

Abstract
Nuclear protein peptidyl-prolyl isomerase Pin1-mediated prolyl isomerization is an essential and novel regulatory mechanism for protein phosphorylation. Therefore, tight regulation of Pin1 localization and catalytic activity is crucial for its normal nuclear functions. Pin1 is commonly dysregulated during oncogenesis and likely contributes to these pathologies; however, the mechanism(s) by which Pin1 catalytic activity and nuclear localization are increased is unknown. Here we demonstrate that mixed-lineage kinase 3 (MLK3), a MAP3K family member, phosphorylates Pin1 on a Ser138 site to increase its catalytic activity and nuclear translocation. This phosphorylation event drives the cell cycle and promotes cyclin D1 stability and centrosome amplification. Notably, Pin1 pSer138 is significantly up-regulated in breast tumors and is localized in the nucleus. These findings collectively suggest that the MLK3-Pin1 signaling cascade plays a critical role in regulating the cell cycle, centrosome numbers, and oncogenesis.
Keywords: breast cancer, JNK
Peptidyl-prolyl isomerase Pin1 plays a critical role in regulating cellular homeostasis by isomerizing the prolyl bond preceded by a phosphorylated Ser or Thr residue (pSer/Thr-Pro) (1). This isomerization by Pin1 regulates the biological function of several target proteins, including cell-cycle regulators, proto-oncogenes, tumor suppressors, and transcription factors (2). Due to its role in controlling the cell cycle, apoptosis, growth, and stress responses, Pin1 has been linked to the pathogenesis of human diseases, including cancer (3, 4), asthma (5), Alzheimer’s disease (AD) (6), and Parkinson disease (PD) (7). It is thus quite likely that tight regulation of Pin1 catalytic activity or expression is important for normal physiology. It is reported that Pin1 is overexpressed in most types of cancer (8), whereas its expression is diminished in AD brains (2).
Accumulating evidence suggests that Pin1 isomerase activity and thus function are regulated by posttranslational modifications (2). Pin1 function is also dependent on its predominant nuclear localization (2), consistent with its substrates being involved in transcription and cell-cycle progression. It was recently reported that Pin1 nuclear import is regulated by a novel nuclear localization sequence in the PPIase domain, composed of basic amino acids (9). Nonetheless, the detailed mechanism that regulates Pin1 nuclear translocation is still not known. It also remains unknown whether any posttranslational modification of Pin1 can regulate its nuclear translocation or catalytic activity, and therefore directly affect its function.
Mixed-lineage kinase 3 (MLK3) is a novel member of the MAP3K superfamily (10, 11), and contains signature sequences of both Ser/Thr and Tyr kinases in the catalytic domain (11). The physiological functions of MLK members, including MLK3, are not fully understood. Recently, MLK members were implicated in PD (12), but the specific function of individual MLK members in PD and other diseases is still unknown. The role of MLK family members in cancer has just started to emerge. We have shown regulation of MLK3 activity and downstream signaling in breast cancer cells by estrogen (13) and involvement of MLK3 in gastric cancer cell migration (14). Despite an established role for MLK3 (15) and Pin1 (16) in cell-cycle progression, a detailed mechanism is yet to be explored.
Here we show that endogenous MLK3 associates with and uniquely phosphorylates S138 within the PPIase domain of Pin1. Phosphorylation at this site increases Pin1’s catalytic activity and nuclear localization, promoting cell-cycle progression and centrosome amplification. Consistent with these data, nuclear Pin1 pS138 is increased in human breast tumor samples. Our results identify an upstream kinase that regulates the catalytic activity, nuclear translocation, and function of Pin1, and likely contributes to oncogenesis.
Results
MLK3 Associates Strongly with Pin1.
