

Abstract
The W chromosome is predicted to be subject to strong female-specific selection stemming from its female-limited inheritance and therefore should play an important role in female fitness traits. However, the overall importance of directional selection in shaping the W chromosome is unknown because of the powerful degradative forces that act to decay the nonrecombining sections of the genome. Here we greatly expand the number of known W-linked genes and assess the expression of the W chromosome after >100 generations of different female-specific selection regimens in different breeds of chicken and in the wild ancestor, the Red Jungle Fowl. Our results indicate that female-specific selection has a significant effect on W chromosome gene-expression patterns, with a strong convergent pattern of up-regulation associated with increased female-specific selection. Many of the transcriptional changes in the female-selected breeds are the product of positive selection, suggesting that selection is an important force in shaping the evolution of gene expression on the W chromosome, a finding consistent with both the importance of the W chromosome in female fertility and the haploid nature of the W. Taken together, these data provide evidence for the importance of the sex-limited chromosome in a female heterogametic species and show that sex-specific selection can act to preserve sex-limited chromosomes from degrading forces.
Keywords: experimental evolution, female heterogamety, sex chromosomes, Y chromosome, gene expression evolution
Y and W sex chromosomes are subject to unique evolutionary pressures because of their sex-limited inheritance, with Y chromosomes subject only to selection for male-specific effects and W chromosomes under selection only for female-specific effects. This sex-specific selection has resulted in genetic complements on Y chromosomes that play a large role in male fitness (1, 2), and an analogous role is predicted for W chromosomes in female fitness.
At the same time, the efficacy of selection on Y and W chromosome-coding regions is severely reduced by the lack of recombination, resulting in several concerted forces that degrade gene content (3–8). The limited evidence of selection on conserved orthologous Y-linked genes has indicated primarily a pattern consistent with purifying selection (9) rather than with positive selection shaping the content of Y chromosome coding content, suggesting that Y chromosome (and by analogy W chromosome) genes are limited in their ability to respond to directional sex-specific selection. However, rates of gene acquisition on the Y chromosome are high in both primates (9) and Drosophila (10), suggesting that the coding content of the Y chromosome is dynamic (11).
In birds, both the Z and the W chromosomes descend from a shared autosomal precursor that was present in the common ancestor of all modern birds (12–14); however, as recombination between the sex chromosomes was curtailed over the course of >120 million years, the W chromosome has degenerated both in size and gene number compared with the Z chromosome (15–17). Theoretically, the genes that have remained on or that have transferred to the W chromosome should play a disproportionately large role in determining female phenotypes, and varying female-specific selection should strongly affect the evolution of the W chromosome. Despite these clear predictions, little is known about the role of the W chromosome in birds as a whole, other than that some W–linked genes are expressed in the developing ovary (18, 19). Even less is known about the relative effect of female-specific selection on the expression and sequence of W-linked genes.
We used the different sex-specific selection regimens that have produced modern chicken breeds as an experimental evolution study of the effects of varying female-specific selection on the expression of W-linked loci. Domestic chickens descend from the Red Jungle Fowl (20, 21), and many chicken breeds have been selected explicitly for increased female fecundity, resulting in layer breeds that produce numerous and large eggs (22). These breeds therefore are the result of elevated female-specific selection compared with the Red Jungle Fowl ancestor. We used two different layer breeds to measure the effects of increased female-specific selection, the White Leghorn (hereafter, “Leghorn”) and the Black Minorca (hereafter, “Minorca”). Other chicken breeds have been selected for male traits such as aggression for fighting or plumage for ornamentation, and these breeds have been subject to relaxed female-specific selection, as evidenced by the reduced number of eggs they produce, variability in egg size, shape, and color, and numerous female fertility problems (23, 24). We used a fighting breed, the Oxford Old English (hereafter, “Old English”) and a plumage breed, the Yokohama, to represent the effects of reduced female-specific selection. Although the exact breed history of chickens is obscure, most modern breeds originated around the turn of the 20th century (25), indicating that sex-specific selection has been ongoing for >100 generations (SI Text). In this time, genetic differentiation has developed among poultry breeds (25).
