

Abstract
Human NEIL2, one of five oxidized base-specific DNA glycosylases, is unique in preferentially repairing oxidative damage in transcribed genes. Here we show that depletion of NEIL2 causes a 6- to 7-fold increase in spontaneous mutation frequency in the HPRT gene of the V79 Chinese hamster lung cell line. This prompted us to screen for NEIL2 variants in lung cancer patients’ genomic DNA. We identified several polymorphic variants, among which R103Q and R257L were frequently observed in lung cancer patients. We then characterized these variants biochemically, and observed a modest decrease in DNA glycosylase activity relative to the wild type (WT) only with the R257L mutant protein. However, in reconstituted repair assays containing WT NEIL2 or its R257L and R103Q variants together with other DNA base excision repair (BER) proteins (PNKP, Polβ, Lig IIIα and XRCC1) or using NEIL2-FLAG immunocomplexes, an ~ 5-fold decrease in repair was observed with the R257L variant compared to WT or R103Q NEIL2, apparently due to the R257L mutant’s lower affinity for other repair proteins, particularly Polβ. Notably, increased endogenous DNA damage was observed in NEIL2 variant (R257L)-expressing cells relative to WT cells. Taken together, our results suggest that the decreased DNA repair capacity of the R257L variant can induce mutations that lead to lung cancer development.
1. INTRODUCTION
Lung cancer is the leading cause of cancer-related death in both men and women, claiming ~1.4 million lives globally (WHO) and 157,300 in the United States in 2010 [1]. In most cases, tumors are detected at advanced stages, and the overall 5-year survival rate is only 15 percent. Lung cancer is broadly classified as either non-small-cell (NSCLC) or small-cell carcinoma (SCLC) [2], [3]. NSCLC, which constitutes ~80% of all cases, grows more slowly; SCLC grows quickly and is always caused by smoking. NSCLC has 3 subtypes: adeno, squamous cell and large-cell carcinoma. Lung cancer is primarily an environmental disease, with cigarette smoke being the primary cause. Cigarette smoke contains thousands of chemicals, many of which are known carcinogens. Exposure to radon gas is another major cause of lung cancer. Radon is an alpha particle emitter; when inhaled, their decay will target cells in the respiratory epithelium, causing damage to their genetic material [4, 5]. One of the very early steps in carcinogenesis due to exposure to these hazards is the generation of reactive oxygen species (ROS). ROS-induced mutations are a known prerequisite for many diseases, particularly cancer [6-8].
ROS-induced oxidation of DNA is complex, leading to single-strand breaks (SSBs) and a multitude of modifications to DNA bases, many of which are highly mutagenic and/or toxic [9, 10]. Oxidized base lesions in DNA are repaired via a highly conserved multistep process, the base excision repair (BER) pathway that is initiated by excision of the damaged base by a member of a class of enzymes called DNA glycosylases [11-13]. Five oxidized base-specific DNA glycosylases have been identified and characterized so far in human cells. 8-oxoguanine-DNA glycosylase (OGG1) and endonuclease III homolog 1 (NTH1) were characterized initially and shown to preferentially excise oxidized purines and pyrimidines, respectively [14, 15]. Several years later we and others identified NEIL (Nei-like) 1-3, whose products share conserved motifs with E. coli MutM or Nei [16-20] and excise both purine and pyrimidines oxidation products. The NEILs are distinct from NTH1 and OGG1 both in their structural features and reaction mechanisms [16, 17]. Upon recognition of an oxidized base, the N-glycosylic bond is cleaved by a DNA glycosylase, releasing the free base, followed by cleavage of the phosphodiester backbone by an associated AP lyase activity, which leaves a blocked 3’-terminus in the resulting nick [21, 22]. The block is removed by the 3′ end-cleaning activity of either AP endonuclease or polynucleotide kinase 3′ phosphatase (PNKP) [23-25]. We have shown that OGG1/NTH1-initiated repair is dependent on APE1, whereas NEIL1- and -2-mediated repair involves PNKP [25, 26]. The resulting gap in the lesion-containing strand is then filled in by DNA polymerase. Finally, the remaining nick in the repaired strand is sealed by DNA ligase [13, 27].
Unlike OGG1 and NTH1, which are active only with duplex DNA, NEIL1 and NEIL2 excise lesions from DNA bubble structures or single-stranded (ss) DNA [28], which are generated transiently during DNA replication and transcription. Our recent studies have indicated that NEIL1 is primarily involved in the repair of replicating genomes [29, 30], and that NEIL2 primarily removes the oxidized bases from transcribing genes via transcription-coupled BER (TC-BER)[31]. This repair pathway is particularly important in terminally differentiated non-dividing cells, the majority of cells in adult mammals. In these cells, mutation fixation by replication is not a concern, so only repair of the transcribed strand in functional genes is necessary to maintain a functional transcriptome and to prevent the synthesis of mutant RNAs and proteins [32, 33].
We thus reasoned that a deficiency in NEIL2-dependent repair pathways would have severely deleterious consequences. Based on this hypothesis, we examined the consequences of NEIL2 depletion in the V79 Chinese hamster lung cell line and observed a significant increase in spontaneous mutation frequency in those cells, implicating NEIL2 in preventing mutations linked to carcinogenesis. This observation prompted us to screen for NEIL2 genetic variations in the genomic DNA from lung cancer patients. We identified two such variants that were common in lung cancer patients. One of these variants had reduced association with NEIL2’s interacting partners for BER, resulting in inefficient repair of oxidized bases and accumulation of endogenous genomic damage. Based on these results we propose that persistent genomic damage due to functional deficiency of this NEIL2 variant could contribute to lung carcinogenesis.
