

Abstract
Background
Gastrointestinal stromal tumors (GIST) mutational status is recognized factor related to the results of tyrosine kinase inhibitors therapy such as imatinib (IM) or sunitinib (SU). Arterial hypertension (AH) is common adverse event related to SU, reported as predictive factor in renal cell carcinoma. The aim of the study was to analyze the outcomes and factors predicting results of SU therapy in inoperable/metastatic CD117(+) GIST patients after IM failure.
Methods
We identified 137 consecutive patients with advanced inoperable/metastatic GIST treated in one center with SU (2nd line treatment). Median follow-up time was 23 months. Additionally, in 39 patients there were analyzed selected constitutive single nucleotide polymorphisms (SNPs) of VEGFA and VEGFR2 genes.
Results
One year progression-free survival (PFS; calculated from the start of SU) rate was 42% and median PFS was 43 weeks. The estimated overall survival (OS, calculated both from start of SU or IM) was 74 weeks and 51 months, respectively. One-year PFS was 65% (median 74 weeks) in 55 patients with AH vs. 22% (median 17 weeks) in patients without AH. Patients with primary tumors carrying mutations in KIT exon 9 or wild-type had substantially better 1-year PFS (68% and 57%; median 65.5 and 50.5 weeks, respectively) than patients having tumors with KIT exon 11 or PDGFRA mutations (34% and 15%; median 36.8 and 9 weeks, respectively). We identified two independent factors with significant impact on PFS and OS in univariate and multivariate analysis: primary tumor genotype and presence of AH. The most common adverse events during therapy were: fatigue, AH, hypothyroidism, hand and foot syndrome, mucositis, skin reactions, dyspepsia, and diarrhea. Two deaths were assessed as related to tumor rupture caused by reaction to SU therapy. The presence of C-allele in rs833061 and the T-allele in rs3025039 polymorphism of VEGFA were associated with significantly higher risk of hypothyroidism (OR: 10.0 p = 0.041 and OR: 10.5; p = 0.015, respectively).
Conclusions
We confirmed that many advanced GIST patients benefit from SU therapy with OS > 1.5 year. Primary tumor KIT/PDGFRA genotype and SU-induced AH, as surrogate of its antiangiogenic activity are two independent factors influencing both PFS and OS.
Note
The preliminary data of this study were presented during Annual Meeting of American Society of Clinical Oncology, 4-8 June 2011 and Connective Tissue Oncology Society Meeting, 26-28 October 2011 in Chicago, IL.
Keywords: Sunitinib, Genotype, GIST, Prognosis, Predictive factors, Arterial hypertension
Background
Unprecedented improvement in advanced gastrointestinal stromal tumors (GIST management has been achieved due to recent recognition of the important biological role of activating mutations in KIT and PDGFRA (platelet-derived growth factor receptor- alpha) genes. Those observations led to the introduction of imatinib mesylate, a small-molecule selective inhibitor of the receptor tyrosine kinases such as stem-cell factor receptor (KIT, CD117), BCR-ABL and platelet-derived growth factor receptors (PDGFRs)-A and -B. Imatinib revolutionized the outcome of patients with advanced CD117-positive GISTs and is currently approved as the first-line treatment in advanced (metastatic and/or inoperable) GISTs [1-5]. However, the spectacular response to imatinib therapy is time-limited and secondary resistance to imatinib therapy (after initial stabilization or response) develops in majority of patients [4].
Currently, the only approved second-line drug is sunitinib malate - a multitargeted agent, an inhibitor of tyrosine kinase, of KIT and PDGFRA/B and of the vascular endothelial growth factor receptors (VEGFRs)-1, -2 and 3, FMS-like tyrosine kinase-3 (FLT3), colony stimulating factor 1 receptor (CSF-1R), and glial cell-line derived neurotrophic factor receptor (REarranged during Transfection; RET) [6-11]. Sunitinib possesses both antiangiogenic and cytostatic properties and by competing with ATP binding prevents multiple receptor tyrosine kinases phosphorylation in vitro and in vivo. Two phase II, one phase III and one "treatment-use" trials have investigated the activity of sunitinib in GIST patients after the failure of prior imatinib treatment, and all these trials have shown the significant activity of sunitinib in this population of patients [11-14]. The objective clinical benefit was achieved in approximately 60% of GIST patients who received sunitinib after failure of prior imatinib treatment [11-14]. Median progression -free survival time on sunitinib is 6-8 months. The adverse events reported during this therapy are frequent. The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, mucositis, arterial hypertension, hand and foot syndrome (palmar-plantar erythrodysesthesia), impairment of left ventricular ejection fraction and hypothyroidism [12,14].
Moreover, arterial hypertension was not only the common adverse event during sunitinib therapy, but it was reported as predictive factor for results of renal-cell carcinoma (RCC) patients [15,16]. This phenomenon has not been yet analyzed in GIST patients.
There is a lack of studies analyzing the outcome of sunitinib in advanced GISTs after imatinib failure therapy in routine practice outside clinical trials. Thus, the aim of our study was to evaluate factors predicting results and toxicity of SU second-line therapy in inoperable/metastatic GISTs. Additionally, we have investigated the impact of the selected single nucleotide polymorphisms (SNPs) in VEGFA and VEGFR2 genes on sunitinib-related toxicity in the subgroup of patients.