Results and Discussion
We used RNA-seq data to correct and expand the known coding content of the chicken W chromosome, as the current W chromosome assembly is known to be incorrect (17). Verification of female-specific genes on genomic DNA samples, using PCR amplification, identified 12 previously unknown W-linked genes and confirmed four suspected W-linked genes (26, 27) that have been misassembled in the current genome draft (WUGSC 2.1/galGal3). The alignment of the sequence data also revealed that three genes currently attributed to the W chromosome (ENSGALG00000013557, ENSGALG 00000013559, and ENSGALG00000022692) are not, in fact, female-limited. Thus, with the expression data on the 10 known W-linked genes, we had a total of 26 genes with which to assess the response of the W chromosome to female-specific selection (Table S1).
Although variation in the coding sequences of the W-linked genes was rare, there was extensive variation in gene expression level among breeds compared with their wild ancestor, Red Jungle Fowl (Fig. 1A), whereby replicate female-selected breeds showed broadly similar expression patterns based on hierarchical Euclidean clustering. Convergent patterns of gene expression were also detected for female-biased autosomal genes (>twofold increase expression in females compared with males) (Fig. 1B). Our experimental design allowed us to look for convergent patterns in replicate sex-selected breeds. On the whole, W-linked genes were expressed more in both replicate layer breeds and less in replicate male-selected breeds, and this pattern holds across the majority of individual W-linked loci. Of the 26 expressed W-linked genes, 19 showed greater expression in both the female-selected breeds than in the Red Jungle Fowl (P < 0.000001; χ2 test). Down-regulation of the W-linked genes is far more common in the replicate male-selected breeds: 11 of the 26 genes were expressed at lower levels (P = 0.08; χ2 test), and only four loci were expressed at higher levels in both the replicate male-selected breeds than in the Red Jungle Fowl. We would expect similar patterns in gene expression in the replicate breeds to have arisen by chance in one out of every four comparisons. The stark contrast in expression of the W-linked genes between female-selected and male-selected breeds suggests that the change in W chromosome gene expression is not a function of domestication per se.
Fig. 1.

Hierarchical gene-expression clustering showing variation in the patterns of gene expression for (A) 26 expressed W-linked genes in females (n = 26); (B) autosomal female-biased genes in females (>twofold change level across all breeds, n = 305); (C) overall genetic similarity among breeds estimated using the pairwise genetic distances (Fst) of 25,000 exonic autosomal SNPs; (D) gene expression clustering of autosomal genes averaged across males and females (n = 8312). Heatmaps are based on the average breed expression compared with the among-breed expression. All trees are rooted using the information from Red Jungle Fowl (Jungle Fowl). The number at each node represents the percentage bootstrap value from 1,000 replicates. The names of breeds subjected to increased female-specific selection are highlighted in red; breeds subject to relaxed female-specific selection are in blue.
To determine whether these similar expression patterns result from convergent selection or from shared ancestry, we generated an Fst tree using 25,000 high-quality autosomal SNPs from highly expressed genes (Fig. 1C). Both the genetic distance analysis and the Euclidean clustering of averaged male and female transcription from autosomal genes (Fig. 1D) indicate that the convergent expression patterns of W-linked and female-biased genes do not result from shared ancestry, because sex-selected replicate breeds do not cluster together. The similarity in the W chromosome and female-biased transcription between the replicate sex-selected breeds therefore is independent of the phylogenetic relationships among the breeds, suggesting that female-specific selection acts convergently on a broad array of genes that are important in female fitness traits, regardless of whether the genes are on the autosomes or the W chromosome. Control analyses of Z-linked and male-limited genes also indicate that the up-regulation of the W chromosome in the replicate layer breeds is the result of female-specific selection rather than some other breed trait (SI Text).