2. MATERIALS AND METHODS
2.1 Study subjects
For our initial study (Table 1), we obtained genomic DNA from 20 lung cancer patients (European-American) from The Human Tumor Bank Core at UTMB. We then received additional samples of 99 European-American and 52 Chinese-American lung cancer patients from the City of Hope (CA). Control genomic DNA from 200 healthy individuals (European-American) was purchased from Sigma.
Table 1.
SNP analysis in NEIL2 in lung cancer patients
| Exon | SNP ID | NT changes | AA changes | Cases (EA, n=119) n (%, MAF) | Controls (EA, n=200) n (%, MAF) | Case (CHA, n=52) n (%, MAF) | (database, dbSNP) MAF
|
|
|---|---|---|---|---|---|---|---|---|
| CEU | Chinese | |||||||
| 2 | NA | A>T | H12L | 1 (0.8, 0.004) | — | — | — | — |
| 3 | NA | G>A | E77K | 1 (0.8, 0.004) | — | — | — | — |
| 3 | rs8191613 | G>A | R103Q | 6 (5, 0.025) | 3(1.5,0.0075) | 25 (48, 0.24) | 0.017 | 0.22 |
| 4 | rs8191666 | A>C | P123T | 1 (0.8, 0.004) | — | — | 0.0013 | — |
| 5 | rs8191664 | G>T | R257L | 10 (8, 0.04) | 4 (2, 0.01) | 26 (50, 0.25) | 0.013 | 0.25 |
NT, nucleotide; AA, amino acid; EA, European-American; CHA, Chinese-American; NA, Not applicable; MAF, Minor allele frequency; —, Not available; CEU, Caucasian
For studies involving the Chinese population (Tables 2-4 and Supplementary Table 1), the case-control study consisted of 670 patients with lung cancer and 666 population controls. All subjects were ethnic Han Chinese living in Beijing and the surrounding regions. Patients were recruited from January 2005 to January 2007 at the Cancer Hospital, Chinese Academy of Medical Sciences (Beijing, China). The response rate of patients was 100%. Control subjects were cancer-free individuals living in the same region as the cancer patients, with a 96% response rate. The cases and controls were frequency-matched by sex and age.
Table 2.
Distributions of select characteristics in lung cancer cases and control subjects (Chinese)
| Variable | Lung cancer cases (n = 670) | Controls (n = 670) | ||
|---|---|---|---|---|
| No. | (%) | No. | (%) | |
| Sex | ||||
| Male | 467 | 69.70 | 467 | 69.70 |
| Female | 203 | 30.30 | 203 | 30.30 |
| Age | ||||
| ≤50 | 125 | 18.66 | 124 | 18.51 |
| 51-60 | 212 | 31.64 | 221 | 32.99 |
| 61-65 | 116 | 17.31 | 112 | 16.72 |
| >65 | 217 | 32.39 | 213 | 31.79 |
| Smoking status | ||||
| Nonsmoker | 284 | 42.39 | 477 | 71.19 |
| Smoker | 386 | 57.61 | 193 | 28.81 |
| Smoking level (pack-years) | ||||
| ≤15 | 78 | 20.21 | 67 | 34.72 |
| 15-30 | 121 | 31.35 | 75 | 38.86 |
| 30-40 | 78 | 20.21 | 35 | 18.13 |
| >40 | 109 | 28.24 | 16 | 8.29 |
| Tumor stage at diagnosis | ||||
| I | 170 | 25.37 | ||
| II | 107 | 15.97 | ||
| III | 249 | 37.16 | ||
| IV | 73 | 10.90 | ||
| Unknown | 71 | 10.60 | ||
| Histological type | ||||
| Adenocarcinoma | 245 | 36.57 | ||
| Squamous-cell carcinoma | 252 | 37.61 | ||
| Small cell carcinoma | 70 | 10.45 | ||
| Others | 60 | 8.96 | ||
| Unknown | 43 | 6.42 | ||
Two-sided χ2 test.
Table 4.
Genotype frequencies of NEIL2 among cases (Chinese) of various histological types
| Genotypes | Controls | AC, (%) | OR* (95% CI) | SC, (%) | OR* (95% CI) | SCC, (%) | OR* (95% CI) | O, (%) | OR* (95% CI) |
|---|---|---|---|---|---|---|---|---|---|
| rs8191664 | |||||||||
| GG (WT/WT) | 461 (69.22) | 160 (65.31) | 1.00 (reference) | 160 (63.49) | 1.00 (reference) | 52 (74.29) | 1.00 (reference) | 31 (51.67) | 1.00 (reference) |
| GT (heterozygous) | 178 (26.73) | 76 (31.02) | 1.36 (0.98-1.91) | 88 (34.92) | 1.50 (1.05-2.14) | 15 (21.43) | 0.89 (0.51-1.56) | 26 (43.33) | 2.11 (1.21-3.67) |
| TT (Var/Var) | 27 (4.05) | 9 (3.67) | 0.89 (0.59-1.36) | 4 (1.59) | 1.08 (0.69-1.61) | 3 (4.29) | 0.99 (0.25-3.67) | 3 (5.00) | 1.62 (0.43-6.10) |
Data were calculated by unconditional logistic regression, adjusted for sex, age, and smoking status.
Histological types: AC, adenocarcinoma; SC, squamous-cell carcinoma; SCC, small cell carcinoma; O, other histological types.
