

Abstract
Atherosclerosis is a chronic inflammatory disease and the number one cause of mortality worldwide. The fundamental causes of atherosclerosis have not been precisely delineated, although pathogenesis clearly involves endothelial dysfunction and both innate and adaptive immunity. Recent evidence suggests that formyl peptide receptor 2 (FPR2), a G protein-coupled receptor (GPCR), mediates a range of inflammatory responses including superoxide production in neutrophils, chemotaxis of monocytes and neutrophils, CCL2 production in endothelial cells (ECs) and monocytes, and increased CXCL8 expression in neutrophils, which are all related with atherogenesis. Therefore, we propose that FPR2 may play a pathogenic role in atherogenesis.
Introduction
Atherosclerosis is a disorder of large and medium-sized arteries and mortality data showed that it accounted for 1/3 of the deaths in the United States in 2008 [1]. The earliest stage of atherosclerosis is thought to involve endothelial dysfunction/activation due to oxidized low-density lipoprotein (oxLDL) stimulation or disturbed blood flow at arterial branching points. This is followed by the adhesion and infiltration of activated platelets and different subsets of leukocytes, including monocytes, neutrophils, dendritic cells, B cells and T cells [2]. Monocytes/macrophages are the predominant cell types that have been identified in atherosclerotic lesions and their uptake of oxLDL leads to the formation of foam cells [3]. Eventually, the atherosclerotic plaque formed in the intimal vessel wall narrows the artery and the rupture of unstable plaques results in myocardial infarction, stroke and other vascular syndromes [1].
Although many risk factors and gene polymorphisms have been identified for atherosclerosis by classical epidemiology and recent genome wide association studies (GWAS), additional work is needed to identify new targets for more effective therapies. FPR2 is a member of the formyl peptide receptor (FPR) family of seven- transmembrane domain, G protein-coupled receptors. FPR2 is expressed mainly by phagocytic leukocytes such as monocytes and neutrophils and is known to be important in both host defense and inflammation [4], thus making it an appealing candidate in promoting atherosclerosis development.
Hypothesis
We hypothesize that FPR2 triggers the pathogenesis of atherosclerosis and it may serve as a potential target for atherosclerosis treatment.
Supporting Evidences
Several recent studies have shown that activation of FPR2 is potentially proatherogenic (Figure 1). The first evidence is that FPR2 activation may contribute to superoxide production when neutrophils are stimulated with N-formyl-Met–Leu–Phe (fMLF), a potent chemoattractant for phagocytes (5). This effect is mediated by NADPH oxidase (NOX) activation and it was found that reactive oxygen species (ROS) production result from NOX activation might accelerate atherogenesis [6].
Figure 1. FPR2 may accelerate atherogenesis through different pathways.
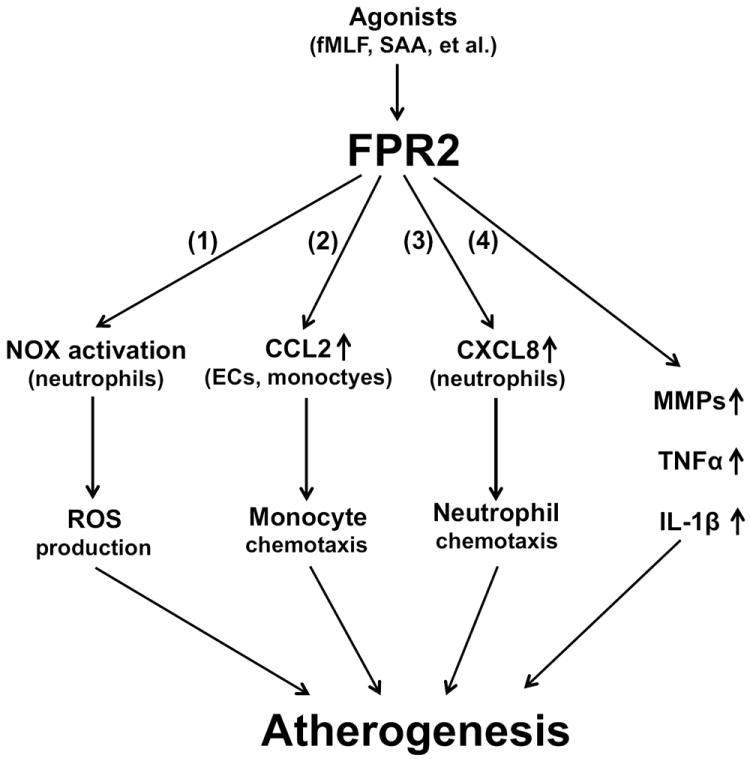
FPR2 activation by its agonists (e.g. fMLF, SAA) lead to (1) NOX activation and ROS production in neutrophils; (2) increased CCL2 expression in ECs and monocytes, thus inducing monocyte chemotaxis; (3) increased CXCL8 expression in neutrophils, thus inducing neutrophil chemotaxis; and (4) increased levels of MMPs, TNFα and IL-1β. All these factors are proatherogenic and FPR2 will probably promote atherogenesis either by one major pathway or by a synergistic effect of different pathways.