Patients and Methods
Patients
We analyzed prospectively collected data of 137 consecutive patients treated with sunitinib maleate because of inoperable and/or metastatic CD117 positive GIST enrolled into therapy between October, 2005 and February, 2011, reviewed in one tertiary cancer center. All patients met the following criteria for sunitinib treatment: 1) histological diagnosis of GIST, confirmed by CD117-immunopositivity (DAKO; Carpintiera, CA), 2) metastatic and/or inoperable lesions after failure on prior treatment with imatinib (confirmed progressive disease or unacceptable toxicity) 3) measurable disease on computed tomography (CT) scans, 4) WHO performance status ≤3, 5) no concomitant therapy for disease, 6) adequate renal, cardiac and liver function.
Each patient provided informed consent for the study and collection of clinical and molecular data prior sunitinib therapy. The study had been approved by the local Bio-Ethics Committee according to Good Clinical Practice Guidelines (approvals from Bio-Ethics Committees from Medical University of Gdansk and from Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw KB/9/2011 and approval for Polish Clinical GIST Registry by Internal Review Board 119/2002). Patients provided additional informed consent for taking the 5 ml blood samples for gene polymorphisms analysis. Patients did not undergo any further selection; 35 patients were initially included in the treatment-use trial A6181036.
All patients were treated with sunitinib in initial licensed dose of 50 mg daily in 6 weeks cycle (4 weeks on/2 weeks off therapy), however the dosing could be reduced (to 37.5 mg or 25 mg) or delayed or modulated to the dosing of 37.5 mg on continuous schedule to optimize the benefit-risk profile according to decision of treating physician. The treatment was continued until confirmed progression of the disease or unacceptable toxicity. All patients were followed carefully with median follow-up time of 23 months (range: 6-68 months). The objective response of GIST to sunitinib therapy was evaluated with serial CT examinations (performed every 2-3 months), according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 [17]. In case of progression, patients were treated with other different tyrosine kinase inhibitors or cytotoxic chemotherapy or best supportive care only. If possible, they were included into clinical trials with new compounds. Toxic effects were graded with National Cancer Institute common toxicity criteria, version 3.0 [18].
Genotyping
Genomic screening was performed for the presence of the KIT (exons 9, 11, 13, and 17) or PDGFRA (exons 12, 14 and 18) genes mutation in randomly selected 89 cases, based on DNA isolated from paraffin-embedded or fresh frozen imatinib-naive tumor tissues, as previously described [19].
SNPs analysis
Genomic DNA was isolated from peripheral blood samples of 39 consenting patients using the standard protocol with proteinase K digestion, phenol - chloroform extraction and ethanol precipitation. Three selected VEGFA SNPs: rs699947 (-2578 C > A), rs3025039 (+936 C > T), and rs2010963 (+405 G > C) and two VEGFR2 (KDR) SNPs: rs1531289 (3405-92A > G) and rs1870377 (+1416 T > A) were genotyped using restriction fragment length polymorphism (RFLP) method. The restriction enzymes which detected - SNPs mentioned above were BsaIII, BsmF1, NlaIII and AluI, respectively. Amplified DNA was digested with endonucleases overnight at optimal temperatures according to the manufacturer (Fermentas, Thermo Scientific) and then electrophoresed on 2% agarose gel. For identification of an additional VEGFA SNP: - rs833061 (460 T > C) direct sequencing was performed using ABI 3100 Genetic Analyser (Applied Biosystems Inc. Foster City, CA). Primers sequences and PCR cycling parameters are available on request.
Statistical analysis
Contingency tables were analyzed by the chi-square test. Progression-free survival (PFS) time was calculated from the date of the start of sunitinib treatment to the date of the most recent follow-up, or progression or death due to the disease. Overall survival (OS) time was calculated either from the date of the start of imatinib and sunitinib treatment to the date of the most recent follow-up or death due to the disease [except cases of two patients, whose death was attributed to adverse event (AE) of sunitinib therapy]. PFS was assessed with respect to the following variables: demographic data (age at the diagnosis ≤ 45 or > 45 years; gender), primary tumor genotype (KIT 11 exon, KIT 9 exon, any PDGFRA mutations and wild-type cases), length of previous therapy on imatinib (≤ 6, > 6-12, > 12 months), and presence of arterial hypertension (defined as occurrence of systolic blood pressure > 140 mm Hg or diastolic blood pressure > 90 mm Hg, or deterioration of preexisting AH during the first three months of therapy with sunitinib). The Kaplan-Meier method was used for analysis of survival curves, compared by log-rank test.
For univariate comparison of the survival between groups, the Kaplan-Meier estimator was used with generalized Wilcoxon and the log-rank tests. For PFS/OS comparisons in relation to presence of arterial hypertension two cases with early death due to tumor perforation were excluded to minimize the lead-time bias of the results. In multivariate analysis of the factors associated with PFS and OS, we used Cox proportional hazards models, applying the stepwise model building procedure that included all covariates significant at 20% level in bivariate analysis. Two-way interactions were then considered in the model. Differences were considered statistically significant if p-values were < 0.05. These statistical computations were performed using Statistica 6.1 software [Statsoft®; Tulsa, OK].
The Hardy-Weinberg equilibrium analysis of VEGFA and VEGFR2 polymorphisms was performed using a chi-square test with one degree of freedom. Multiplicative, dominant and recessive models were tested in every SNP for the associations with OS, PFS and treatment AEs. Odds ratios (ORs) with their exact 95% confidence intervals (CI) were calculated and the exact test for OR was used. The polymorphisms analyses were performed using StatXact-3 software, version 3.1.
Results
Clinicopathological data
The distribution of clinical and pathological data of patients included in the study is listed in Table 1. There were 74 male and 63 female patients, with median age at the start of sunitinib therapy 55 years (range: 15 - 82). The majority of primary tumors were located in the small intestine (57.6%), followed by the stomach (33.5%). All but two patients had documented progression on imatinib. Majority of patients (70.1%) were pre-treated with imatinib for more than one year. Almost 90% of patients started sunitinib therapy being in relatively good performance status (0-1).