Given that both Pin1 and MLK3 are necessary for cell-cycle progression, we assessed whether endogenous Pin1 and MLK3 interacted in breast cancer cell lines. We observed that both endogenous proteins interacted robustly in all cell lines tested (Fig. 1A). MLK3 is a 97-kDa protein, and contains an SH3 domain, kinase domain, LZ domain, and a long C-terminal domain (Fig. S1). As these domains have distinct functions (17, 18), we mapped the Pin1 interaction site to the C-terminal domain of MLK3, whereas the other regions failed to interact with Pin1 (Fig. 1B). In addition, we also mapped the Pin1 domain that interacts with MLK3. Pin1 primarily contains an N-terminal WW domain, which reportedly interacts with substrates, and a C-terminal PPIase domain, which is necessary for isomerase activity (Fig. S1). Quite contrary to the reported function of the WW domain of Pin1 for substrate interaction, we observed that the Pin1 PPIase domain interacted robustly with MLK3, whereas the WW domain failed to interact (Fig. 1 C and D), confirming that the MLK3 C-terminal domain specifically interacts with the Pin1 PPIase domain.
Fig. 1.

MLK3 associates with Pin1. (A) Endogenous MLK3 from breast cancer cell lines was immunoprecipitated, and associated endogenous Pin1 was detected by anti-Pin1 antibody. The total expression of Pin1 and MLK3 was detected in whole-cell extracts (WCEs). IB, immunoblotting; IP, immunoprecipitation. (B) HEK293 cells were transfected with GFP-Pin1 (WT) along with either full-length or different deletion mutants of GST-MLK3, as indicated. The MLK3 proteins were pulled down with GSH beads and blotted with anti-GFP antibody to detect associated GFP-Pin1. The WCEs were blotted with anti-GFP for Pin1 and anti-GST for MLK3 expression. (C and D) HEK293 cells were transfected with FLAG-tagged MLK3 along with full-length (FL) or GFP-tagged different domains (i.e., WW and PPI) of Pin1. MLK3 (C) and Pin1 (D) were immunoprecipitated using anti-FLAG and anti-GFP antibodies, respectively, and blotted with anti-GFP for Pin1 and anti-FLAG for MLK3 detection.
MLK3 Phosphorylates Pin1.
The interaction between MLK3 and Pin1 (Fig. 1) suggests a mutual regulatory relationship. Somewhat unexpectedly, we observed a strong phosphorylation of bacterially expressed Pin1 by kinase-active MLK3 (Fig. 2A). Given that we observed interaction between MLK3 and the Pin1 PPIase domain, we examined any phosphorylation of this domain by highly purified MLK3. As shown in Fig. 2B, MLK3 phosphorylated the PPIase domain, but not the WW domain. The phosphorylation of the isolated PPIase domain, however, was less than that of the full-length Pin1 protein, suggesting that there could be another MLK3 phosphorylation site(s) specifically targeted in the full-length Pin1 protein.
Fig. 2.

MLK3 directly phosphorylates Pin1. (A) In vitro phosphorylation of bacterially expressed Pin1 (5 μg) was performed using increasing concentrations of either recombinant WT or kinase-dead (K-A) MLK3, expressed in HEK293 cells. (B) Direct phosphorylation of bacterially expressed full-length or different domains of Pin1 was performed in the presence of purified recombinant GST-MLK3 (GST-rMLK3), produced in baculovirus. GST-Pin1 and GST-rMLK3 were blotted with anti-GST antibody. (C and D) Serine 138 on Pin1 was identified as an MLK3-specific phosphorylation site by mass spectroscopy and mutated to alanine to make phospho-deficient protein in bacteria. The purified Pin1 proteins were used in an in vitro phosphorylation assay with purified GST-rMLK3 protein (C), and these phosphorylated Pin1 proteins were analyzed by 2D peptide mapping (D). (E) Pin1 (WT) or S138A expression vectors were coexpressed either with WT or kinase-dead MLK3 in HEK293 cells. The recombinant GFP-Pin1 was blotted with Pin1 pSer138 antibody. The kinase activities are expressed as phosphorimager units (PI-Units).
Next, we identified the precise location of the Pin1 phosphorylation site by mass spectroscopy. These studies revealed that the S138 residue in the PPIase domain of Pin1 was phosphorylated by MLK3 in vitro (Fig. 2 C and D). Mutant Pin1 (S138 to A) was almost 10- to 12-fold less phosphorylated (Fig. 2C), and tryptic peptides containing A138 were absent after in vitro phosphorylation of mutant Pin1 protein (Fig. 2D). We further verified phosphorylation of the S138 site on Pin1 by using purified anti-pS138 antibody. The specificity of the antibody was tested by using in vitro phosphorylated Pin1, either by purified MLK3 (Fig. S2, lane 2) or by MLK3 protein expressed in mammalian cells (Fig. S2, lane 3). In both cases, the antibody recognized phosphorylated Pin1 WT and not Pin1 S138A mutant protein. In the mammalian cells, basal S138 phosphorylation of exogenous wild-type Pin1 was increased upon overexpression of kinase-active (Fig. 2E, lane 2) but not -inactive (Fig. 2E, lane 3) MLK3. Conversely, recombinant mutant Pin1 S138A protein was not recognized by this antibody under similar conditions (Fig. 2E, lanes 4–6). These results collectively show that MLK3 phosphorylates Pin1 on S138, both under in vivo and in vitro conditions, and can be specifically detected by our anti-Pin1 pS138 antibody.
Phosphorylation of Pin1 Is Necessary for Its Interaction with MLK3.
Pin1 interacts with its substrates in a phosphorylation-dependent manner; however, how phosphorylation of Pin1 might affect its interaction with upstream kinases is not known. To determine the interaction between phosphorylated Pin1 and MLK3, mammalian cells were transfected with Pin1 WT, S138A, or S138E, along with WT MLK3. As shown in Fig. 3, Pin1 WT and S138E interacted strongly with MLK3, which was minimal with phospho-deficient Pin1 S138A. Interestingly, the interaction between phospho-mimetic Pin1 S138E and MLK3 was highest (Fig. 3A), which was reconfirmed by reverse immunoprecipitation (Fig. 3B). These results demonstrate that phosphorylation of the Pin1 S138 site is necessary for a strong interaction with MLK3.
Fig. 3.
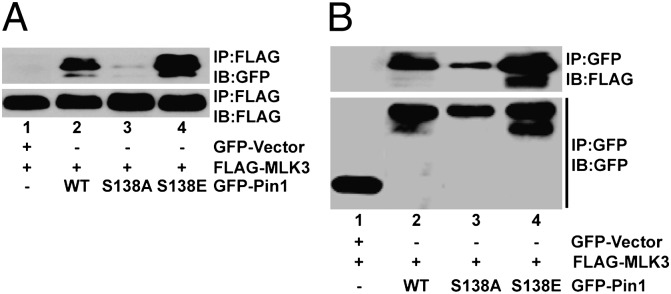
Phosphorylated Pin1 interacts with MLK3. HEK293 cells were transfected with FLAG-tagged MLK3 along with GFP-tagged WT or different mutants of Pin1, as indicated. MLK3 (A) and Pin1 (B) were immunoprecipitated using anti-FLAG or anti-GFP antibodies, respectively, and blotted with anti-GFP for Pin1 and anti-FLAG for MLK3.
MLK3-Induced Phosphorylation of Pin1 Increases Its Catalytic Activity and Nuclear Translocation.
The biological impact of MLK3-mediated phosphorylation of the Pin1 S138 site is unknown. To determine whether this phosphorylation affected Pin1 PPIase activity, recombinant WT, S138A, and S138E mutant Pin1 proteins were expressed in Pin1-null (Pin1−/−) murine embryonic fibroblasts (MEFs) prior to a protease-coupled isomerase assay (19). Our results show that Pin1 S138E was about fourfold more active compared with WT, whereas the activity of the S138A mutant was diminished about twofold (Fig. 4A). The catalytic activity of Pin1 WT and S138A mutant proteins was also measured upon in vitro phosphorylation by purified MLK3. Again the activity of Pin1 WT was increased by MLK3, whereas that of the S138A mutant was about fourfold lower (Fig. 4B). These results clearly demonstrate that MLK3-mediated phosphorylation of S138 increases Pin1 isomerase activity.
Fig. 4.