2.2 Genotyping
For genotyping of samples available from UTMB and the City of Hope, all four NEIL2 exons were PCR-amplified and sequenced in UTMB’s core facilities. Electropherograms were aligned with STADEN software to identify the mutations. However, for the Chinese population genomic DNA was extracted from blood samples. The classical PCR-based RFLP method was used to genotype (rs8191664) the R257L variant. The forward and reverse primers for PCR were 5’-CCCCGCTTTATTTCAAGGAACATCATT-3’ and 5’-CACCACGTGATCCACTAGGACCTGC-3’, respectively, yielding a product of 123bp. A 10 μl reaction mixture was used comprising 100 ng DNA, 0.2 μM each primer, 0.3 mM each deoxynucleotide triphosphate, 2.0 mM MgCl2, and 0.5 units of Taq DNApolymerase with 1.0 μl 1×Reaction Buffer (TAKARA). The PCR was performed beginning with an initial melting step of 5 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 57°C, and 30 s at 72°C, and a final elongation step of 7 min at 72°C. The PCR product was then digested with the restriction enzyme PstI (MBI). Genotype TT was completely digested into two fragments as 22bp and 101bp respectively.
2.3 Statistical analysis
We used the χ2 test to examine differences in demographic variables, smoking, and distribution of genotypes between case and control groups. The associations between the genotype and risk of lung cancer were estimated by calculating odds ratios (ORs) and 95% confidence intervals (CIs) with unconditional logistic regression models. The ORs were adjusted for age, sex, and smoking status. The number of pack-years was used to represent the cumulative cigarette dose level (pack-years = cigarettes per day/20 × years of smoking). The multiplicative gene-smoking joint effect was tested. Stratification analysis in terms of the histological types of lung cancer was also performed. All analyses were done with Statistical Analysis System software (version 9.0; SAS Institute, Cary, NC.)
2.4 DNA damage estimation by long amplicon quantitative PCR (LA-QPCR)
Human lung epithelial BEAS-2B (purchased from ATCC) and HEK293 (human embryonic kidney) cells stably expressing WT NEIL2-FLAG or its R257L variant-FLAG were generated as described previously [31, 34] and maintained in DMEM/F12 (1:1) and DMEM media, respectively. The cells were harvested for genomic DNA extraction using the QIAGEN Genomic-tip 20/G kit (Qiagen) per the manufacturer’s directions. This kit is particularly useful, as it minimizes DNA oxidation during the isolation step and has been previously used for LA-QPCR assays [35, 36]. After quantitation by Pico Green (Molecular Probes) in a 96-well plate, the genomic DNA was digested with the E. coli enzymes Fpg and Nei to induce strand breaks at the sites of unrepaired oxidized base lesion sites. Gene-specific LA-QPCR assays for measuring DNA damage were performed as described earlier [35] using LongAmp Taq DNA Polymerase (New England BioLabs) to amplify a 10.4 kb region of the HPRT or 12.2 kb of the POLB gene in human genomic DNA using the following primers: 5′-TGG GAT TAC ACG TGT GAA CCA ACC-3′ and 5′-GCT CTA CCC TCT CCT CTA CCG TCC-3′ for HPRT and 5′-CAT GTC ACC ACT GGA CTC TGA AC-3′ and 5′-CCT GGA GTA GGA ACA AA ATT GCT-3′ for POLB [35]. Preliminary assays were carried out to ensure the linearity of PCR amplification with respect to the number of cycles and DNA concentration. Since amplification of a small region would be independent of DNA damage, a small DNA fragment for each gene (HPRT and POLB) was also amplified for normalization of amplification of large fragments using the following primers: 5′-TGC TCG AGATGT GAT GAA GG-3′ and 5′-CTG CAT TGT TTT GCC AGT GT-3′ for HPRT and 5′-AGT GGG CTG GAT GTA ACCTG-3′ and 5′-CCA GTA GAT GTG CTG CCA GA-3′ for POLB. The amplified products were then visualized on gels and quantitated with UN-SCAN-IT gel (Version 6.1) automated digitizing system.
2.5 Mutation analysis at the hprt Locus in V79 Cells
The ability of 6-thioguanine (6-TG)-resistant (TGR) cells to synthesize DNA in the presence of 6-TG is a reliable measure of hprt mutation. For mutant frequency analysis, the sense or antisense oligo-treated cells are washed three times with PBS, subcultured into 100-mm dishes (5×105 cells/dish) in growth medium containing 6-TG (7 μg/ml) to identify presumptive mutants, or in nonselective medium to determine the plating efficiency. Colonies of 6-TG-resistant cells after growth for 6 to 8 days are fixed in 3.7% formalin, and stained with 0.1% crystal violet. The hprt mutant frequency is calculated from the number of 6-TG-resistant colonies relative to the total colonies in non-selecting medium [37, 38].
3. RESULTS
3.1 Enhanced endogenous mutation frequency in NEIL2-deficient cells
To test the role of NEIL2 in preventing endogenous mutations, we examined the consequences of NEIL2 deficiency on the HPRT locus in Chinese hamster V79 lung cells. These cells are hemizygous for the HPRT gene, permitting a screen for resistance to 6-thioguanine (6-TG) resulting from mutation of the HPRT gene [39]. To decrease the NEIL2 level specifically, the cells were treated with antisense DNA (to NEIL2 mRNA) or control DNA (Fig 1A; see also Supplementary Methods). The cells were then treated with 6-TG and the mutation frequencies were calculated from the number of 6-TG-resistant colonies relative to the number of cells seeded, after correcting for the plating efficiency without the selective agent [38]. Fig 1B shows that NEIL2 depletion induced an ~6-fold increase in mutation frequency in cultured cells, suggesting a critical role for NEIL2 in maintaining genomic integrity, consistent with its function in TC-BER of endogenous oxidatively damaged bases [31].
Fig 1.

