

Abstract
Hepatocellular carcinoma (HCC) is the third most common cause of cancer mortality. Short-term prognosis of patients with HCC has improved recently due to advances in early diagnosis and treatment, but long-term prognosis is still unsatisfactory. Therefore, obtaining a further understanding of the molecular carcinogenic mechanisms and the unique pathogenic biology of HCC is important. The most characteristic process in hepatocarcinogenesis is underlying chronic liver injury, which leads to repeated cycles of hepatocyte death, inflammation, and compensatory proliferation and subsequently provides a mitogenic and mutagenic environment leading to the development of HCC. Recent in vivo studies have shown that the stress-activated mitogen-activated protein kinase (MAPK) cascade converging on c-Jun NH2-terminal kinase (JNK) and p38 plays a central role in these processes, and it has attracted considerable attention as a therapeutic target. However, JNK and p38 have complex functions and a wide range of cellular effects. In addition, crosstalk with each other and the nuclear factor-kappaB pathway further complicate these functions. A full understanding is essential to bring these observations into clinical settings. In this paper, we discuss the latest findings regarding the mechanisms of liver injury and hepatocarcinogenesis focusing on the role of the stress-activated MAPK pathway.
1. Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most common cause of cancer mortality worldwide [1]. The age-adjusted incidence rate of HCC varies geographically, with high rates in East Asia and moderate rates in Europe, North/South America, and Oceania. In addition, a definite increase in HCC incidence has recently been reported in Europe and North America [2].
Although the etiology of background liver disease varies geographically, chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection is the main cause of HCC in most areas. Other major etiologies include alcoholic hepatitis, hemochromatosis, and nonalcoholic steatohepatitis (NASH) [1, 3]. Accumulating evidence indicates that a sustained inflammatory reaction in the liver is the major contributing factor to the development of HCC. For example, in chronic hepatitis C, the host immune responses to HCV are often not strong enough to completely clear the infection, resulting in chronic stimulation of an antigen-specific immune response [4]. Hepatocyte damage is induced by the continued expression of cytokines and recruitment of activated inflammatory cells to the liver, which is followed by hepatocyte regeneration. This persistent cycle of necroinflammation and hepatocyte regeneration is thought to provide a mitogenic and mutagenic environment leading to the development of HCC [5–7].
Effective treatments for HCC include surgical resection, percutaneous ablation, and liver transplantation. Although liver transplantation results in a better survival rate [8], its application is limited by the scarcity of donor organs. Surgical resection and percutaneous ablation also provide a high rate of complete responses [9, 10], but long-term prognosis is not satisfactory, as indicated by the low overall survival of 22%–35% at 10 years [2]. This is due to frequent recurrence and lack of effective treatment for advanced HCC. Transarterial chemoembolization, radiotherapy, and conventional chemotherapy have been performed for advanced HCC, but their efficacies are limited [11].
Recently, molecular-targeted therapy was developed for the treatment of HCC. Sorafenib is a small molecule multikinase inhibitor targeting the Raf serine/threonine kinases, vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) pathway. In the SHARP (Sorafenib HCC Assessment Randomized Protocol) trial, sorafenib significantly prolonged median overall survival compared to placebo (10.7 versus 7.9 months, resp.) [12]. However, median survival was not satisfactory even in the sorafenib group. Although other molecular-targeting agents are currently under investigation, only limited improvements in survival benefit have been reported [13].
Eradication of the cause of chronic inflammation, such as hepatitis viruses, is the most beneficial means of preventing HCC [14]. Recent advances in antiviral treatment have made the eradication of HCV or significant suppression of HBV replication possible [15–17]. However, these therapies are not effective for all patients, and the development of HCC will not be prevented completely, especially in patients with advanced fibrosis. In addition, in patients with HCC based on metabolic syndrome including NASH, the incidence rate of which is increasing, dietary modifications and exercise are fundamental, but these approaches are sometimes ineffective and genetic background may also be important in such cases. Furthermore, once HCC has developed, the recurrence rate does not decline with time regardless of background liver disease, suggesting that most cases of late-phase recurrence are due to metachronous multicentric carcinogenesis [11]. Thus, gaining further understanding of the molecular mechanisms of carcinogenesis in HCC is important to discover new molecular targets, especially for preventing the occurrence or recurrence of HCC and systemic therapy for patients in the advanced stage.
Several molecular pathways have been reported to play important roles in hepatocarcinogenesis. As mentioned above, inflammation-mediated liver injury and compensatory hepatocyte proliferation are indispensable components during HCC development. Recent high-quality in vivo studies have shown that stress-activated mitogen-activated protein kinase (MAPK) signaling converging on c-Jun NH2-terminal kinase (JNK) and p38 plays a central role in these processes, and it has attracted considerable attention as a therapeutic target [18–24]. In this paper, we discuss the mechanisms of liver injury and hepatocarcinogenesis focusing on the role of the stress-activated MAPK pathway.
2. General Role of JNK in the Liver
2.1. Stress-Activated MAPK
MAPK cascades are signaling systems that transmit stimuli from outside the cell to the nucleus [25, 26]. Three major MAPK cascades have been characterized in detail, converging on ERKs, JNKs, and p38 MAPKs; each consists of three classes of serine/threonine kinases, MAPK, MAPK kinase (MAPKK), and MPKK kinase (MAP3K). MAP3K phosphorylates and thereby activates MAPKK, and activated MAPKK in turn phosphorylates and activates MAPK (Figure 1). Among the three MAPKs, ERKs are activated by various cytokines and growth factors and play a central role in cell growth and differentiation. JNKs and p38 MAPKs, also called stress-activated MAPKs, are preferentially activated by proinflammatory cytokines, such as tumor necrosis factor α (TNFα) and environmental and genotoxic stresses. After activation, stress-activated MAPKs phosphorylate specific serine/threonine residues of target substrates and exert a variety of cellular functions, such as cell death, survival, proliferation, migration, and inflammation.
Figure 1.
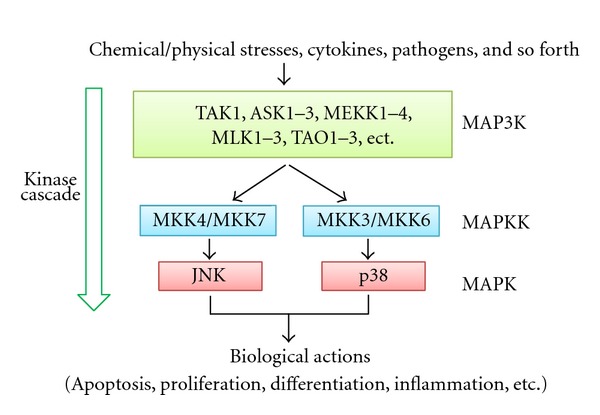
Stress-activated MAPK signaling pathway. This pathway consists of three classes of protein kinase—MAP3K, MAPKK, and MAPK—and converges on JNK and p38 through a kinase cascade. Only representative molecules are shown in MAP3K.
2.2. JNK Signaling
JNK has three isoforms (JNK1, JNK2, and JNK3) encoded by three different genes. JNK1 and JNK2 are expressed in almost all organs, including the liver, whereas JNK3 is a neuronal-specific isoform [27]. JNK is phosphorylated and activated by two MAPKKs, MKK4, and MKK7. Activated JNK phosphorylates and thereby activates c-Jun, JunD, ATF, and other transcription factors, which are involved in the formation and activation of the AP-1 complex [28]. In addition, JNK also modulates the function of other proteins by phosphorylation, such as Bcl-2 family members, which are closely related to apoptotic cell death factors [29]. Furthermore, although JNK1 and JNK2 isoforms play redundant roles in many physiological processes, they also have distinct biological activities in some situations [30]. For example, JNK1-deficient CD8+ T cells are unable to undergo antigen-stimulated expansion, whereas JNK2-deficient CD8+ T cells are hyperproliferative [31]. Therefore, the role of JNK in controlling diverse cellular functions, such as cell proliferation, differentiation, and apoptosis, is based on the phosphorylation and functional modification of downstream molecular targets in a stimulus- and cell-type-dependent manner.
2.3. Role of JNK in TNFα-Mediated Liver Injury
Underlying liver injury, which leads to repeated cycles of hepatocyte death, inflammation, and compensatory proliferation, is an indispensable factor in the development of most liver cancers. Therefore, determining the mechanism of liver injury is a basic requirement for understanding hepatocarcinogenesis.