The variation in W chromosome gene expression is in stark contrast to the amount of coding sequence polymorphism. Of the 20 W-linked genes present in a single copy, corresponding to roughly 38 kb, we detected very little interbreed polymorphism on the basis of SNP data, with only one SNP present in a UTR of a putatively single-copy gene (ENSGALG00000014441). Earlier reports have documented little standing polymorphism in intronic regions of the W chromosome in chickens (28), despite the overall variability of the chicken genome as a whole (25, 29). This broad-scale sequence homogeneity can be the result of the strength of female-specific selection expected on the W chromosome (10), Hill-Robertson effects selective sweeps resulting from suppressed recombination (5, 6), or the reduced effective population size of the W chromosome. To differentiate these possible causes of reduced polymorphism, we compared sequence variation in a set of similarly expressed female-limited genes located on the autosomes (Table 1). Of the 13 autosomal female-limited genes, corresponding to ∼25 kb of sequence data, we identified 105 polymorphic sites. Because female-limited autosomal and W-linked genes are subject to similar levels of female-specific selection, the difference between these regions in polymorphism for similar types of genes can be explained best by differences in other characteristics, such as linkage effects or effective population size, that differentiate W-linked and autosomal loci.
Table 1.
Observed polymorphisms in W-linked and in autosomal female-limited genes
| Chromosome | No. of genes | No. of polymorphic genes | Exonic length (kb) | Coverage | Functional class | No. of SNPs |
| Autosomes | 13 | 13 | 25 | 106X | Synonymous | 77 |
| Nonsynonymous | 24 | |||||
| Untranslated region | 4 | |||||
| W | 26 | 6 | 48 | 88X | Synonymous | 4 |
| Nonsynonymous | 4 | |||||
| Untranslated region | 1 |
We examined the 26 W-linked genes for evidence of multiple copy number among breeds, because copy number variation (CNV) could explain transcriptional differences, and CNV has been documented for W chromosome loci (30). Our nucleotide polymorphism data indicate CNV in six W-linked genes; however, few of these loci showed discrete expression classes. Therefore we also examined our data for patterns of discontinuous variation in gene expression as a likely indicator of CNV. We identified seven genes with potential CNV between breeds and were able to design primers that amplified solely in females for four of these genes (Table S2). Quantitative PCR (qPCR) on genomic DNA samples, using a Z-linked gene that lacks an intragenomic paralog as a calibrating control, was used to verify CNV for these W-linked genes. One of these four genes is present in at least two copies, and two have a minimum of three variants. The other gene displayed no CNV across individuals (Table S1). Of the loci with CNV, only one showed a significant association between copy number and expression (P = 0.037; two-tailed t test) (Table S2), with higher copy number associated with higher transcription rates. The Y chromosome in mammals has been shown to undergo nonhomologous intrachromosomal recombination, producing CNV that in turn is implicated in male fertility (1). If the chicken W chromosome recombines in a similar manner, it could explain the CNV we observe. Therefore CNV may prove to be a useful genetic character for quantitative trait locus analyses, which previously have not been incorporated into the W chromosome architecture while mapping for egg-production traits (22, 31, 32), or for marker-assisted selection for industrial egg production.
However, CNV does not explain the majority of variation in expression. Additionally, the low amount of sequence polymorphism on the W chromosome indicates that cis-regulatory variation is an unlikely source of the majority of the variation in expression we observe, because, aside from CNV, the W chromosome sequence is nearly identical in all our samples. The lack of W-linked diversity suggests that transregulation or epigenetic and/or epistatic interactions between the W chromosome and the remainder of the genome are the primary mode of expression evolution for W-linked genes. The latter has been documented for the Y chromosome of Drosophila (33).
To investigate further whether changes in gene expression were the result of selective or neutral forces, we calculated the degree of directional selection using Δx, a statistic that assesses the divergence in expression level for a focal breed in relation to the variation in expression level seen across replicates. As with the McDonald–Kreitman test, when divergence does not exceed polymorphism, we can assume that neutral processes or stabilizing selection are the dominant forces, although in this instance on the gene expression level rather than on coding sequence evolution (34). However, when Δx is greater than 1 or is less than −1, divergence exceeds polymorphism, therefore suggesting that directional selection for expression is acting at that locus. This method differs from other estimates of gene expression evolution in that it does not assume that selection acts on a similar proportion of genes across all genomic regions; further, it provides a conservative but meaningful cutoff value (>1 or < −1) that can be used to indicate when directional selection has occurred, and it is not confounded by expression level (35, 36).
Δx was calculated for all genes expressed in females, independent of whether they are expressed in males. For three of the breeds, the number of W-linked genes that are up-regulated and under selection is not significantly less than the number of autosomal up-regulated genes under selection (Minorca, P = 0.441; Leghorn, P = 0.425; Yokohama P = 0.169; Z-test, Fig. 2)). This result indicates that selection for up-regulation on W-linked genes is as effective as that acting on autosomal genes, despite the interfering processes related to the lack of recombination (SI Text and Figs. S3 and S4).
