

Abstract
Alzheimer’s Disease (AD) is often accompanied by changes in mood as well as increases in circulating cortisol levels, suggesting that regulation of the stress responsive Hypothalamic-Pituitary-Adrenal (HPA) axis is disturbed. Here, we show that APP is endogenously expressed in important limbic, hypothalamic, and midbrain nuclei that regulate HPA axis activity. Furthermore, in a knock-in mouse model of AD that expresses familial AD (FAD) mutations of both APP with humanized Aβ, and PS1, in their endogenous patterns (APP/hAβ/PS1 animals), Corticotropin Releasing Factor (CRF) levels are increased in key stress-related nuclei, resting corticosteroid levels are elevated and animals display increased anxiety-related behavior. Endocrine and behavioral phenotypes can be normalized by loss of one copy of Corticotropin Releasing Factor Receptor type-1 (Crfr1), consistent with a perturbation of central CRF signaling in APP/hAβ/PS1 animals. However, reductions in anxiety and corticosteroid levels conferred by hemizygosity of Crfr1 do not improve a deficit in working memory observed in APP/hAβ/PS1 mice, suggesting that perturbations of the CRF system are not the primary cause of decreased cognitive performance.
Keywords: Corticotropin Releasing Factor, CRF, CRH, CRFR1, CRHR1, HPA axis, Alzheimer’s Disease, corticosteroids, APP, Presenilin, anxiety, depression
1. Introduction
Alzheimer’s Disease (AD) is characterized by the progressive loss of cognitive ability and eventual dementia. On autopsy, AD patients present with amyloid plaques and neurofibrillary tangles in addition to neurodegeneration, together considered the pathological hallmarks of AD. The most well characterized outcome of these neuropathologies is the progressive loss of memory, however, other brain systems have been shown to mis-function with the onset of AD. For example, AD patients exhibit elevated cortisol levels [10,23], indicating a progressive imbalance in the function of the Hypothalamic-Pituitary Adrenal (HPA) axis, the endocrine axis that is initiated by Corticotropin Releasing Factor (CRF) release by the hypothalamus and coordinates the release of corticosteroids by the adrenal glands [63]. Imbalance in HPA axis function can directly impact emotional status, responses to stress, and cognitive ability, and increased HPA axis activity in many cases accompanies depression [44]. Interestingly, changes in mood, anxiety, and depression often precede or present concomitantly with the earliest signs of memory loss or mild cognitive impairment (MCI) in AD, and are found to precede dementia in many families carrying FAD mutations in APP and PS1, leading many to propose that depression is perhaps one of the earliest signs of progressing AD pathology [14,17,30,42,45,51]. Indeed, patients with higher levels of corticosteroids suffer more rapid progression of dementia [9,10,39,67]. However, it remains unclear whether stress and increased cortisol accelerate dementia or are in fact a symptom of progressing AD pathogenesis.
While the cause of increased anxiety and corticosteroid levels in AD is still unknown, the impact of stress and corticosteroids on the development and progression of AD pathogenesis is more thoroughly understood. In transgenic animal models of AD, increased stress leads to higher Aβ and Aβ oligomer levels, increased phosphorylated tau, and accelerated amyloid plaque deposition [12,49,50,62]. This may be due to stress induced elevations in corticosteroid levels, which have been shown to have many of the same effects [6,8,20]. These studies suggest that animals pre-disposed to succumb to AD are sensitized to the effects of stress and stress hormones, which can severely provoke the progression of AD pathogenesis. However, reports on the status of stress responsive circuitry and the HPA axis at a baseline state, before stress has been given have been inconsistent. In various AD transgenic mouse models, anxiety-related behavior has been reported to be reduced [19,22,28,33], increased [13,35,58,62], or not affected [1]. Changes in corticosteroid levels have also varied in reports using different AD model animals [13,19,62]. These differences might be partially explained by differences between the heterologous promoters used to mis-express APP, which do not necessarily recapitulate the endogenous expression pattern of APP, and might lead to APP gain-of-function phenotypes not typically present in human AD.
To examine stress responsive neural and endocrine pathways in the context of endogenous expression of APP mutations, we have taken advantage of a knock-in AD mouse model (APP/hAβ/PS1) in which a mutant APP allele carrying the disease causing Familial Alzheimer’s Disease (FAD) Swedish (K670N/M671L) and London (V717F) mutations with a humanized Aβ sequence has replaced mouse APP [31]. Wildtype PS1 has been replaced by mouse PS1 carrying the M146V FAD mutation [21]. APP/hAβ/PS1 mice display endogenous temporal and spatial expression of mutant forms of APP and PS1, and model AD in the absence of high levels of mis-expression.
2. Methods
2.1 Mouse maintenance and breeding
Mice were housed up to five per cage with ad libitum access to food and water in a room with a 12 h light/dark cycle in a specific pathogen-free mouse facility. All experimental procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC) and performed in accordance with National Institutes of Health (NIH) guidelines. All mice used for behavioral and endocrine analysis have been backcrossed for at least 6 generations onto a C57/B6 background. APPsl/Aβ/sl/Aβ mice and PS1M146V/M146V mice were bred together to generate double heterozygous APPsl/Aβ/+; PS1M146V/+ animals which were intercrossed to generate homozygous littermates including wildtype mice with the genotype APP+/+; PS1+/+ (wt), and double knock-in mice with the genotype APPsl/Aβ/sl/Aβ; PS1M146V/M146V (APP/hAβ/PS1) animals. To generate large cohorts of animals for behavioral studies, we subsequently set up wt x wt and APP/hAβ/PS1 x APP/hAβ/PS1 crosses to obtain age-matched male mice. For Crfr1 interaction studies, APP/hAβ/PS1 mice were crossed with Crfr1+/− mice to generate APP/hAβ/PS1; Crfr1+/− animals. APP/hAβ/PS1; Crfr1+/− animals were subsequently intercrossed to give APP/hAβ/PS1; Crfr1+/+, APP/hAβ/PS1; Crfr1+/−, and APP/hAβ/PS1; Crfr1−/−, male offspring. Control Crfr1+/+, Crfr1+/−, and Crfr1−/− animals were obtained by intercrossing Crfr1+/− animals. The Crfr1 allele was obtained courtesy of Wylie Vale (Salk Institute, San Diego), where it was backcrossed for more than 10 generations to C57/B6 mice.
2.2 Behavior
Elevated Plus Maze (EPM)
Mice were transferred to the testing room, which was at a light level of 700 lux and a sound level of 60db generated by a white noise generator. The EPM apparatus consists of four arms placed at right angles to each other and is elevated 50 cm from the ground. Two of the arms had 20 cm high walls (enclosed arms), whereas two had no walls (open arms). After 30 minutes of acclimatization to the testing room in their home cages, mice were placed in the center of the elevated plus maze facing the open arms and allowed to explore the maze for 10 minutes. All parameters of movement were scored by an automated ANY-maze software system (Stoelting Co., Wood Dale, IL) including time in open, center, and closed areas of the maze, as well as number and duration of each entrance into an arm. After testing, animals were returned to their home cage. Animals tested on the EPM were naïve to all behavioral testing at the time of the test. Animals were grouped depending on genotype and analyzed for statistical differences in all parameters of movement using a two-tailed Student’s T-test. 2–3 month old males were used for all behavioral tests, with n=18–24 per genotype. EPM was always the first behavioral test performed and was never repeated on an individual animal.