Catalytic activity and nuclear translocation of Pin1 is increased upon phosphorylation by MLK3. (A) Activity of recombinant WT, S138A, and S138E Pin1 mutants expressed in Pin1−/− MEFs was measured by taking 2 μg of whole-cell extracts. pNA, p-nitroanilide. (B) Activity of in vitro phosphorylated WT or S138A Pin1 proteins was measured upon phosphorylation by purified MLK3. Data shown are mean ± SEM, (n = 4). (C and D) The nuclear (N) and cytoplasmic (C) distributions of recombinant WT or Pin1 mutants in HeLa cells were determined, either expressed alone (C) or coexpressed with MLK3 (D). The nuclear and cytoplasmic fractions were also blotted with GAPDH and lamin A/C as cytoplasmic and nuclear markers. (E) The localization of GFP-tagged WT, S138A, and S138E Pin1 was visualized in HeLa cells by confocal microscopy.
We next examined whether phosphorylation of Pin1 by MLK3 somehow regulates its nuclear translocation. GFP-tagged Pin1 WT, S138A, and S138E vectors were either expressed alone (Fig. 4C) or in combination with MLK3 (Fig. 4D) in HeLa cells. The nuclear and cytoplasmic fractions from these cells were prepared and blotted for GFP-Pin1. Interestingly, the Pin1 S138E mutant was highly enriched in the nuclear fraction in the absence (Fig. 4C and Fig. S3A) or presence (Fig. 4D and Fig. S3B) of MLK3. Pin1 WT or S138A proteins were normally enriched in the cytoplasm in the absence of MLK3 (Fig. 4C and Fig. S3A). However, upon MLK3 coexpression, Pin1 WT translocated to the nucleus, whereas the S138A mutant was still mostly in the cytoplasm (Fig. 4D and Fig. S3B). We also examined the localization of Pin1 phospho mutants in HeLa cells by confocal microscopy. As shown in Fig. 4E, the S138E mutant was primarily in the nuclear compartment, compared with the S138A mutant. Taken together, these results strongly suggest that phosphorylation of Pin1 by MLK3 promotes its translocation to the nucleus.
Pin1 Phosphorylation by MLK3 Promotes Cell-Cycle Progression.
Pin1 has been shown to control cell-cycle progression and is required for G2/M transition (16). To know whether Pin1 phosphorylated at S138 altered cell-cycle progression, HeLa cells transfected either with GFP-Pin1 WT, S138A, or S138E vectors were synchronized at G1/S by double-thymidine block and then released into the cell cycle by addition of serum for 5 h. As shown in Fig. 5A, in the GFP-positive (i.e., transfected) population, cells expressing the Pin1 S138A mutant had the fewest numbers of cells in G2/M phase (13.3%), compared with cells transfected either with S138E (30.1%) or wild type (23.4%). Notably, cells transfected with the phospho-mimetic Pin1 S138E mutant showed the highest numbers of cells undergoing G2/M transition. To confirm further, we also measured the activity of cyclin B1-associated kinase, Cdc2, which is known to be elevated during G2/M phase of the cell cycle (20). Cdc2 kinase activity was highest in Pin1 S138E-transfected cells (Fig. 5B). The activity of Cdc2 was also determined indirectly by estimating the phosphorylation of Tyr15 on Cdc2, whose dephosphorylation is necessary for Cdc2 activation (21). Cdc2 Tyr15 phosphorylation was significantly decreased in cells transfected with the Pin1 S138E mutant, compared with cells expressing S138A (Fig. S4). Collectively, these results establish that the MLK3-induced phosphorylation of Pin1 is physiologically important and might perturb normal cell-cycle progression under pathological conditions.
Fig. 5.

The cell-cycle function of Pin1 is regulated via MLK3-mediated phosphorylation. (A) HeLa cells were transfected with either empty GFP vector or the indicated GFP-Pin1 expression vectors. The cells were synchronized using double-thymidine block and released into the cell cycle with 5% (vol/vol) FBS. The cell-cycle progression of GFP-positive HeLa cells was determined after PI staining by FACS analysis. (B) HeLa cells were transfected with the indicated Pin1 expression vectors, and cyclin B1-associated Cdc2 kinase activity was determined from cyclin B1 immunoprecipitates, using histone H1 as substrate. The WCEs were blotted with the indicated antibodies for the total expression of endogenous Cdc2, MLK3, and cyclin B1 and recombinant GFP-Pin1 proteins.
Pin1 Phosphorylation by MLK3 Regulates Cyclin D1 and Centrosome Amplification.
Pin1 has been shown to positively regulate cyclin D1 stabilization (4) and expression (22). To determine the effect of Pin1 phosphorylation on cyclin D1 levels, NIH 3T3 cells were transfected either with vector control or wild type, S138A, or S138E Pin1 expression vectors followed by determination of cyclin D1 protein stability. Cyclin D1 protein was rapidly degraded in cells expressing vector, wild type, or S138A, but was stable in cells expressing Pin1 S138E (Fig. 6A and Fig. S5A). Consistent with its longer half-life, the promoter activity of cyclin D1 was significantly up-regulated by Pin1 S138E compared with the S138A mutant (Fig. 6B).
Fig. 6.

Cyclin D1 expression and centrosome duplication are regulated by phospho-mimetic Pin1. (A) Cyclin D1 protein stability was determined in the NIH 3T3 cell line, expressing indicated GFP-Pin1 expression vectors. The cells were treated with cyclohexamide and harvested at the times indicated. The total cell lysates were blotted with cyclin D1, β-actin, and GFP antibodies (for GFP-Pin1). (B) Cyclin D1 reporter activity was determined in HeLa cells, and the reporter was transfected with the indicated Pin1 plasmids along with cyclin D1 luciferase reporter vector. The activity of the reporter luciferase is expressed relative to vector-transfected cells. Data shown are mean ± SEM, (n = 3). (C) The centrosome duplication assay in NIH 3T3 cells, transfected with the indicated Pin1 or vector plasmids, was performed upon arresting them at G1/S phase. The centrosomes were stained with anti–γ-tubulin and analyzed by fluorescence microscopy.
Pin1 has also been reported to regulate centrosome amplification, which is necessary for proper cell division (23). We examined whether phosphorylation of Pin1 regulates centrosome amplification during G1/S transition. NIH 3T3 cells transfected with wild type and Pin1 mutants were arrested in G1/S phase, and centrosomes and nuclei were stained with γ-tubulin and DAPI, respectively, before counting. WT Pin1 overexpression caused centrosome amplification, which was partially blocked by Pin1 S138A but enhanced by the S138E mutant (Fig. 6C and Fig. S5B), suggesting that MLK3-induced phosphorylation of Pin1 promotes genomic instability via dysregulation of centrosome division.
Phosphorylated Pin1 Is Elevated in Breast Tumors and Localized in the Nucleus.
Role of phosphorylated Pin1 in the development of breast or other types of cancer is not known. To probe this important question, we blotted extracts from primary breast tumors and control normal breast tissues with our Pin1 pS138 antibody. Pin1 expression was up-regulated in almost all of the breast tumors examined (Fig. 7A and Fig. S6B); therefore, Pin1 total protein expression in tissue lysates was normalized between each pair (tumor and normal) and blotted with Pin1 pS138 antibody. Expression of Pin1 pS138 was significantly up-regulated in tumors, compared with matching normal breast tissues (Fig. 7A, normalized expression lanes). Interestingly, MLK3 protein was also up-regulated specifically in tumors (Fig. 7A and Fig. S6A), showing that both Pin1 pS138 and MLK3 proteins are up-regulated in breast tumors.
Fig. 7.

Phosphorylated Pin1 is elevated in breast tumors and localizes in the nucleus. (A) Normalized Pin1 expression from six pairs of breast tumors (T) and matching normal (N) breast tissue extracts that were blotted, either with Pin1 pSer138 or MLK3 or with Pin1 antibodies. (B and C) Commercial tissue microarrays containing normal and cancer tissues (B) or normal, benign, and cancer tissues (C) were stained with Pin1 pSer138 antibodies and counted. (D) Representative normal and cancerous breast tissue stained with Pin1 pSer138 showing nuclear localization (arrowheads).
To understand the physiological significance of nuclear Pin1 pS138 in breast tumorigenesis, we examined expression of Pin1 pS138 in two breast cancer tissue microarrays. The first array contained 50 paired breast carcinomas and adjacent normal breast tissues, and the second array contained 72 breast tissue samples at different stages of breast cancer progression (i.e., progression array). These arrays were stained with Pin1 pS138 antibody and the levels of Pin1 pS138 were scored. Overall, the data indicated that Pin1 pS138 was significantly higher (P ≤ 0.0001) in carcinomas in general (Fig. 7 B and C), and localized primarily in the nuclei as puncta (Fig. 7D). The differential expression of Pin1 pS138, in normal and benign tumor samples, was statistically significant (P ≤ 0.0001); however, differences between benign and cancer samples were not statistically significant (Fig. 7C). These results suggest that possibly Pin1 is phosphorylated by MLK3 on Ser138 during the early stages of breast cancer.
Discussion
Pin1 regulates large numbers of phospho proteins having diverse cellular functions, including in the cell cycle, cell transformation, and apoptosis. Therefore, tight regulation of Pin1’s catalytic activity is likely important to maintain normal homeostasis. Despite the importance of Pin1 in regulating various cellular pathways, the precise mechanism that regulates Pin1 catalytic activity is currently unknown. Here we demonstrate that an MLK family member, MLK3, increases the catalytic activity and downstream signaling of Pin1 via direct phosphorylation of the S138 residue in the PPIase domain. Interestingly, Pin1 pS138 was highly enriched in the nuclei of breast tumors, where many relevant Pin1 targets reside. Phosphorylation of Pin1 on S138 also promoted G2/M cell-cycle progression, cyclin D1 stability, and centrosome amplification.
It was reported that MLK3 resembles never in mitosis A (NIMA) protein within its noncatalytic domain and that MLK3 activity was increased during G2/M transition (15). Because both Pin1 and MLK3 can regulate the cell cycle, we therefore decided to examine any functional interaction between Pin1 and MLK3 in breast cancer cell lines. The interaction between the C-terminal domain of MLK3 and Pin1 was robust (Fig. 1B), and the PPIase domain of Pin1 interacted strongly with MLK3 whereas the WW domain failed to show any interaction (Fig. 1 C and D). These results were surprising, given the fact that most Pin1 substrates interact through the WW domain (2). Nevertheless, our observation is supported by a recent crystal structure of Pin1 showing two possible β1/α1 flexible domains, indicating that both the WW and PPIase domains can bind to its substrates (2). Our initial assumption was that Pin1 binding might regulate the kinase activity of MLK3, due to Pin1’s known role in altering the catalytic activity of several enzymes (1). Our repeated attempts with either overexpression or knockdown of Pin1 in mammalian cells failed to yield any alteration of MLK3 activity. To explore any potential impact of MLK3 on Pin1, we examined whether MLK3 phosphorylates Pin1. Indeed, kinase-active MLK3 specifically phosphorylated bacterially expressed Pin1 protein in a dose-dependent manner (Fig. 2A), on the Ser138 site in the Pin1 PPIase domain (Fig. 2 C and D), in vitro (Fig. 2B), and in vivo (Fig. 2E). Pin1 has been shown to be posttranslationally modified via phosphorylation by PKA (2), PLK1 (2), and PKMζ (24). However, no posttranslational modification of Pin1 has been reported to increase its catalytic activity. Our result demonstrates that MLK3-induced phosphorylation of the Pin1 S138 site can induce Pin1’s catalytic activity (Fig. 4 A and B). We also observed that Pin1 protein was significantly enriched in nuclear fractions in the presence of kinase-active MLK3 (Fig. 4D) and that the Pin1 S138E mutant was primarily localized in the nuclei (Fig. 4 C–E). This S138E mutant was further enriched in the nucleus upon MLK3 overexpression (Fig. 4D), suggesting that perhaps Pin1 contains an additional MLK3 phosphorylation site(s) that is necessary for complete nuclear localization.
Pin1 has also been shown to control cell-cycle progression and is required for G2/M transition (16). It was thus conceivable that any change in Pin1’s catalytic activity might impact on the cell cycle. Indeed, our results suggest that MLK3-induced phosphorylation of the Pin1 S138 site promotes G2/M transition (Fig. 5A). Pin1 targets various cell-cycle regulatory proteins, including cyclin D1 (22). Interestingly, Pin1 and cyclin D1 knockout animals show similar phenotypic characteristics, suggesting a functional cross-talk between these two proteins (4). Pin1 levels have been reported to be correlated with cyclin D1 mRNA and protein levels in human cancer tissues (3, 22). Moreover, Pin1 can activate the cyclin D1 promoter via binding to phosphorylated c-Jun (22) and β-catenin (25). Because MLK3 specifically activates JNK (10), it is conceivable that MLK3 might induce cyclin D1 expression by two means: (i) via activating the JNK/c-Jun axis and (ii) by activating the Pin1/c-Jun axis, ultimately increasing cyclin D1 transcription. Indeed, we observed that Pin1 S138E increased the promoter activity (Fig. 6B) and protein stability (Fig. 6A and Fig. S5A) of cyclin D1. Proper duplication of centrosomes is an essential step for appropriate cell division (26), and centrosome multiplication is seen in many cancer tissues (26). Pin1 has been linked to centrosome duplication, where overexpression of Pin1 caused multiplication of centrosomes and genome instability (23). Our data showed that phosphorylation of Pin1 by MLK3 promotes centrosome multiplication, which was partially blocked by Pin1 S138A (Fig. 6C and Fig. S5B). Collectively, these results point toward a strong possibility that MLK3-induced phosphorylation of Pin1 could promote oncogenesis. This notion was supported by the fact that Pin1 pS138 levels were increased in the nuclei of breast cancer tissues (Fig. 7 B–D). In breast cancer tissue microarrays, there was a significant difference in Pin1 pS138 expression between normal and cancer tissue, although there was no statistically significant difference between benign and cancer samples. These results suggest that MLK3-induced phosphorylation of Pin1 could be an early event in oncogenesis, a notion that was also suggested previously for Pin1 (3).
Based on our current data and published results, we propose a model for MLK3-induced Pin1 phosphorylation and its impact on cellular homeostasis (Fig. 8). Upon activation of MLK3 by known agonists ceramide and TNFα (27) or other unidentified agonists, MLK3 could phosphorylate Pin1 on the S138 site (Fig. S7) and promote its nuclear translocation. MLK3 is reported to specifically activate JNK in response to its agonists (27) and, thus activated, JNK could then phosphorylate its downstream targets, c-Jun and c-Fos, which are initially inactive but which upon isomerization by phospho-Pin1 in the nucleus might attain the active conformation. These activated transcription factors could act on the cyclin D1 promoter to induce its transcription. The cyclin D1 protein initially remains unstable until phospho-Pin1 in the nucleus isomerizes cyclin D1 to a stable conformation. Stabilized cyclin D1 now up-regulates Cdk activity, which ultimately promotes cell-cycle progression (Fig. 8).
Fig. 8.

Proposed model for the regulation of Pin1 by MLK3.
In conclusion, our data provide an insight into the role of MLK3 in Pin1 regulation via direct phosphorylation that regulates Pin1 localization and activation, leading to G2/M cell-cycle transition. Thus, it is tempting to speculate that therapeutics that target MLK3 or Pin1 could prove beneficial for a subset of cancers where the MLK3-Pin1 pathway is dysregulated.
Materials and Methods
Cell Lines and Plasmids.
Breast cancer, HeLa, and Pin1 MEF cells were cultured as described previously (13, 28). Pin1 constructs were made in pGEX and pEGFP vectors, and the deletion mutants of MLK3 were constructed in pEBG vector (SI Materials and Methods).
Recombinant Pin1 Proteins, in Vitro Phosphorylation, and Peptide Mapping.
Pin1 proteins were made in bacteria, and in vitro phosphorylation of Pin1 proteins was carried out by purified recombinant MLK3 from baculovirus, as described (29). Phosphorylated Pin1 proteins were digested with trypsin, and peptides were analyzed by 2D electrophoresis as described (30) (SI Materials and Methods).
Mass Spectroscopy Analysis and Generation of Pin1 pS138 Antibody.
The bacterially expressed wild-type Pin1 was phosphorylated with purified MLK3 (29). Phosphorylated and nonphosphorylated Pin1 were analyzed by MS for phosphorylation-site identification. Phosphorylated Pin1 S138 peptides were used to generate Pin1 pS138 antibody in rabbit (SI Materials and Methods).
Isomerase Activity Determination.
Pin1 isomerase activity was determined as described (5), with slight modifications (SI Materials and Methods).
Supplementary Material
Acknowledgments
We acknowledge financial support from Veterans Affairs Merit awards and National Institutes of Health (NIH) Grant R01 GM55835 (to A.R.), NIH Grant R01 NS074443 (to A.K.), and Veterans Affairs Merit and Department of Defense awards (to B.R.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1200804109/-/DCSupplemental.
References
- 1.Lu KP, Zhou XZ. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Uchida C, Shin RW, Shimazaki K, Uchida T. Prolyl isomerase, Pin1: New findings of post-translational modifications and physiological substrates in cancer, asthma and Alzheimer’s disease. Cell Mol Life Sci. 2008;65:359–375. doi: 10.1007/s00018-007-7270-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liou YC, et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen ZJ, Esnault S, Malter JS. The peptidyl-prolyl isomerase Pin1 regulates the stability of granulocyte-macrophage colony-stimulating factor mRNA in activated eosinophils. Nat Immunol. 2005;6:1280–1287. doi: 10.1038/ni1266. [DOI] [PubMed] [Google Scholar]
- 6.Pastorino L, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-β production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 7.Ryo A, et al. Prolyl-isomerase Pin1 accumulates in Lewy bodies of Parkinson disease and facilitates formation of α-synuclein inclusions. J Biol Chem. 2006;281:4117–4125. doi: 10.1074/jbc.M507026200. [DOI] [PubMed] [Google Scholar]
- 8.Bao L, et al. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lufei C, Cao X. Nuclear import of Pin1 is mediated by a novel sequence in the PPIase domain. FEBS Lett. 2009;583:271–276. doi: 10.1016/j.febslet.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 10.Rana A, et al. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem. 1996;271:19025–19028. doi: 10.1074/jbc.271.32.19025. [DOI] [PubMed] [Google Scholar]
- 11.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 12.Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: A new therapeutic target in Parkinson’s disease. Mov Disord. 2005;20:653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 13.Rangasamy V, et al. Estrogen suppresses MLK3-mediated apoptosis sensitivity in ER+ breast cancer cells. Cancer Res. 2010;70:1731–1740. doi: 10.1158/0008-5472.CAN-09-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol. 2010;24:598–607. doi: 10.1210/me.2009-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swenson KI, Winkler KE, Means AR. A new identity for MLK3 as an NIMA-related, cell cycle-regulated kinase that is localized near centrosomes and influences microtubule organization. Mol Biol Cell. 2003;14(1):156–172. doi: 10.1091/mbc.E02-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nat Rev Cancer. 2007;7:381–388. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 17.Pawson T, Raina M, Nash P. Interaction domains: From simple binding events to complex cellular behavior. FEBS Lett. 2002;513(1):2–10. doi: 10.1016/s0014-5793(01)03292-6. [DOI] [PubMed] [Google Scholar]
- 18.Pawson T, Kofler M. Kinome signaling through regulated protein-protein interactions in normal and cancer cells. Curr Opin Cell Biol. 2009;21(2):147–153. doi: 10.1016/j.ceb.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Yaffe MB, et al. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 20.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat Rev Mol Cell Biol. 2008;9:910–916. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 21.Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67(1):197–211. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- 22.Wulf GM, et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suizu F, Ryo A, Wulf G, Lim J, Lu KP. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol Cell Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westmark PR, et al. Pin1 and PKMζ sequentially control dendritic protein synthesis. Sci Signal. 2010;3:ra18. doi: 10.1126/scisignal.2000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of β-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 26.Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005;230(1):6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 27.Sathyanarayana P, et al. Activation of the Drosophila MLK by ceramide reveals TNF-α and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527–1533. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- 28.Barthwal MK, et al. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278:3897–3902. doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- 29.Mishra R, et al. Glycogen synthase kinase-3β induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem. 2007;282:30393–30405. doi: 10.1074/jbc.M705895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balan V, et al. Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol Biol Cell. 2006;17:1141–1153. doi: 10.1091/mbc.E04-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.