The proinflammatory cytokine TNFα plays a pivotal role in various inflammatory liver diseases [32]. Serum and hepatic TNFα levels are increased in patients with acute and chronic viral hepatitis, alcoholic hepatitis, and NASH [33–36]. Although TNFα can kill hepatocytes during such liver diseases, the healthy liver usually has defense systems against TNFα-induced cell death. In fact, TNFα stimulates proliferation rather than death through DNA synthesis in the normal hepatocyte, which is required for liver regeneration after partial hepatectomy [37, 38]. However, once antiapoptotic response is inhibited, TNFα signaling is converted from proliferation to apoptosis. Thus, TNFα has the capacity to induce hepatocyte death as well as hepatocyte proliferation, and plays a crucial role in preserving liver homeostasis [39]. Recent studies have uncovered the important role of the cross talk between JNK and nuclear factor-kappaB (NF-κB) pathways in these processes, as described later.
TNFα is produced mainly by Kupffer cells in the liver and exerts biological effects by binding to two plasma membrane receptors, TNF-receptor 1 (TNF-R1) and TNF-R2 [40]. The majority of the biological effects of TNFα are mediated by TNF-R1. Upon binding of TNFα to TNF-R1, the signaling molecule TNF-R-associated death domain (TRADD), TNF-R-associated factor 2 (TRAF2), and receptor interacting protein 1 (RIP1) are recruited to form the so-called complex I [41] (Figure 2). Assembly of this complex is essential to activate two key players, JNK and NF-κB pathways.
Figure 2.
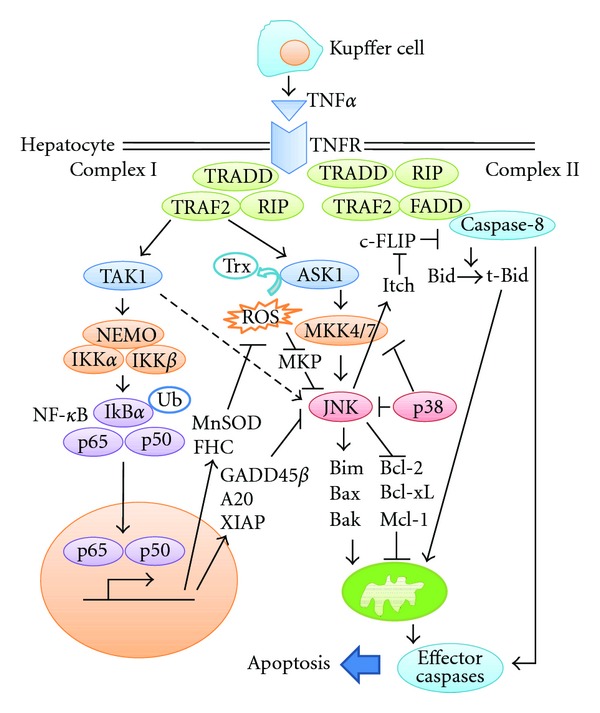
Signaling pathways of TNFα-induced hepatocyte death. After binding of TNFα to TNF-R1, complexes I and II are formed. As the amount of caspase-8 activation in complex II is not sufficient to induce cell death of hepatocytes, activation of the mitochondrial apoptotic pathway is essential. Activated caspase-8 initiates the mitochondrial apoptotic pathway by cleaving Bid to tBid. In addition, JNK can also activate the mitochondrial apoptotic pathway though modulation of Bcl-2 family members or degradation of c-FLIP. These pathways cooperate to induce cell death, while NF-κB and p38 inhibit TNFα-induced hepatocyte death by suppressing JNK activation through various mechanisms.
In addition to the formation of complex I, TNFα binding to TNF-R1 simultaneously recruits Fas-associated death domain (FADD) and caspase-8, thus forming another complex, the so-called complex II [41] (Figure 2). Here, caspase-8 is activated through autocatalytic cleavage and active caspase-8 is then able to proteolytically activate several effector caspases such as caspase-3. However, in the case of hepatocytes, the amount of active caspase-8 generated by complex II is too small for activation of effector caspases, requiring an amplification loop through mitochondria [42]. Caspase-8 cleaves Bid to a truncated form, tBid, which translocates to the mitochondria, causing activation of Bak/Bax and subsequent activation of the mitochondrial apoptotic pathway. This mitochondrial pathway involves the release of numerous apoptogenic factors from the mitochondrial intermembrane space into the cytosol, including cytochrome c, which activates effector caspases and then induces cell death and amplification of caspase-8 activation.
The importance of JNK in TNFα-mediated liver injury has been confirmed by many in vitro and in vivo studies. In an in vitro study, TNFα-induced apoptosis was blocked by the small molecule JNK inhibitor SP600125 in mouse and rat primary hepatocytes [43]. In vivo, NF-κB-deficient embryos, which normally die due to TNFα-mediated hepatocyte apoptosis, were shown to survive longer when the JNK1 gene was also disrupted [44]. In addition, JNK1 knockout (JNK1−/−) and JNK2 knockout (JNK2−/−) mice were resistant to concanavalin-A-(Con-A-) induced liver injury, which is primarily TNFα dependent [45]. In another study, JNK2−/− mice, but not JNK1−/−, mice were shown to be resistant to lipopolysaccharide (LPS)/d-galactosamine (GalN)-induced liver injury, which is also a TNFα-dependent liver injury model [46]. A recent study showed that both JNK1−/−, and JNK2−/−, mice are resistant to LPS/GalN-induced liver injury, and JNK2 plays a more prominent role in TNFα-induced hepatocyte death [47]. Despite the discrepancies among studies concerning which of the JNK isoforms is more important in hepatocyte death, they all support the concept that JNK plays a key role in controlling TNFα signaling toward cell death.
As TNFα-mediated hepatocyte death is also important for acute liver injury, JNK is considered to be a promising therapeutic target for acute liver injury. In fact, SP600125 and another JNK inhibitor D-JNKi, which is a peptide inhibitor of JNK, exert protective effects on LPS/GalN-induced fulminant hepatitis [44, 48].
2.4. Reactive Oxygen Species as Upstream Activators of JNK in TNFα Signaling
TNFα binding to TNF-R1 leads to the formation of complex I and then activates a member of the MAP3K family, transforming growth factor β-activated kinase 1 (TAK1). Activated TAK1 activates MKK4 and MKK7, which leads to JNK activation [49]. However, transient activation of JNK is not sufficient to induce cell death. Reactive-oxygen-species-(ROS-) mediated prolongation of JNK activation is considered important for the proapoptotic function of JNK. This concept was supported by the higher levels of intracellular ROS observed in TNFα-treated cells and the observation that antioxidants, such as N-acetyl-l-cysteine which is precursor of glutathione and butylated hydroxyanisole which is synthetic antioxidant authorized as a food additive, can suppress TNFα-induced prolonged JNK activation and subsequent apoptosis [30].
In TNFα signaling, two mechanisms have been proposed for ROS-mediated prolongation of JNK activation. One study indicated that ROS extended JNK activation by inactivating MAPK phosphatases (MKPs), which are essential for dephosphorylation of activated JNK [50]. TNFα-induced ROS oxidize critical Cys residues in the catalytic sites of various MKPs, leading to their inactivation, and ROS-mediated oxidation of MKP-1 rapidly leads to its degradation by the ubiquitin-proteasome pathway. Another study demonstrated an important role of apoptosis signal-regulating kinase 1 (ASK1), one of the major MAP3Ks, in the regulation of stress-activated MAPK [51]. TNFα-induced sustained activation of JNK is lost in ASK1−/− embryonic fibroblasts, and ASK1−/− cells are resistant to TNFα-induced apoptosis. While TAK1 is activated directly by TNF-R1-induced signaling, activation of ASK1 is secondary to the generation of ROS. The involvement of ASK1 in ROS-mediated JNK activation is based on links between ASK1 and thioredoxin (Trx), a reduction/oxidation regulatory protein [52]. In the absence of oxidative stress, Trx inhibits ASK1 kinase activity via direct binding to the N-terminal region of ASK1. Excess oxidative stress converts Trx to its oxidized form, resulting in its dissociation from ASK1 and subsequent ASK1 kinase activation. This system has been confirmed to function in the liver in vivo using an acetaminophen-induced liver injury model, which is a typical ROS-mediated liver injury model [53]. In addition, a recent study showed that ASK1−/− mice are resistant to LPS/GalN-induced liver injury [54]. Furthermore, Trx transgenic mice are also resistant to LPS/GalN-induced liver injury [55]. Thus, TNFα-induced ROS prolongs JNK activity by blocking the inhibitory MKPs and simultaneously promoting the activation of ASK1, then shifting the balance from cell survival toward cell death.