Light-Dark (LD) box
The LD tests were done in the same room conditions described above for the EPM test. The LD apparatus has two compartments, an open transparent light chamber and a covered dark chamber. To begin the test, the animal was placed in the center of the light chamber. The animals were allowed to explore both chambers for 10min and all movements were recorded by a computer. The time spent in each chamber and the number of transitions between chambers were scored by the ANY-maze software system (Stoelting Co., Wood Dale, IL). Animals were grouped by genotype and analyzed for statistical differences in all parameters of movement.
Conditioned Fear (CF)
On the first day (training day) the mice were first transferred to a holding room next to the CF testing room in their home cages and allowed to acclimate for 30min. Mice were then transferred in a sound-attenuated cage to the testing room and placed in a testing chamber (Med Associates, St. Albans, VT). After a delay period, an auditory cue was played which was immediately followed by a mild footshock (0.7mA) for 2 sec. On the next day (testing day), mice were placed back in the same chamber for 5 min without any sound stimulus (context conditioning), during which time the movements of the mice were recorded by a video camera and the freezing frequency was scored by FreezeFrame software (San Diego Instruments, San Diego, CA). Two hours after the context test, the mice were placed in a testing box in which the environment in the chamber was changed (different floor, shape and odor). Movements of the mice were first recorded for 3 min without any sound stimulus to establish a baseline freezing level and then recorded during a 3 minute presentation of the sound (cued conditioning). Computer generated percentages of freezing behavior were grouped according to genotype and statistically compared for significance.
Hotplate nociception test
Mice were acclimated to the testing room for 30 minutes and the hotplate was pre-warmed to 55° C before the test. Mice were placed on the hotplate one at a time. The time before the mouse showed its first hindlimb response, including jumping, shaking and licking, was recorded and statistically compared between groups.
Spontaneous Alternation T-maze
Mice were acclimated to the testing room for 30 min before the start of the test. The T-maze apparatus is a T-shaped maze with a removable door at the base of the long arm of the maze. At the beginning of each trial, the mouse was placed behind the removable door at the base of the maze. After 30 seconds, the door was removed and the mouse was allowed to explore the maze and enter one of two arms freely. Once the animal entered an arm (first choice), the arm entrance was blocked by a transparent plastic panel and the animal was restricted to the chosen arm for 5 sec. The animal was then transferred back to the base with the door closed. After a 45 second waiting period, the door was removed, and the mouse was again allowed to explore the maze and choose an arm (second choice). If on the second choice the animal enters the alternate arm from the first choice, this is scored as an alternating trial. The mice were tested on the T-maze by an observer blind to the genotype for 3 trials a day for 3 test days with a 3-day interval between each testing day. Animals were grouped by genotype and compared for statistical differences.
2.3 Corticosterone measurement
For measuring plasma corticosteroids at rest, animals were moved to a quiet procedure room and allowed to calm down and acclimatize to the room for 2 hours, during which time no entries or exits were made from the room, and all noise was minimized. After 2 hours, an experimenter entered the room and drew blood via retro-orbital eye bleed. All mice in the room were bled in under 5 minutes. For high stress corticosteroid measurements, animals were placed into restraining tubes for 20 minutes before being bled. For dexamethasone suppression studies, animals were given an injection of 30μg/kg (i.p.) dexamethasone 2 hours before being bled. Blood samples were mixed with EDTA (final concentration 2mM) and centrifuged to remove red blood cells. Plasma samples were collected and frozen before shipment to the UVA Center for Research in Reproduction Ligand Assay and Analysis Core, where corticosteroid levels were measured using a rat corticosterone RIA. For corticosteroid measurements, n=14–16 per genotype. Animals were not used in behavioral testing after being bled for corticosterone measurement.
2.4 Immunohistochemistry
IHC was performed according to standard procedures as described previously (Justice et al, 2008). Briefly, separate cohorts of 2–3 month old animals were perfused with 4% Paraformaldehyde, brains were removed and sucrose protected overnight, and then sectioned on a frozen sliding microtome at 30μM. Sections were rinsed in PBS and incubated in primary antibodies overnight in PBS with 0.4% Triton X-100, and 2% normal donkey serum, rinsed in PBS, incubated in secondary antibodies for 2 hours in PBS + 0.4% Triton X-100, rinsed in PBS and mounted on gelatin coated slides. For human tissue, we used PBS + 0.4% Triton X-100 + 5% normal donkey serum (tissue courtesy of Yong Shen). For APP immunohistochemistry, we used a rabbit monoclonal antibody directed against the C-terminal NPXY motif (APP Y188, 1:1000, Epitomics, CA), followed by incubation in biotin labeled donkey anti-rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA), and the vectastain elite DAB kit with nickel intensification (Vector Labs, Burlingame, CA). Brightfield images were generated using and EVOS brightfield microscope (Advanced Microscopy Group, Bothell, WA). To detect CRF, we used rabbit anti-rat/human CRF antibodies provided by W. Vale [rC70, 1:2000; 60]. Secondary Goat anti-rabbit antibodies conjugated to Alexa-Fluor 555 (Invitrogen, San Diego) were used for detection. Images of each area were acquired on a Leica AOBS Confocal Microscope using identical setting for acquisition of all images. Quantification of the intensity of CRF immunolabeling was performed using the Nikon NIS-Elements software package by an observer blind to the genotype. A single section of a confocal stack containing the peak level of staining was used for quantification. Each image was thresholded using identical settings to remove any saturated background signal. The structure was then outlined using the Bezier tool, thereby defining an ROI. Within this outline, three non-overlapping 205 pixel square ROIs were defined. Average luminosity was calculated for each of the ROIs. The luminosity from each of the ROIs was averaged to determine a value for the intensity of CRF staining for that area. CRF staining intensities were generated for the BSTov, rostral pericommisural BST, subcommisural BST, PVN, and CeA. Because staining in the subcommisural BST was consistently low and evenly distributed, we used this area to normalize within each animal. This allowed for variance between animals in luminosity due to the quality of staining to be reduced. After normalization, luminosity scores for each area were grouped by genotype and compared for statistically significant differences using a two-tailed Student’s T-test. n=8–10 per genotype.
2.5 Data Analysis and Statistics
For all quantifications, measurements were made blind to the genotype. Upon data analysis, the genotype of each animal was revealed. Animal scores were grouped by genotype, averaged and compared pair-wise for statistically significant differences by performing two-tailed Student’s T-tests. A difference was determined to be statistically significant if p<0.05.