Table 1.
Characteristics of 137 patients treated with sunitinib due to advanced GIST
| Clinicopathological features | No of patients (%) | |
|---|---|---|
| Total number of patients | 137 (100) | |
| Age [years] at the start of therapy with sunitinib | Median (range) | 55 (15-82) |
| ≤45 | 14 (10) | |
| > 45 | 123 (88) | |
| Gender | Female | 63 (46) |
| Male | 74 (54) | |
| Primary tumor site | Stomach | 46 (33.5) |
| small bowel | 79 (57.6) | |
| large bowel/rectum | 4 (2.9) | |
| other or intraperitoneally with unknown primary origin | 8 (5.8) | |
| Time on imatinib therapy | ≤ 6 months (early resistance) | 25 (18.2) |
| 6-12 months | 16 (11.7) | |
| > 12 months | 96 (70.1) | |
| Primary reason for stop of imatinib therapy | Disease progression | 135 (98.5) |
| Imatinib intolerance | 2 (1.5) | |
| ECOG Performance Status | 0 | 48 (35) |
| 1 | 72 (52.6) | |
| ≥2 | 17 (12.4) | |
| Tumor genotype* | Exon 11 KIT mutation | 52 (58.4) |
| Exon 9 KIT mutation | 15 (16.9) | |
| PDGFRA mutation | 12 (13.5) | |
| Wild-type | 10 (11.2) | |
| Data not available | 48 | |
* mutational status was evaluated in 89 cases (65%)
Mutational analysis data
The distribution of patients according to the initial tumor mutational status is shown in Table 1. In the group of 89 patients, whose initial tumor mutational status was evaluated, 58.4% of GISTs had an exon 11 KIT mutation, 16.9% had an exon 9 KIT mutation, 13.5% had PDGFRA gene mutation (11 of 12 cases had D842V mutation) and in 11.2% of tumors we have not detected any mutations (wild-type).
Treatment toxicity
Adverse events were common during sunitinib treatment (127/137 evaluated patients; 93%), and in 31.4% of patients they were assessed as grade 3/4 (Table 2). The most common non-hematological adverse events were: fatigue (65%), arterial hypertension (43%), hand-foot syndrome (40%), hypothyroidism (31%), skin/hair discoloration (30%), diarrhea (28%) and mucositis (25.5%). The frequency of reported hematological toxicity was as follows: anemia (37%), neutropenia (36%) and thrombocytopenia (13%). Two deaths (grade 5) due to the tumor rupture and hemorrhage were assessed as related to reaction to sunitinib therapy (during 1st month of therapy). Additionally, three patients were operated due to tumor perforation (bleeding/bowel perforation) also attributed to reaction to sunitinib. For management of drug toxicity sunitinib dose was reduced to 37.5/25 mg in 44% of cases (60 patients).
Table 2.
The most common adverse events (AEs) during sunitinib therapy in the entire analyzed group of GIST patients
| Any grade | Grade 3/4 | |||
|---|---|---|---|---|
| AEs | n | % | N | % |
| Any treatment-related AE | 127 | 92.7 | 43 | 31.4 |
| Fatigue | 89 | 65 | 12 | 8.7 |
| Diarrhea | 37 | 28 | 4 | 2.9 |
| Hand-foot syndrome | 55 | 40 | 3 | 2.2 |
| Decreased appetite/dysgeusia | 25 | 18.2 | 0 | 0 |
| Mucositis | 35 | 25.5 | 2 | 1.5 |
| Hypertension | 59 | 43 | 4 | 2.9 |
| Neutropenia | 49 | 36 | 7 | 5.1 |
| Anemia | 51 | 37 | 8 | 5.8 |
| Skin/hair discoloration | 41 | 30 | 0 | 0 |
| Dyspepsia | 43 | 31.4 | 0 | 0 |
| Thrombocytopenia | 18 | 13.1 | 3 | 2.2 |
| Hypothyroidism | 42 | 31 | 2 | 1.5 |
Outcomes of sunitinib treatment
Median PFS was 43 weeks and estimated 1-year PFS rate was 42%. Progression of disease during sunitinib therapy was observed in 105 cases (77%). At the time of the analysis, 45 patients (33%) were alive. Estimated 2-year OS rate was 40% and median OS was 73.5 weeks. Estimated 5-year OS in this group of patients was 45% (when calculated from the date of imatinib start) and median OS - 51 months.
The best responses observed during sunitinib therapy and estimated by CT imaging (two consecutive examinations) according to RECIST criteria were as follows: none complete responses (CRs), 21 (15%) partial responses (PRs), 62 (45%) stable disease (SD) at least four months, 51 (37%) progressive disease (PD) and three patients were not assessable for response. Overall clinical benefit of sunitinib therapy (counted as the sum of CR, PR and SD rates) was 60%.
Correlations between mutational status of primary GISTs and response to sunitinib therapy
We have found a strong relationship (p < 0.001) between the primary tumor genotype and best observed, confirmed response to sunitinib according to RECIST criteria: the best outcomes were observed for KIT exon 9 mutants (six PR - 40%, seven SD - 47%, two PD - 13%), followed by wild-type GISTs (seven SD - 70% and three PD - 30%) and KIT exon 11 mutants (10 PR - 19%, 23 SD - 44%, 19 PD - 37%); the worst results of sunitinib therapy were found in patients with PDGFRA mutated GISTs (two SD - 17% and 10 PD - 82%).