A. Antisense oligonucleotide-mediated depletion of NEIL2 in Chinese hamster V79 cells. Total RNA was isolated from cells treated with antisense or control oligonucleotide or from non-treated cells (NT), and NEIL2 mRNA levels were measured by Q-RTPCR. A.U., arbitrary units. B. Increased mutation frequency at the hprt locus in V79 cells, either non-treated (NT) or treated with NEIL2 control or antisense oligo. The bar graphs represent the means ± standard error from 4 independent experiments.
3.2 Identification of nonsynonymous SNPs in lung cancer patients
The NEIL2 gene, which is located on chromosome 8 at 8p23.1, is comprised of 4 coding exons. Sequencing of the PCR-amplified products of the 4 exons from genomic DNA isolated from 171 lung cancer patients (119 European-American and 52 Chinese-American) and 200 healthy controls (European-American) enabled us to identify various nonsynonymous polymorphisms in the coding regions of NEIL2 gene (Table 1). Of these, R103Q, R257L and P123T are present in the dbSNP databases; however, H12L and E77K have not been reported thus far. Using information from several other databases (such as dbSNPs), the ethnic population-based Minor Allele Frequency (MAF) of these nonsynonymous SNPs (Table 1) indicated that all these SNPs are rare (MAF <0.01) in the majority of ethnic groups, except R103Q and R257L, which have a higher MAF (>0.25) in the Chinese population. We further assessed the inheritance of these 2 SNPs in the Chinese population, using data retrieved from the 1000 Genomes Project (http://www.1000genomes.org/). The physical distance between rs8191613 (R103Q) and rs8191664 (R257L) is 6218bp. Haplotype analysis suggested that these 2 SNPs are tightly linked (r2>0.95 for rs8191613 and rs8191664) and inherited together in the Chinese population (Fig S1).
3.3 Poor BER activity of R257L is due to its lack of association with downstream repair proteins
Since we could not detect any major functional (biochemical) difference in the activity of H12L, E77K and P123T (data not shown), we focused on the other 2 variants (R103Q and R257L) for further studies. For biochemical characterization of the variants, we purfied the recombinant proteins (Fig S2, Supplementary Methods) and examined their DNA glycosylase and total repair activity. R257L showed a modest decrease (~1.5- fold) in DNA glycosylase activity vs. that of WT NEIL2 (Fig 2A, lane 2 vs. 3), while R103Q (lane 4) showed no change in activity. To further examine R257L’s role, we also tested total BER by the variants. Repair was monitored by analyzing the incorporation of α32P-dCMP into a duplex oligo (51-mer) containing a single AP-site. We have shown previously that NEIL2 has poor DNA glycosylase activity with duplex DNA containing oxidized lesions (e.g., 5-OHU) [28]; however, AP site-containing duplex DNA is an excellent substrate for NEIL2, which is the reason for using an AP site-containing oligo in our complete BER assay. We found that the repair involving WT and R103Q (Fig 2B, lanes 2 and 4) was comparable, but that with R257L (lane 3) was markedly decreased (by ~5-fold). This large difference in total BER cannot be explained based on the modest (~1.5-fold) decrease in the DNA glycosylase activity of R257L (Fig 2A). We then generated human HEK293 cells stably expressing C-terminally FLAG-tagged, enzymatically active WT or R257L NEIL2 with comparable expression levels. We have previously shown that a NEIL2-FLAG immunocomplex (IC) is repair-proficient [31]. We therefore isolated ICs from the nuclear extracts of WT and R257L; the FLAG-complexes were eluted using FLAG peptides and tested for complete BER. For repair assays, total protein (0.5μg) and the level of FLAG in the WT vs. R257L IC were adjusted to contain equal amounts. Fig 2C shows that the WT NEIL2-FLAG complex was repair-proficient (lane 1), but R257L-FLAG was not (lane 2). However, when the latter complex was supplemented with PNKP, Polβ, Lig IIIα and XRCC1 (lane 3, necessary for repair completion), total repair was comparable to that of WT (lane 1 vs. 3).
Fig 2.

A. A 5’ 32P-labeled 51-mer oligo (5-OHU.B11, 1 pmol), was used for DNA glycosylase/AP lyase assay [28] with purified (0.2 pmol) WT (lane 2) or R257L (lane 3) or R103Q (lane 4) NEIL2. Lane 1, no protein. Quantitation of the radioactive bands (lanes 2-4) is represented in a histogram (bottom), with lane 2 arbitrarily set as 1. B. Reconstitution of BER. Complete repair of AP site-containing duplex oligo (10 pmol, top) was measured by incorporation of α32P-dCMP using purified (0.25 pmol) WT or the NEIL2 variants and other BER proteins (50 fmol each) as indicated. Lane 1 = control, non-damaged oligo. C. BER using NEIL2-FLAG IP. Complete repair of AP site-containing duplex oligo (10 pmol, top) was measured by incorporation of α32P-dCMP using a FLAG-pulldown complex (0.5μg) of WT (lane 1), R257L (lane 2) or R257L supplemented with a mix of purified PNKP, Pol β, Lig IIIα and XRCC1 (lane 3, 50 fmol of each). Lane 4, mix only.