3. Results
3.1 APP is abundantly expressed in the Central Autonomic System
A highly interconnected system of brain nuclei that are coordinately activated by stress and by CRF have been collectively termed the Central Autonomic System [CAS; 52,60]. This system includes limbic structures such as the Central Nucleus of the Amygdala (CeA) and the lateral dorsal Bed nucleus of the Stria Terminalis (also known as the BSTov). The CAS additionally includes the paraventricular nucleus of the hypothalamus (PVN), which contains CRF neurons that project to the median eminence and activate the HPA axis, the Locus Coereleus (LC) which coordinates autonomic activity, and two visceral sensory nuclei, the Parabrachial nucleus (PB) and the nucleus of the Solitary Tract (NTS). Using an antibody that specifically recognizes APP, we stained wildtype mouse brain and examined APP expression in the CAS. We found that APP is abundantly expressed in nuclei of the CAS. Specifically, we found high levels of expression in multiple regions of the BST, including the BSTov (Fig. 1A), and in the amygdala (Fig. 1B). In the amygdala, expression is highest in the Basolateral nucleus (BLA), with moderate levels of expression in the Lateral Amygdala (LA), CeA and Medial Amygdala (MeA). The PVN expresses particularly high levels of APP (Fig. 1C), while the PB, LC and NTS all express moderate to high levels of APP (Fig. 1D, E). Labeling was specific for APP because in APP null mutant animals [70], all staining in the PVN (Fig. 1F) and other areas (data not shown) is absent. Using the same antibody on human amygdala sections from non-diseased brains, we saw prominent APP labeling of neurons in the Basolateral and Cortical nuclei with lower levels of staining in the Central nucleus (Fig. 1G-I), similar to the APP expression pattern observed in the mouse amygdala. Many transgenic AD model animals express mutant forms of APP from heterologous promoters which often lack expression in these important stress responsive nuclei. Endogenous expression of APP in neurons of key limbic, autonomic, and sensory nuclei suggest that APP may be important to the function of these neurons, and that disease causing mutations may directly impact the excitability of the CAS.
Figure 1.

APP is abundantly expressed in nuclei of the Central Autonomic System (CAS). Immunohistochemical staining for APP reveals high levels of expression in the Bed Nucleus of the Stria Terminalis (BST; A), Amygdala (LA-lateral, BLA-Basolateral, CeA-Central, MeA-Medial, B), Paraventricular Nucleus of the Hypothalamus (PVN; C), , Locus Coereleus (LC) and Parabrachial Nucleus (PB; D), and the nucleus of the Solitary Tract (NTS; E). (F) Lack of staining in a section of the PVN from an APP null mutant (APP−/−) demonstrates the specificity of the antibody used against APP. A (G, H, I) Staining of human amygdala sections for APP reveals prominent staining in the Basolateral (G), Cortical (H) and Central (I) nuclei. Scale Bar: A–F = 200μm; G-I = 100μm
3.2 Increased central CRF levels in APP/hAβ/PS1 animals
Release of corticosteroids is initiated by the release of CRF into the portal system by hypophysiotropic CRF neurons which reside in the PVN [63]. CRF also functions centrally, where it modulates activity of stress responsive neuronal circuitry and drives anxiety-related behavior [32,60]. We examined the expression of CRF in key stress responsive nuclei by performing immunohistochemistry for CRF peptide and quantifying relative fluorescence levels. Because staining intensity may vary depending on the quality of fixation of the tissue and the efficacy of staining, within each sample we normalized fluorescence levels with CRF levels in the medial-ventral BST (BSTmv) which expresses moderate to low levels of CRF, and where CRF expression is relatively consistent between animal groups (data not shown). CRF is expressed at the highest levels in the BSTov, PVN, and CeA (Fig. 2). Within the BSTov, which has been proposed to be critical for the expression of chronic anxiety [65], CRF levels were found to be significantly higher in APP/hAβ/PS1 animals compared to wildtype controls (Fig 2A,B; p<0.05). Similarly, in the PVN we saw that CRF levels were higher in APP/hAβ/PS1 animals compared to wildtype controls (Fig.2A,B; p<0.05).In the CeA, we found no statistical difference in CRF levels between APP/hAβ/PS1 and wildtype groups (Fig. 2A,B, p=0.32). We also measured CRF levels in APP single knock-in and PS1 single knock-in animals, and found that CRF is elevated in the BSTov of APP single knock-ins but is at wildtype levels in PS1 single knock-in animals, suggesting that this increase is primarily due to mutations in APP (Figure S1E). Higher levels of CRF in key limbic, stress responsive, nuclei such as the BSTov and PVN suggest that these circuits are perturbed by APP, which led us to investigate endocrine and behavioral outputs of this circuitry.
Figure 2.

APP/hAβ/PS1 animals have increased CRF levels and increased circulating corticosterone. (A) CRF labeling in wt (top row) and APP/hAβ/PS1 (second row) shows increased CRF levels in the BSTov and the PVN of APP/hAβ/PS1 animal, while CRF levels in the CeA were comparable between genotypes. Scale bar = 100μm. (B) Quantification of fluorescence levels revealed a statistically significant difference in signal intensity between wildtype (white bars) and APP/hAβ/PS1 (black bars) in the BSTov (p<0.05) and the PVN (p<0.05). (C, D) Corticosterone was measured in blood samples from wildtype (white bars) and APP/hAβ/PS1 mice (black bars) at rest (C) and at the peak of corticosterone release in response to stress (D). APP/hAβ/PS1 had higher resting corticosterone levels (p<0.05) and similar peak corticosterone levels. (E) After injection with dexamethasone, both wildtype and APP/hAβ/PS1 animals suppressed corticosterone release to a similar degree.
3.3 Elevated resting corticosterone levels in APP/hAβ/PS1 animals
Given elevated CRF levels in important stress responsive central nuclei including the PVN, we next measured corticosteroid levels, which have been shown to be elevated in elderly patients with MCI in the early stages of AD progression [10,23]. We found that APP/hAβ/PS1 animals have higher resting levels of circulating corticosteroids compared to control animals (Fig. 2C), indicating basal activity of the HPA axis is perturbed. Next, we tested corticosteroid levels after 20 minutes of restraint stress when corticosteroid levels peak in response to this stressor. We found that APP/hAβ/PS1 animals on average have elevated peak corticosteroid levels compared to wildtype animals, however, this failed to reach statistical significance (Fig. 2D; p=0.09). Finally, we administered dexamethasone, a potent glucocorticoid receptor agonist which suppresses corticosterone release by inhibiting CRF and Adenocorticotropic Hormone (ACTH) release, and measured circulating corticosterone levels two hours later. In wildtype and APP/hAβ/PS1 animals we saw an equivalent reduction in circulating corticosterone levels, indicating that the efficacy of corticosteroid negative feedback on the HPA axis is normal in APP/hAβ/PS1 animals (Fig 2E).
3.4 Enhanced anxiety-related behavior in APP/hAβ/PS1 animals
Given changes in CRF levels in key stress responsive nuclei, and elevated resting corticosteroid levels, we next examined anxiety-related behavior in APP/hAβ/PS1 animals. We saw that on the Elevated Plus Maze (EPM), which measures anxiety-related behavior by determining how much time an animal is willing to investigate exposed arms of an elevated plus maze versus how much time the animal stays in the closed arms of the maze [46], APP/hAβ/PS1 animals spent significantly more time in the closed arms and significantly less time on the open arms of the maze compared to wildtype control animals, indicating higher levels of anxiety (Fig. 3A). In addition, APP/hAβ/PS1 animals made fewer entries into the open arms and navigated less distance on the open arms compared to wildtype animals, indicating less willingness to explore the exposed arms (Fig. 3B, C), a sign of increased anxiety [46]. APP/hAβ/PS1 animals also displayed elevated anxiety-related behavior on the Light-Dark box (LD) test. Similar to the EPM, the LD tests how much time the animal spends in the open and lighted side or in the closed and covered area of a two chambered box [4]. APP/hAβ/PS1 animals spent significantly more time in the closed portion of the box, and significantly less time in the open side (Fig. 3D). APP/hAβ/PS1 animals made fewer entries to the light side of the box, and spent on average more time on each visit to the dark side (Fig. 3E, F), suggesting increased hiding behavior and reduced exploring, which suggests higher levels of anxiety. Animals carrying only APP mutations or PS1 mutations were also tested for anxiety-related behavior. APP single knock-in animals displayed decreased open time and increased closed time on the EPM, similar to APP/hAβ/PS1 animals, while PS1 single knock-in animals did not display the anxiety phenotype (Fig. S1A,B). Additionally, we tested APP/hAβ/PS1 animals that were over 12 months of age and found that the anxiety phenotype persists at this stage, after the commencement of amyloid plaque deposition [Fig. S1C,D; 15]. The observed increases in anxiety-related behavior displayed by APP/hAβ/PS1 animals is consistent with overactive central CRF signaling and increased corticosteroid levels [24].