Factors influencing PFS and OS during sunitinib therapy
In univariate analysis two factors significantly correlated with shorter PFS and OS that were: tumor genotype: exon 11 KIT or PDGFRA mutation (p = 0.04 and p = 0.04, respectively) (Figure 1), and absence of arterial hypertension during sunitinib therapy (p = 0.0001 and p = 0.001) (Figure 2). We did not find any significant correlation between PFS/OS and patients gender and age at diagnosis, GIST primary tumor location, WHO performance status 0 vs. 1, or time on previous imatinib treatment. Estimated 1-year PFS and 2-year OS according to primary tumor genotype were as follows: KIT exon 9 mutations - 68%/73% (median 65.5/151.5 weeks), wild type - 57%/70% (median 50.5/121 weeks), KIT exon 11 mutations - 34%/34% (median 36.8/65.5 weeks) and PDGFRA mutations - 15%/25% (median 9/40 weeks). Patients with presence of arterial hypertension during first three months of sunitinib therapy had substantially better 1-year PFS and 2-year OS than patients without this adverse event (65% vs. 22%, and 63% vs. 18%; median 77 vs. 25.5 weeks, and 128.5 vs. 43 weeks, respectively).
Figure 1.
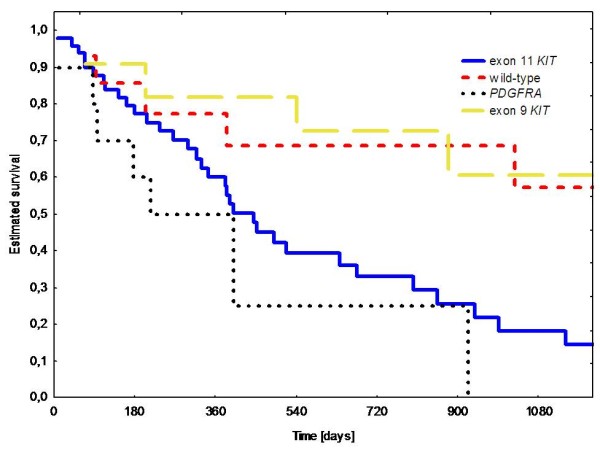
Overall survival during sunitinib therapy according to primary tumor mutational status.
Figure 2.
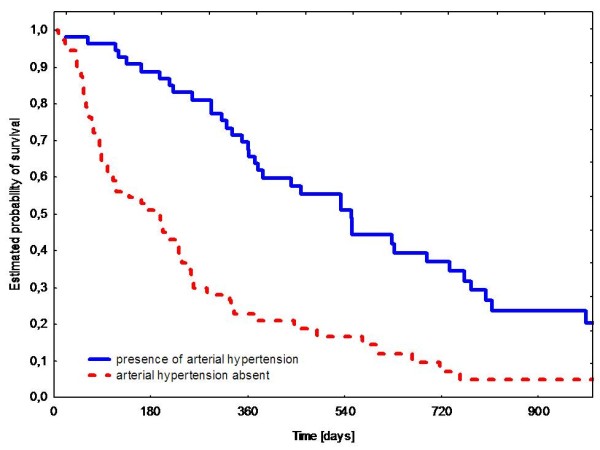
Progression-free survival during sunitinib therapy according to presence of arterial hypertension.
In the multivariate analysis two factors (tumor genotype and arterial hypertension) had independent predictive value for PSF and OS (Tables 3 and 4).
Table 3.
Multivariate analysis of prognostic factors for progression-free survival
| Hazard ratio | 95% CI | Standard Error | z | p value | |
|---|---|---|---|---|---|
| Arterial hypertension Yes |
0.2025 | 0.1097 - 0.3739 | 0.0633 | -5.11 | 0.000 |
| Primary tumor mutation Wild type |
0.3292 | 0.1447 - 0.7491 | 0.1381 | -2.65 | 0.008 |
| PDGFRA mutation | 1.8753 | 0.9096 - 3.8661 | 0.0692 | 1.70 | 0.049 |
Table 4.
Multivariate analysis of prognostic factors for overall survival
| Hazard ratio | 95% CI | Standard Error | z | p value | |
|---|---|---|---|---|---|
| Primary tumor mutation Exon 9 KIT mutation |
0.7491 | 0.3308 - 1.6968 | 0.3125 | -0.69 | 0.489 |
| PDGFRA mutation | 1.2678 | 0.5487 - 2.9327 | 0.5425 | 0.55 | 0.579 |
| Wild-type | 0.1029 | 0.0317 - 0.3336 | 0.0618 | -3.79 | 0.000 |
| Arterial hypertension Yes |
0.2056 | 0.1006 - 0.4201 | 0.0749 | -4.34 | 0.000 |
CI confidence interval
Genetic variations of VEGFA and VEGFR2 genes
The genotype frequency of VEGFA/VEGFR2 gene SNPs are presented in Table 5. All analyzed genotypes were in Hardy-Weinberg equilibrium. The higher risk of hypothyroidism during sunitinib therapy was associated with: C-allele in rs833061 (OR: 10.l; p = 0.041) and T-allele in rs3025039 (OR: 10.5; p = 0.015) (Table 6). We did not find the correlation between the presence of SNPs and arterial hypertension, hand-foot-syndrome, skin toxicity/mucositis or diarrhea (data not shown).
Table 5.