To further examine the cause of decreased repair activity with the R257L variant, we analyzed its IC for the presence of NEIL2-associated proteins by Western blotting, and found that Lig IIIα, PNKP and Polβ are indeed all present in lower amounts than in the WT complex (Fig 3A). We have shown previously that NEIL2 associates with RNAP II and is involved in TC-BER [31], so we also tested the association of RNAP II with the variant. Fig 3A shows that the association of RNAP II with both WT and variant NEIL2 was similar. To further confirm the association of those proteins with WT or variant NEIL2, we used an in situ proximity ligation assay (PLA) in which the close physical association of two proteins is visualized by a fluorescent signal. This is a new approach to study interaction of endogenous proteins [40, 41]. We used the Duolink kit per the manufacturer’s protocol (Olink Bioscience, Uppsala, Sweden), and found that WT NEIL2 associated closely with RNAP II, Polβ, PNKP and Lig IIIα. However, while the association of the variant with RNAP II was comparable to that of WT, it was decreased with Lig IIIα and PNKP, and more significantly with Polβ (Fig 3B). As a control we have also tested the interaction between WT or variant NEIL2 (mouse) with IgG (rabbit); no fluorescent signal was observed. We have shown previously that the NEIL2 IC isolated from cell extract is proficient in repair, and all the necessary proteins for repair completion, including PNKP, are present in the complex [26, 31]. PLA thus both confirmed those previous data and was also consistent with the IP results in Fig 3A. Taken together, these data clearly indicate that R257L-mediated repair is mostly decreased due to a lack of association with other downstream BER proteins.
Fig 3.

A. Representative Western analysis showing the levels of FLAG and association of other proteins in the eluted complexes (WT vs. R257L variant). B. Detection of WT vs. variant NEIL2-FLAG (mouse Ab) interaction with RNAP II /Lig IIIα/PNKP/Polβ (rabbit Ab) in cultured cells by Proximity Ligation Assay. WT or variant NEIL2 (mouse Ab) with IgG (rabbit Ab), as controls.
3.4 The association between individual polymorphisms and lung cancer risk
Because R257L showed a significant functional impairement and had a higher MAF for the Chinese population, we further analyzed ~670 cancer samples and an equal number of matched controls to confirm an association of R257L with lung cancer in the Chinese population, and found that R257L was indeed more frequent in lung cancer patients. The characteristics of case and control subjects are summarized in Table 2, showing that the two groups were frequency-matched by sex and age. The difference between patients and controls in terms of smoking status and smoking levels is statistically significant. The genotyping results (Table 3) show that the percentages of the three genotypes in the cancer group were 64.33 (GG, homozygous WT), 32.39 (GT, heterozygous) and 3.28 (TT, homozygous R257L), respectively, compared with 69.22 (GG), 26.73 (GT) and 4.05 (TT) in the control group. The result in the control group was confirmed by the Hardy–Weinberg equilibrium (Phwe>.05). The unconditional logistic regression model was used to estimate the association between genotypes and risk of lung cancer (Table 3). It was observed that the OR of the rs8191664 GT genotype in lung cancer was 1.33 (95% CI: 1.03-1.71) compared with the GG genotype. However the OR of rs8191664 TT genotype compared with GG genotype did not suggest it as a risk factor, perhaps due to a limited statistical power because of the relatively low frequency of the TT genotype. Stratification analysis showed that genotype GT was a risk factor compared with genotype GG for lung squamous-cell carcinoma, but not for adenocarcinoma or small-cell carcinoma (Table 4). A joint effect of genotype and smoking was not observed (Supplementary Table 1). The unusually high percentage of non-smokers having lung cancer may be due to their exposure to second-hand smoke which has an established linkage with lung cancer. It has been reported that living with a smoker increases a nonsmoker’s chances of developing lung cancer by 20 to 30 percent [42]; these data come from Americans, while a majority of our subjects are from China, where the percentage of smokers is much higher than in the U.S. Also, lung cancer in non-smokers could result from exposure to other environmental carcinogens or pollutants, whose levels are again higher in the Beijing and surrounding areas than in most parts of the U.S.
Table 3.
Genotype frequencies of NEIL2 among cases and controls (Chinese) and their association with lung cancer
| Genotype | Cases (n = 670, MAF = 0.19) | Controls (n = 666, MAF = 0.17) * | OR(95% CI) † |
|---|---|---|---|
| n (%) | n (%) | ||
| rs8191664 | |||
| GG (WT/WT) | 431 (64.33) | 461 (69.22) | reference |
| GT (heterozygous) | 217 (32.39) | 178 (26.73) | 1.33 (1.03,1.71) |
| TT (Var/Var) | 22 (3.28) | 27 (4.05) | 0.99 (0.54,1.84) |
Genotyping results of 4 subjects in control group are missing due to PCR failure.
ORs and 95% CIs were calculated using logistic regression model with genotype GG as the reference and adjusted for sex, age, and smoking status.
3.5 The R257L variant accumulates a higher amount of endogenous oxidative DNA damage in the transcribed genes
To examine whether the decreased repair capacity of the variant affects the accumulation of oxidative base damage, we depleted endogenous NEIL2 (by ~70%) using 3′-UTR-specific siRNA (Fig 4A) in both BEAS-2B and HEK293 cells. The cellular DNA was then isolated, and the DNA samples were treated with E. coli Fpg/Nei before PCR to excise oxidized bases and generate SSBs after excision of the damaged bases, preventing PCR amplification. The levels of base damage in the HPRT and POLB genes in WT- vs. R257L-expressing cells were compared using long amplicon quantitative PCR (LA-QPCR). We selected these genes because the LA-QPCR conditions for amplification of a long region (~10-12kb) with the proper set of primers had already been well standarized by Van Houten’s group [35]. A decrease in the PCR product will reflect a higher DNA damage level, and we indeed consistently found accumulation of a higher level of oxidative DNA damage in the genomic DNA of the R257L variant-expressing cells than in DNA of the WT-expressing cells (Fig 4B) when a long region (~10-12 kb) was amplified. However, amplification of a smaller fragment for each gene was similar between the samples, because the probability of damage in a small fragment is low. Hence, the relative PCR amplification after normalization to the small fragment for each gene was found to be significantly lower in both cell lines (HEK 293 and BEAS-2B) expressing the variant. These data thus indicated that the variant accumulated a higher amount of endogenous genome damage, further supporting our previous data.