Figure 3.

APP/hAβ/PS1 animals display elevated anxiety-related behavior. Results of behavioral testing of wt animals (white bars) and APP/hAβ/PS1 animals (black bars) on the Elevated Plus Maze (EPM; A,B,C) and Light-Dark box (LD; D,E,F). (A) On the EPM, APP/hAβ/PS1 spent less time on the open arms (p<0.01) and more time on the closed arms of the maze (p<0.05). (B) APP/hAβ/PS1 animals made fewer entries into the open arms (p<0.05). (C) APP/hAβ/PS1 animals traveled less distance on the open arms of the maze (p<0.001). (D) In the LD box, APP/hAβ/PS1 animals spent less time in the light area of the box (p<0.01), and more time in the dark area (p<0.01). (E) APP/hAβ/PS1 animals made fewer entries into the light area (p<0.05) and fewer entries into the dark area (p<0.05). (F) On each visit to the dark area of the box, APP/hAβ/PS1 animals stayed for a longer period of time (p<0.05).
To further examine the anxiety phenotype of APP/hAβ/PS1 mice, we tested the ability of mice to pair a painful footshock with a sound or with the context in which they received the shock. This Conditioned Fear (CF) paradigm has been used extensively to dissect the function of the amygdala in establishing the association between a sound and a painful stimulus [34]. 24 hours after two exposures to a paired sound and footshock, animals were returned to the original box in which they were shocked, or to a different box with altered cage floor, walls, and scent. Freezing was measured in the context of the shock in the absence of the sound (contextual fear), or in the new context during presentation of the sound (cued fear). APP/hAβ/PS1 mice displayed increased freezing in the context in which they received the shock compared to wildtype animals but displayed similar levels of freezing to wildtype animals in response to the sound (Fig 4A). Interestingly, during the cued trial, we measured freezing levels in the 3 minutes immediately before presentation of the sound, and found that APP/hAβ/PS1 animals displayed increased baseline freezing levels, suggesting that higher levels of amygdalar activity are present in the absence of the context or the sound (Fig 4B). Increased baseline freezing was not present before the animals were shocked (Fig 4B, pre-shock). Changes in baseline freezing are not likely due to a pain threshold difference because we detected no statistical difference in nociception by the hotplate assay (3.68 vs. 3.92 seconds to twitch, p=0.38). Increased anxiety-related behavior on these three behavioral paradigms indicates a perturbed stress-reactive circuitry, due in part to increased levels of CRF, which enhance fear and anxiety-like responses.
Figure 4.

APP/hAβ/PS1 animals display anxiety-like phenotypes in conditioned fear testing. (A) When placed back in the context where they were shocked (Context), APP/hAβ/PS1 animals displayed higher levels of freezing (p<0.05). In response to the sound that was paired with the shock (Cue), APP/hAβ/PS1 did not have higher rates of freezing compared to wildtype animals. (B) Both wildtype (white bars) and APP/hAβ/PS1 (black bars) animals had very low freezing rates before being shocked in the conditioned fear box (Pre-Shock). After receiving sound-shock pairings, in a novel context but before the sound was presented (Post-Shock), APP/hAβ/PS1 animals displayed higher freezing rates (p<0.05).
3.5 Crfr1 hemizygosity reverses elevated corticosterone levels and anxiety-related behavior in APP/hAβ/PS1 animals
CRFR1 is required for the activation of ACTH release by pituitary corticotropes, and also mediates anxiogenic actions of CRF in the central nervous system [48,56]. Therefore, we intercrossed the APP/hAβ/PS1 double knock-in mouse with a null allele of Crfr1 to generate APP/hAβ/PS1; Crfr1+/+ animals, APP/hAβ/PS1; Crfr1+/− animals that lack one copy of Crfr1, and APP/hAβ/PS1; Crfr1−/− animals that are null for Crfr1, and measured resting and peak circulating corticosteroid levels. Loss of one copy of Crfr1 (Crfr1+/−) in APP/hAβ/PS1 animals returned resting corticosteroid levels to levels comparable to wildtype animals (Fig. 5A, p<0.001), suggesting that a reduction in CRFR1 signaling can reverse the HPA axis perturbation seen in APP/hAβ/PS1 animals. We did not see a decrease in the resting corticosteroid levels in Crfr1+/− control animals, consistent with previous work indicating that heterozygous Crfr1 mutations do not decrease resting corticosteroid levels [Fig. 5A; 36,61]. This suggests that reduced CRF signaling only mitigates corticosteroid release in the case of CRF system perturbation, as we see in the APP/hAβ/PS1 mouse. We also measured corticosteroid levels as they peak in response to restraint stress. We saw no difference in peak levels of corticosteroids in heterozygous Crfr1 animals (Crfr1+/−) of either genotype, suggesting that the HPA axis can be activated to a similar degree in response to stress (Fig. 5B). We also measured resting and peak corticosteroid levels in APP/hAβ/PS1 animals carrying homozygous mutant copies of Crfr1 (Crfr1−/−). As has been shown previously [56,61], both resting and peak corticosteroid levels were at or below the limits of detection, indicating a near complete block of HPA axis activity in Crfr1 homozygous mutants (not shown).
Figure 5.

Crfr1 hemizygosity normalizes corticosteroid levels and reduces anxiety-related behavior. Circulating corticosteroid levels were measured at rest (A) and at peak levels of stress (B) in wildtype control (white bars) and APP/hAβ/PS1 animals (black bars) carrying wildtype (Crfr1+/+) or heterozygous (Crfr1+/−) alleles of Crfr1. (A) At rest, APP/hAβ/PS1 animals have higher resting corticosterone levels. This increase is normalized after removal of one copy of Crfr1 (Crfr1+/−). Loss of one copy of Crfr1 has no impact on resting corticosterone levels in a wildtype background. (B) At peak stress, wildtype and APP/hAβ/PS1 animals carrying wildtype or heterozygous mutant alleles of Crfr1 have similar peak coricosterone levels. (C,D) Anxiety-related behavior was measured as time spent in the closed arms (C) and open arms (D) of the EPM, in animals wildtype (white bars) or mutant for APP/hAβ/PS1 (black bars). (C) APP/hAβ/PS1 animals spent more time in the closed arms of the EPM compared to wildtype animals (p<0.01). When one copy of Crfr1 was mutant (Crfr1+/−), APP/hAβ/PS1; Crfr1+/− animals spent less time in the closed arms compared to APP/hAβ/PS1; Crfr1+/+ animals (p<0.01), displaying similar closed arm times to Crfr1+/− control animals. (D) APP/hAβ/PS1 animals wildtype for Crfr1 (Crfr1+/+) spent less time on the open arms (p<0.01) compared to wildtype animals. When one copy of Crfr1 is mutant (Crfr1+/−), APP/hAβ/PS1; Crfr1+/− animals increased the time spent on the open arms (p<0.05). Crfr1+/− control animals still spent more time on the open arms than APP/hAβ/PS1; Crfr1+/− animals (p<0.05).