The genotype frequency of VEGFA/VEGFR2 genes SNPs and Hardy-Weinberg equilibrium (HWE) test results
| Polymorphism | rs number | Genotype (%) | HWE exact p value | Frequency of risk allele | ||
|---|---|---|---|---|---|---|
| Homozygous (wild) | Heterozygou | Homozygous (mutant) | ||||
| VEGFA | ||||||
| -2578 C > A | rs699947 | 32.4 | 54.1 | 13.5 | 0.73 | 0.41 |
| -460 T > C | rs833061 | 29.7 | 45.9 | 24.3 | 0.74 | 0.47 |
| 936 C > T | rs3025039 | 75.7 | 24.3 | - | 1.00 | 0.12 |
| 405 G > C | rs2010963 | 24.3 | 40.5 | 35.1 | 0.32 | 0.55 |
| VEGFR2 (= KDR) | ||||||
| 3405-92A > G | rs1531289 | 48.6 | 45.7 | 5.7 | 0.69 | 0.29 |
| 1416 T > A | rs1870377 | 53.1 | 40.6 | 6.3 | 1.00 | 0.27 |
Table 6.
Relationship between VEGFA/VEGFR2 gene polymorphisms and sunitinib-induced hypothyroidism grade ≥2.Statistically significant correlations are marked in bold
| Polymorphism | Reference/risk genotype | OR | 95%CI | p value |
|---|---|---|---|---|
| VEGFA | ||||
| rs699947 (C > A) | CC → AC → AA | 2.38 | 0.82 - 6.99 | 0.125 |
| rs833061 (T > C) | TT vs CT + CC | 10.00 | 1.07 - 466.60 | 0.041 |
| rs3025039 (C > T) | CC vs CT + TT | 10.50 | 1.42 - 117.40 | 0.015 |
| rs2010963 (G > C) | GG vs CG + CC | 1.29 | 0.22 - 9.62 | 1.000 |
| VEGFR2 (= KDR) | ||||
| rs1531289 (A > G) | GG vs AG + AA | 1.60 | 0.33 - 8.29 | 0.758 |
| rs1870377 (T > A) | TT → AT → AA | 0.74 | 0.19 - 2.64 | 0.822 |
CI confidence interval, OR odds ratio
Discussion
A majority of patients with advanced GISTs ultimately stop responding to imatinib and unquestionably management of disease resistant to first-line treatment represents a clinical challenge [4]. Insights into resistance mechanisms have allowed developing several strategies in patients with progression during imatinib treatment. In case of generalized progression (or intolerance to imatinib) the main option is using monotherapy with alternative multi-tyrosine kinase inhibitor - sunitinib, which remains the only approved second line drug for the treatment of advanced GISTs after imatinib therapy failure [20]. Sunitinib has demonstrated robust clinical effectiveness in imatinib-resistant or -intolerant GIST as shown in randomized, placebo-controlled phase III trial in which the median time to tumor progression for patients treated with sunitinib was more than four times longer than that for patients receiving placebo (27.3 vs. 6.4 weeks) [12]. Present study, according to our best knowledge, represents of the largest series of GIST patients after imatinib failure analyzed for the outcome of sunitinib treatment in routine clinical practice outside randomized, controlled clinical trial.
We have also attempted to prove tumor genotype implications and to find new predictive factors in this group of patients. We have confirmed that many advanced GIST patients benefit from sunitinib therapy (mainly due to stabilization of disease according to RECIST, not Choi criteria [21]) with OS exceeding 1.5 years. The median PFS longer than seven months is almost equal to the results of the Korean one-institution study [22]. We have also confirmed in more detailed way and on the larger group of patients, than ever published, data regarding the correlation between primary tumor mutational status and sunitinib treatment outcomes [22-24]. As for imatinib, KIT mutation status appears to serve as a predictor of tumor response to sunitinib. We have proven that, contrary to imatinib, tumors initially (pre-imatinib treatment) bearing KIT exon 9 mutation or with wild-type genotype have a higher chance to respond to sunitinib. Moreover, GISTs harboring KIT exon 9 mutations appear to be more sensitive to sunitinib than those with primary KIT exon 11 mutations (however we have observed some objective responses also in this group of patients). The clinical benefit of sunitinib in wild-type cases is also clear. We have not observed any response to sunitinib in group of patients with PDGFRA mutations (mainly D842V), which has been also shown in preclinical data. We did not analyze the impact of secondary mutations, although patients from clinical trials with tumors harboring a secondary mutation in exon 13 or exon 14 KIT have a longer PFS than patients with exon 17 or 18 mutations [23,25-27]. On the other hand, utility of analysis of secondary mutations is very challenging because imatinib-resistant GISTs are very heterogeneous with multiple clones having different secondary mutations within the same or different nodules [28-30].
Sunitinib therapy is associated with several adverse events, which were generally mild to moderate and could be managed by dose modulation (including continuous administration of lower dose) [20,22,24]. The toxicity profile reported in our study is similar to that observed in clinical trials, with exception of hypothyroidism, which occurred in more than 30% of patients (it has been reported outside clinical trials [31,32]). However, up to one third of cases were classified as more severe toxicity (and two deaths due to tumor hemorrhage were classified as related to sunitinib therapy). Our own experience with patients with unresectable or metastatic GISTs, treated with tyrosine kinase inhibitors, suggested the higher incidence of emergency operations for gastrointestinal bleeding, bowel obstruction, or abscess, occur during second-line therapy with sunitinib than during first-line therapy with imatinib [33,34]. This increased incidence of complications leading to surgical interventions with sunitinib could be associated with the presence of more advanced and drug-resistant disease, or to the direct mechanism of action of sunitinib, i.e., the combination of cytotoxic and antiangiogenic activity, leading to dramatic tumor response.