Fig. 2.

The proportion of genes where Δx >1 (up-regulated genes under putative positive selection, black bars) or <−1 (down-regulated genes under putative positive selection, gray bars) are shown for W-linked genes (W) (n = 26), Z-linked genes expressed in females (Z) (n = 515), and autosomal genes expressed in females (A) (n = 10,281) for each of the four breeds compared with the ancestral Red Jungle Fowl: (A) Minorca, (B) Leghorn, (C) Old English, and (D) Yokohama. Gene categories in which the proportion of genes under selection differed from that expected are indicated by an asterisk and the corresponding P value (Z-test).
Although W-linked gene expression appears to respond to directional selection, it is clear that selection has acted differently on the replicate female- and male-selected breeds. Specifically, the number of W-linked genes under selection for up-regulation in the Old English breed is lower than expected (P = 0.028; Z-test, Fig. 2C), and the number under selection for down-regulation in the Yokohama breed is higher than expected (P = 0.009; Z-test, Fig. 2D). Taken together, the data suggest increased female-specific selection has caused the up-regulation of W-linked genes. However, in the male-selected breeds, where female-specific selection has been relaxed, there has been either little selection, as seen in the Old English breed, or selection for both up- and down-regulation, as seen in the Yokohama breed.
Δx also was calculated for male-specific genes and for female- and male-expressed Z-linked genes (Fig. S1). For all gene categories there was no support for convergent directional selection on the two female-specific selection lines. However, there is convergence in the male-specific selection lines, with both breeds having fewer Z-linked genes under directional selection than expected compared with autosomes (Fig. S1). This result suggests that selection for both female and male expression is less effective on the Z chromosome.
Collectively, the analyses of gene-expression evolution suggest that selection explains much of the variation in gene expression for W-linked genes and that the W chromosome remains a target for gene expression evolution despite the neutral processes that decrease selection efficacy (Table S3). Taken together, our results show that selection for female egg-laying, a major potential component of female fitness, has a major influence on the evolution of gene expression on the W chromosome because of the role of the W chromosome in female fecundity. Independent female-specific selection has produced convergent results in replicate sex-selected breeds. The divergence in gene expression that we observe could result from genetic drift, given that the effective population size of the W chromosome is only one-fourth that of an autosome and one-third that of the Z chromosome. Also, in theory, any recent bottleneck event or extensive inbreeding that reduces variation in gene expression will lead to a lower expression range across replicates (r value) and hence will raise Δx values artificially. To determine whether any of the breeds do show evidence of inbreeding or a dramatic reduction in population size we took the average number of heterozygous sites across all autosomal genes expressed in males or in females for each breed (Table S4). Although there is variation in the amount of polymorphism within each breed, the effect of reduced heterozygosity is not evident when gene-expression variation is compared across the breeds (Table S4). Therefore genetic diversity does not appear to correlate with diversity of expression level, and there is little evidence that increased homozygosity would skew the Δx analysis.
Another factor possibly contributing to the strong and convergent response of the W chromosome to female-specific selection is that the W chromosome is effectively haploid. Although paralogs for many W-linked genes exist on the Z chromosome, their function and gene sequence have diverged, leaving females with only a single copy of each Z and W chromosome locus. Selection is more effective in haploid than in diploid genes (10).
In this study, we used chicken breeds as proxies for sex-specific experimental evolution lines, and this approach has both advantages and disadvantages. This tactic makes possible experimental evolution studies in a long-lived vertebrate that would otherwise not be tractable. It also allows the leveraging of the wealth of diversity extant in chicken breeds. However, some concerns are worth mentioning. First, modern chicken breeds have been under artificial selection that is much stronger than the selection regimens that usually are in place in natural systems (37). Therefore, the patterns of convergence in the evolution of gene expression might not be as readily evident in wild populations over similarly short time-spans. However, limited data on Drosophila and human Y-linked genes are consistent with strong evolutionary forces acting on these chromosomes (10, 38), suggesting that our results are not outside the bounds of natural populations. Second, chicken breeds are assessed primarily by their conformity with defined breed standards rather than by their position in a known pedigree; thus there may be some admixture among breeds by breeders seeking to increase genetic diversity upon which to select, and it is not possible to know the exact pedigree at early stages in the breed histories. However, whole-genome resequencing efforts have identified genetic distinctions among breeds (25, 29), and our Fst analysis indicates that the replicate breeds are independent of shared ancestry. Additionally, we cannot control for different generation times or exact details of artificial selection regimens among breeds.
Although these concerns should be taken seriously, several lines of evidence suggest that they have not influenced our results unduly. First, we used replicate breeds with similar sex-specific selection histories to identify loci that have responded convergently to increased female-specific selection. Our data suggest that the convergent patterns in gene expression have emerged in roughly 100 years of artificial selection. However, considering low levels of potential admixture after the origin of breeds, it is possible that these convergent patterns have emerged in a shorter time span, making our results, in fact, conservative. Our observation of such a strong pattern across replicates indicates that the small variations in generation time and selection regimen among breeds have not produced radically different results. At the same time, the results in the replicate breeds are broadly similar but not identical, suggesting that if admixture among breeds has occurred, it occurred long ago, and there has been sufficient breed isolation and selection to result in independent samples.
In summary, our results show the utility of in silico subtraction of RNA-seq data for identifying sex-limited genes. Given the difficulty of sequencing Y and W chromosomes, this method provides a quick and efficient way to detect coding sequences from these chromosomes. More importantly, this study indicates the important role of the W chromosome in female fertility traits. Additionally, the rapid and convergent response of the W chromosome to female-specific selection also may indicate that genes on the W chromosome are more responsive than genes of similar function in the remainder of the genome. Several thousand genes are expressed in the ovary, of which only a small proportion are located on the W chromosome. Of the genes expressed in the ovary that are located on the autosomes and Z chromosome, the great majority also are expressed in males, and male functionality may represent a constraint on the response of these loci to female-specific selection. Because the W chromosome is confined to females, W-linked genes are free of constraints arising from male functionality, possibly leaving the W chromosome better able to respond to female-specific selection (39).
Materials and Methods
We obtained fertilized eggs from four chicken breeds. Two breeds, the Minorca (from R. Walker, Herefordshire, United Kingdom) and Leghorn (from J. Taylor, Kent, United Kingdom) were selected independently for egg number and size and represent female-selected breeds, because they are classified as highly productive egg layers, delivering >250 large eggs per 52 wk in lay We also obtained samples from two male-selected breeds, the Yokohama (from E. Bootheman, Yorkshire, United Kingdom), which has been selected for male plumage characters, and the Old English Game (from D. Sims, Oxfordshire, United Kingdom), which has been selected for male fighting ability. Both these breeds lay 50–100 eggs per 52 wk in lay, and the eggs are far smaller than those from the layer breeds. We also obtained fertilized eggs of Red Jungle Fowl (from T. Pizzari, Oxford University, Oxford, United Kingdom), which produce on average 20–50 eggs per year. All eggs were kept under standard incubator conditions, and the left gonad was collected at embryonic day (ed)19 and stored in RNAlater (Qiagen) until preparation.
RNA was prepared with the Animal Tissue RNA Kit (Qiagen). Library and RNA-sequence samples were prepared by the Wellcome Trust Centre for Human Genetics Facility at Oxford University using standard methods. Four samples per sex were prepared for each breed, and each individual was barcoded so that individual variation in coding sequence and expression level could be tracked. The only exception was the Yokohama breed, for which high inviability rates resulted in only three female samples.
All samples were sequenced using Illumina Genome Analyzer II or Illumina HiSeq as paired-end 51-bp reads, resulting in 16 million paired-end mappable reads per sample, on average. Sequences were mapped to the chicken reference genome (WUGSC 2.1/galGal3) using Bowtie (v.0.12.7) (40) and Tophat (v.1.1.1) (41). Up to three mismatches in each alignment were allowed, and all reads with multiple “best hits” were discarded from the later analysis.
Transcript abundances for the Ensembl-annotated genes were estimated using cufflinks (v.0.9.3) (42), where relative expressions are expressed as fragments per kilobase of exon per million mapped reads (FPKM) values. Putative W-linked genes were identified first through in silico analysis of the gene expression profiles in males and females and were validated further by PCR genotyping of the genomic DNA from an array of 29 individuals representing both sexes from five different breeds (Table S5). Single-nucleotide variations were called using the Genome Analysis Toolkit (43), in which at least three separate unique reads, with a minimum quality score of 30, were required to support the alternative allele.
Evidence for directional selection was assessed by calculating the ratio Δx, which is the divergence in mean gene expression between the focal breed and Red Jungle Fowl (d), divided by the expression level range for the focal breed (r). Divergence, d, is calculated as 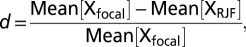 where X = log2[relative gene expression]. Range, r, is calculated as
where X = log2[relative gene expression]. Range, r, is calculated as 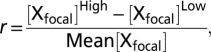 where [Xfocal]High is the highest value for X, and [Xfocal]Low is the lowest. To correct for different sample sizes, we multiplied r by f, where
where [Xfocal]High is the highest value for X, and [Xfocal]Low is the lowest. To correct for different sample sizes, we multiplied r by f, where 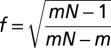 , m is the sample size and n = 4 (the most common sample size) (35). Therefore,
, m is the sample size and n = 4 (the most common sample size) (35). Therefore, 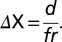
Values for Δx greater than 1 or less than −1 indicate that divergence (up or down respectively) from Red Jungle Fowl is greater than the standing variation in gene expression within the breed. Hence divergence exceeds polymorphism, and the basic tenet of the McDonald–Kreitman test is fulfilled, suggesting that these genes are under putative positive selection for gene-expression change. Δx was calculated using a relaxed filtered dataset; any genes that were not expressed in at least one individual in all breeds were removed. In this instance, zero expression can be informative, providing evidence for down-regulation from the ancestral state in Red Jungle Fowl.
To explore the amount of genetic differentiation among breeds, we examined pairwise fixation indices (Fst) by analyzing the allelic frequencies of 25,000 autosomal SNPs selected from genes showing high levels of expression across all individuals from all the breeds investigated in this study. Genetic divergence between breeds was measured using Genepop (v.4.1) (44), and the distance phylogenetic tree was constructed using the neighbor-joining method implemented in Phylip v. 3.2 (45).
Analyses of similarity in patterns of gene expression between breeds were performed by hierarchical clustering using Euclidean distance with complete linkage, as implemented in Cluster 3.0 (46) and visualized by TreeView (v.1.1.6) (47). The reliability of the inferred trees was tested by bootstrap resampling (1,000 replicates) of the expression values using the R package, Pvclust (48). To define a female-biased gene, we used cutoff threshold >twofold comparing male and female expression within breeds and required that the gene show the same bias across all breeds.
For CNV analysis, DNA was extracted from the liver samples using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s protocol. Primer sequences were designed for four W-linked genes and for a Z-linked control gene present in a single copy in the current Ensembl Chicken Genome Assembly using Primer3 and GenScript Real-Time PCR Primer design (Table S2). Real-time qPCR was carried out in a Mastercycler EP Realplex (Eppendorf) with SYBR Green (Qiagen) under standard PCR conditions followed by a melting curve analysis. Each W-linked gene was run in quadruplicate on every female individual for which we had expression data as well as for two males of every breed to serve as negative controls. Each plate included the Z-linked control gene run in triplicate on every female. Anomalous CT values were removed where appropriate, and averages were calculated but were included in the analysis only if the SE was ≤ 0.5. CT values were normalized against the Z-linked control gene run on a single Red Jungle Fowl sample and were compared to determine relative copy number differences. Amplification curves run for all primers using a 10-fold serial dilution showed that reaction efficiencies were all >80%.
Supplementary Material
Acknowledgments
We thank Geoff Parker, who provided invaluable expertise and advice on chicken breeds and chicken breeders; Tommaso Pizzari (Oxford University), who provided Red Jungle Fowl samples and the use of incubation facilities; Lorna Gregory and the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust Grant reference 090532/Z09/Z and MRC Hub Grant G0900747 91070 for the generation of the sequencing data); two anonymous reviewers for helpful comments; the breeders who provided samples; and E. Bootheman, J. Taylor, R. Walker, D. Sims. We also thank P. Harrison, D. Hosken, S. Montgomery, J. Perry, J. Walters, and N. Wedell for helpful suggestions on a previous draft. Preliminary work leading to this study was funded by the Royal Society and the John Fell Fund. The work described here was funded by the Biotechnology and Biological Sciences Research Council and the European Research Council under the Framework 7 Agreement (Grant 260233).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1202721109/-/DCSupplemental.
References
- 1.Lange J, et al. Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell. 2009;138:855–869. doi: 10.1016/j.cell.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemos B, Araripe LO, Hartl DL. Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science. 2008;319:91–93. doi: 10.1126/science.1148861. [DOI] [PubMed] [Google Scholar]
- 3.Bachtrog D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics. 2008;179:1513–1525. doi: 10.1534/genetics.107.084012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachtrog D, Charlesworth B. Reduced adaptation of a non-recombining neo-Y chromosome. Nature. 2002;416:323–326. doi: 10.1038/416323a. [DOI] [PubMed] [Google Scholar]
- 5.Berlin S, Tomaras D, Charlesworth B. Low mitochondrial variability in birds may indicate Hill-Robertson effects on the W chromosome. Heredity (Edinb) 2007;99:389–396. doi: 10.1038/sj.hdy.6801014. [DOI] [PubMed] [Google Scholar]
- 6.Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haddrill PR, Halligan DL, Tomaras D, Charlesworth B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol. 2007;8:R18. doi: 10.1186/gb-2007-8-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaiser VB, Charlesworth B. Muller’s ratchet and the degeneration of the Drosophila miranda neo-Y chromosome. Genetics. 2010;185:339–348. doi: 10.1534/genetics.109.112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes JF, et al. Conservation of Y-linked genes during human evolution revealed by comparative sequencing in chimpanzee. Nature. 2005;437:100–103. doi: 10.1038/nature04101. [DOI] [PubMed] [Google Scholar]
- 10.Koerich LB, Wang X, Clark AG, Carvalho AB. Low conservation of gene content in the Drosophila Y chromosome. Nature. 2008;456:949–951. doi: 10.1038/nature07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mank JE. Small but mighty: The evolutionary dynamics of W and Y sex chromosomes. Chromosome Research. 2012;20:21–33. doi: 10.1007/s10577-011-9251-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fridolfsson AK, et al. Evolution of the avian sex chromosomes from an ancestral pair of autosomes. Proc Natl Acad Sci USA. 1998;95:8147–8152. doi: 10.1073/pnas.95.14.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pigozzi MI, Solari AJ. The ZW pairs of two paleognath birds from two orders show transitional stages of sex chromosome differentiation. Chromosome Res. 1999;7:541–551. doi: 10.1023/a:1009241528994. [DOI] [PubMed] [Google Scholar]
- 14.Mank JE, Ellegren H. Parallel divergence and degradation of the avian W sex chromosome. Trends Ecol Evol. 2007;22:389–391. doi: 10.1016/j.tree.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Berlin S, Ellegren H. Fast accumulation of nonsynonymous mutations on the female-specific W chromosome in birds. J Mol Evol. 2006;62:66–72. doi: 10.1007/s00239-005-0067-6. [DOI] [PubMed] [Google Scholar]
- 16.van Tuinen M, Hedges SB. Calibration of avian molecular clocks. Mol Biol Evol. 2001;18:206–213. doi: 10.1093/oxfordjournals.molbev.a003794. [DOI] [PubMed] [Google Scholar]
- 17.International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 18.Reed KJ, Sinclair AH. FET-1: A novel W-linked, female specific gene up-regulated in the embryonic chicken ovary. Mech Dev. 2002;119(Suppl 1):S87–S90. doi: 10.1016/s0925-4773(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 19.Hori T, Asakawa S, Itoh Y, Shimizu N, Mizuno S. Wpkci, encoding an altered form of PKCI, is conserved widely on the avian W chromosome and expressed in early female embryos: Implication of its role in female sex determination. Mol Biol Cell. 2000;11:3645–3660. doi: 10.1091/mbc.11.10.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fumihito A, et al. Monophyletic origin and unique dispersal patterns of domestic fowls. Proc Natl Acad Sci USA. 1996;93:6792–6795. doi: 10.1073/pnas.93.13.6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisby DP, Weiss RA, Roussel M, Stehelin D. The distribution of endogenous chicken retrovirus sequences in the DNA of galliform birds does not coincide with avian phylogenetic relationships. Cell. 1979;17:623–634. doi: 10.1016/0092-8674(79)90270-8. [DOI] [PubMed] [Google Scholar]
- 22.Kerje S, et al. The twofold difference in adult size between the red junglefowl and White Leghorn chickens is largely explained by a limited number of QTLs. Anim Genet. 2003;34:264–274. doi: 10.1046/j.1365-2052.2003.01000.x. [DOI] [PubMed] [Google Scholar]
- 23.Ekarius C. Storey's Illustrated Guide to Poultry Breeds. Marceline, MO: Walsworth Publishing Company; 2007. [Google Scholar]
- 24.Lewis C. The Illustrated Guide to Chickens: How to Choose Them - How to Keep Them. Oswestry, UK: Scotprint; 2010. [Google Scholar]
- 25.Rubin CJ, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 26.Wahlberg P, et al. A high-resolution linkage map for the Z chromosome in chicken reveals hot spots for recombination. Cytogenet Genome Res. 2007;117:22–29. doi: 10.1159/000103161. [DOI] [PubMed] [Google Scholar]
- 27.Nam K, Ellegren H. The chicken (Gallus gallus) Z chromosome contains at least three nonlinear evolutionary strata. Genetics. 2008;180:1131–1136. doi: 10.1534/genetics.108.090324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berlin S, Ellegren H. Chicken W: A genetically uniform chromosome in a highly variable genome. Proc Natl Acad Sci USA. 2004;101:15967–15969. doi: 10.1073/pnas.0405126101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong GK, et al. International Chicken Polymorphism Map Consortium A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature. 2004;432:717–722. doi: 10.1038/nature03156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backström N, Ceplitis H, Berlin S, Ellegren H. Gene conversion drives the evolution of HINTW, an ampliconic gene on the female-specific avian W chromosome. Mol Biol Evol. 2005;22:1992–1999. doi: 10.1093/molbev/msi198. [DOI] [PubMed] [Google Scholar]
- 31.Sasaki O, et al. Genetic mapping of quantitative trait loci affecting body weight, egg character and egg production in F2 intercross chickens. Anim Genet. 2004;35:188–194. doi: 10.1111/j.1365-2052.2004.01133.x. [DOI] [PubMed] [Google Scholar]
- 32.Schreiweis MA, Hester PY, Settar P, Moody DE. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Anim Genet. 2006;37:106–112. doi: 10.1111/j.1365-2052.2005.01394.x. [DOI] [PubMed] [Google Scholar]
- 33.Lemos B, Branco AT, Hartl DL. Epigenetic effects of polymorphic Y chromosomes modulate chromatin components, immune response, and sexual conflict. Proc Natl Acad Sci USA. 2010;107:15826–15831. doi: 10.1073/pnas.1010383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 35.Ometto L, Shoemaker D, Ross KG, Keller L. Evolution of gene expression in fire ants: The effects of developmental stage, caste, and species. Mol Biol Evol. 2011;28:1381–1392. doi: 10.1093/molbev/msq322. [DOI] [PubMed] [Google Scholar]
- 36.Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. Sex-specific and lineage-specific alternative splicing in primates. Genome Res. 2010;20:180–189. doi: 10.1101/gr.099226.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kingsolver JG, Diamond SE. Phenotypic selection in natural populations: What limits directional selection? Am Nat. 2011;177:346–357. doi: 10.1086/658341. [DOI] [PubMed] [Google Scholar]
- 38.Bachtrog D. Evidence that positive selection drives Y-chromosome degeneration in Drosophila miranda. Nat Genet. 2004;36:518–522. doi: 10.1038/ng1347. [DOI] [PubMed] [Google Scholar]
- 39.Bachtrog D, et al. Are all sex chromosomes created equal? Trends Genet. 2011;27:350–357. doi: 10.1016/j.tig.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rousset F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 45.Felsenstein J. Phylip (Phylogeny inference package v3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 46.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 47.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki R, Shimodaira H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.