Next, we measured anxiety-related behavior on the EPM in cohorts of APP/hAβ/PS1; Crfr1+/+ and APP/hAβ/PS1; Crfr1+/− animals, along with cohorts of Crfr1+/+ or Crfr1+/− animals as controls. Animals homozygous mutant for Crfr1 (Crfr1−/−) were excluded from the behavioral analysis because of potential non-specific effects of chronically low corticosteroid levels. In APP/hAβ/PS1 animals wildtype for Crfr1 (Crfr1+/+), we again saw an increase in anxiety-related behavior measured as an increase in time spent in the closed arms (Fig. 5C, p<0.01) and a decrease in time spent in the open arms (Fig. 5D, p<0.01). In APP/hAβ/PS1 animals heterozygous for Crfr1 (Crfr1+/−), closed time decreased compared to APP/hAβ/PS1; Crfr1+/+ animals (Fig. 5C, p<0.01). Similarly, APP/hAβ/PS1; Crfr1+/− animals displayed increased open time compared to APP/hAβ/PS1; Crfr1+/+animals (Fig. 5D, p<0.05). These results suggest that reducing CRF signaling levels by removing a single copy of Crfr1 can reduce anxiety levels in APP/hAβ/PS1 animals without changing anxiety levels in a wildtype background. However, while anxiety as measured by closed arm time was equivalent between Crfr1+/− and APP/hAβ/PS1; Crfr1+/− (Fig. 5C), APP/hAβ/PS1; Crfr1+/− animals still displayed more anxiety than Crfr1+/− controls measured by the time they spent on the open arms (Fig. 5D, p<0.05). Therefore, even in the context of Crfr1 hemizygosity, where resting corticosteroid levels have returned to normal levels (Fig., 5A), APP/hAβ/PS1 animals have elevated anxiety, suggesting additional central anxiogenic mechanisms that act in the APP/hAβ/PS1 mice.
3.6 Crfr1 hemizygosity does not improve working memory in APP/hAβ/PS1 animals
To determine whether high anxiety levels in APP/hAβ/PS1 animals affect cognitive performance, we tested working memory in APP/hAβ/PS1 animals with and without Crfr1 mutations using the spontaneous alternation T-maze test. Rodents spontaneously alternate between two arms of a T-maze in consecutive trials [11]. Working memory is required for the animal to successfully alternate because a memory of which arm was selected on the previous trial must be maintained for a duration period between trials. We tested APP/hAβ/PS1 animals on the T-maze and found that they display decreased alternation compared to wildtype control animals, suggesting reduced working memory (Fig. 6). In APP/hAβ/PS1 animals carrying a heterozygous mutation of Crfr1 (Crfr1+/−), alternation rates were similar to APP/hAβ/PS1; Crfr1+/+ animals (Fig. 6), suggesting that working memory is not improved by the reduction in the levels of anxiety-related behavior conferred by loss of one allele of Crfr1.
Figure 6.
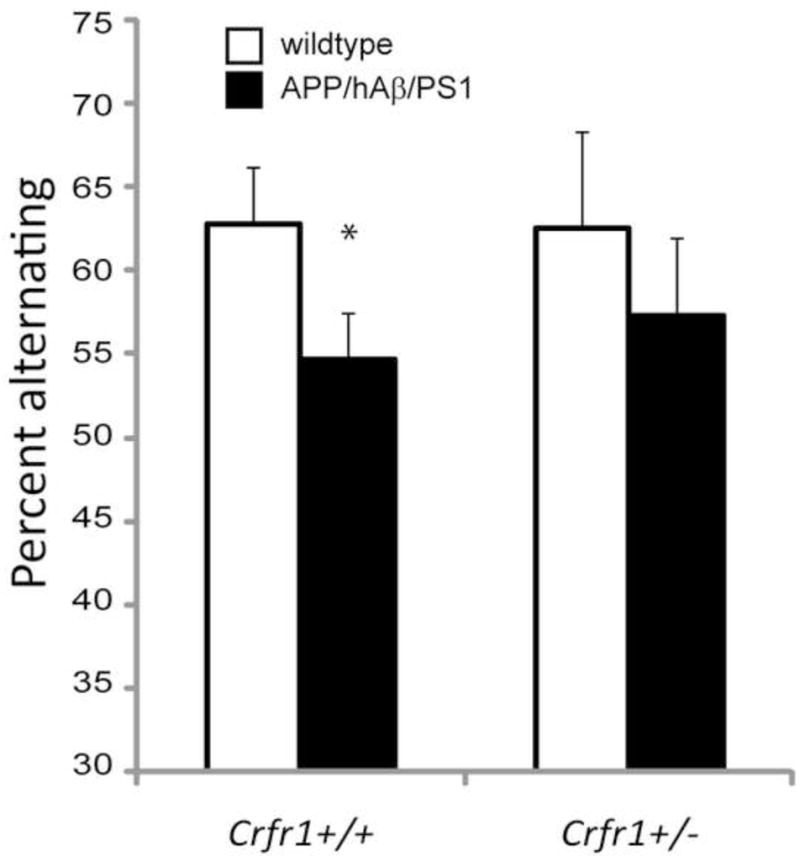
Reduced cognitive performance is not reversed by Crfr1 hemizygosity. Working memory was measured using a spontaneous alternation task in wildtype (white bars) and APP/hAβ/PS1 (black bars) animals. APP/hAβ/PS1 animals displayed reduced alternation rates compared to wildtype animals (p<0.05). Removal of one copy of Crfr1 in APP/hAβ/PS1 animals did not increase alternation rates. APP/hAβ/PS1; Crfr1+/− animals alternated at a similar frequency as APP/hAβ/PS1; Crfr1+/+ animals. Similarly, Crfr1+/− control animals alternated at a similar frequency as Crfr1+/+ wildtype animals.
4. Discussion
4.1 Expression of APP in CAS nuclei
APP is highly expressed in the central nervous system as well as in many peripheral tissues [71]. Early immunohistochemical studies in rat brain revealed that although APP is expressed in all neuronal populations, the expression level of APP varies in different brain regions [7,43]. We took advantage of a highly specific APP antibody and found high levels of APP expression in important stress-related areas, particularly the BST, Basolateral amygdala (BLA), and PVN. Expression of APP in these nuclei suggests that APP may function directly in limbic pathways to modulate the activity or connectivity of neurons that respond to stress. Previously used models of AD have relied on transgenic expression of APP from heterologous promoters. One of the most commonly used APP transgenic AD mouse model animals expresses mutant forms of APP from a hamster PrP promoter [tg2576; 27]. Although this promoter is believed to be expressed in all neurons, transgenic studies using the PrP promoter to express marker proteins have found that expression is exaggerated in the cortex and hippocampus and there is little or no expression in the amygdala, hypothalamus, and other nuclei of the CAS [5]. Other transgenic model animals have used the Thy-1 or PDGF promoters, and have been selected for high levels of cortical expression, but may lack significant expression in limbic structures [18,41,59]. Consistent with different patterns of expression of mutant APP in transgenic models, reported anxiety-related phenotypes in these models have been varied [1,13,19,22,28,33,35,58,62]. In the APP/hAβ/PS1 knock-in AD model animal, endogenous expression of mutant forms of APP in key nuclei of the CAS may lead to a direct perturbation of stress responsive circuits, perhaps more accurately modeling how the stress system becomes disrupted in human cases of AD.
4.2 Direct and indirect actions of APP/PS1 mutations on HPA axis activation
APP/hAβ/PS1 animals have elevated central CRF levels and elevated circulating corticosteroid levels at a young age, before any amyloid plaque deposition has taken place. How do APP and PS1 mutations lead to increased CRF release and HPA axis activity? One possibility is that altered APP and PS1 function leads to changes in the excitability of neural circuits that regulate HPA axis activity. Loss of APP function has been shown to increase levels of the L-type calcium channel subunit CaV1.2, leading to increased excitability of GABAergic hippocampal neurons [69]. FAD mutations in APP might disrupt APP function to alter the excitability of the predominantly GABAergic CeA or BST. The excitability of neurons might also be perturbed by elevated levels of toxic species of Aβ being produced in cells of the CAS. FAD mutations in APP and PS1 skew cleavage of APP towards production of the more amyloidogenic and toxic Aβ42 species [3]. Oligomers of Aβ42 have been shown to block long-term potentiation and synapse remodeling and to interfere with NMDA receptor function and glutamate uptake [16,38,54,55,66]. Production of these toxic species in CAS nuclei would directly alter regulation of the HPA axis. In support of an Aβ based model of stress system perturbation, APP single knock-in animals displayed a similar anxiety phenotype and elevated CRF levels, whereas this phenotype was not present in PS1 single knock-in animals (Fig. S1A,B, E). PS1 knock-in mutations confer only a mild increase in total Aβ levels in this genetic context, further supporting an Aβ toxicity model [37]. Alternatively, APP and PS1 FAD mutations might lead to reduced inhibition of the HPA axis by hippocampal and cortical structures, thereby increasing corticosteroid levels and promoting anxiety-related behavior. However, when mutant forms of APP are transgenically expressed selectively in the entorhinal cortex, leading to amyloid plaque formation in the hippocampus, anxiety-related behavior is reduced, suggesting that cortical and hippocampal APP mis-expression alone does not drive anxiety but in fact might reduce anxiety levels [22]. Consistent with this finding, tg2576 mice display reduced anxiety-related behavior at an advanced age [33]. Increased levels of corticosteroids have been reported in tg2576 and other transgenic AD mouse model animals, suggesting that high levels of mis-expression can perturb the HPA axis, although whether this occurs through the same mechanism as in the APP/hAβ/PS1 mutant animal remains unclear [13,50,62].
4.3 The role of CRF in anxiety-related behavior and AD pathogenesis
We have shown that CRF levels are increased in APP/hAβ/PS1 mice. Increased CRF can have a direct impact on AD pathogenesis. CRF and stress have been shown to cause elevated release of Aβ [29]. CRF also acts on hippocampal neurons to increase levels of tau phosphorylation [49]. Whether APP and PS1 mutations are the initial cause of increased CRF release or serve to exacerbate stress responses remains an open question. We abrogated activity of the CRF system by genetically removing copies of Crfr1 in the background of APP/hAβ/PS1 mutations and saw that removing a single copy of Crfr1 reduced anxiety-related behavior and normalized resting corticosteroid levels, while hemizygosity for Crfr1 alone does not alter resting corticosteroid levels [Fig. 5A; 36,61]. Therefore, reduction of CRF signaling through CRFR1 alleviates perturbation of the CAS by early stage AD pathogenesis caused by mutations in APP and PS1, consistent with elevated CRF and corticosteroid levels in part causing altered anxiety-related behavior. However, we cannot exclude the possibility that a reduction of CRF signaling functions in parallel to reduce anxiety-related behavior, caused by APP/PS1 mutations through CRF independent mechanisms. High levels of stress and corticosteroids can also negatively impact cognitive performance. Subjecting mice to a mild chronic stress protocol has been shown to produce depressive-like features, increase resting corticosteroid levels, and decrease cognitive performance [2,57,68]. However, we do not see a benefit to cognitive performance when we reduce anxiety levels in APP/hAβ/PS1 mutants by including a heterozygous Crfr1 mutation (Fig. 6). This suggests that observed reductions in working memory in APP/hAβ/PS1 mice are not caused by perturbations to the stress circuitry. We would predict that events affecting working memory are localized to the hippocampus, because this structure is required for the spontaneous alternation working memory task [11]. It remains formally possible that disruptions at the level of the hippocampus also account for activation of the HPA axis and elevated anxiety-like behavior, as the hippocampus provides important inhibitory tone to stress responsive limbic circuits. However, given high levels of expression of APP in these circuits, and reductions in anxiety-related behavior observed when APP mutants are selectively mis-expressed in hippocampal pathways [22], we favor a scenario in which mutant forms of APP and PS1 directly activate these circuits.
4.4 CRF independent anxiogenic mechanisms
Although we can normalize resting corticosteroid levels in APP/hAβ/PS1 animals by removing one copy of Crfr1, this did not normalize all aspects of anxiety-related behavior. Additionally, removing both copies of Crfr1 (Crfr1−/−), which blocks HPA axis function and renders the animals profoundly corticosteroid deficient [56], displayed mild increases in anxiety-related behavior, perhaps due to reduced robustness and sickness (data not shown). Therefore, there are most likely other, CRF-independent, mechanisms driving increases in anxiety. One potential source of this anxiogenic activity is LC. The LC has been shown to degenerate preferentially in AD, and LC function has been shown to be perturbed in AD model animals [25,40]. Another alternative is anxiogenic actions of CRF through its second receptor, CRFR2 [47], which is expressed in distinct subsets of neurons from CRFR1 [64]. CRFR2 has been implicated in some CRF mediated anxiogenic activity through actions in the lateral septum, which maintains strong connectivity with the CAS [26]. Identifying this additional source of anxiety may further our understanding of the mechanism by which the central CRF system becomes perturbed, due to the high degree of cross-talk between different stress responsive circuits.
4.5 The influence of the HPA axis on AD susceptibility and progression
Activation of the HPA axis causes the release of corticosteroids which have been shown to be detrimental to the health of neurons [53]. Because of this and other findings, much of the focus on the role of corticosteroids in AD has been on the negative influence that increased levels of corticosteroids have on amyloid plaque formation. For example, injection of dexamethasone, a glucocorticoid receptor agonist, has been shown to increase APP and BACE1 expression, increase Aβ production, and increase tau pathologies in the 3x-FAD AD mouse model [20]. In the AD mouse model tg2576, isolation stress leads to elevated corticosteroid levels and exacerbates plaque formation [12,13]. Recently, it has been shown that chronic mild stress in the 3x-FAD model animal can lead to prolonged exacerbation of features of anxiety including anxiety related behavior and elevated corticosteroid levels, leading to higher Aβ levels and decreased BDNF [50]. We have described an endogenous APP/PS1 knock-in mouse model with heightened stress responses before any stress manipulations, suggesting that ongoing AD pathogenesis sensitizes these mice to perturbation of central and endocrine stress circuitry. Enhanced stress responses can then impact AD progression through mechanisms involving elevated CRF, corticosteroids, and other consequences of elevated stress.
Supplementary Material
Figure S1. Anxiety-related behavior in single mutants and aged animals. (A,B) Animals were tested on the EPM for time on the open arms (A), and time in the closed arms (B). APP/hAβ/PS1 (black bars) animals displayed reduced time on the open arms compared to wildtype animals (white bars). APP single knock-in animals showed similar open arm time as APP/hAβ/PS1 animals (p<0.05 compared to wildtype) whereas PS1 knock-in mutants displayed wildtype levels of anxiety. (C, D) Aged animals were tested for anxiety-related behavior on the EPM. Both wildtype (white bars) and APP/hAβ/PS1 animals displayed decreased open arm time at 12 months of age compared to the same genotype at 3 months of age. At 12 months APP/hAβ/PS1 animals spent significantly less time in the open arms compared to age-matched control animals (p<0.05). (E) CRF levels were measured in the BSTov, PVN, and CeA of wildtype (white bars), APP single knock-in mice (light grey bars), PS1 single knock-in mice (dark grey bars), and APP/hAβ/PS1 mice (black bars). CRF levels are higher in APP single knock-in and APP/hAβ/PS1 mice in the BSTov. In the PVN, CRF levels were higher in APP/hAβ/PS1 mice but not in single knock-in mice. In the CeA, CRF levels were similar in all lines, with APP/hAβ/PS1 having the highest levels of CRF signal.
Acknowledgments
We thank all members of the Zheng lab for helpful and stimulating discussion of these results. We thank C. Spencer and the Baylor College of Medicine IDDRC Administrative, Mouse Neurobehavior, and Mouse Physiology cores (HD24064) for their assistance. We thank Dr.Yong Shen (Roskamp Institute) for kindly providing post-mortem human tissue samples. We thank the UVA Center for Research and Reproduction Ligand Assay and Analysis Core, (supported by the Eunice Kennedy Shriver NICHD/NIH Grant U54-HD28934), for RIA analysis. This work was supported by NIH K01AG036738 and an ADMDC Mitchell Pilot Grant (N.J.J.); and R01AG020670, R01AG032051, and R01AG035467 (H.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891(1–2):42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- 2.Ayensu WK, Pucilowski O, Mason GA, Overstreet DH, Rezvani AH, Janowsky DS. Effects of chronic mild stress on serum complement activity, saccharin preference, and corticosterone levels in Flinders lines of rats. Physiol Behav. 1995;57(1):165–9. doi: 10.1016/0031-9384(94)00204-i. [DOI] [PubMed] [Google Scholar]
- 3.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005–13. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 4.Bourin M, Hascoet M. The mouse light/dark box test. Eur J Pharmacol. 2003;463(1–3):55–65. doi: 10.1016/s0014-2999(03)01274-3. [DOI] [PubMed] [Google Scholar]
- 5.Boy J, Leergaard TB, Schmidt T, Odeh F, Bichelmeier U, Nuber S, Holzmann C, Wree A, Prusiner SB, Bujard H, Riess O, Bjaalie JG. Expression mapping of tetracycline-responsive prion protein promoter: digital atlasing for generating cell-specific disease models. Neuroimage. 2006;33(2):449–62. doi: 10.1016/j.neuroimage.2006.05.055. [DOI] [PubMed] [Google Scholar]
- 6.Budas G, Coughlan CM, Seckl JR, Breen KC. The effect of corticosteroids on amyloid beta precursor protein/amyloid precursor-like protein expression and processing in vivo. Neurosci Lett. 1999;276(1):61–4. doi: 10.1016/s0304-3940(99)00790-9. [DOI] [PubMed] [Google Scholar]
- 7.Card JP, Meade RP, Davis LG. Immunocytochemical localization of the precursor protein for beta-amyloid in the rat central nervous system. Neuron. 1988;1(9):835–46. doi: 10.1016/0896-6273(88)90131-6. [DOI] [PubMed] [Google Scholar]
- 8.Catania C, Sotiropoulos I, Silva R, Onofri C, Breen KC, Sousa N, Almeida OF. The amyloidogenic potential and behavioral correlates of stress. Mol Psychiatry. 2009;14(1):95–105. doi: 10.1038/sj.mp.4002101. [DOI] [PubMed] [Google Scholar]
- 9.Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry. 2006;163(12):2164–9. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis KL, Davis BM, Greenwald BS, Mohs RC, Mathe AA, Johns CA, Horvath TB. Cortisol and Alzheimer's disease, I: Basal studies. Am J Psychiatry. 1986;143(3):300–5. doi: 10.1176/ajp.143.3.300. [DOI] [PubMed] [Google Scholar]
- 11.Deacon RM, Rawlins JN. T-maze alternation in the rodent. Nat Protoc. 2006;1(1):7–12. doi: 10.1038/nprot.2006.2. [DOI] [PubMed] [Google Scholar]
- 12.Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG. Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience. 2004;127(3):601–9. doi: 10.1016/j.neuroscience.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 13.Dong H, Yuede CM, Yoo HS, Martin MV, Deal C, Mace AG, Csernansky JG. Corticosterone and related receptor expression are associated with increased beta-amyloid plaques in isolated Tg2576 mice. Neuroscience. 2008;155(1):154–63. doi: 10.1016/j.neuroscience.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman H, Scheltens P, Scarpini E, Hermann N, Mesenbrink P, Mancione L, Tekin S, Lane R, Ferris S. Behavioral symptoms in mild cognitive impairment. Neurology. 2004;62(7):1199–201. doi: 10.1212/01.wnl.0000118301.92105.ee. [DOI] [PubMed] [Google Scholar]
- 15.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23(3):335–48. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- 16.Freir DB, Fedriani R, Scully D, Smith IM, Selkoe DJ, Walsh DM, Regan CM. Abeta oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gabryelewicz T, Styczynska M, Pfeffer A, Wasiak B, Barczak A, Luczywek E, Androsiuk W, Barcikowska M. Prevalence of major and minor depression in elderly persons with mild cognitive impairment--MADRS factor analysis. Int J Geriatr Psychiatry. 2004;19(12):1168–72. doi: 10.1002/gps.1235. [DOI] [PubMed] [Google Scholar]
- 18.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein [see comments] Nature. 1995;373(6514):523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 19.Gil-Bea FJ, Aisa B, Schliebs R, Ramirez MJ. Increase of locomotor activity underlying the behavioral disinhibition in tg2576 mice. Behav Neurosci. 2007;121(2):340–4. doi: 10.1037/0735-7044.121.2.340. [DOI] [PubMed] [Google Scholar]
- 20.Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J Neurosci. 2006;26(35):9047–56. doi: 10.1523/JNEUROSCI.2797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5(1):101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 22.Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu GQ, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68(3):428–41. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer's and Parkinson's disease compared to normal controls: ultradian secretory pulsatility and diurnal variation. Neurobiol Aging. 1997;18(3):285–9. doi: 10.1016/s0197-4580(97)80309-0. [DOI] [PubMed] [Google Scholar]
- 24.Heinrichs SC, Menzaghi F, Merlo Pich E, Britton KT, Koob GF. The role of CRF in behavioral aspects of stress. Ann N Y Acad Sci. 1995;771:92–104. doi: 10.1111/j.1749-6632.1995.tb44673.x. [DOI] [PubMed] [Google Scholar]
- 25.Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer's disease. J Neurosci. 2002;22(7):2434–42. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry B, Vale W, Markou A. The effect of lateral septum corticotropin-releasing factor receptor 2 activation on anxiety is modulated by stress. J Neurosci. 2006;26(36):9142–52. doi: 10.1523/JNEUROSCI.1494-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice [see comments] Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 28.Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 29.Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104(25):10673–8. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasuga K, Ohno T, Ishihara T, Miyashita A, Kuwano R, Onodera O, Nishizawa M, Ikeuchi T. Depression and psychiatric symptoms preceding onset of dementia in a family with early-onset Alzheimer disease with a novel PSEN1 mutation. J Neurol. 2009;256(8):1351–3. doi: 10.1007/s00415-009-5096-4. [DOI] [PubMed] [Google Scholar]
- 31.Kohler C, Ebert U, Baumann K, Schroder H. Alzheimer's disease-like neuropathology of gene-targeted APP-SLxPS1mut mice expressing the amyloid precursor protein at endogenous levels. Neurobiol Dis. 2005;20(2):528–40. doi: 10.1016/j.nbd.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Koob GF, Thatcher-Briton K, Tazi A, Le Moal M. Behavioral pharmacology of stress: Focus on CNS corticotropin-releasing factor. Adv Exp Med Biol. 1988;245:25–34. doi: 10.1007/978-1-4899-2064-5_3. [DOI] [PubMed] [Google Scholar]
- 33.Lalonde R, Lewis TL, Strazielle C, Kim H, Fukuchi K. Transgenic mice expressing the betaAPP695SWE mutation: effects on exploratory activity, anxiety, and motor coordination. Brain Res. 2003;977(1):38–45. doi: 10.1016/s0006-8993(03)02694-5. [DOI] [PubMed] [Google Scholar]
- 34.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–84. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 35.Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, Han PL. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res. 2004;76(4):572–80. doi: 10.1002/jnr.20127. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Smith GW, Vale W, Lee KF, Rivier C. Mice that lack corticotropin-releasing factor (CRF) receptors type 1 show a blunted ACTH response to acute alcohol despite up-regulated constitutive hypothalamic CRF gene expression. Alcohol Clin Exp Res. 2001;25(3):427–33. [PubMed] [Google Scholar]
- 37.Li H, Wang Z, Wang B, Guo Q, Dolios G, Tabuchi K, Hammer RE, Sudhof TC, Wang R, Zheng H. Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. J Biol Chem. 2010;285(40):30598–605. doi: 10.1074/jbc.M110.137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62(6):788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1(1):69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- 40.Matthews KL, Chen CP, Esiri MM, Keene J, Minger SL, Francis PT. Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biol Psychiatry. 2002;51(5):407–16. doi: 10.1016/s0006-3223(01)01235-5. [DOI] [PubMed] [Google Scholar]
- 41.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20(11):4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullan M, Tsuji S, Miki T, Katsuya T, Naruse S, Kaneko K, Shimizu T, Kojima T, Nakano I, Ogihara T, et al. Clinical comparison of Alzheimer's disease in pedigrees with the codon 717 Val-->Ile mutation in the amyloid precursor protein gene. Neurobiol Aging. 1993;14(5):407–19. doi: 10.1016/0197-4580(93)90099-w. [DOI] [PubMed] [Google Scholar]
- 43.Ouimet CC, Baerwald KD, Gandy SE, Greengard P. Immunocytochemical localization of amyloid precursor protein in rat brain. J Comp Neurol. 1994;348(2):244–60. doi: 10.1002/cne.903480207. [DOI] [PubMed] [Google Scholar]
- 44.Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43(4):425–73. [PubMed] [Google Scholar]
- 45.Panza F, Frisardi V, Capurso C, D'Introno A, Colacicco AM, Imbimbo BP, Santamato A, Vendemiale G, Seripa D, Pilotto A, Capurso A, Solfrizzi V. Late-life depression, mild cognitive impairment, and dementia: possible continuum? Am J Geriatr Psychiatry. 18(2):98–116. doi: 10.1097/JGP.0b013e3181b0fa13. [DOI] [PubMed] [Google Scholar]
- 46.Pellow S, Chopin P, File SE, Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods. 1985;14(3):149–67. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- 47.Perrin M, Donaldson C, Chen R, Blount A, Berggren T, Bilezikjian L, Sawchenko P, Vale W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc Natl Acad Sci U S A. 1995;92(7):2969–73. doi: 10.1073/pnas.92.7.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perrin MH, Donaldson CJ, Chen R, Lewis KA, Vale WW. Cloning and functional expression of a rat brain corticotropin releasing factor (CRF) receptor. Endocrinology. 1993;133(6):3058–61. doi: 10.1210/endo.133.6.8243338. [DOI] [PubMed] [Google Scholar]
- 49.Rissman RA, Lee KF, Vale W, Sawchenko PE. Corticotropin-releasing factor receptors differentially regulate stress-induced tau phosphorylation. J Neurosci. 2007;27(24):6552–62. doi: 10.1523/JNEUROSCI.5173-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rothman SM, Herdener N, Camandola S, Texel SJ, Mughal MR, Cong WN, Martin B, Mattson MP. 3xTgAD mice exhibit altered behavior and elevated Abeta after chronic mild social stress. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rozzini L, Vicini Chilovi B, Conti M, Delrio I, Borroni B, Trabucchi M, Padovani A. Neuropsychiatric symptoms in amnestic and nonamnestic mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;25(1):32–6. doi: 10.1159/000111133. [DOI] [PubMed] [Google Scholar]
- 52.Saper CB. Central autonomic system. In: Paxinos G, editor. The Rat Nervous System. 2. San Diego: Academic Press; 1995. pp. 107–28. [Google Scholar]
- 53.Sapolsky RM. Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol. 1999;34(6):721–32. doi: 10.1016/s0531-5565(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 54.Selkoe DJ, Wolfe MS. In search of gamma-secretase: presenilin at the cutting edge [comment] Proc Natl Acad Sci U S A. 2000;97(11):5690–2. doi: 10.1073/pnas.97.11.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27(11):2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20(6):1093–102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 57.Song L, Che W, Min-Wei W, Murakami Y, Matsumoto K. Impairment of the spatial learning and memory induced by learned helplessness and chronic mild stress. Pharmacol Biochem Behav. 2006;83(2):186–93. doi: 10.1016/j.pbb.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 58.Sterniczuk R, Antle MC, Laferla FM, Dyck RH. Characterization of the 3xTg-AD mouse model of Alzheimer's disease: part 2. Behavioral and cognitive changes. Brain Res. 2010;1348:149–55. doi: 10.1016/j.brainres.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 59.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94(24):13287–92. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36(3):165–86. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 61.Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19(2):162–6. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 62.Touma C, Ambree O, Gortz N, Keyvani K, Lewejohann L, Palme R, Paulus W, Schwarze-Eicker K, Sachser N. Age- and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2004;25(7):893–904. doi: 10.1016/j.neurobiolaging.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 63.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213(4514):1394–7. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 64.Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428(2):191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 65.Walker DL, Miles LA, Davis M. Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(8):1291–308. doi: 10.1016/j.pnpbp.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 67.Weiner MF, Vobach S, Olsson K, Svetlik D, