are from three independent experiments; *P < 0.05 relative to untreated control sample.
Figure 4.
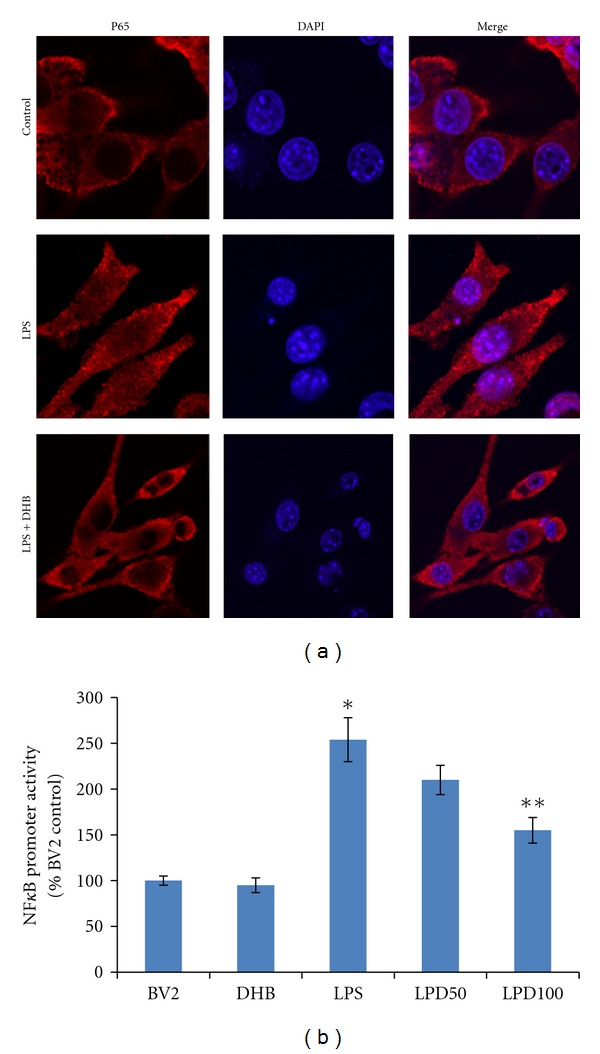
(a) After pretreatment with DHB (100 μM) for 1 h, BV2 cells were stimulated with 100 ng/mL LPS for 1 h and nuclear translocation of the NFκB p65 subunit assessed by confocal fluorescence microscopy using a fluorescent anti-p65 antibody. Representative laser confocal microcopy images of p65 (red stain) and nuclear DAPI staining (blue) in cells exposed to LPS ± DHB are shown; pink, merged. (b) BV2 cells were transiently transfected with NFκB-Luc for 24 h and treated with 100 ng/mL LPS for 4 h ± DHB for 1 h. Cell lysates were assayed for luciferase activity (mean ± SE, n = 4). *P < 0.05 versus control, **P < 0.05 versus LPS.
3.3. DHB Induces Upregulation of Hemoxygenase-1 (HO-1) Expression and Protein Levels in BV-2 Microglia Cells
We demonstrated in our previous study that PHD inhibition via DHB results in upregulation of the enzyme HO-1, likely via a HIF-1α transcription-dependent process [21]. HO-1 has recently emerged as a key molecule in the resolution of oxidative stress-mediated microglial activation including that induced by LPS [31]. BV2 microglia treated with DHB for 24 h were found to have both elevated HO-1 expression and protein levels and this was found to occur in a concentration-dependent manner (Figures 5(a) and 5(b)). Pretreatment with ZnPPIX, a potent HO-1 inhibitor, was found to abrogate the inhibitory effects of DHB on LPS-induced NO production (Figure 5(c)) have a small but significant effects on NFkB activity (Figure 5(d)).
Figure 5.