Arterial hypertension is one of the most common complications of sunitinib therapy, occurring usually early after treatment initiation. Serial monitoring of blood pressure is recommended during therapy with sunitinib. Sunitinib-induced arterial hypertension may also serve as biomarker of antitumor efficacy (probably by antiangiogenic mechanism), because it was an independent factor influencing patient both progression-free and overall survival. Antiagiogenic activity may play an important role in therapy of sarcomas, what has been recently confirmed by positive results of phase III trial with pazopanib in pre-treated soft tissue sarcoma patients [35]. Similar relationships between arterial hypertension induced by VEGF inhibitors (including sunitinib) and oncological outcomes have been reported in renal cell carcinoma patients [15,16,36-39]. Treatment-induced persistent hypertension was associated with frequent tumor response, a long time to disease progression and longer overall survival [39]. Clinical outcomes are not compromised by treatment with anti-hypertension medications, moreover, patients who required at least three antihypertensive drugs had the longest PFS and OS [38]. There are proposed some hypothetical mechanisms leading to hypertension related to sunitinib, e.g. presence of less-perfused microvessels and/or diminished number of microvessels, decreasing nitric oxide production and activation of the endothelin-1 pathway leading to vasoconstriction [40,41].
In the subgroup of patients we have analyzed some possible pharmacogenetical relationships with sunitinib tolerance. It has been shown that single nucleotide polymorphisms of VEGF and VEGFR2 genes has some potential as biomarkers for clinical outcomes and toxicity of VEGF pathway targeted therapy [42-46]. We have not studied correlation between SNPs of VEGFA/VEGFR genes and outcomes of therapy due to limited number of cases, but we have found clear associations between two SNPs of VEGFA gene and sunitinib-induced hypothyroidism. The molecular mechanisms of hypothyroidism induced by sunitinib are unknown, but recent studies have suggested that VEGFR inhibition can induce vasculature regression in various organs, predominantly in thyroid, what can be linked to different properties of VEGF protein caused by gene polymorphisms and sunitinib sensitivity [47,48].
Conclusions
To summarize, we confirmed that many advanced GIST patients benefit from sunitinib therapy with overall survival exceeding 1.5 year. Exploring the toxicity of multi-kinase targeting agents in GISTs may allow better adjusted therapy as well as to define novel pharmacodynamics markers. Primary tumor genotype and sunitinib-induced arterial hypertension (as surrogate of its antiangiogenic activity) are two independent factors influencing the progression-free survival and OS. The mechanism of side effects and its correlation with pharmacogenetic data during sunitinib therapy need further studies.
Competing interests
P. Rutkowski, C. Osuch, Z. Nowecki and A. Wozniak have received honoraria and travel grants form Novartis, P. Rutkowski served in advisory board for Novartis, J. Kroc is employed by Pfizer; P.Rutkowski and Z. Nowecki have received honoraria and travel grants from Pfizer.
Authors' contributions
Study concepts: PR, EB, AW, JK, ZIN, IL, JL. Study design: PR, AW, CO, IL, JL. Data acquisition: PR, EB, AK, TŚ, SF, JK, IŁ, WM, CO, AW, UG, ZIN. Data analysis and interpretation: PR, EB, IŁ, MB, EM, KW, AW, UG, JS, JL. Statistical analysis: PR, IL, EM. Manuscript preparation: PR, AW, JL, MB, IŁ. Manuscript review: PR, EB, AK, TŚ, SF, JK, IŁ, MB, WM, CO, EM, KW, AW, UG, ZIN, JS, JL. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Piotr Rutkowski, Email: rutkowskip@coi.waw.pl.
Elżbieta Bylina, Email: elab@coi.waw.pl.
Anna Klimczak, Email: aniaklimczak@poczta.onet.pl.
Tomasz Świtaj, Email: tomek89@yahoo.co.uk.
Sławomir Falkowski, Email: sfalcon@neostrada.pl.
Jacek Kroc, Email: jacek.kroc@pfizer.com.
Iwona Ługowska, Email: iwonalugowska@coi.waw.pl.
Magdalena Brzeskwiniewicz, Email: magda_brzeskwiniewicz@interia.pl.
Wojciech Melerowicz, Email: wojmel5@wp.pl.
Czesław Osuch, Email: msosuch@cyf-kr.edu.pl.
Ewa Mierzejewska, Email: emierzejewska@imid.med.pl.
Kacper Wasielewski, Email: k.wasielewski@gumed.edu.pl.
Agnieszka Woźniak, Email: agnieszka.wozniak@med.kuleuven.be.
Urszula Grzesiakowska, Email: ugrzesiakowska@yahoo.com.
Zbigniew I Nowecki, Email: nowecki@coi.waw.pl.
Janusz A Siedlecki, Email: jas@coi.waw.pl.
Janusz Limon, Email: jlimon@gumed.edu.pl.
Acknowledgements and funding
We thank Katarzyna Szamotulska and Magdalena Rosinska for statistical assistance; and Dr Hanna Kosela for linguistic assistance. We thank Pfizer for unrestricted educational grant for Polish Clinical GIST Registry. This work was supported by the Polish Ministry of Science grant No NN402 2092 35.