Fig 4.
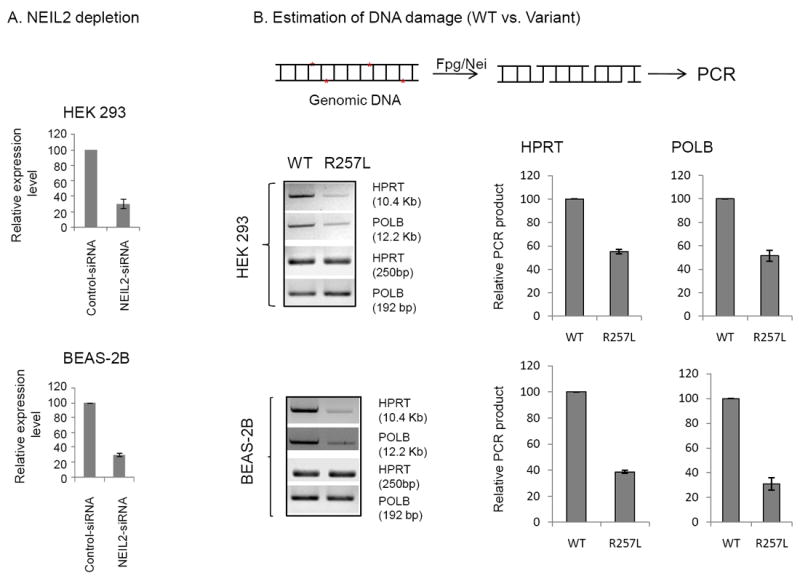
A. NEIL2 transcript levels in control vs. 3’ UTR-specific siRNA-treated cells, quantitated by qPCR. B. Long-range qPCR was used to evaluate genomic DNA damage levels in the WT- and the R257L-expressing cells. Representative gel showing PCR-amplified fragments of HPRT and POLB genes. Amplification of the large fragment was normalized to the amplification product of a small fragment of the corresponding gene. Quantitation of the amplified products is represented in a histogram with the WT arbitrarily set as 100. Upper panel, HEK 293; lower panel, BEAS-2B cells. Other details are described in the Materials and Methods section.
4. DISCUSSION
Although the etiological basis of lung cancer is not fully understood, genetic predisposition and environmental factors are thought to play critical roles. Lung cancer is primarily viewed as an environmental disease, with cigarette smoke being the primary cause. However, ~15-20% of lung cancer cases worldwide are not attributable to tobacco use [43] which suggests that individuals might differ in their susceptibility to environmental risk factors. Genetic variations, including single nucleotide polymorphisms, have the potential to exert profound effects on gene function and/or expression and consequent phenotypes in the human population. One of the strategies to prevent lung cancer should be to screen and identify “at risk” individuals and encourage them to change their lifestyles and enter enhanced clinical surveillance and care. Many studies showed the association of polymorphic variants of various genes with lung cancer; in addition, associations of mutations in DNA repair genes with specific cancers are now being discovered [44, 45]. Several variants (R103Q, R103W, P123T and R257L) in NEIL2 gene have been identified in colorectal cancer [46], suggesting that they are risk factors for colon cancer. A SNP in the 5′ regulatory region of the NEIL2 gene has also been shown to be associated with oral cancer [47]. However, the functional properties of those modifications were not investigated.
Here we initially identified two polymorphic variants of NEIL2 (R103Q and R257L) that occur more frequently in lung cancer patients (European-American) than normal individuals (Table 1). We characterized these variants biochemically in vitro and in cells, and found that significantly less repair is initiated by the R257L variant (~5- to 6-fold) than by WT NEIL2 or R103Q. We then extended our studies further with the Han Chinese population, and found that the R257L variant is indeed more frequent in lung cancer patients in that population group as well (Tables 2-4). The poor repair activity of R257L can be attributed to lower association of the variant with downstream BER proteins. This caused accumulation of higher level of endogenous genomic damage in R257L-expressing cells than in those expressing WT NEIL2. Thus NEIL2’s genetic alterations may influence the level of persistent genomic damage, which is an early and critical event in inducing mutations in tumor suppressor and/or oncogenes, leading to oncogenesis. How changing a single amino acid in this NEIL2 variant affects its interactions with other BER proteins is an enigma. Solving the crystal structures of both the WT and variant NEIL2 will help our understanding of the molecular mechanism of the protein’s DNA damage recognition, and its interaction with other proteins.
Most of the coding nonsynonymous SNPs of NEIL2 reported here are rare in the the normal populations of major ethnic groups. However, R103Q and R257L showed higher levels even in the normal Chinese population. Hence, the strong association of the R257L variant with Chinese lung cancer patients clearly indicates that heterozygous G/T is a significant risk factor for lung cancer. Moreover, stratification analysis using logistic regression suggests that G/T is more significantly associated with squamous cell carcinoma (OR 1.50) and other histological lung carcinomas (OR 2.11) than with adenocarcinoma and non- small-cell carcinoma. Thus R257L may be a risk factor for certain histologically different types of lung cancer in the Chinese population, where occurrence of this SNP is much higher in the population. No alteration in the known molecular functions of R103Q NEIL2 variant was observed. However, other unknown molecular function of R103Q cannot be ruled out. One important point to make here is that the smokers carrying the R257L variant did not show any increase in their overall risk for developing lung carcinoma. Other risk factors (e.g. environmental, nutritional, etc) may be involved in this population group for lung pathogenesis. Generation of knock-in mice may provide a deeper understanding of the physiological significance of the variant in inducing lung cancer.