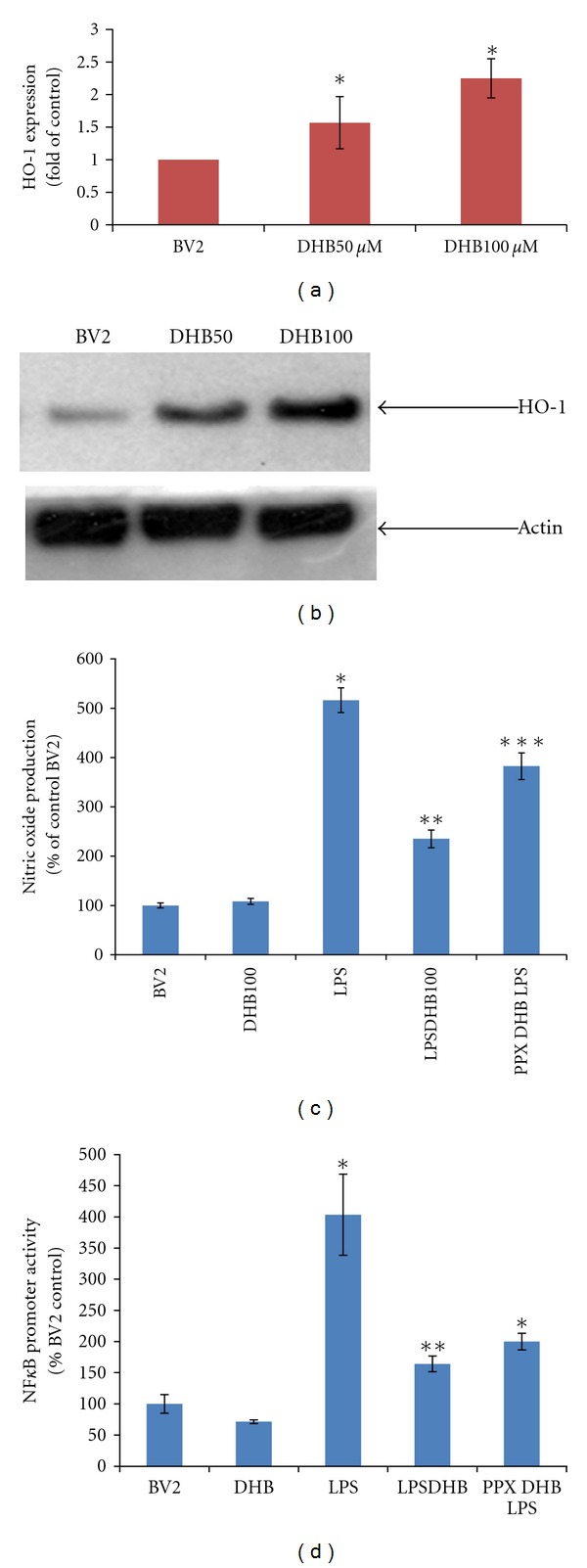
(a) Cells were stimulated with given concentrations of DHB for 6 h and mRNA levels of HO-1 analyzed via real-time PCR as described. (b) BV2 cells were stimulated with various concentrations of DHB for 24 h and whole cell lysates subjected to western blot analysis using antibody against HO-1 protein; *P < 0.05 versus control. (c) Cells were pretreated with ZnPPIX (20 μM) for 30 min followed by co-treatment with DHB (100 μM) for another 1 h before LPS (100 ng/mL) addition. Conditioned media (CM) was collected after 24 h of LPS treatment and nitrite levels were determined using the Griess reagent. Each bar represents mean ± SEM from at least four independent experiments. *P < 0.05 compared with the control group, **P < 0.05 compared with the LPS treated group, ***P < 0.05 compared with the LPSDHB100 treated group. (d) BV2 NFκB-Luc cells were pretreated with HO-1 inhibitor ZnPPIX (20 μM) for 30 min followed by co-treatment with DHB (100 μM) for another 1 h before LPS (100 ng/mL) addition for 4 h. Cell lysates were assayed for luciferase activity (mean ± SE, n = 4). *P < 0.05 versus control, **P < 0.05 versus LPS.