Viral proteins, including HCV core proteins, are also capable of ROS accumulation in hepatocytes, and ROS-mediated JNK activation in the liver is linked not only to liver disease but also to systemic disorders, such as insulin resistance and metabolic syndrome. Therefore, further elucidation of this process is very important [56–58].
2.5. Downstream Targets of JNK in TNFα-Induced Cell Death
Several targets of JNK in TNFα-induced cell death have been proposed. The primary question is whether JNK is or is not involved in caspase-8 activation. JNK was initially suggested to induce caspase-8-independent cleavage of Bid at a site distinct from the Bid cleavage product jBid, which translocates to mitochondria and subsequently amplifies caspase-8 activation [59]. However, this finding has not been confirmed in an in vivo liver injury model. In the LPS/GalN-induced liver injury model, JNK2−/− mice showed significant attenuation of caspase-8 activation and subsequent Bid cleavage, suggesting that JNK2 acts at the level of caspase-8 activation [46]. However, the detailed mechanism remains unclear and attenuated caspase-8 activation may be the indirect result of a lack of amplification by the mitochondrial release of proapoptotic factors. JNK may also be involved in caspase-8 activation through regulation of the caspase-8 inhibitor c-FLIPL [44]. Prolonged activation of JNK1 leads to phosphorylation of Itch, thereby activating its ubiquitin ligase activity, which results in polyubiquitination and subsequent degradation of c-FLIPL. As c-FLIPL competes with caspase-8 for binding to complex II and impairs caspase-8 activation, JNK1-mediated c-FLIPL degradation results in full activation of caspase-8 and cell death. These studies support the suggestion that JNK is located upstream of caspase-8 activation.
Other studies support the alternative suggestion that JNK plays an apoptotic role at a level farther downstream through the regulation of Bcl-2 family activation. Activated JNK has been shown to phosphorylate and inactivate the antiapoptotic proteins Bcl-2 and Bcl-xL [60–62]. Furthermore, activated JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3, a cytoplasmic anchor of Bax. Phosphorylation of 14-3-3 leads to dissociation of Bax from this protein [63]. A more recent study showed that JNK contributes to TNFα-induced cell death through phosphorylation of the pro-apoptotic BH-3-only protein Bim, rather than Bid [64]. JNK-mediated Bim phosphorylation triggers the proapoptotic activity of Bim by causing its release from sequestration to the microtubular dynein motor complex and then activates the mitochondrial apoptotic pathway [65, 66]. These studies indicated that JNK acts in parallel and independent of caspase-8. Thus, although the downstream targets of JNK in TNFα signaling are not fully understood, most studies have demonstrated that JNK is required for activation of the mitochondrial apoptotic pathway.
2.6. Crosstalk between JNK and NF-κB
As prolonged JNK activation is closely related to cell death, systems for the regulation of JNK activity are mandatory for tissue homeostasis. The crosstalk between NF-κB and JNK is considered to be a major determinant in the fate of hepatocytes, including TNFα signaling.
NF-κB is a transcription factor that consists of homodimers and heterodimers composed of five members, that is, p65 (RelA), c-Rel, RelB, p50, and p52 [67]. Each NF-κB dimer has a different DNA-binding affinity and controls specific targets. Additionally, transcriptional activity of NF-κB is regulated by transcription coactivators such as SRC family and corepressors such as HDAC family [68]. The p50/p65 heterodimer, the most abundant form of the NF-κB, is regulated by so-called canonical pathway. Without stimulation, this heterodimer is bound to specific inhibitory factors, such as inhibitor of NF-κB (IκB) proteins in the cytoplasm. TNFα signaling complex I activates canonical NF-κB pathway via the IKK complex, which consists of two catalytic subunits, IKKα and IKKβ, and a regulatory component, IKKγ/NEMO (Figure 2). The IKK complex phosphorylates and subsequently induces degradation of IκB. Once activated, NF-κB dimers translocate into the nucleus and stimulate transcription of various genes, such as those encoding cytokines and antiapoptotic factors.
Hepatocyte-specific IKKβ or NEMO knockout mice are more sensitive to the hepatotoxic effects of injecting Con-A or LPS, respectively, suggesting a crucial role of NF-κB activation in protection from TNFα-induced liver injury [45, 69]. Also note that the extent of TNFα-induced JNK activation is strengthened and prolonged in these mice.
Several mechanisms have been proposed by which NF-κB activation controls the level of JNK activation. At least three NF-κB-dependent genes have been identified as potential candidates, that is, growth arrest DNA damage-inducible gene 45β (Gadd45β), X-linked inhibitor of apoptosis (XIAP), and the zinc finger protein A20 [70–76]. However, the precise implications and detailed mechanisms of action of these molecules in the regulation of TNFα-induced JNK activation and cell death remain unclear, especially in vivo. In addition to the expression of JNK inhibitory proteins, NF-κB also inhibits JNK activation by controlling intracellular ROS levels. NF-κB activation leads to expression of the antioxidants ferritin heavy chain (FHC) and manganese-dependent superoxide dismutase (MnSOD), both of which prevent accumulation of ROS, thus inhibiting prolongation of JNK activation [77, 78].
2.7. Crosstalk between JNK and Other MAPK Signaling Molecules
p38α also plays an important role in inhibiting TNFα-induced JNK activation and liver injury. Hepatocyte-specific p38α knockout mice showed much stronger JNK activation after administration of LPS [79]. Although p38α ablation is not sufficient to induce hepatocyte death after LPS administration, hepatocyte-specific p38α, and IKKβ double-knockout mice show severe liver injury, suggesting that p38α and IKKβ act synergistically to protect the liver from TNFα-induced toxicity. Unlike in the case of NF-κB, pretreatment with antioxidants could not reduce LPS-induced JNK activation in hepatocyte-specific p38α knockout mice. Furthermore, the levels of MKPs were similar between hepatocyte-specific p38α knockout mice and wild-type controls. Therefore, the detailed mechanisms of action of p38α in regulating TNFα-induced JNK activation and cell death remain unclear. However, hepatocyte-specific p38α knockout mice showed increased MKK4 activation, so targets of p38α are considered to be upstream of JNK, such as MAPKKs and MAP3Ks. Interestingly, not only MKK4 but also MKK3 and MKK6 activation, all of which are involved in p38 activation, are increased in hepatocyte-specific p38α knockout mice, suggesting that there might be negative feedback mechanism in p38 signaling pathway in the liver. In fact, p38 has been reported to negatively regulate upstream MAP3Ks such as TAK1 and MLK3 in other cells [80, 81].
Two recent studies have introduced MAP3K TAK1 into the complex field of NF-κB/JNK interaction in the liver [82, 83]. In TNFα signaling, TAK1 is recruited into complex I and phosphorylated through TRAFs. Then, phosphorylated TAK1 activates IKK and MKK4/7. Therefore, TAK1 can activate both NF-κB and JNK, which play opposing roles in cell death. Inokuchi et al. reported that hepatocyte-specific TAK1 knockout mice generated by intercrossing TAK1 floxed mice with Alb-Cre mice showed spontaneous hepatocyte death, which was partially canceled by further genetic deletion of TNF-R1, suggesting that TAK1-deficient hepatocytes are sensitive to endogenous TNFα [82]. They also reported that hepatocyte-specific TAK1-deficient mice showed enhanced JNK activation in vivo. Primary hepatocytes derived from these mice also demonstrated enhanced cell death by TNFα stimulation, and not only NF-κB activation but also JNK activation were inhibited ex vivo. Bettermann et al. reported that liver-specific TAK1 knockout mice generated by intercrossing TAK1 floxed mice with Alfp-Cre mice showed strong JNK activation, lack of NF-κB activation, and massive hepatocyte apoptosis after LPS injection [83]. This enhanced JNK activation may occur through activation of other MAP3K TAO2. Furthermore, they showed that TAK1 is required for the prevention of cholangiocyte apoptosis. Thus, although the role of TAK1 in the regulation of TNFα-mediated JNK activation has not been fully elucidated, TAK1 plays an antiapoptotic role in the liver mainly through NF-κB activation.
2.8. Implication of JNK in Other Types of Liver Injury
JNK signaling has also been implicated in hepatocyte injury due to other causes, including lipotoxicity, ER stress, ischemia-reperfusion, and drug toxicity, such as from acetaminophen [53, 84–87]. Although the role of JNK in these types of hepatocyte death cannot be discussed here in detail due to space limitations, JNK is generally considered to play an important role in cell death induction through similar mechanisms as seen in TNFα signaling. Therefore, JNK is a possible candidate therapeutic target for various types of liver injury.