The second evidence is that several agonists for FPR2 are associated with chronic inflammation (e.g. serum amyloid A [SAA] and amyloid β peptide 42 [Aβ42]), which is the major cause of atherosclerosis [2, 4]. For example, SAA is an excellent marker for inflammation and is positively associated with prevalent cardiovascular diseases in humans [7]. A recent study showed that elevated plasma levels of SAA directly accelerate disease progression in a mouse model of atherosclerosis, indicating that it is also an active participant in atherogenesis [8]. FPR2 activation by SAA results in a range of inflammatory responses: chemotaxis of monocytes, neutrophils, mast cells and T lymphocytes, production of proinflammatory cytokines such as TNFα and IL-1β, and increased expression of matrix metalloproteinases (MMPs), which may all accelerate the progression of atherosclerosis (Figure 1) [4, 9].
During atherosclerosis, a critical step is the recruitment of leukocytes by chemokines [10]. CCL2 is one of the best-studied chemokines in this regard and is a key mediator of monocyte migration into early atherogenic lesions [3]. Genetic inactivation of either CCL2 or its receptor CCR2 has been reported to significantly reduce the development of atherosclerosis in mouse models [10]. Moreover, there is a significant correlation in humans of CCL2 plasma levels with peripheral arterial disease and acute coronary syndrome [11]. Recently it has been reported that SAA can induce CCL2 production in human monocytes in a time- and concentration-dependent manner, and the induction is mediated by FPR2 since a FPR2 antagonist can dramatically inhibit this process [12]. Also, SAA can induce CCL2 production in human endothelial cells through FPR2 and FPR2 knockdown by short interfering RNA almost completely blocked the induction [13]. Both endothelial cells and monocytes play important roles in atherosclerosis because endothelial dysfunction is considered the trigger of atherogenesis, whereas monocyte recruitment is critical for both the initiation and progression of atherosclerosis. FPR2 activation by SAA in endothelial cells may increase expression of CCL2, which can recruit monocytes to the endothelium and promote transendothelial migration. Monocytes can then secrete additional CCL2 through FPR2 activation, recruiting more circulating monocytes into the atherosclerotic lesions, thus form a pro-atherogenic positive feedback loop in the vessel wall.
Another important chemokine involved in atherogenesis is CXCL8, which targets monocytes and neutrophils [10]. CXCL8 has been reported to induce firm adhesion of rolling monocytes to endothelial cells but the receptors for CXCL8, CXCR1 and CXCR2, are mainly expressed on neutrophils [3, 11]. Mice depleted of neutrophils or transferred with CXCR2-/- bone marrow are protected from atherosclerotic lesion development, suggesting that the CXCL8-CXCR2 axis may play a pro-atherogenic role [10, 14]. In neutrophils, SAA can induce rapid secretion of CXCL8 through activation of FPR2; moreover, overexpression of FPR2 increased CXCL8 secretion while an antibody against FPR2 inhibited CXCL8 expression [15]. Considering that SAA can also induce chemotaxis of neutrophils through FPR2 [4], it is possible that FPR2 is an important bridge between SAA and CXCL8 during the recruitment of neutrophils into atherosclerotic plaques (Figure 1).
The role of FPR2 in inflammation is complicated by the fact that some of its agonists (e.g. LXA4 and its analogs) also exert anti-inflammatory effects [16]. The deletion of FPR2 in mice resulted in either increased inflammatory response or reduced inflammation depending on the specific animal model [17, 18]. Thus, it is still not clear how FPR2 can respond to both pro-inflammatory and anti-inflammatory ligands, but it suggests at least that FPR2 is critical in the modulation of immune and inflammatory responses.