3.4. The Neuroprotective Effects of DHB Coincides with Inhibition of Microglial Activation Both In Vitro and In Vivo
A number of studies have demonstrated that activated microglia can induce neural toxicity [32–34]. Our in vitro results suggest that DHB could possibly exert neuroprotective effects via its ability to suppress microglial activation. To assess this possibility, the neuronal toxicity of conditioned media (CM) from LPS-treated BV2 cells grown in the absence and presence of DHB treatment was evaluated in dopaminergic N27 cells. CM from LPS-stimulated microglia (LPS-CM) was found to produce significant toxicity in N27 cells, which was attenuated in CM from BV2 cells grown in the additional presence of co-DHB treatment (Figure 6). This suggested that the toxicity of the CM derived from LPS-treated microglia could be dependent on release of soluble neurotoxic factors and be prevented by presence of DHB. We cannot, however, rule out the possibility that part of the neuroprotective effects may be due to residual DHB in the CM from the microglial cells that could have a direct impact on neuronal survival.
Figure 6.
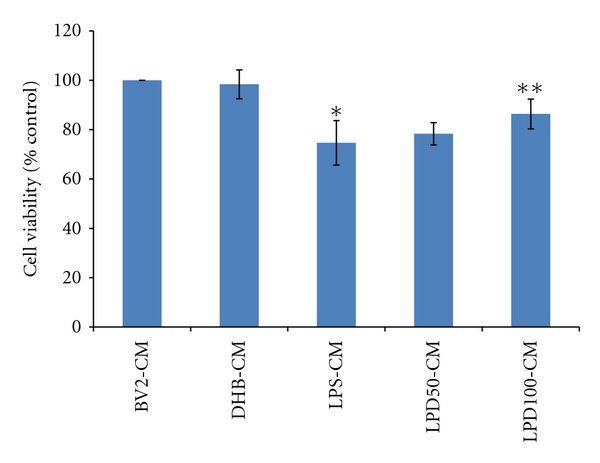
BV2 cells were stimulated with LPS (100 ng/mL) ± DHB (100 μM) for 24 h. CM from control (BV2-CM), DHB treated (DHB-CM), LPS-treated (LPS-CM), and LPS/DHB treated (50 & 100 μM; LPD50-CM, LDP100-CM) BV2 cells was added to dopaminergic N27 cells plated in 96 well plates. After 48 h, N27 cell viability was assessed via the MTT assay. Data is expressed as mean ± SD, n = 4. *P < 0.01, compared with control-CM group, **P < 0.01, compared with LPS-CM group.
Based on our in vitro results, we next assessed the ability of DHB to prevent microglial activation in MPTP-treated mice, a commonly used toxin model of PD. Our previously published data demonstrated that DHB treatment prevented nigrostriatal neurotoxicity associated with this model [21]. We found that MPTP treatment resulted in a significant increase in striatal IL-6 levels that was significantly attenuated by co-DHB administration (Figure 7(a)). We next evaluated the effects of DHB treatment on striatal expression of various cytokine mRNAs (TNFα, IL6, and iNOS) following MPTP intoxication. qPCR analysis revealed significant induction of proinflammatory gene expression in the presence of MPTP that was blocked by pretreatment with DHB (Figure 7(b)). To assess the impact of DHB on microglial activation in MPTP-treated mice [28], immunohistochemistry was performed on sections from control and MPTP mice in the absence and presence of DHB cotreatment using the microglial activation marker Iba1 (Figure 8). Prior to activation, microglia normally exhibits a highly ramified morphology (as observed in both control and DHB-treated mice). In response to activation by MPTP, microglia begins to withdraw their ramified branches and became amoeboid-like. This activated morphological phenotype was found to be significantly attenuated in the presence of DHB pretreatment, demonstrating the ability of DHB to in part attenuate microglial activation resulting from MPTP treatment.
Figure 7.