The overall frequency of both R257L and R103Q is quite low in the Caucasian control population (MAF =0.018). However, our preliminary analysis of a limited sample suggests R257L’s strong association with lung cancer among Caucasians (Table 1). A large number of cases and controls need to be evaluated to draw any further conclusion as to whether this variant also poses a risk factor for squamous cell lung carcinoma in the Caucasian or other ethnic populations.
Finally, we are aware that most of the variations found in our studies are heterozygous. A major question is the dominant phenotype of the variant in vivo. It is possible that the mutant allele exerts its effect either through haplo-insufficiency or by interfering with the function of WT protein. While both alleles in heterozygous cells are usually expressed equally, several reports have documented that differential expression of alleles is common in the human genome [48-50]. Allele-specific expression is modulated by both genetic and epigenetic changes. A link between asymmetric expression of alleles and disease susceptibility is now well recognized [51]. We will investigate this complex issue in our future studies. In conclusion, it appears that many genes etiologically linked to lung pathogenesis, which is highly complex, could be identified by systematic genomic analysis.
Supplementary Material
Highlights.
R257L and R103Q, the two variants of NEIL2, predominantly present in patients with squamous cell lung carcinoma, are linked and inherited together in the population.
R257L shows only a modest decrease (~1.5 fold) in DNA glycosylase activity; however, the total BER activity is significantly decreased (~5 fold) compared to the wild type enzyme.
Decreased interaction of the R257L variant with downstream proteins in the BER pathway, particularly with DNA polymerase β, contributes to reduced repair.
R257L-expressing cells accumulate significant amounts of endogenous DNA damage.
Depletion of NEIL2 in Chinese hamster lung V79 cells induced higher spontaneous mutation frequency, indicating the role of NEIL2 in repairing endogenous, oxidative genome damage.
Acknowledgments
This research was supported by USPHS grants CA102271 and ES017353 (T.K.H.), and CA81063 (Sankar Mitra.), and ES 012512 and CA92584 (A.E.T), CA94160 (J.X.) and R21CA143583 (B.S). We acknowledge the generous help of Drs. Chandrasekha Yallampalli for allowing us to use Fluorescent Microscope and Michael Weinfeld for giving us PNKP Ab. We thank Dr. David Konkel for critically editing this manuscript.
ABBREVIATIONS
- AP site
apurinic/apyrimidinic site
- BER
Base excision repair
- Fpg
Formamidopyrimidine [fapy]-DNA glycosylase
- HPRT
Hypoxanthine phosphoribosyltransferase
- IC
Immunocomplex
- LA QPCR
Long amplicon quantitative PCR
- Lig IIIα
Ligase IIIα
- MAF
Minor Allele Frequency
- NEIL2
Nei-like 2
- NSCLC
non-small-cell lung carcinoma
- NTH1
Endonuclease III homolog 1
- OGG1
8-oxoguanine-DNA glycosylase
- OR
Odd ratio
- PLA
Proximity ligation assay
- PNKP
polynucleotide kinase 3’-phosphatase
- Polβ
Polymerase β
- RFLP
Restriction fragment length polymorphism
- RNAPII
RNA polymerase II
- SCLC
small-cell lung carcinoma
- SNP
Single nucleotide polymorphism
- SSBs
Single-strand breaks
- TC-BER
Transcription-coupled BER
- 6-TG
6-thioguanine
- UTR
Untranslated region
- WT
Wild Type
- XRCC1
X-ray repair cross-complementing protein 1
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Marchetti A, Martella C, Felicioni L, Barassi F, Salvatore S, Chella A, Camplese PP, Iarussi T, Mucilli F, Mezzetti A, Cuccurullo F, Sacco R, Buttitta F. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J Clin Oncol. 2005;23:857–865. doi: 10.1200/JCO.2005.08.043. [DOI] [PubMed] [Google Scholar]
- 3.Zochbauer-Muller S, Gazdar AF, Minna JD. Molecular pathogenesis of lung cancer. Annu Rev Physiol. 2002;64:681–708. doi: 10.1146/annurev.physiol.64.081501.155828. [DOI] [PubMed] [Google Scholar]
- 4.Samet JM. Radiation and cancer risk: a continuing challenge for epidemiologists. Environ Health. 10(Suppl 1):S4. doi: 10.1186/1476-069X-10-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagoner JK, Archer VE, Lundin FE, Jr, Holaday DA, Lloyd JW. Radiation as the Cause of Lung Cancer among Uranium Miners. N Engl J Med. 1965;273:181–188. doi: 10.1056/NEJM196507222730402. [DOI] [PubMed] [Google Scholar]
- 6.Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radical Biology and Medicine. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 7.Ames BN, Gold LS, Willett WC. The causes and prevention of cancer. Proc Natl Acad Sci U S A. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Rev Physiol Biochem Pharmacol. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 9.Wallace SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res. 1998;150:S60–79. [PubMed] [Google Scholar]
- 10.Ward JF. The complexity of DNA damage: relevance to biological consequences. International Journal of Radiation Biology. 1994;66:427–432. doi: 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- 11.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochemical Journal. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 13.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington,DC: 2006. [Google Scholar]
- 14.Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, Sarker AH, Seki S, Mitra S. Purification and charcterization of human hNTH1, a homolog of Escherichia coli endonuclease III: Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem. 1998;273:21585–21593. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- 15.Lu R, Nash HM, Verdine GL. A DNA repair enzyme that excises oxidatively damaged guanines from the mammalian genome is frequently lost in lung cancer. Current Biology. 1997;7:397–407. doi: 10.1016/s0960-9822(06)00187-4. [DOI] [PubMed] [Google Scholar]
- 16.Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 18.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA repair. 2002:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 19.K S, Takao M, Kobayashi K, Zhang QM, Yonei S, Van Der Horst GT, Yasui A. A back-up glycosylase in Nth1-knockout mice is a functional Nei (endonuclease VIII) homologue. J Biol Chem. 2002 doi: 10.1074/jbc.M206884200. in press. [DOI] [PubMed] [Google Scholar]
- 20.Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, Wallace SS. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A. 2010;107:4925–4930. doi: 10.1073/pnas.0908307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dodson ML, Michaels ML, Lloyd RS. Unified catalytic mechanisms for DNA glycosylases. J Biol Chem. 1994;269:32709–32712. [PubMed] [Google Scholar]
- 22.Hazra TK, Izumi T, Maidt L, Floyd RA, Mitra S. The presence of two distinct 8-oxoguanine repair enzymes in human cells: Their potential complementary roles in preventing mutation. Nucleic Acids Research. 1998;26:5116–5122. doi: 10.1093/nar/26.22.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demple B, Harrison L. Repair of oxidative damage to DNA: Enzymology and Biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 24.Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 25.Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Das A, Wiederhold L, Leppard JB, Kedar P, Prasad R, Wang H, Boldogh I, Karimi-Busheri F, Weinfeld M, Tomkinson AE, Wilson SH, Mitra S, Hazra TK. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair (Amst) 2006;5:1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomkinson AE, Mackey ZB. Structure and function of mammalian DNA ligases. Mutat Res. 1998;407:1–9. doi: 10.1016/s0921-8777(97)00050-5. [DOI] [PubMed] [Google Scholar]
- 28.Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 29.Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J Biol Chem. 2008;283:3130–3140. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- 30.Hegde ML, Theriot CA, Das A, Hegde PM, Guo Z, Gary RK, Hazra TK, Shen B, Mitra S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J Biol Chem. 2008;283:27028–27037. doi: 10.1074/jbc.M802712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of Mammalian cells. J Biol Chem. 2011;286:6006–6016. doi: 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bregeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell. 2003;12:959–970. doi: 10.1016/s1097-2765(03)00360-5. [DOI] [PubMed] [Google Scholar]
- 33.Saxowsky TT, Doetsch PW. RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev. 2006;106:474–488. doi: 10.1021/cr040466q. [DOI] [PubMed] [Google Scholar]
- 34.Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH, Hazra TK. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem. 2007;282:28474–28484. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 36.Jung D, Cho Y, Meyer JN, Di Giulio RT. The long amplicon quantitative PCR for DNA damage assay as a sensitive method of assessing DNA damage in the environmental model, Atlantic killifish (Fundulus heteroclitus) Comp Biochem Physiol C Toxicol Pharmacol. 2009;149:182–186. doi: 10.1016/j.cbpc.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradley MO, Bhuyan B, Francis MC, Langenbach R, Peterson A, Huberman E. Mutagenesis by chemical agents in V79 chinese hamster cells: a review and analysis of the literature. A report of the Gene-Tox Program. Mutat Res. 1981;87:81–142. doi: 10.1016/0165-1110(81)90029-4. [DOI] [PubMed] [Google Scholar]
- 38.Maiti AK, Boldogh I, Spratt H, Mitra S, Hazra TK. Mutator phenotype of mammalian cells due to deficiency of NEIL1 DNA glycosylase, an oxidized base-specific repair enzyme. DNA Repair (Amst) 2008;7:1213–1220. doi: 10.1016/j.dnarep.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thacker J. The chromosomes of a V79 Chinese hamster line and a mutant subline lacking HPRT activity. Cytogenet Cell Genet. 1981;29:16–25. doi: 10.1159/000131547. [DOI] [PubMed] [Google Scholar]
- 40.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 41.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 42.U.S. Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Rockville, MD: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006. http://m.cancer.gov/topics/factsheets/ETS. [Google Scholar]
- 43.Thun MJ, Hannan LM, Adams-Campbell LL, Boffetta P, Buring JE, Feskanich D, Flanders WD, Jee SH, Katanoda K, Kolonel LN, Lee IM, Marugame T, Palmer JR, Riboli E, Sobue T, Avila-Tang E, Wilkens LR, Samet JM. Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 2008;5:e185. doi: 10.1371/journal.pmed.0050185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka Y, Maniwa Y, Bermudez VP, Doi T, Nishio W, Ohbayashi C, Okita Y, Hurwitz J, Hayashi Y, Yoshimura M. Nonsynonymous single nucleotide polymorphisms in DNA damage repair pathways and lung cancer risk. Cancer. 2010;116:896–902. doi: 10.1002/cncr.24850. [DOI] [PubMed] [Google Scholar]
- 45.Kiyohara C, Takayama K, Nakanishi Y. Lung cancer risk and genetic polymorphisms in DNA repair pathways: a meta-analysis. J Nucleic Acids. 2010;2010:701760. doi: 10.4061/2010/701760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broderick P, Bagratuni T, Vijayakrishnan J, Lubbe S, Chandler I, Houlston RS. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer. 2006;6:243. doi: 10.1186/1471-2407-6-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhai X, Zhao H, Liu Z, Wang LE, El-Naggar AK, Sturgis EM, Wei Q. Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clin Cancer Res. 2008;14:4345–4352. doi: 10.1158/1078-0432.CCR-07-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- 49.Khatib H. Is it genomic imprinting or preferential expression? Bioessays. 2007;29:1022–1028. doi: 10.1002/bies.20637. [DOI] [PubMed] [Google Scholar]
- 50.Bell CG, Beck S. Advances in the identification and analysis of allele-specific expression. Genome Med. 2009;1:56. doi: 10.1186/gm56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knight JC. Allele-specific gene expression uncovered. Trends Genet. 2004;20:113–116. doi: 10.1016/j.tig.2004.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.