Testing the hypothesis
In order to test the hypothesis, we suggest the following research directions:
Compare the development of atherosclerosis in wild type and FPR2 knockout mice. Considering that normal mice do not develop atherosclerosis spontaneously, those mice may need to be backcrossed with ApoE-/- or Ldlr-/- mice, which are prone to atherosclerosis.
Test the effect of FPR2 agonists/antagonists on the development of atherosclerosis in mouse models.
Check the function of FPR2 on other human leukocytes such as T cells and dendritic cells to see whether they play similar roles as in monocytes and neutrophils.
Identify FPR2 polymorphisms in humans and test their associations with cardiovascular disease.
Implications of the hypothesis
Although atherosclerosis has been studied for decades, and there has been major progress in the diagnosis and treatment of patients, there is still only a limited range of treatment options. New targets are necessary for more effective therapies and here we propose FPR2 as a potential target worth studying. Agonists and antagonists are already available for FPR2 and if it can be validated as a pro-atherogenic factor in mouse models and in humans, there will be a rationale for moving them forward in clinical trials of atherosclerotic cardiovascular disease.
Acknowledgments
We thank Dr. Philip M. Murphy for critical reading of the manuscript.
Funding
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Abbreviations
- fMLF
N-formyl-Met–Leu–Phe
- SAA
serum amyloid A
- FPR2
formyl peptide receptor 2
- NOX
NADPH oxidase
- ROS
reactive oxygen species
- ECs
endothelial cells
- MMPs
matrix metalloproteinases
- TNα
tumor necrosis factor-alpha
Footnotes
Conflicts of interest statement
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, et al. Heart Disease and Stroke Statistics--2012 Update: A Report From the American Heart Association. Circulation. 2012;125:e2–220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Zernecke A, Weber C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res. 2010;86:192–201. doi: 10.1093/cvr/cvp391. [DOI] [PubMed] [Google Scholar]
- 4.Ye RD, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–61. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavigne MC, Murphy PM, Leto TL, Gao JL. The N-formylpeptide receptor (FPR) and a second G(i) -coupled receptor mediate fMet-Leu-Phe-stimulated activation of NADPH oxidase in murine neutrophils. Cell Immunol. 2002;218:7–12. doi: 10.1016/s0008-8749(02)00564-6. [DOI] [PubMed] [Google Scholar]
- 6.Madamanchi NR, Runge MS. NADPH oxidases and atherosclerosis: unraveling the details. Am J Physiol Heart Circ Physiol. 2010;298:H1–2. doi: 10.1152/ajpheart.01020.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosuge M, et al. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ J. 2007;71:186–90. doi: 10.1253/circj.71.186. [DOI] [PubMed] [Google Scholar]
- 8.Dong Z, et al. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med. 2011;17:1357–64. doi: 10.2119/molmed.2011.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, Wang JM. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koenen RR, Weber C. Chemokines: established and novel targets in atherosclerosis. EMBO Mol Med. 2011;3:713–25. doi: 10.1002/emmm.201100183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apostolakis S, Amanatidou V, Spandidos DA. Therapeutic implications of chemokine-mediated pathways in atherosclerosis: realistic perspectives and utopias. Acta Pharmacol Sin. 2010;31:1103–10. doi: 10.1038/aps.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee HY, et al. Serum amyloid A induces CCL2 production via formyl peptide receptor like 1-mediated signaling in human monocytes. J Immunol. 2008;181:4332–9. doi: 10.4049/jimmunol.181.6.4332. [DOI] [PubMed] [Google Scholar]
- 13.Lee HY, Kim SD, Shim JW, Yun J, Kim K, Bae YS. Activation of formyl peptide receptor like-1 by serum amyloid A induces CCL2 production in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2009;380:313–7. doi: 10.1016/j.bbrc.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 14.Boisvert WA, Curtiss LK, Terkeltaub RA. Interleukin-8 and its receptor CXCR2 in atherosclerosis. Immunol Res. 2000;21:129–37. doi: 10.1385/ir:21:2-3:129. [DOI] [PubMed] [Google Scholar]
- 15.He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–81. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 16.Chiang N, et al. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–87. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 17.Dufton N, et al. Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–9. doi: 10.4049/jimmunol.0903526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen K, et al. A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol. 2010;184:3331–5. doi: 10.4049/jimmunol.0903022. [DOI] [PMC free article] [PubMed] [Google Scholar]