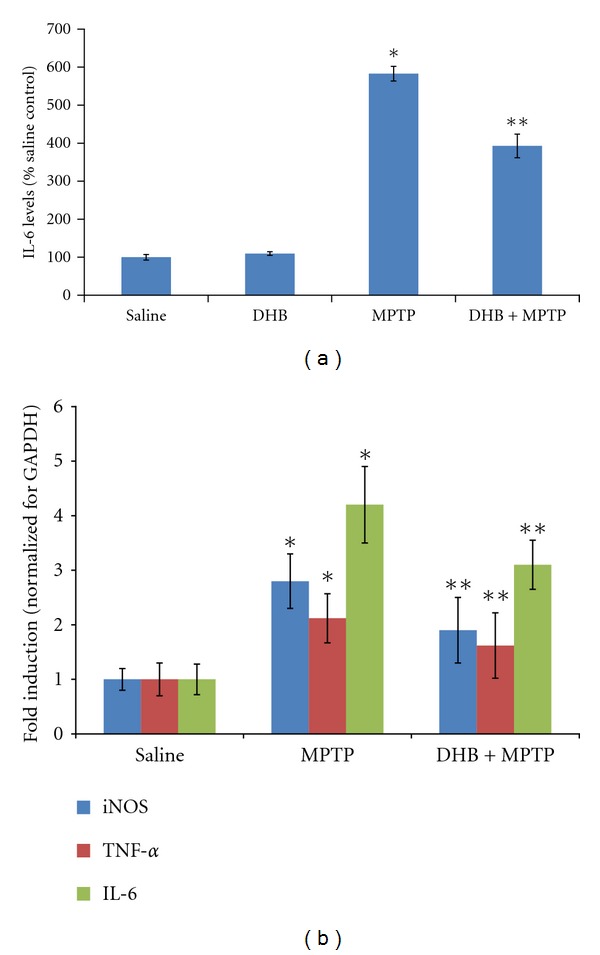
Mice were pretreated with DHB (100 mg/kg, intraperitoneally) 6 h prior to administration of either saline (Sal) or MPTP (20 mg/kg x2, 12 h apart). Animals were sacrificed 2 days later. (a) Levels of striatal IL-6 were measured using an ELISA kit from BD Biosciences. Each bar represents mean + SEM for 4 animals in each group. *P < 0.05 compared with the Sal group, **P < 0.01 compared with the MPTP-treated group. (b) DHB pretreatment decreases the mRNA levels of MPTP upregulated proinflammatory cytokines. *P < 0.05 compared with the Sal group, **P < 0.05 compared with the MPTP-treated group.
Figure 8.
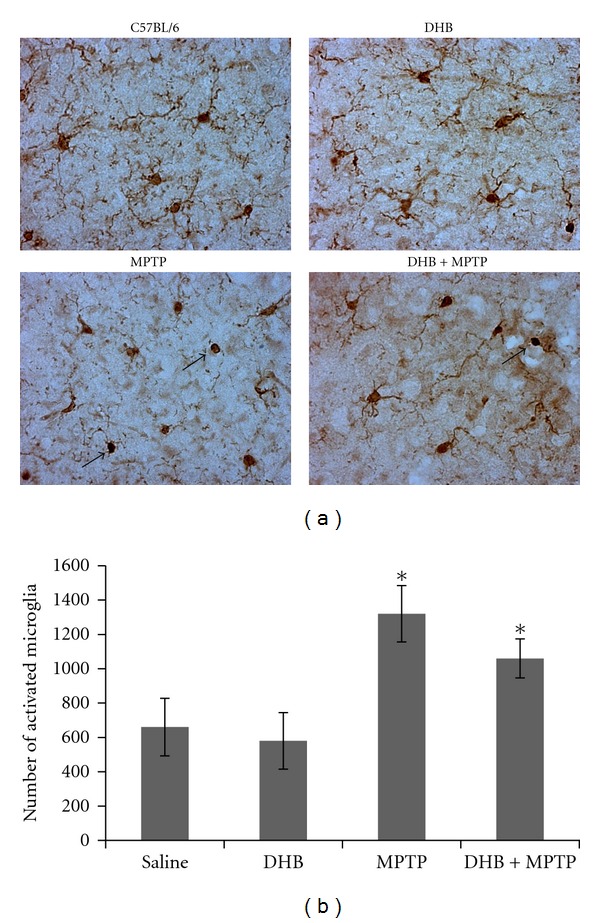
Iba1 immunostaining of SN sections of mice treated with either saline (Control), DHB, MPTP or DHB + MPTP. (a) Microglia in MPTP-treated mice display a morphological transformation from resting ramified microglia to activated amoeboid like forms with withdrawn processes (indicated by arrows) which is inhibited in the presence of DHB treatment. (b) MPTP treatment leads to a significant increase in the number of activated microglia relative to saline and DHB treated mice. Activated microglia were bilaterally counted under a 40x objective. Data are means ± SEM for 4 mice per group, *P < 0.05 compared to saline group.
4. Discussion
Previous studies have demonstrated that the pharmacological PHD inhibitor 3,4-DHB can directly protect cultured neuronal cells against both oxidative stress [20] and ischemic injury [19]. Our current study suggests that in addition to these direct effects, DHB could also elicit neuroprotection via a heretofore unknown action of the drug—its capacity to dampen microglial activation via its ability to prevent oxidative induction of the MAPK and NFκB signaling pathways. These pathways can in turn elicit the synthesis and release of proinflammatory factors from activated microglia, indirectly impacting on neighboring neurons [35–37].
LPS is known to induce oxidative phosphorylation of p38 and JNK within microglia, stimulating activation of NFκB via phosphorylation of its p65 subunit. Subsequent nuclear translocation of p65 results in increased expression of several proinflammatory genes [28]. In our current study, DHB treatment was found to partially inhibit LPS-induced increases in microglial ROS levels as well as phosphorylation of p38 and JNK, subsequent p65 translocation and mediation of transcriptional activation. This was associated with reduced expression of proinflammatory genes in microglial cells induced either directly in vitro by LPS or in vivo by MPTP along with reduced release of IL-6. In vitro, DHB also partially prevented both increased NO release and had a small but significant effect on NFκB activation. Pharmacological inhibition of iNOS has been shown to prevent dopaminergic neurodegeneration as a consequence of microglial activation and transgenic mice lacking iNOS are more resistant to MPTP-mediated dopaminergic neurotoxicity [38–41].
The anti-inflammatory effects of DHB could be due to its ability to upregulate HO-1 expression. HO-1 has recently been demonstrated to inhibit oxidatively induced microglial activation such as that elicited by LPS [31]. In the present study, inhibition of HO-1 using the pharmacological inhibitor ZnPPIX was found to attenuate DHB-mediated inhibition of microglial NO production as well as having a small but significant effect on NFκB activity. HO-1 is a microsomal enzyme that catalyzes oxidative cleavage of the porphyrin ring of the heme molecule leading to the formation of biliverdin, carbon monoxide (CO), and free iron [42]. The beneficial protective effects of HO-1 in inflammation are mediated not only via enzymatic degradation of proinflammatory free heme, but also via production of the antiinflammatory compounds bilirubin and carbon monoxide [43, 44]. Recent reports have suggested that upregulation of HO-1 may have both anti-oxidant and anti-inflammatory effects [30, 45–47] and that therefore the HO-1 system may be an important therapeutic target for inflammation associated with neurodegeneration. The concomitant enhancement of HO-1 expression and reduction in LPS-induced NO production by DHB is consistent with previous reports using other anti-inflammatory agents [48, 49].
Based on our current results, we propose that the neuroprotective effects of DHB may be in part due to its ability to inhibit the inflammatory NFκB/cytokine pathway. However, this does not definitively rule out that it may alternatively be acting via direct neuroprotective effects on the neurons themselves. Future studies using in vivo LPS-induced nigrostriatal degeneration models will be helpful in delineating the relative contributions of anti-inflammatory versus direct antioxidant in terms of the neuroprotective effects of DHB.
Microglial activation has long been associated with dopaminergic neuropathology in PD [9, 32]. DHB administration was found to reduce microglial activation and the release of soluble inflammatory factors in association with reduced neurotoxicity both in vitro and in vivo. DHB's ability to dampen microglial activation may suggest a novel mechanism of action for the drug that mechanistically could involve HO-1 induction. This hypothesis will require further validation, but presents a possible novel mode of action for the drug.
Acknowledgments
These studies were funded by R01 NS045615 (J. K. Andersen) and a grant from the MJFF to JKA. The authors thank Mr. Anand Rane for help with immunohistochemistry experiments.
References
- 1.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nature Medicine. 2004;10(supplement):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 2.Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Movement Disorders. 2011;26(6):1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- 3.Thomas B, Beal MF. Parkinson's disease. Human Molecular Genetics. 2007;16(2):R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 4.Gupta A, Dawson VL, Dawson TM. What causes cell death in Parkinson’s disease? Annals of Neurology. 2008;64(supplement 2):S3–S15. doi: 10.1002/ana.21573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minghetti L. Role of inflammation in neurodegenerative diseases. Current Opinion in Neurology. 2005;18(3):315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 6.Kuhn W, Müller T, Nastos I, Poehlau D. The neuroimmune hypothesis in Parkinson’s disease. Reviews in the Neurosciences. 1997;8(1):29–34. doi: 10.1515/revneuro.1997.8.1.29. [DOI] [PubMed] [Google Scholar]
- 7.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 8.Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson’s disease. Progress in Neurobiology. 2002;68(5):325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- 9.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Experimental Neurology. 2007;208(1):1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 11.Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Molecular and Cellular Neuroscience. 2000;16(6):724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- 12.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 13.Qian L, Flood PM, Hong JS. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. Journal of Neural Transmission. 2010;117(8):971–979. doi: 10.1007/s00702-010-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JK, Tran T, Tansey MG. Neuroinflammation in Parkinson’s disease. Journal of Neuroimmune Pharmacology. 2009;4(4):419–429. doi: 10.1007/s11481-009-9176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. Journal of Pharmacology and Experimental Therapeutics. 2003;304(1):1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 16.Epstein ACR, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 17.Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. The Journal of Biological Chemistry. 2003;278(33):30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107(1):1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 19.Hurn PD, Koehler RC, Blizzard KK, Traystman RJ, Beckman JS. Deferoxamine reduces early metabolic failure associated with severe cerebral ischemic acidosis in dogs. Stroke. 1995;26(4):688–695. doi: 10.1161/01.str.26.4.688. [DOI] [PubMed] [Google Scholar]
- 20.Siddiq A, Ayoub IA, Chavez JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition: a target for neuroprotection in the central nervous system. The Journal of Biological Chemistry. 2005;280(50):41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee DW, Rajagopalan S, Siddiq A, et al. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. Model for the potential involvement of the hypoxia-unducible factor pathway in Parkinson disease. The Journal of Biological Chemistry. 2009;284(42):29065–29076. doi: 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. Journal of Neuroimmunology. 1990;27(2-3):229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 23.Chinta SJ, Rajagopalan S, Butterfield DA, Andersen JK. In vitro and in vivo neuroprotection by γ-glutamylcysteine ethyl ester against MPTP: relevance to the role of glutathione in Parkinson’s disease. Neuroscience Letters. 2006;402(1-2):137–141. doi: 10.1016/j.neulet.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 24.Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. NeuroMolecular Medicine. 2008;10(4):333–342. doi: 10.1007/s12017-008-8047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sriram K, Miller DB, O’Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-α . Journal of Neurochemistry. 2006;96(3):706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- 26.Kaku K, Shikimi T, Kamisaki Y, et al. Elevation of striatal interleukin-6 and serum corticosterone contents in MPTP-treated mice. Clinical and Experimental Pharmacology and Physiology. 1999;26(9):680–683. doi: 10.1046/j.1440-1681.1999.03113.x. [DOI] [PubMed] [Google Scholar]
- 27.Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. The Journal of Biological Chemistry. 2004;279(31):32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- 28.Wang S, Wang H, Guo H, Kang L, Gao X, Hu L. Neuroprotection of Scutellarin is mediated by inhibition of microglial inflammatory activation. Neuroscience. 2011;185:150–160. doi: 10.1016/j.neuroscience.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Mallajosyula JK, Kaur D, Chinta SJ, et al. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One. 2008;3(2) doi: 10.1371/journal.pone.0001616. Article ID e1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung JS, Shin JA, Park EM, et al. Anti-inflammatory mechanism of ginsenoside Rh1 in lipopolysaccharide- stimulated microglia: critical role of the protein kinase A pathway and hemeoxygenase-1 expression. Journal of Neurochemistry. 2010;115(6):1668–1680. doi: 10.1111/j.1471-4159.2010.07075.x. [DOI] [PubMed] [Google Scholar]
- 31.Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochemical Pharmacology. 2010;80(12):1895–1903. doi: 10.1016/j.bcp.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 32.Long-Smith CM, Sullivan AM, Nolan YM. The influence of microglia on the pathogenesis of Parkinson’s disease. Progress in Neurobiology. 2009;89(3):277–287. doi: 10.1016/j.pneurobio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Li F, Zhu S, Wu C, Yan C, Liu Y, Shugan L. Neuroinflammation and cell therapy for Parkinson's disease. Frontiers in Bioscience. 2011;3:1407–1420. doi: 10.2741/232. [DOI] [PubMed] [Google Scholar]
- 34.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease. 2010;37(3):510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen- activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. Journal of Neuroscience. 1998;18(5):1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Z, Smith CJ, Van Eldik LJ. glias induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45(2):170–179. doi: 10.1002/glia.10314. [DOI] [PubMed] [Google Scholar]
- 37.Koistinaho M, Kettunen MI, Goldsteins G, et al. β-amyloid precursor protein transgenic mice that harbor diffuse Aβ deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1610–1615. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibata H, Katsuki H, Nishiwaki M, Kume T, Kaneko S, Akaike A. Lipopolysaccharide-induced dopaminergic cell death in rat midbrain slice cultures: role of inducible nitric oxide synthase and protection by indomethacin. Journal of Neurochemistry. 2003;86(5):1201–1212. doi: 10.1046/j.1471-4159.2003.01929.x. [DOI] [PubMed] [Google Scholar]
- 39.Iravani MM, Kashefi K, Mander P, Rose S, Jenner P. Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience. 2002;110(1):49–58. doi: 10.1016/s0306-4522(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 40.Liberatore GT, Jackson-Lewis V, Vukosavic S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nature Medicine. 1999;5(12):1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 41.Nowis D, Bugajski M, Winiarska M, et al. Zinc protoporphyrin IX, a heme oxygenase-1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer. 2008;8, article 197 doi: 10.1186/1471-2407-8-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiological Reviews. 2006;86(2):583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 43.Scott JR, Chin BY, Bilban MH, Otterbein LE. Restoring HOmeostasis: is heme oxygenase-1 ready for the clinic? Trends in Pharmacological Sciences. 2007;28(5):200–205. doi: 10.1016/j.tips.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 44.Jeong GS, Li B, Lee DS, et al. Lavandulyl flavanones from Sophora flavescens protect mouse hippocampal cells against glutamate-induced neurotoxicity via the induction of heme oxygenase-1. Biological and Pharmaceutical Bulletin. 2008;31(10):1964–1967. doi: 10.1248/bpb.31.1964. [DOI] [PubMed] [Google Scholar]
- 45.Lu DY, Tsao YY, Leung YM, Su KP. Docosahexaenoic acid suppresses neuroinflammatory responses and induces heme oxygenase-1 expression in BV-2 microglia: implications of antidepressant effects for omega-3 fatty acids. Neuropsychopharmacology. 2010;35(11):2238–2248. doi: 10.1038/npp.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. Journal of Neuroscience. 2006;26(6):1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitamura Y, Kakimura JI, Matsuoka Y, Yasuyuki Nomura, Gebicke-Haerter PJ, Taniguchi T. Activators of peroxisome proliferator-activated receptor-γ (PPARγ) inhibit inducible nitric oxide synthase expression but increase heme oxygenase-1 expression in rat glial cells. Neuroscience Letters. 1999;262(2):129–132. doi: 10.1016/s0304-3940(99)00055-5. [DOI] [PubMed] [Google Scholar]
- 48.Tsoyi K, Kim HJ, Shin JS, et al. HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride. Cellular Signalling. 2008;20(10):1839–1847. doi: 10.1016/j.cellsig.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Vareille M, Rannou F, Thélier N, et al. Heme oxygenase-1 is a critical regulator of nitric oxide production In Enterohemorrhagic Escherichia coli-infected human enterocytes1. The Journal of Immunology. 2008;180(8):5720–5726. doi: 10.4049/jimmunol.180.8.5720. [DOI] [PubMed] [Google Scholar]