References
- Demetri GD, von Mehren M, Blanke CD, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. New Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- Joensuu H. Gastrointestinal stromal tumor (GIST) Ann Oncol. 2006;17(Suppl 10):x280–x286. doi: 10.1093/annonc/mdl274. [DOI] [PubMed] [Google Scholar]
- Rutkowski P, Debiec-Rychter M, Ruka W. Gastrointestinal stromal tumors: key to diagnosis and choice of therapy. Mol Diagn Ther. 2008;12(3):131–143. doi: 10.1007/BF03256278. [DOI] [PubMed] [Google Scholar]
- Rutkowski P, Nowecki ZI, Dębiec-Rychter M, Grzesiakowska U, Michej W, Woźniak A, Siedlecki JA, Limon J, Jerzak vel Dobosz A, Kąkol M, Osuch C, Ruka W. Predictive factors for long term effects of imatinib therapy in patients with inoperable/metastatic CD117(+) gastrointestinal stromal tumors (GISTs) J Canc Res Clin Oncol. 2007;133:589–597. doi: 10.1007/s00432-007-0202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–478. [PubMed] [Google Scholar]
- O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–3605. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
- Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, Keast PK, Brassard JA, O'Farrell AM, Cherrington JM, Pryer NK. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis. 2003;20:757–766. doi: 10.1023/b:clin.0000006873.65590.68. [DOI] [PubMed] [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- Kim DW, Jo YS, Jung HS, Chung HK, Song JH, Park KC, Park SH, Hwang JH, Rha SY, Kweon GR, Lee SJ, Jo KW, Shong M. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91:4070–4076. doi: 10.1210/jc.2005-2845. [DOI] [PubMed] [Google Scholar]
- Maki RG, Fletcher JA, Heinrich MC, Morgan JA, George S, Desai J, Scheu K, Fletcher CD, Baum C, Demetri GD. Results from a continuation trial of SU11248 in patients (pts) with imatinib (IM)-resistant gastrointestinal stromal tumor (GIST) J Clin Oncol. 2005;23(16S):Abs 9011.. [Google Scholar]
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- George S, Blay JY, Casali PG, Le Cesne A, Morgan JA, Pokela J, Quigley MT, Tassell V, Baum CM, Demetri GD. Continuous daily dosing (CDD) of sunitinib malate (SU) compares favorably with intermittent dosing in pts with advanced GIST. J Clin Oncol. 2007;25(18S):Abs 10015.. [Google Scholar]
- Reichardt P, Kang YK, Ruka W, Seddon B, Baum CM, Demetri GD. Subpopulation analyses in a worldwide treatment-use trial of sunitinib (SU) in GIST patients (pts) with resistance or intolerance to prior imatinib (IM) therapy. J Clin Oncol. 2007;25(18S):Abs 10022.. [Google Scholar]
- Levy BI. Blood pressure as a potential biomarker of the efficacy angiogenesis inhibitor. Ann Oncol. 2009;20:200–203. doi: 10.1093/annonc/mdp018. [DOI] [PubMed] [Google Scholar]
- Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, Figlin RA, Baum MS, Motzer RJ. Hypertension as a Biomarker of Efficacy in Patients With Metastatic Renal Cell Carcinoma Treated With Sunitinib. J Natl Cancer Inst. 2011;103:763–773. doi: 10.1093/jnci/djr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- Cancer therapy evaluation program. Common terminology criteria for adverse events version 3.0 (CTCAE) Bethesda, MD: National Cancer Institute; 2003. (Available at http://ctep.cancer.gov/forms/CTCAEv3.pdf) [Google Scholar]
- Wozniak A, Rutkowski P, Piskorz A, Ciwoniuk M, Osuch C, Bylina E, Sygut J, Chosia M, Rys J, Urbanczyk K, Kruszewski W, Sowa P, Siedlecki J, Debiec-Rychter M, Limon J, on behalf of Polish Clinical GIST Registry. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol. 2011. in press . [DOI] [PubMed]
- Blay J-Y. Pharmacological management of gastrointestinal stromal tumours: an update on the role of sunitinib. Ann Oncol. 2010;21:208–215. doi: 10.1093/annonc/mdp291. [DOI] [PubMed] [Google Scholar]
- Dudeck O, Zeile M, Reichardt P, Pink D. Comparison of RECIST and Choi criteria for computed tomographic response evaluation in patients with advanced gastrointestinal stromal tumor treated with sunitinib. Ann Oncol. 2011;22(8):1828–1833. doi: 10.1093/annonc/mdq696. [DOI] [PubMed] [Google Scholar]
- Yoon DH, Ryu M-H, Ryoo B-Y, Beck M, Choi D-R, Cho Y, Lee Y-L, Chang H-M, Kim TW, Kang Y-K. Sunitinib as a second-line therapy for advanced GISTs after failure of imatinib: relationship between efficacy and tumor genotype in Korean patients. Invest New Drugs. 2011. doi 10.1007/s10637-010-9593-1 S2. [DOI] [PubMed]
- Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou W-B, Fletcher JA, Fletcher CDM, Huang X, Cohen DP, Baum CM, Demetri GD. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J Clin Oncol. 2008;26:5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD, Heinrich MC, Fletcher JA, Fletcher CDM, Van den Abbeele AD, Corless CL, Antonescu CR, George S, Morgan JA, Chen MH, Bello CL, Huang X, Cohen DP, Baum CM, Maki RG. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15(18):5902–5909. doi: 10.1158/1078-0432.CCR-09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Hajdu M, Agaram NP, Shinoda H, Veach D, Clarkson BD, Maki RG, Singer S, Dematteo RP, Besmer P, Antonescu CR. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res. 2009;15(22):6862–6870. doi: 10.1158/1078-0432.CCR-09-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T, Takahashi T, Nishitani A, Doi T, Shirao K, Komatsu Y, Nakajima K, Hirota S. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol. 2009;14(2):143–149. doi: 10.1007/s10147-008-0822-y. [DOI] [PubMed] [Google Scholar]
