

Abstract
Recent experimental and theoretical studies on the dynamics of the reactions of methane with F and Cl atoms have modified our understanding of mode-selective chemical reactivity. The O + methane reaction is also an important candidate to extend our knowledge on the rules of reactivity. Here, we report a unique full-dimensional ab initio potential energy surface for the O(3P) + methane reaction, which opens the door for accurate dynamics calculations using this surface. Quasiclassical trajectory calculations of the angular and vibrational distributions for the ground state and CH stretching excited O + CHD3(v1 = 0,1) → OH + CD3 reactions are in excellent agreement with the experiment. Our theory confirms what was proposed experimentally: The mechanistic origin of the vibrational enhancement is that the CH-stretching excitation enlarges the reactive cone of acceptance.
Keywords: mode-specific dynamics, reactive scattering computations, state-to-state dynamics, permutationally invariant potential energy surfaces, benchmark ab initio data
The simplest chemical reaction is the reactive collision of an atom with a diatomic molecule. This class of reactions was extensively studied during the past couple of decades (1). Recently, attention has turned toward more complex systems, such as the abstraction reactions of methane (CH4, CHD3, etc.) with different atoms (H, F, Cl, and O), as well as the substitution reactions like X- + CH3Y (X, Y = F, Cl, OH, etc.) (2–18). The former set of reactions can utilize the same amount of energy with different efficiency depending on how the energy is initially distributed and this initial condition can have dramatic effects on the rate of reaction, branching ratio, etc. (11, 12) Many experimental and theoretical studies of atom-plus-diatom reactions showed that the early barrier reactions can be efficiently accelerated by increasing the relative translational energy of the reactants, whereas the excitation of the diatomic vibration activates the late-barrier reactions more efficiently (19). However, recent experiments on the early barrier F + CHD3 (3) and late-barrier Cl + CHD3 (11) reactions uncovered departures from the above rules. Crossed-beam experiments on F + CHD3 showed that vibrational energy can control early barrier reactions in an unexpected way, i.e., CH-stretching excitation enhanced the D atom abstraction channel (3). On the other hand, translational energy can be more efficient than vibrational energy to activate the late-barrier Cl + CHD3 reaction (11). Our recent theoretical work showed the importance of the weak van der Waals (vdW) forces in the entrance channel of these reactions (5, 12). These attractive intermolecular vdW forces are considered as secondary interactions in chemistry, but our studies showed that they play a major role in orienting the reactants in a way that results in an unexpected outcome of the dynamics. One of the keys of these calculations is the development of an accurate full-dimensional potential energy surface (PES), which governs the motion of the nuclei. In 2009 and 2011 we reported high-quality PESs for the F and Cl + methane reactions, respectively (4, 12). The dynamics computations using these PESs reproduced and explained many experimental features for these two fundamental polyatomic reactions.
The reaction of methane with the electronic ground state oxygen atom, O(3P), has also been studied experimentally (20, 21); however, a full-dimensional ab initio PES has not been available for this system. Instead, previous theoretical work used direct-dynamics or semiempirical PESs (8, 13–16, 22). In the present paper we describe an ab initio PES for the O(3P) + methane reaction, following our previous work on the F and Cl + methane reactions (4, 12). Two features of the PESs ensure their high quality. First, the ab initio energy points are computed by an efficient composite electronic structure method, which outperforms the orthodox ab initio methodology. Second, these accurate energies are represented by a full-dimensional mathematical function, which is invariant under the permutation of identical atoms (23). Having an accurate PES at hand, we focus on the dynamics of the ground state and CH-stretch excited O(3P) + CHD3(v1 = 0,1) reactions, which were recently studied experimentally by Wang and Liu (20). The experiment found substantial vibrational enhancement upon CH-stretching excitation (20). Based on the measured angular distributions the authors suggested that the CH-stretching excitation enlarges the reactive cone of acceptance, thereby promoting the OH channel (20). In the present study we investigate this suggestion theoretically after we describe the uniquely developed PES and its high-level ab initio characterization.
Results and Discussions
Full-Dimensional ab Initio Potential Energy Surface.
We have developed permutationally invariant PESs by fitting more than 10,000 electronic energies (4, 12, 23) For the F and Cl + CH4 reactions we used composite ab initio methods, which combine different methods and bases, thereby improving the accuracy/computational-cost ratio (4, 12). These composite methods are not fully black-box procedures; therefore, we have carefully tested the method applied to the O(3P) + CH4 reaction. As Fig. 1 shows, second-order Møller–Plesset perturbation theory (MP2) yields a more than 800 cm-1 rms error relative to accurate reference results (see Fig. 1), and this error cannot be improved by increasing the size of the basis set. The often-used coupled-cluster singles and doubles with perturbative triples [CCSD(T)] method with the correlation-consistent double zeta basis (aug-cc-pVDZ) also performs very poorly (about 1,200 cm-1 rms error). The first level that provides chemical accuracy, considered as 1 kcal/mol (350 cm-1), is CCSD(T)/aug-cc-pVTZ (rms error of 282 cm-1). The use of the large aug-cc-pVQZ basis further decreases the rms to 116 cm-1; however, these calculations cannot be employed to compute more than 10,000 energies with affordable computational time. Therefore, we used a composite method, which calculates the energies as ECCSD(T)/aug-cc-pVDZ + EMP2/aug-cc-pVQZ - EMP2/aug-cc-pVDZ, providing an rms error of only 133 cm-1, i.e., CCSD(T)/aug-cc-pVQZ quality results without performing these very expensive computations. We have used this efficient composite method to compute 17,212 points (energies go up to 50,000 cm-1 above the global minimum) and fit using 3,262 terms to get a full-dimensional analytical PES for the O(3P) + CH4 reaction.
Fig. 1.

Accuracy of the different ab initio methods and bases based on computations at 15 configurations along the reaction coordinate of the O(3P) + CH4 reaction. The rms errors are relative to high-quality reference results obtained at the all-electron CCSD(T)/aug-cc-pCVQZ level of theory. The composite energy is defined as ECCSD(T)/aug-cc-pVDZ + EMP2/aug-cc-pVQZ - EMP2/aug-cc-pVDZ.
Ab Initio Characterization and the Accuracy of the PES.
In 2011 Zhang and Liu (21) wrote, “the saddle-point geometry is somewhat uncertain. The predictions of the relative order of the lengths of the breaking C-H bond and the forming O-H bond obtained at various ab initio levels are not consistent.” We have determined the structure of the abstraction saddle point and that of the complex in the exit channel at the all-electron CCSD(T)/aug-cc-pCVXZ [X = D,T,Q] levels of theory. These results, shown in Fig. 2, are the best estimates to date and serve as benchmark values. At the saddle point the C-Hb distance (see Fig. 2) is 1.288 Å, stretched by 0.201 Å relative to the CH bond length in CH4. This distortion is roughly midway between the C-Hb displacements of 0.023 and 0.309 Å at the abstraction saddle points of the F and Cl + CH4 reactions, respectively (4, 24). On the basis of these distances, the location of the O + CH4 barrier is between saddle-point positions of the early-barrier F + CH4 and the late-barrier Cl + CH4 reactions, thus O + CH4 can be called as central-barrier reaction.
Fig. 2.

Structural parameters for the saddle point (Left) and the product-channel complex (Right) of the O(3P) + CH4 reaction obtained at the all-electron CCSD(T)/aug-cc-pCVXZ [X = D,T,Q] levels of theory. aCVQZ denotes the benchmark results and the PES values are also shown for comparison. The distances are given in angstrom and the angles are in degrees. The saddle point is slightly bent (Cs symmetry and 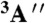 electronic state) with C-Hb-O angle of 179.6, 179.2, and 179.3 for X = D,T, and Q, respectively. The product-channel complex has C3v symmetry (E electronic state). The equilibrium {aCVDZ,aCVTZ,aCVQZ,PES} bond lengths in CH4, CH3, and OH are {1.101,1.088,1.087,1.089}, {1.092,1.078,1.077,1.079}, and {0.979,0.972,0.970,0.973}, respectively.
electronic state) with C-Hb-O angle of 179.6, 179.2, and 179.3 for X = D,T, and Q, respectively. The product-channel complex has C3v symmetry (E electronic state). The equilibrium {aCVDZ,aCVTZ,aCVQZ,PES} bond lengths in CH4, CH3, and OH are {1.101,1.088,1.087,1.089}, {1.092,1.078,1.077,1.079}, and {0.979,0.972,0.970,0.973}, respectively.
We have computed the best technically feasible barrier height, reaction energy, and dissociation energy of the (CH3---HO) complex using the so-called focal-point analysis (FPA) approach (25, 26). The FPA study systematically increases the accuracy of the ab initio methods, thereby providing better and better description of the correlated motion of the electrons. The CCSD(T) method is called the gold standard of electronic structure theory, but we even go beyond this level by performing CCSDT and CCSDT(Q) computations. The FPA also considers extrapolation to the complete basis set limit using large aug-cc-pCVXZ [X = 5 and 6] bases. Furthermore, we have correlated all the electrons (core as well as valence); thus, these benchmark computations do not use the usual frozen-core approximation.
A schematic of the PES, showing the energetics at different levels of theory, is given in Fig. 3. The benchmark classical barrier height is 4,925 cm-1, relative to O(3P) + CH4(eq), about twice as large as that of the 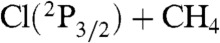 reaction (2,670 cm-1) (12). The barrier height on the O + CH4 PES is 5,116 cm-1, similar to the CCSD(T)/aug-cc-pVTZ result of 5,103 cm-1 and much better than the CCSD(T)/aug-cc-pVDZ value of 5,403 cm-1. The reactions of CH4 with F, Cl, and O have complexes in the exit channels, stabilized by dipole—induced dipole interactions, with benchmark dissociation energies (De) of 1,070 (4), 820 (24), and 771 cm-1, respectively. The O + CH4 PES exit channel De is 772 cm-1, fortuitously in near perfect agreement with the benchmark value. The benchmark reaction endoergicity is 1,861 cm-1, very similar to that of Cl + CH4. The CCSD(T) method with aug-cc-pVDZ and aug-cc-pVTZ bases seriously overestimates this key energy by 913 and 457 cm-1, respectively, whereas the endoergicity on the PES is 2,035 cm-1, only a deviation of 174 cm-1 (0.5 kcal/mol) from the highly accurate benchmark data (see Fig. 3). Finally, it is important to note that there is a shallow vdW well in the entrance channel with depths, in reciprocal centimeters, of (140, 90, 65) and (230, 185, 170) for the CH---O and HC---O collinear bond arrangements, respectively. The three numbers in parentheses show the all-electron CCSD(T) results with larger and larger basis sets (aug-cc-pCVDZ, aug-cc-pCVTZ, aug-cc-pCVQZ). Because O is less polarizable than Cl, this well is shallower than that of the Cl + CH4 reaction, where the benchmark values of the depths are 100 and 300 cm-1 (without spin-orbit correction) for CH---Cl and HC---Cl orientations, respectively (12, 24). The corresponding O + CH4 benchmark values are only 65 and 170 cm-1, as shown above. The O + CH4 PES provides depths of 103 and 150 cm-1, respectively, which is reasonably good considering the basis set dependence of these values discussed above. Furthermore, we expect a minor effect of this vdW well here, because the barrier height of O + CH4 is about twice as large as that of the Cl + CH4 reaction.
reaction (2,670 cm-1) (12). The barrier height on the O + CH4 PES is 5,116 cm-1, similar to the CCSD(T)/aug-cc-pVTZ result of 5,103 cm-1 and much better than the CCSD(T)/aug-cc-pVDZ value of 5,403 cm-1. The reactions of CH4 with F, Cl, and O have complexes in the exit channels, stabilized by dipole—induced dipole interactions, with benchmark dissociation energies (De) of 1,070 (4), 820 (24), and 771 cm-1, respectively. The O + CH4 PES exit channel De is 772 cm-1, fortuitously in near perfect agreement with the benchmark value. The benchmark reaction endoergicity is 1,861 cm-1, very similar to that of Cl + CH4. The CCSD(T) method with aug-cc-pVDZ and aug-cc-pVTZ bases seriously overestimates this key energy by 913 and 457 cm-1, respectively, whereas the endoergicity on the PES is 2,035 cm-1, only a deviation of 174 cm-1 (0.5 kcal/mol) from the highly accurate benchmark data (see Fig. 3). Finally, it is important to note that there is a shallow vdW well in the entrance channel with depths, in reciprocal centimeters, of (140, 90, 65) and (230, 185, 170) for the CH---O and HC---O collinear bond arrangements, respectively. The three numbers in parentheses show the all-electron CCSD(T) results with larger and larger basis sets (aug-cc-pCVDZ, aug-cc-pCVTZ, aug-cc-pCVQZ). Because O is less polarizable than Cl, this well is shallower than that of the Cl + CH4 reaction, where the benchmark values of the depths are 100 and 300 cm-1 (without spin-orbit correction) for CH---Cl and HC---Cl orientations, respectively (12, 24). The corresponding O + CH4 benchmark values are only 65 and 170 cm-1, as shown above. The O + CH4 PES provides depths of 103 and 150 cm-1, respectively, which is reasonably good considering the basis set dependence of these values discussed above. Furthermore, we expect a minor effect of this vdW well here, because the barrier height of O + CH4 is about twice as large as that of the Cl + CH4 reaction.
Fig. 3.

Schematic of the potential energy surface of the O(3P) + CH4 → OH + CH3 reaction showing the benchmark energetics and the PES values relative to O(3P) + CH4(eq). The accurate relative energies (red numbers) are all-electron CCSDT(Q)/complete-basis-set quality results obtained by the focal-point analysis approach as described in the text.
Dynamics of the O(3P) + CHD3 Reaction.
We performed quasiclassical trajectory (QCT) calculations for the ground state and CH-stretch excited O + CHD3(v1 = 0,1) → OH + CD3 reactions using the new PES. More than 4 million trajectories were analyzed with and without zero-point energy (ZPE) constraint (24) applied to the products. All the qualitative features discussed below were found insensitive to the ZPE treatment (for more details see figure legends). The cross-sections as a function of collision energy are given in Fig. 4. Significant enhancement of reactivity is seen upon CH-stretching excitation, in agreement with the experiment (20). Based on the measured product state-specific angular distributions, Wang and Liu recently suggested that the mechanistic origin of the vibrational enhancement is that the CH-stretching excitation enlarges the reactive cone of acceptance, thereby promoting the OH channel (20). The accurate computation of these state-to-state angular distributions for a six-atom system is extremely challenging to theory. In 2011 Zhang and coworkers (27) carried out computations for the HD + OH reaction on an accurate PES that yielded differential cross-sections (DCS) in unprecedented agreement with experiment for a four-atom reaction. Here, we report DCSs for the O + CHD3(v1 = 0,1) → OH(v = 0,1) + CD3(v = 0) reactions at collision energy of 4,000 cm-1 as shown in Fig. 5. Note that the harmonic ZPE of CHD3 is 7,877 cm-1, whereas the ZPE at the D3C-H-O saddle point is only 6,481 cm-1. (These values correspond to the PES.) Thus, the ground state adiabatic barrier height is about 5,100 - 1,400 = 3,700 cm-1. Because the collision energy of 4,000 cm-1 is above the adiabatic barrier, we can expect that QCT is reliable at this region. As also shown in Fig. 5, the agreement between theory and experiment (20) is very good for this six-atom reaction. One can clearly observe the significant qualitative difference between the DCSs of the ground state and CH-stretch excited reactions. For the O + CHD3(v = 0) reaction the DCS shows the dominance of backward scattering, whereas in the case of the O + CHD3(v1 = 1) reaction the products are scattered more sideways. This finding indicates that the ground state reaction occurs at small impact parameters with a direct rebound mechanism, whereas the CH-stretching excitation opens the reaction at larger impact parameters, thereby enlarging the reactive cone of acceptance as suggested by Wang and Liu (20). Theory can further investigate this statement by examining the opacity function [P(b)], i.e., the impact parameter (b) dependence of the reaction probabilities. P(b) cannot be directly measured, but it can be straightforwardly computed. Fig. 6 shows the opacity functions for the O + CHD3(v1 = 0,1) reactions. Indeed, CH-stretch excitation has a substantial effect on the shape of P(b). It is best seen when the P(b) of the ground state reaction is scaled to have the same probability at b = 0 as the CH-stretch excited reaction has (see Fig. 6). The CH-stretch excitation clearly enhances the reactivity at larger impact parameters. The maximum impact parameter (bmax) is only 3 bohr for O + CHD3(v = 0), whereas bmax is about 5 bohr for O + CHD3(v1 = 1). In the case of the Cl + CHD3 reaction the picture is quite different as also shown in Fig. 6. The shift of P(b) toward larger impact parameters upon CH-stretch excitation is not significant at collision energy of 4,400 cm-1. (Note that at a lower collision energy of 2,450 cm-1, the CH-stretching effect is more substantial, because this collision energy is below the classical barrier height of the Cl + CHD3 reaction.) Furthermore, bmax is much larger for Cl + CHD3 than that for O + CHD3, i.e., about 5.5 and 6.5 bohr for Cl + CHD3(v = 0) and Cl + CHD3(v1 = 1), respectively (see Fig. 6). Note that the above reported bmax values are almost independent on the collision energy. These results show that at the same collision energy the mechanistic origin of vibrational enhancement in the O and Cl + CHD3 reactions is different, for O + CHD3 the extension of the range of the reactive impact parameters plays a major role.
Fig. 4.

Cross-sections as a function of collision energy for the ground state and CH-stretching excited O(3P) + CHD3(v1 = 0,1) → OH + CD3 reactions. The excitation functions obtained with and without ZPE constraint are very similar; here we show the ZPE-constrained results using the same technique as in ref. 24.
Fig. 5.

Computed and measured differential cross-sections of the ground state and CH-stretching excited O(3P) + CHD3 reactions at collision energy of 4,000 cm-1. In the QCT analysis the correlated product-state assignment was done using histogram binning as described in ref. 6. Gaussian binning (6) gave similar results with larger statistical uncertainty. The experimental data are taken from ref. 20.
Fig. 6.

Reaction probabilities as a function of impact parameter for the ground state and CH-stretching excited O(3P) + CHD3 and 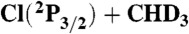 reactions at collision energies of 4,000 cm-1 and 4,400 cm-1, respectively. Although the ZPE-constraint decreases the reaction probabilities especially for the ground state reactions, the shapes of the opacity functions obtained with and without ZPE constraint are very similar; here we show the ZPE-constrained results using the same technique as in ref. 24.
reactions at collision energies of 4,000 cm-1 and 4,400 cm-1, respectively. Although the ZPE-constraint decreases the reaction probabilities especially for the ground state reactions, the shapes of the opacity functions obtained with and without ZPE constraint are very similar; here we show the ZPE-constrained results using the same technique as in ref. 24.
We computed the OH(v = 1)/OH(v = 0) vibrational branching ratios from the O + CHD3(v1 = 0,1) reactions. The ground state reaction produces exclusively OH(v = 0) products, in agreement with the experiment (20). As noted by Wang and Liu, this finding is expected, because the formation of OH(v = 1) requires at least 4,300 cm-1 of energy, which is at the top of the experimental collision energy range (20). Our calculations extend the collision energy range up to 7,000 cm-1, but the O + CHD3(v = 0) reaction produces OH(v = 0) molecules, indicating that the ground state reaction is strongly vibrationally adiabatic. For O + CHD3(v1 = 1) experiment found that the population of OH(v = 1), correlated to CD3(v = 0), is about 90% (20). Wang and Liu wrote (20), “the observed OH vibrational distribution is highly inverted, in sharp contrast to the theoretical prediction” (8). The present calculations now support the experiment; we find the percentage of OH(v = 1) is around 80–100% in a collision energy range of 2,100 to 4,200 cm-1 [this range is similar to that of the experiment (20)]. It is important to note that we get this inverted OH vibrational distribution if we follow the experiment (20) and correlate the results to CD3(v = 0). The QCT calculations show that CD3(v = 0) is the dominant product state; however, significant populations of the bending-excited CD3 products are also seen. Summing over all the states of CD3, the fraction of OH(v = 1) drops to 25–60%. We can conclude that the O + CHD3(v1 = 1) reaction is vibrationally more adiabatic than the Cl + CHD3(v1 = 1) reaction, as the OH(v = 1) population is 80–100% or 25–60%, whereas the fraction of HCl(v = 1) is only 30–50% or around 10% (12) if the results are correlated to CD3(v = 0) or CD3 (all states), respectively.
Conclusions
We reported a highly accurate full-dimensional ab initio potential energy surface for the O(3P) + methane reaction and unique benchmark structures and energies for a number of important properties of this reaction. The high accuracy was achieved by using a large basis set and high-level electron correlation method to compute about 17,000 energy-points. Our fitting method based on permutationally invariant polynomials allows representing the accurate energy points by an analytical function making the evaluation of the potential energies computationally highly efficient. The dynamics computations on the new PES provided results in excellent agreement with the experiment. We showed that the impact parameter dependence of the reactions of O and Cl atoms with CHD3 is quite different; O + CHD3 reacts at a much narrower impact parameter range, and at the same collision energy the CH-stretching excitation extends the reactive impact parameter range of O + CHD3 more efficiently.
The present work can motivate and help future experimental and theoretical investigations of the O + methane reaction. Theory may study additional aspects of the dynamics using quasiclassical and/or quantum computations on the present PES. Quantum computations may reveal the effects of tunneling and reactive resonances. One may further improve the PES to accurately describe the high-energy region, where many product channels open, which can be important for fast O atom collisions at low Earth orbit conditions (15). Finally, we note that the present study used the assumption that the dynamics can be well-described on a single triplet PES. The early work on the prototypical O(3P) + H2 reaction (28) and the nice agreement between theory and experiment in the present work support this statement. Of course, a fruitful direction of future research could be the development of a global PES for the singlet electronic state allowing surface hopping study on multiple coupled PESs.
Materials and Methods
The PES was represented by a polynomial expansion in variables yij = exp(-rij/a), where rij are the interatomic distances and a = 2 bohr, using a polynomial basis that is invariant under permutations of like atoms. Including all terms up to total degree six, 3,262 coefficients were determined by a weighted linear least-squares fit of 17,212 energy points, obtained by the composite approach defined in Fig. 1. The rms fitting errors are 87, 211, and 560 cm-1 for energy intervals (0, 11,000), (11,000, 22,000), and (22,000,50,000) cm-1, respectively.
The benchmark ab initio computations were performed by Molpro up to coupled-cluster (CC) singles and doubles with perturbative triples [CCSD(T)]. The Mrcc program, interfaced to Cfour, was used for the CCSDT and CCSDT(Q) computations. The CCSD(T) computations utilized restricted open-shell Hartree–Fock orbitals and an unrestricted CC formalism, whereas unrestricted Hartree–Fock references were used for the CCSDT and CCSDT(Q) methods. The present study followed an FPA (25, 26) procedure described in refs. 4 and 24.
QCT calculations of the O(3P) + CHD3(v1 = 0,1) → OH + CD3 reactions were performed using the full-dimensional ab initio PES of the present study. We employed standard normal mode sampling (29) to prepare the quasiclassical vibrational ground state (v = 0) and CH-stretch excited state (v1 = 1) of the reactant. The impact parameter was scanned from 0 to 5 bohr with a step size of 0.5 bohr. At each b, 25,000 trajectories were computed; thus, the total number of trajectories was 275,000 for each collision energy. (At collision energy of 4000 cm-1, where detailed experimental DCSs are available, the impact parameter was scanned by a smaller step of 0.25 bohr; thus, the total number of trajectories was 525,000.) We have run QCTs at several collision energies in the rage of 2,100–7,000 cm-1. For other computational details see ref. 24.
Acknowledgments.
The authors thank Dr. Kopin Liu for sending the experimental data shown in Fig. 5. The work was funded by the National Science Foundation (CHE-0625237) (to G.C.); the Scientific Research Fund of Hungary (OTKA, NK83583) (to G.C.); and the European Union and the European Social Fund (TÁMOP-4.2.1/B-09/1/KMR-2010-0003) (to G.C.); and the Department of Energy (DE-FG02-97ER14782) (to J.M.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. H.G. is a guest editor invited by the Editorial Board.
References
- 1.Clary DC. Quantum dynamics of chemical reactions. Science. 2008;321:789–791. doi: 10.1126/science.1157718. [DOI] [PubMed] [Google Scholar]
- 2.Hu W, et al. H + CD4 abstraction reaction dynamics: Product energy partitioning. J Phys Chem A. 2006;110:3017–3027. doi: 10.1021/jp055017o. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W, Kawamata H, Liu K. CH stretching excitation in the early barrier F + CHD3 reaction inhibits CH bond cleavage. Science. 2009;325:303–306. doi: 10.1126/science.1175018. [DOI] [PubMed] [Google Scholar]
- 4.Czakó G, Shepler BC, Braams BJ, Bowman JM. Accurate ab initio potential energy surface, dynamics, and thermochemistry of the F + CH4 → HF + CH3 reaction. J Chem Phys. 2009;130:084301. doi: 10.1063/1.3068528. [DOI] [PubMed] [Google Scholar]
- 5.Czakó G, Bowman JM. CH stretching excitation steers the F atom to the CD bond in the F + CHD3 reaction. J Am Chem Soc. 2009;131:17534–17535. doi: 10.1021/ja906886z. [DOI] [PubMed] [Google Scholar]
- 6.Czakó G, Bowman JM. Quasiclassical trajectory calculations of correlated product distributions for the F + CHD3(v1 = 0,1) reactions using an ab initio potential energy surface. J Chem Phys. 2009;131:244302. doi: 10.1063/1.3276633. [DOI] [PubMed] [Google Scholar]
- 7.Palma J, Clary DC. A quantum model Hamiltonian to treat reactions of the type X + YCZ3 → XY + CZ3: Application to O(3P) + CH4 → OH + CH3. J Chem Phys. 2000;112:1859–1867. [Google Scholar]
- 8.Yu H-G, Nyman G. Quantum dynamics of the O(3P) + CH4 → OH + CH3 reaction: An application of the rotating bond umbrella model and spectral transform subspace iteration. J Chem Phys. 2000;112:238–247. [Google Scholar]
- 9.Murray C, Orr-Ewing EJ. The dynamics of chlorine-atom reactions with polyatomic organic molecules. Int Rev Phys Chem. 2004;23:435–482. [Google Scholar]
- 10.Yoon S, Holiday RJ, Crim FF. Vibrationally controlled chemistry: Mode- and bond-selected reaction of CH3D with Cl. J Phys Chem B. 2005;109:8388–8392. doi: 10.1021/jp0463565. [DOI] [PubMed] [Google Scholar]
- 11.Yan S, Wu Y-T, Zhang B, Yue X-F, Liu K. Do vibrational excitations of CHD3 preferentially promote reactivity toward the chlorine atom? Science. 2007;316:1723–1726. doi: 10.1126/science.1142313. [DOI] [PubMed] [Google Scholar]
- 12.Czakó G, Bowman JM. Dynamics of the reaction of methane with chlorine atom on an accurate potential energy surface. Science. 2011;334:343–346. doi: 10.1126/science.1208514. [DOI] [PubMed] [Google Scholar]
- 13.Corchado JC, Espinosa-García J, Roberto-Neto O, Chuang Y-Y, Truhlar DG. Dual-level direct dynamics calculations of the reaction rates for a Jahn–Teller reaction: Hydrogen abstraction from CH4 or CD4 by O(3P) J Phys Chem A. 1998;102:4899–4910. [Google Scholar]
- 14.Espinosa-García J, García-Bernáldez JC. Analytical potential energy surface for the CH4 + O(3P) → CH3 + OH reaction. Thermal rate constants and kinetic isotope effects. Phys Chem Chem Phys. 2000;2:2345–2351. [Google Scholar]
- 15.Troya D, Pascual RZ, Schatz GC. Theoretical studies of the O(3P) + methane reaction. J Phys Chem A. 2003;107:10497–10506. [Google Scholar]
- 16.Troya D, García-Molina E. Quasiclassical trajectory study of the O(3P) + CH4 → OH + CH3 reaction with a specific reaction parameters semiempirical Hamiltonian. J Phys Chem A. 2005;109:3015–3023. doi: 10.1021/jp044304+. [DOI] [PubMed] [Google Scholar]
- 17.Mikosch J, et al. Imaging nucleophilic substitution dynamics. Science. 2008;319:183–186. doi: 10.1126/science.1150238. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, et al. F- + CH3I → FCH3 + I- reaction dynamics. Nontraditional atomistic mechanisms and formation of a hydrogen-bonded complex. J Phys Chem Lett. 2010;1:2747–2752. [Google Scholar]
- 19.Polanyi JC. Some concepts in reaction dynamics. Science. 1987;236:680–690. doi: 10.1126/science.236.4802.680. [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Liu K. Enlarging the reactive cone of acceptance by exciting the C-H bond in the O(3P) + CHD3 reaction. Chem Sci. 2010;1:126–133. [Google Scholar]
- 21.Zhang J, Liu K. Imaging the reaction dynamics of O(3P) + CH4 → OH + CH3. Chem Asian J. 2011;6:3132–3136. doi: 10.1002/asia.201100414. [DOI] [PubMed] [Google Scholar]
- 22.Yang M, Lee S-Y, Zhang DH. Seven-dimensional quantum dynamics study of the O(3P) + CH4 reaction. J Chem Phys. 2007;126:064303. doi: 10.1063/1.2434171. [DOI] [PubMed] [Google Scholar]
- 23.Braams BJ, Bowman JM. Permutationally invariant potential energy surfaces in high dimensionality. Int Rev Phys Chem. 2009;28:577–606. doi: 10.1146/annurev-physchem-050317-021139. [DOI] [PubMed] [Google Scholar]
-
24.Czakó G, Bowman JM. Accurate ab initio potential energy surface, thermochemistry, and dynamics of the
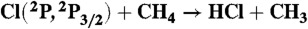 and H + CH3Cl reactions. J Chem Phys. 2012;136:044307. doi: 10.1063/1.3679014. [DOI] [PubMed] [Google Scholar]
and H + CH3Cl reactions. J Chem Phys. 2012;136:044307. doi: 10.1063/1.3679014. [DOI] [PubMed] [Google Scholar] - 25.Allen WD, East ALL, Császár AG. Ab initio anharmonic vibrational analyses of non-rigid molecules. In: Laane J, Dakkouri M, van der Veken B, Oberhammer H, editors. Structures and Conformations of Non-Rigid Molecules. Dordrecht, The Netherlands: Kluwer; 1993. pp. 343–373. [Google Scholar]
- 26.Császár AG, Allen WD, Schaefer HF. In pursuit of the ab initio limit for conformational energy prototypes. J Chem Phy. 1998;108:9751–9764. [Google Scholar]
- 27.Xiao C, et al. Experimental and theoretical differential cross sections for a four-atom reaction: HD + OH → H2O + D. Science. 2011;333:440–442. doi: 10.1126/science.1205770. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann MR, Schatz GC. Theoretical studies of intersystem crossing effects in the O + H2 reaction. J Chem Phys. 2000;113:9456–9465. [Google Scholar]
- 29.Hase WL. Encyclopedia of Computational Chemistry . New York: Wiley; 1998. pp. 399–407. [Google Scholar]