3. JNK in Hepatocarcinogenesis
3.1. General Function of JNK in Carcinogenesis
Although this review has mainly discussed the pro-apoptotic function of JNK, this molecule has various functions in carcinogenesis. First, JNK plays an important role in cell proliferation. JNK is considered to be involved in cell cycle progression mostly through interaction with the transcription factor c-Jun, which is a well-established cell cycle regulator. A previous study using JNK1−/− or JNK2−/− fibroblasts showed that JNK2−/− fibroblasts proliferate faster with increased c-Jun expression, whereas JNK1−/− fibroblasts proliferate slower with decreased c-Jun expression, suggesting that JNK1 might play a major role in c-Jun-mediated cell cycle progression [88]. However, another study using fibroblasts from mice with a knockin mutation in the JNK2 gene showed that JNK2 is also involved in c-jun expression and cellular proliferation [89]. Thus, in general, both JNK1 and JNK2 play important roles in cell cycle progression.
In the liver, JNK1 has been shown to be a major player in cell cycle regulation using a partial hepatectomy model. Hui et al. reported that the number of Ki67-positive proliferating hepatocytes in JNK1−/− mice was significantly reduced compared to wild-type controls after partial hepatectomy [20]. The impaired proliferation in JNK1−/− mice was caused by increased expression of p21, a cell cycle inhibitor, and reduced expression of c-Myc, a negative regulator of p21. In contrast, JNK2−/− mice showed similar hepatocyte proliferation rates to wild-type controls. Another study showed that administration of the pan-JNK inhibitor SP600125 inhibited hepatocyte proliferation after partial hepatectomy through reduced expression of cyclin D1 [90]. Thus, JNK plays an important role in hepatocyte proliferation in vivo.
In addition to its roles in cell proliferation, JNK has been shown to also have various oncogenic functions. For example, the JNK pathway is involved in HCC cell migration and tumor invasion through matrix metalloproteinase (MMP) production [91]. A recent study showed that JNK is required for Ras-induced suppression of contact growth inhibition by regulating e-cadherin expression [92]. Furthermore, JNK facilitates cancer progression by interacting with other oncogenic pathways, such as Wnt/β-catenin signaling [93–95]. Thus, although JNK is generally considered to play oncogenic roles, it also plays roles in tumor suppression in some cases through p53 activation and induction of apoptosis [96–98].
3.2. Role of JNK in Hepatocarcinogenesis
In general, induction of cell death acts as a tumor-suppressing function. However, this function does not always act as a tumor suppressor, but instead acts as a tumor promoter in the liver, because the loss of hepatic mass due to hepatocyte death activates the proliferation of residual hepatocytes that may provide a promoting environment for tumor formation. Recent in vivo studies strongly implicated JNK as a key regulator in this process.
Diethylnitrosamine (DEN) is a chemical carcinogen commonly used to induce HCC in rodents. Administration of DEN causes acute liver injury and DNA damage in the hepatocytes, and subsequently leads to inflammation, liver regeneration, and neoplastic lesions in the liver. A study using hepatocyte-specific IKKβ knockout mice demonstrated a critical concept with regard to the role of cross talk between NF-κB and JNK in hepatocytes in DEN-induced HCC [78] (Figure 3). Hepatocyte-specific knockout of IKKβ markedly enhanced DEN-induced HCC. The ablation of hepatocyte IKKβ induces greater DEN-induced JNK activation and greater cell death during the acute reaction to DEN administration. The absence of NF-κB induces prolonged JNK activation by enhancing ROS production, and prolonged JNK activation plays a critical role in DEN-induced hepatocyte death, as seen in TNFα-mediated liver injury. Subsequently, the elevated hepatocyte death rate enhances compensatory proliferation. Thus, the hepatocyte-specific deletion of IKKβ augments DEN-induced hepatocyte death, compensatory proliferation, and increased tumorigenesis, probably through enhanced JNK activation. Similar findings were obtained in mice lacking NEMO, the hepatocyte-specific deletion of which results in spontaneous liver damage, hepatosteatosis, fibrosis, and the development of HCC [69]. To further investigate the role of JNK in this process, JNK1−/− mice were interbred with hepatocyte-specific IKKβ knockout mice, and the double-knockout mice developed significantly fewer tumors compared to IKKβ knockout mice [18]. In addition, JNK1−/− mice showed a significant reduction of tumor development compared to wild-type controls. In addition to the involvement in DEN-induced acute liver injury, JNK1 has tumor-promoting functions by enhancing cancer cell proliferation and neovascularization through the increased expression of cyclin D1 and VEGF, respectively. Another group also reported the critical role of JNK1 in hepatocarcinogenesis using the DEN and phenobarbital-induced HCC model [20]. They showed that cancer cell proliferation decreases significantly in JNK1−/− mice due to increased p21 expression and reduced c-Myc expression. In addition, pharmacological inhibition of JNK by D-JNKi reduced the growth of xenografted human HCC cells and chemically induced mouse liver cancers, suggesting that JNK is a promising therapeutic target for HCC. Their study, however, also showed that JNK2 is not involved in hepatocarcinogenesis. About 50%–60% of human HCC show strong activation of JNK1 compared to adjacent nontumor tissue, whereas JNK2 activation is similar between HCC and non-tumor tissue [20, 99]. These results suggest that JNK, especially JNK1, plays an important role in the development of HCC.
Figure 3.
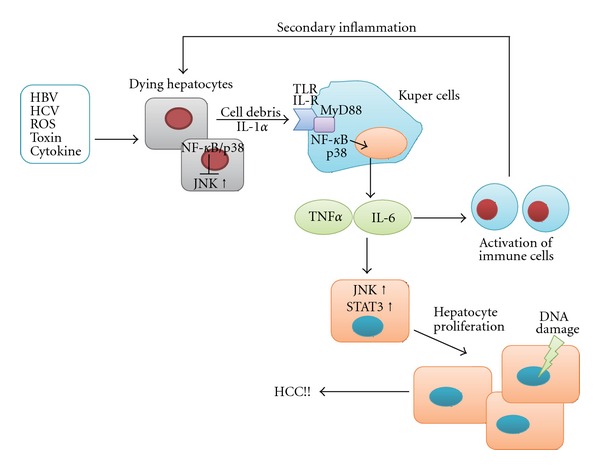
Current model of the role of stress-activated MAPK and NF-κB in inflammation-mediated hepatocarcinogenesis. In chronic liver injury, hepatitis virus, ROS, toxins, and cytokines kill hepatocytes. JNK promotes cell death, while NF-κB and p38 promote cell survival. Dying hepatocytes activate Kupffer cells via TLR or IL-1R. Activated Kupffer cells secrete cytokines, such as IL-6 and TNFα, through a mechanism mediated by NF-κB and p38. Subsequently, these cytokines induce recruitment of activated inflammatory cells to the liver, which is followed by hepatocyte regeneration. JNK and STAT3 promote cytokine-mediated hepatocyte proliferation. This persistent cycle of necroinflammation and hepatocyte regeneration provides a mitogenic and mutagenic environment, leading to the development of HCC.
JNK activation is also implicated in HCC based on viral hepatitis. In vitro studies showed that several HBV and HCV proteins can activate the JNK pathway [100, 101]. In addition, JNK-mediated enhanced cell proliferation is involved in hepatocarcinogenesis of HCV core protein transgenic mice and HBx protein transgenic mice, both of which spontaneously develop HCC [102, 103].
However, a more recent study using conditional knockout mice of both JNK isoforms, JNK1 and JNK2, suggested that JNK may play dual roles in hepatocarcinogenesis [104]. In their study, JNK deficiency in hepatocytes increased DEN-induced HCC. In contrast, JNK deficiency in both hepatocytes and myeloid cells reduced hepatic inflammation and the development of HCC. Although the detailed mechanisms remain unclear, this study suggested that JNK in hepatocytes may act as a tumor suppressor, whereas that in myeloid cells may act as a tumor promoter in the development of HCC.
JNK is also wellknown as a critical regulator of obesity and metabolic syndrome through inhibition of insulin signaling in the liver and muscle, and modulation of adipokine secretion from adipocytes [105, 106]. Recent studies have suggested that obesity-induced insulin resistance and dysregulation of adipokines play important roles in hepatocarcinogenesis [107–110]. As the incidence rate of this metabolic syndrome-associated hepatocarcinogenesis is likely to increase in the near future, investigating whether JNK activation also contributes to hepatocarcinogenesis under this condition is critical.
4. p38 and MAP3Ks in Hepatocarcinogenesis
4.1. p38 Signaling
The p38 MAPK group consists of four members: p38α, p38β, p38γ, and p38δ [111]. p38α and p38β are universally expressed and closely related proteins that have overlapping functions. Whereas p38α is highly abundant in most cell types, p38β seems to be expressed at very low levels. p38γ and p38δ have more restricted expression patterns and are likely to have specialized functions [23]. Most of the published literature on p38 MAPKs refers to p38α, and we mainly discuss p38α in this paper. MKK3 and MKK6 are the primary upstream MAPKKs of p38, but MKK4 has also been shown to activate p38 in response to certain stimuli. p38 can also be activated independently of the MAP3K/MAPKK cascade by autophosphorylation [112]. Substrates of p38 include protein kinases, such as MAPKAP kinase 2 (MK2) and MK5, and transcription factors, such as ATF2, p53, and Mitf [113]. These diverse targets mediate the activated p38 signal to various types of cellular functions such as differentiation, apoptosis, cytokine production, and cell cycle control.
4.2. General Function of p38 in Carcinogenesis
Although p38 was first recognized as a regulator of inflammatory cytokine production, recent studies uncovered a role of p38α as an essential inhibitor of cell proliferation. p38α negatively regulates cell cycle progression both at the G1/S and G2/M transitions by several mechanisms, including the downregulation of cyclins, upregulation of cyclin-dependent kinase (CDK) inhibitors, and modulation of the tumor suppressor p53 [114]. p38α also acts as an inhibitor of hepatocyte proliferation after partial hepatectomy, and inactivation of p38α is seen during liver regeneration [115, 116]. This effect of p38α is partially mediated by negative regulation of the JNK/c-Jun pathway [19].
In addition to the suppression of cell proliferation, p38 exerts tumor-suppressing effects through oncogene-induced senescence (OIS), contact inhibition, and DNA damage responses [111]. In particular, recent studies demonstrated a major role of p38 in OIS caused by oncogenic Ras or its downstream effector Raf-1. The Ras-Raf-MEK-ERK signaling cascade increases the intracellular ROS level and subsequently activates the p38 pathway [117]. Activated p38 phosphorylates multiple residues on p53, leading to increased transcriptional activity of p53 and induction of p21 [111, 118]. Active p38 also induces the expression of p16 and p14/p19, which together with the p53-p21 cascade, cause senescence that serves as a tumor-suppressing defense mechanism [119]. Thus, a great deal of evidence supports a role of p38 as a tumor suppressor. However, p38 may also have oncogenic effects by facilitating cell invasion, inflammation, and angiogenesis [120, 121].
4.3. Role of p38 in Hepatocarcinogenesis
The role of p38 in hepatocarcinogenesis has also been investigated using the DEN-induced mouse HCC model. Similar to hepatocyte-specific IKKβ knockout mice, hepatocyte-specific p38α knockout mice showed enhanced DEN-induced ROS accumulation, JNK activation, liver damage, and subsequent hepatocyte proliferation, which resulted in enhanced carcinogenesis [21]. In addition, hepatocyte-specific p38α ablation leads to enhanced IL-1α release from necrotic hepatocytes. IL-1α can activate Kupffer cells, and activated Kupffer cells produce various cytokines, such as TNFα, IL-6, and hepatocyte growth factor, which promote proliferation of residual hepatocytes (Figure 3). The activation of p38 and NF-κB in Kupffer cells plays an important role in this process because mice with p38α or IKKβ knockout in both hepatocytes and myeloid cells, including Kupffer cells, showed markedly reduced hepatic expression of these cytokines after DEN challenge, resulting in reduced future HCC development [21, 78]. Thus, p38α and NF-κB in the hepatocytes act as tumor suppressors by conferring protection against DEN-induced cell death, whereas p38α and NF-κB in the Kupffer cells act as tumor promoters by enhancing cytokine production. Another study using hepatocyte-specific p38α knockout mice indicated its role in tumor suppression by focusing on the antiproliferative function at the advanced stage [19]. Ablation of p38α in the hepatocytes results in upregulation of the JNK/c-Jun pathway, which plays an important role in the increased proliferation of tumor cells.
In human samples, the activity of the MKK6-p38 pathway is decreased in HCC tissues compared to adjacent non-tumor tissues [122]. In particular, its activity is significantly lower in larger HCC tissues, suggesting that the p38 pathway may play an antiproliferative role in human HCC.
4.4. Role of MAP3Ks in Hepatocarcinogenesis
The evidence presented to date suggests that JNK generally acts as a tumor promoter and p38 generally acts as a tumor suppressor in hepatocarcinogenesis. However, as JNK and p38 have complex functions and modulate a wide range of cellular effect, some studies showed dual roles of these molecules in hepatocarcinogenesis. Furthermore, cross talk among JNK and p38, and molecules involved in other signaling pathways such as NF-κB, further complicate their roles. Several recent studies have uncovered the roles of MAP3Ks in the regulation of stress-activated MAPK signaling in hepatocarcinogenesis.
One player is TAK1, which can activate NF-κB as well as JNK and p38 in response to the signaling of Toll-like receptors, IL-1 receptor, TNF receptor, and TGF-β receptor. As mentioned above, hepatocyte-specific TAK1 knockout mice show spontaneous hepatocyte death, which is partially mediated by endogenous TNFα [82, 83]. This spontaneous cell death subsequently causes compensatory hepatocyte proliferation, inflammation, and fibrosis. Furthermore, about 80%–90% of these mice spontaneously develop HCC at 9 months of age. These phenomena, which resembled the phenotype observed in hepatocyte-specific NF-κB knockout mice, are considered to occur through attenuated NF-κB activation and elevated JNK activation. The p38 pathway, however, seems not to be involved in this phenotype. Thus, TAK1 in the hepatocytes acts as a tumor suppressor mainly by regulating activation of the NF-κB pathway. Unexpectedly, crossing hepatocyte-specific TAK1 knockout mice with NEMO knockout mice attenuates JNK activation and prevents hepatocyte death and the development of HCC, suggesting that NEMO has a tumor-promoting function in the setting of TAK1 deletion [83]. Furthermore, this function of NEMO is considered to be independent of NF-κB.
Another player is ASK1, which selectively activates JNK and p38 signaling in response to a variety of stimuli, including ROS and cytokines [123]. The role of ASK1 in hepatocarcinogenesis has been investigated using the DEN-induced HCC model [54]. The number of tumors in ASK1−/− mice is three times higher than that in wild-type controls, and cancer cell apoptosis is significantly suppressed in ASK1−/− HCC. JNK activation and hyperphosphorylation of BimEL, which are required for death receptor-mediated apoptosis, including TNF-R and Fas, are attenuated in ASK1−/− HCC but not in non-tumor tissues, suggesting escape from the antitumor immune response. In contrast, tumor-promoting functions of JNK, such as cell proliferation and neovascularization, are preserved in ASK1−/− mice. Therefore, ASK1 is considered to play a major part in the tumor-suppressing role of JNK in hepatocarcinogenesis. In addition, activation of the p38 pathway is also attenuated in ASK1−/− mice, and DNA damage-induced p21 upregulation via the p38 pathway is inhibited. Thus, ASK1 controls the tumor-suppressing function of stress-activated MAPK signaling through induction of apoptosis and the DNA damage response, and acts as a tumor suppressor in hepatocarcinogenesis (Figure 4). Furthermore, another recent study indicated that ASK1 and Bim are also required for sorafenib-induced apoptosis in HCC cells [124]. Therefore, ASK1 may play important roles in not only hepatocarcinogenesis but also in the therapeutic effects of anticancer drugs.
Figure 4.
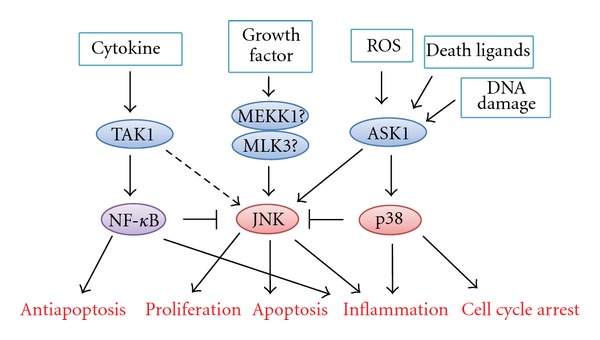
Roles of MAP3Ks in controlling the function of stress-activated MAPK and NF-κB in hepatocarcinogenesis. TAK1 can activate NF-κB and JNK, but TAK1 mainly regulates NF-κB activation and acts as a tumor suppressor in hepatocarcinogenesis. ASK1 controls the tumor-suppressing function of JNK and p38 through apoptosis induction and DNA damage response, and thus acts as a tumor suppressor. MEKK1 and MLK3 may be involved in JNK-mediated tumor cell proliferation in hepatocarcinogenesis, but no supporting in vivo evidence exists to support this contention at present. Furthermore, the roles of these molecules may differ according to the disease model and timing.
In some cases, MAP3K may be a better therapeutic target than MAPK. To date, more than ten molecules have been identified as MAP3K in stress-activated MAPK signaling, which is much more than the number of the downstream MAPK (Figure 1) [125]. This fact indicates that the MAP3Ks are activated by various stimuli, and function as signaling hubs, integrating these signals into a unique pattern of MAPK activation, and eventually leading to a specific function of MAPK to the stimulus. Thus, blocking MAP3K may be able to inhibit specific function of MAPK. In addition, inhibition of JNK or p38 causes compensatory activation of another stress-activated MAPK, whereas inhibition of some MAP3Ks such as ASK1 does not cause such compensation, which may be advantageous in the clinical settings as discussed in the next section. However, which MAP3K is important in growth factor-mediated JNK activation in HCC remains unclear, especially in vivo, although MEKK1 and MLK3 have been reported to be involved in this role of JNK in non-HCC cells such as colon cancer [126, 127]. Therefore, further understanding of the regulatory system of stress-activated MAPK signaling by MAP3Ks is important.
5. Future Perspectives
These recent studies have gradually clarified the roles and regulation systems of stress-activated MAPK in hepatocarcinogenesis, but some issues remain to be elucidated. One issue is that the role of these molecules may change according to the disease model. A good example is NF-κB. As mentioned above, DEN-induced HCC is increased in hepatocyte-specific IKKβ knockout mice and spontaneous HCC development is seen in hepatocyte-specific NEMO knockout mice, suggesting a role of NF-κB as a tumor suppressor [69, 78]. However, experiments crossing transgenic mice expressing a nondegradable IκBα mutant with MDR2 knockout mice, which show chronic inflammation in the portal area and subsequent cancer development, indicated a tumor-promoting role of NF-κB [128]. These results indicate the dual roles of NF-κB in hepatocarcinogenesis depending on the disease model. Thus, more appropriate models mimicking human chronic hepatitis and hepatocarcinogenesis are required. In this sense, TAK1 knockout mice may be a promising candidate because TAK1-deficient HCC develops in the context of chronic inflammation and fibrosis, as commonly found in human HCC [82, 83]. Furthermore, the time manipulation is also an important problem. For example, studies in TAK1 knockout and ASK1 knockout mice confirmed that too much apoptosis in the chronic inflammatory phase facilitates hepatocarcinogenesis, whereas too little apoptosis in already initiated cells also aggravates cancer progression, suggesting that the apoptotic function plays dual roles in a stage-dependent manner [54, 82, 83]. Therefore, therapeutic targets may need to be redefined according to the carcinogenic stage.
The critical role of stress-activated MAPK signaling in hepatocarcinogenesis prompts their introduction (especially JNK1) into the clinical settings as therapeutic targets. Several inhibitors targeting JNK or p38 have entered clinical trials for other diseases. In particular, p38 inhibitors for inflammatory diseases, such as rheumatoid arthritis and inflammatory bowel disease, have been studied extensively [129, 130]. However, few have progressed beyond phase I clinical trials and none have yet appeared in clinical settings. One reason for this is the occurrence of side effects, such as liver toxicity. Although whether these are due to target inhibition or to the effects of the drugs on additional targets is unclear, cross talk among signaling pathways is considered to be one explanation because p38 inhibition induces compensatory JNK activation. Therefore, alternative strategies, such as the targeting of particular MAPK isoforms, including JNK1, upstream regulators, including MAP3K, and downstream effectors, including Bcl-2 family members, will be more beneficial than targeting the whole pathway. Note that the roles of these molecules differ among cell types, such as hepatocytes and myeloid cells, including Kupffer cells. Therefore, one must take into account its total function in disease progression. Furthermore, combination therapies are likely to be more important in the future. Hence, further understanding of these pathways will help to develop novel treatment strategies for advanced HCC or prevention of HCC development.
6. Conclusion
Recent studies have indicated that MAPK signaling pathways play key roles and have potential as therapeutic targets in liver injury and hepatocarcinogenesis. Among them, JNK signaling is a promising therapeutic target. However, these pathways have a wide range of functions and show complex cross talk. In addition, their roles differ between cell types and disease processes. Our final goal is to establish them as new diagnostic markers and targets for therapies or prevention of liver diseases. In fact, recent report showed that activation of JNK in noncancerous liver tissue predicted a high risk of HCC recurrence after surgical resection, suggesting that testing JNK activation in the liver might be useful for the risk assessment of HCC occurrence or recurrence [131]. Thus, further understanding of these very important and complicated pathways, including surrounding modulators, is essential for application in clinical settings.
Acknowledgments
This work was supported by a fellowship from the Daiichi Sankyo Foundation of Life Science (to H. Nakagawa).
Abbreviations
- ASK1:
Apoptosis signal-regulating kinase 1
- DEN:
Diethylnitrosamine
- GalN:
Galactosamine
- HBV:
Hepatitis B virus
- HCC:
Hepatocellular carcinoma
- HCV:
Hepatitis C virus
- JNK:
c-Jun NH2-terminal kinase
- LPS:
Lipopolysaccharide
- MAPK:
Mitogen-activated protein kinase
- MAP3K:
Mitogen-activated protein kinase kinase kinase
- NASH:
Nonalcoholic steatohepatitis
- NF-κB:
Nuclear factor-kappa B
- OIS:
Oncogene-induced senescence
- ROS:
Reactive oxygen species
- TAK1:
Transforming growth factor β-activated kinase 1
- TNF-α:
Tumor necrosis factor-α
- TNF-R1:
TNF-receptor 1
- Trx:
Thioredoxin.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Masuzaki R, Yoshida H, Tateishi R, Shiina S, Omata M. Hepatocellular carcinoma in viral hepatitis: improving standard therapy. Best Practice and Research. 2008;22(6):1137–1151. doi: 10.1016/j.bpg.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Yu MC, Yuan JM. Environmental factors and risk for hepatocellular carcinoma. Gastroenterology. 2004;127:S72–S78. doi: 10.1016/j.gastro.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Budhu A, Xin WW. The role of cytokines in hepatocellular carcinoma. Journal of Leukocyte Biology. 2006;80(6):1197–1213. doi: 10.1189/jlb.0506297. [DOI] [PubMed] [Google Scholar]
- 5.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25(27):3834–3847. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 6.Elsharkawy AM, Mann DA. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology. 2007;46(2):590–597. doi: 10.1002/hep.21802. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa H, Maeda S, Yoshida H, et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: an analysis based on gender differences. International Journal of Cancer. 2009;125(10):2264–2269. doi: 10.1002/ijc.24720. [DOI] [PubMed] [Google Scholar]
- 8.Mazzaferro V, Regalia E, Doci R, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. The New England Journal of Medicine. 1996;334(11):693–699. doi: 10.1056/NEJM199603143341104. [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM, Schwartz M, Mazzaferro V. Resection and liver transplantation for hepatocellular carcinoma. Seminars in Liver Disease. 2005;25(2):181–200. doi: 10.1055/s-2005-871198. [DOI] [PubMed] [Google Scholar]
- 10.Tateishi R, Shiina S, Teratani T, et al. Percutaneous radiofrequency ablation for hepatocellular carcinoma: an analysis of 1000 cases. Cancer. 2005;103(6):1201–1209. doi: 10.1002/cncr.20892. [DOI] [PubMed] [Google Scholar]
- 11.Masuzaki R, Yoshida H, Omata M. Does chemotherapy prevent HCV-related hepatocellular carcinoma? Pros. Digestive and Liver Disease. 2010;42(supplement 3):281–286. doi: 10.1016/S1590-8658(10)60517-8. [DOI] [PubMed] [Google Scholar]
- 12.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. The New England Journal of Medicine. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 13.Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140(5):1410–1426. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida H, Shiratori Y, Moriyama M, et al. Interferon therapy reduces the risk for hepatocellular carcinoma: national surveillance program of cirrhotic and noncirrhotic patients with chronic hepatitis C in Japan. IHIT Study Group. Inhibition of Hepatocarcinogenesis by Interferon Therapy. Annals of Internal Medicine. 1999;131:174–181. doi: 10.7326/0003-4819-131-3-199908030-00003. [DOI] [PubMed] [Google Scholar]
- 15.Fried MW, Shiffman ML, Rajender Reddy K, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. The New England Journal of Medicine. 2002;347(13):975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 16.Chang TT, Gish RG, De Man R, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. The New England Journal of Medicine. 2006;354(10):1001–1010. doi: 10.1056/NEJMoa051285. [DOI] [PubMed] [Google Scholar]
- 17.McHutchison JG, Everson GT, Gordon SC, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. The New England Journal of Medicine. 2009;360(18):1827–1838. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]
- 18.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(28):10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hui L, Bakiri L, Mairhorfer A, et al. p38α suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nature Genetics. 2007;39(6):741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 20.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. The Journal of Clinical Investigation. 2008;118(12):3943–3953. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai T, He G, Matsuzawa A, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14(2):156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maeda S. NF-κB, JNK, and TLR signaling pathways in hepatocarcinogenesis. Gastroenterology Research and Practice. 2010;2010:10 pages. doi: 10.1155/2010/367694. Article ID 367694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Reviews Cancer. 2009;9(8):537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 24.Min L, He B, Hui L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Seminars in Cancer Biology. 2011;21(1):10–20. doi: 10.1016/j.semcancer.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological Reviews. 2001;81(2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 26.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 27.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 28.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nature Cell Biology. 2002;4(5):E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 29.Weston CR, Davis RJ. The JNK signal transduction pathway. Current Opinion in Genetics and Development. 2002;12(1):14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 30.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radical Biology and Medicine. 2006;40(6):928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 31.Conze D, Krahl T, Kennedy N, et al. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. Journal of Experimental Medicine. 2002;195(7):811–823. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-α-induced liver injury: role of IKK, JNK, and ROS pathways. American Journal of Physiology. 2006;290(4):G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 33.Nelson DR, Lim HL, Marousis CG, et al. Activation of tumor necrosis factor-α system in chronic hepatitis C virus infection. Digestive Diseases and Sciences. 1997;42(12):2487–2494. doi: 10.1023/a:1018804426724. [DOI] [PubMed] [Google Scholar]
- 34.Sheron N, Lau J, Daniels H, et al. Increased production of tumour necrosis factor alpha in chronic hepatitis B virus infection. Journal of Hepatology. 1991;12(2):241–245. doi: 10.1016/0168-8278(91)90945-8. [DOI] [PubMed] [Google Scholar]
- 35.Bird GLA, Sheron N, Goka AKJ, Alexander GJ, Williams RS. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Annals of Internal Medicine. 1990;112(12):917–920. doi: 10.7326/0003-4819-112-12-917. [DOI] [PubMed] [Google Scholar]
- 36.Jarrar MH, Baranova A, Collantes R, et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Alimentary Pharmacology and Therapeutics. 2008;27(5):412–421. doi: 10.1111/j.1365-2036.2007.03586.x. [DOI] [PubMed] [Google Scholar]
- 37.Feingold KR, Soued M, Grunfeld C, Moser AH, Verdier JA, DoVale HG. Tumor necrosis factor stimulates DNA synthesis in the liver of intact rats. Biochemical and Biophysical Research Communications. 1988;153(2):576–582. doi: 10.1016/s0006-291x(88)81134-3. [DOI] [PubMed] [Google Scholar]
- 38.Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(4):1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wullaert A, Van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-κB: effects on liver homeostasis and beyond. Endocrine Reviews. 2007;28(4):365–386. doi: 10.1210/er.2006-0031. [DOI] [PubMed] [Google Scholar]
- 40.Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: cross-talk between the NF-κB and JNK pathways. Biological Chemistry. 2009;390(10):965–976. doi: 10.1515/BC.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 42.Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ. The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Molecular and Cellular Biology. 1998;18(11):6353–6364. doi: 10.1128/mcb.18.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. The FASEB Journal. 2004;18(6):720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 44.Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIPL turnover. Cell. 2006;124(3):601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 45.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα . Immunity. 2003;19(5):725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. The Journal of Biological Chemistry. 2006;281(22):15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ni HM, Chen X, Shi YH, et al. Genetic delineation of the pathways mediated by bid and JNK in tumor necrosis factor-α-induced liver injury in adult and embryonic mice. The Journal of Biological Chemistry. 2009;284(7):4373–4382. doi: 10.1074/jbc.M807259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takamura M, Matsuda Y, Yamagiwa S, et al. An inhibitor of c-Jun NH2-terminal kinase, SP600125, protects mice from d-galactosamine/lipopolysaccharide-induced hepatic failure by modulating BH3-only proteins. Life Sciences. 2007;80(14):1335–1344. doi: 10.1016/j.lfs.2006.12.034. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Deng L, Hong M, Akkaraju GR, Inoue JI, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 50.Kamata H, Honda SI, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 51.Tobiume K, Matsuzawa A, Takahashi T, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Reports. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO Journal. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakagawa H, Maeda S, Hikiba Y, et al. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology. 2008;135(4):1311–1321. doi: 10.1053/j.gastro.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa H, Hirata Y, Takeda K, et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology. 2011;54(1):185–195. doi: 10.1002/hep.24357. [DOI] [PubMed] [Google Scholar]
- 55.Okuyama H, Nakamura H, Shimahara Y, et al. Overexpression of thioredoxin prevents acute hepatitis caused by thioacetamide or lipopolysaccharide in mice. Hepatology. 2003;37(5):1015–1025. doi: 10.1053/jhep.2003.50203. [DOI] [PubMed] [Google Scholar]
- 56.Moriya K, Nakagawa K, Santa T, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Research. 2001;61(11):4365–4370. [PubMed] [Google Scholar]
- 57.Kaneto H, Nakatani Y, Miyatsuka T, et al. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nature Medicine. 2004;10(10):1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa H, Isogawa A, Tateishi R, et al. Serum gamma-glutamyltransferase level is associated with serum superoxide dismutase activity and metabolic syndrome in a Japanese population. Journal of Gastroenterology. 2012;47(2):187–194. doi: 10.1007/s00535-011-0477-8. [DOI] [PubMed] [Google Scholar]
- 59.Deng Y, Ren X, Yang L, et al. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Molecular and Cellular Biology. 1999;19(12):8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng X, Xiao L, Lang W, et al. Novel role for JNK as a stress-activated Bcl2 kinase. The Journal of Biological Chemistry. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 62.Fan M, Goodwin M, Vu T, Brantley-Finley C, Gaarde WA, Chambers TC. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-X(L) is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. The Journal of Biological Chemistry. 2000;275(39):29980–29985. doi: 10.1074/jbc.M003776200. [DOI] [PubMed] [Google Scholar]
- 63.Tsuruta F, Sunayama J, Mori Y, et al. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. The EMBO Journal. 2004;23(8):1889–1899. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaufmann T, Jost PJ, Pellegrini M, et al. Fatal hepatitis mediated by tumor necrosis factor TNFalpha requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30(1):56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Reviews Molecular Cell Biology. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 67.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunological Reviews. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 68.Gao Z, Chiao P, Zhang X, et al. Coactivators and corepressors of NF-κB in IκBα gene promoter. The Journal of Biological Chemistry. 2005;280(22):21091–21098. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 70.De Smaele E, Zazzeroni F, Papa S, et al. Induction of gadd4β by NF-κB downregulates pro-apoptotic JNK signalling. Nature. 2001;414(6861):308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 71.Papa S, Zazzeroni F, Bubici C, et al. Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nature Cell Biology. 2004;6(2):146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 72.Papa S, Zazzeroni F, Fu YX, et al. Gadd45β promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. The Journal of Clinical Investigation. 2008;118(5):1911–1923. doi: 10.1172/JCI33913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaur S, Wang F, Venkatraman M, Arsura M. X-linked inhibitor of apoptosis (XIAP) inhibits c-Jun N-terminal kinase 1 (JNK1) activation by transforming growth factor β1 (TGF-β1) through ubiquitin-mediated proteosomal degradation of the TGF-β1-activated kinase 1 (TAK1) The Journal of Biological Chemistry. 2005;280(46):38599–38608. doi: 10.1074/jbc.M505671200. [DOI] [PubMed] [Google Scholar]
- 74.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lademann U, Kallunki T, Jäättelä M. A20 zinc finger protein inhibits TNF-induced apoptosis and stress response early in the signaling cascades and independently of binding to TRAF2 or 14-3-3 proteins. Cell Death and Differentiation. 2001;8(3):265–272. doi: 10.1038/sj.cdd.4400805. [DOI] [PubMed] [Google Scholar]
- 76.Arvelo MB, Cooper JT, Longo C, et al. A20 protects mice from D-galactosamine/lipopolysaccharide acute toxic lethal hepatitis. Hepatology. 2002;35(3):535–543. doi: 10.1053/jhep.2002.31309. [DOI] [PubMed] [Google Scholar]
- 77.Pham CG, Bubici C, Zazzeroni F, et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119(4):529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 78.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 79.Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38α MAPK inhibits JNK activation and collaborates with IκB kinase 2 to prevent endotoxin-induced liver failure. EMBO Reports. 2008;9(10):1048–1054. doi: 10.1038/embor.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheung PCF, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α . The EMBO Journal. 2003;22(21):5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muniyappa H, Das KC. Activation of c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: a MLK-3-MKK7-dependent mechanism. Cellular Signalling. 2008;20(4):675–683. doi: 10.1016/j.cellsig.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Inokuchi S, Aoyama T, Miura K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(2):844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bettermann K, Vucur M, Haybaeck J, et al. TAK1 suppresses a NEMO-dependent but NF-kappaB-independent pathway to liver cancer. Cancer Cell. 2010;17(5):481–496. doi: 10.1016/j.ccr.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 84.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. The Journal of Biological Chemistry. 2006;281(17):12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 85.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Seminars in Liver Disease. 2008;28(4):360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Uehara T, Bennett B, Sakata ST, et al. JNK mediates hepatic ischemia reperfusion injury. Journal of Hepatology. 2005;42(6):850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 87.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131(1):165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 88.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Molecular Cell. 2004;15(5):713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 89.Jaeschke A, Karasarides M, Ventura JJ, et al. JNK2 is a positive regulator of the cJun transcription factor. Molecular Cell. 2006;23(6):899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 90.Schwabe RF, Bradham CA, Uehara T, et al. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37(4):824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 91.Sugioka Y, Watanabe T, Inagaki Y, et al. c-Jun NH2-terminal kinase pathway is involved in constitutive matrix metalloproteinase-1 expression in a hepatocellular carcinoma-derived cell line. International Journal of Cancer. 2004;109(6):867–874. doi: 10.1002/ijc.20095. [DOI] [PubMed] [Google Scholar]
- 92.Cellurale C, Sabio G, Kennedy NJ, et al. Requirement of c-Jun NH2-terminal kinase for Ras-initiated tumor formation. Molecular and Cellular Biology. 2011;31(7):1565–1576. doi: 10.1128/MCB.01122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature. 2005;437(7056):281–285. doi: 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- 94.Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell. 2008;133(2):340–353. doi: 10.1016/j.cell.2008.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sancho R, Nateri AS, De Vinuesa AG, et al. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. The EMBO Journal. 2009;28(13):1843–1854. doi: 10.1038/emboj.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cellurale C, Weston CR, Reilly J, et al. Role of JNK in a Trp53-dependent mouse model of breast cancer. PLoS One. 2010;5(8) doi: 10.1371/journal.pone.0012469. Article ID e12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schramek D, Kotsinas A, Meixner A, et al. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nature Genetics. 2011;43(3):212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- 98.Saxena NK, Fu PP, Nagalingam A, et al. Adiponectin modulates C-Jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology. 2010;139(5):1762–e1765. doi: 10.1053/j.gastro.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang Q, Zhang Y, Beezhold KJ, et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. Journal of Hepatology. 2009;50(2):323–333. doi: 10.1016/j.jhep.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Erhardt A, Hassan M, Heintges T, Häussinger D. Hepatitis C virus core protein induces cell proliferation and activates ERK, JNK, and p38 MAP kinases together with the MAP kinase phosphatase MKP-1 in a HepG2 Tet-Off cell line. Virology. 2002;292(2):272–284. doi: 10.1006/viro.2001.1227. [DOI] [PubMed] [Google Scholar]
- 101.Tanaka Y, Kanai F, Ichimura T, et al. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene. 2006;25(4):633–642. doi: 10.1038/sj.onc.1209093. [DOI] [PubMed] [Google Scholar]
- 102.Tsutsumi T, Suzuki T, Moriya K, et al. Alteration of intrahepatic cytokine expression and AP-1 activation in transgenic mice expressing hepatitis C virus core protein. Virology. 2002;304(2):415–424. doi: 10.1006/viro.2002.1702. [DOI] [PubMed] [Google Scholar]
- 103.Murata M, Matsuzaki K, Yoshida K, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49(4):1203–1217. doi: 10.1002/hep.22765. [DOI] [PubMed] [Google Scholar]
- 104.Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes and Development. 2011;25(6):634–645. doi: 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 106.Sabio G, Das M, Mora A, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322(5907):1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ohki T, Tateishi R, Shiina S, et al. Visceral fat accumulation is an independent risk factor for hepatocellular carcinoma recurrence after curative treatment in patients with suspected NASH. Gut. 2009;58(6):839–844. doi: 10.1136/gut.2008.164053. [DOI] [PubMed] [Google Scholar]
- 108.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50(3):957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 109.Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arano T, Nakagawa H, Tateishi R, et al. Serum level of adiponectin and the risk of liver cancer development in chronic hepatitis C patients. International Journal of Cancer. 2011;129(9):2226–2235. doi: 10.1002/ijc.25861. [DOI] [PubMed] [Google Scholar]
- 111.Han J, Sun P. The pathways to tumor suppression via route p38. Trends in Biochemical Sciences. 2007;32(8):364–371. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 112.Ge B, Gram H, Di Padova F, et al. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α . Science. 2002;295(5558):1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- 113.Hui L, Bakiri L, Stepniak E, Wagner EF. p38α: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007;6(20):2429–2433. doi: 10.4161/cc.6.20.4774. [DOI] [PubMed] [Google Scholar]
- 114.Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. International Journal of Biological Sciences. 2009;5(1):44–52. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stepniak E, Ricci R, Eferl R, et al. c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes and Development. 2006;20(16):2306–2314. doi: 10.1101/gad.390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Campbell JS, Argast GM, Yuen SY, Hayes B, Fausto N. Inactivation of p38 MAPK during liver regeneration. International Journal of Biochemistry and Cell Biology. 2011;43(2):180–188. doi: 10.1016/j.biocel.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11(2):191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 118.Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128(2):295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 119.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19 Arf pathway. Nature Genetics. 2004;36(4):343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 120.Hsieh YH, Wu TT, Huang CY, Hsieh YS, Hwang JM, Liu JY. p38 mitogen-activated protein kinase pathway is involved in protein kinase Cα-regulated invasion in human hepatocellular carcinoma cells. Cancer Research. 2007;67(9):4320–4327. doi: 10.1158/0008-5472.CAN-06-2486. [DOI] [PubMed] [Google Scholar]
- 121.Kim MS, Lee EJ, Kim HRC, Moon A. p38 kinase is a key signaling molecule for H-ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Research. 2003;63(17):5454–5461. [PubMed] [Google Scholar]
- 122.Iyoda K, Sasaki Y, Horimoto M, et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003;97(12):3017–3026. doi: 10.1002/cncr.11425. [DOI] [PubMed] [Google Scholar]
- 123.Ichijo H, Nishida E, Irie K, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 124.Huynh H, Choo SP, Toh HC, et al. Comparing the efficacy of sunitinib with sorafenib in xenograft models of human hepatocellular carcinoma: mechanistic explanation. Current Cancer Drug Targets. 2011;11(8):944–953. doi: 10.2174/156800911797264716. [DOI] [PubMed] [Google Scholar]
- 125.Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxidants and Redox Signaling. 2011;15(1):205–218. doi: 10.1089/ars.2010.3733. [DOI] [PubMed] [Google Scholar]
- 126.Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. The EMBO Journal. 1997;16(16):4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nature Cell Biology. 2004;6(8):770–776. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- 128.Pikarsky E, Porat RM, Stein I, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 129.Mayer RJ, Callahan JF. p38 MAP kinase inhibitors: a future therapy for inflammatory diseases. Drug Discovery Today. 2006;3(1):49–54. [Google Scholar]
- 130.Kumar S, Boehm J, Lee JC. P38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nature Reviews Drug Discovery. 2003;2(9):717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 131.Hagiwara S, Kudo M, Chung H, et al. Activation of c-Jun N-terminal kinase in non-cancerous liver tissue predicts a high risk of recurrence after hepatic resection for hepatocellular carcinoma. Hepatology Research. 2012;42(4):394–400. doi: 10.1111/j.1872-034X.2011.00932.x. [DOI] [PubMed] [Google Scholar]