- DiNitto JP, Deshmukh GD, Zhang Y, Jacques SL, Coli R, Worrall JW, Diehl W, English JM, Wu JC. Function of activation loop tyrosine phosphorylation in the mechanism of c-Kit auto-activation and its implication in sunitinib resistance. J Biochem. 2010;147(4):601–609. doi: 10.1093/jb/mvq015. [DOI] [PubMed] [Google Scholar]
- Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, English JM, Greig MJ, He YA, Jacques SL, Lunney EA, McTigue M, Molina D, Quenzer T, Wells PA, Yu X, Zhang Y, Zou A, Emmett MR, Marshall AG, Zhang HM, Demetri GD. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci USA. 2009;106(5):1542–1547. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, JeVrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, Speidel N, Pietsch T, Buettner R, Pink D, Reichardt P, Hohenberger P. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–1749. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- Desai J, Yassa L, Marqusee E, George S, Frates MC, Chen MH, Morgan JA, Dychter SS, Larsen PR, Demetri GD, Alexander EK. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med. 2006;145(9):660–664. doi: 10.7326/0003-4819-145-9-200611070-00008. [DOI] [PubMed] [Google Scholar]
- Wolter P, Stefan C, Decallonne B, Dumez H, Bex M, Carmeliet P, Schoeffski P. The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br J Cancer. 2008;99:448–454. doi: 10.1038/sj.bjc.6604497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruka W, Rutkowski P, Nowecki Z, Dziewirski W. Emergency surgery due to complications during molecular targeted therapy in advanced gastrointestinal stromal tumors (GIST) Ann Surg Oncol. 2008;15(Suppl 2):27. abstr 82. [Google Scholar]
- Rutkowski P, Ruka W. Emergency surgery in the era of molecular treatment of solid tumors. Lancet Oncol. 2009;10(2):157–163. doi: 10.1016/S1470-2045(09)70017-8. [DOI] [PubMed] [Google Scholar]
- Van Der Graaf WT, Blay JY, Chawla SP, Kim D, Nguyen BB, Casali PG, Schöffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, Araki N, Ouali M, Marreaud S, Hodge R, Dewji M, Dei Tos AP, Hohenberger P. PALETTE: A randomized, double-blind, phase III trial of pazopanib versus placebo in patients (pts) with soft-tissue sarcoma (STS) whose disease has progressed during or following prior chemotherapy-An EORTC STBSG Global Network Study (EORTC 62072) J Clin Oncol. 2011. p. 29. abstr LBA10002.
- Zhu X, Stergiopoulos K, Wu S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: systematic review and meta-analysis. Acta Oncol. 2009;48(1):9–17. doi: 10.1080/02841860802314720. [DOI] [PubMed] [Google Scholar]
- Ravaud A, Sire M. Arterial hypertension and clinical benefit of sunitinib, sorafenib and bevacizumab in first and second-line treatment of metastatic renal cell cancer. Ann Oncol. 2009;20:966–967. doi: 10.1093/annonc/mdp201. [DOI] [PubMed] [Google Scholar]
- Szmit S, Langiewicz P, Zołnierek J, Nurzyński P, Zaborowska M, Filipiak KJ, Opolski G, Szczylik C. Hypertension as a predictive factor for survival outcomes in patients with metastatic renal cell carcinoma treated with sunitinib after progression on cytokines. Kidney Blood Press Res. 2011. in press . [DOI] [PubMed]
- Bono P, Rautiola J, Utriainen T, Joensuu H. Hypertension as predictor of sunitinib treatment outcome in metastatic renal cell carcinoma. Acta Oncol. 2011;50(4):569–73. doi: 10.3109/0284186X.2010.543696. Epub 2011 Jan 5. [DOI] [PubMed] [Google Scholar]
- Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol. 1998;274(3, pt 2):H1054–H1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- van der Veldt AA, de Boer MP, Boven E, Eringa EC, van der Eertwegh AJ, van Hinsbergh VW, Smudlers YM, Serne EH. Reduction in skin microvascular density and changes in vessel morphology in patients treated with sunitinib. Anticancer Drugs. 2010;21(4):439–446. doi: 10.1097/CAD.0b013e3283359c79. [DOI] [PubMed] [Google Scholar]
- Vaziri SA, Kim J, Ganapathi MK, Ganapathi R. Vascular endothelial growth factor polymorphisms: role in response and toxicity of tyrosine kinase inhibitors. Curr Oncol Rep. 2010;12:102–108. doi: 10.1007/s11912-010-0085-4. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Vaziri SA, Rini BI, Elson P, Garcia JA, Wirka R, Dreicer R, Ganapathi MK, Ganapathi R. Association of VEGF and VEGFR2 single nucleotide polymorphisms with hypertension and clinical outcome in metastatic clear cell renal cell carcinoma patients treated with sunitinib. Cancer. 2011. in press doi: 10.1002/cncr.26491. [DOI] [PMC free article] [PubMed]
- Schneider BP, Radovich M, Miller KD. The role of vascular endothelial growth factor genetic variability in cancer. Clin Cancer Res. 2009;15:5297–5302. doi: 10.1158/1078-0432.CCR-08-2576. [DOI] [PubMed] [Google Scholar]
- Jain L, Vargo CA, Danesi R, Sissung TM, Price DK, Venzon D, Venitz J, Figg WD. The role of vascular endothelial growth factor SNPs as predictive and prognostic markers for major solid tumors. Mol Cancer Ther. 2009;8:2496–2508. doi: 10.1158/1535-7163.MCT-09-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, Flockhart DA, Hancock B, Davidson N, Gralow J, Dickler M, Perez EA, Cobleigh M, Shenkier T, Edgerton S, Miller KD, ECOG 2100. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26:4672–4678. doi: 10.1200/JCO.2008.16.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D, McDonald DM. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol. 2006;290:H547–H559. doi: 10.1152/ajpheart.00616.2005. [DOI] [PubMed] [Google Scholar]
- Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O'Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–H576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]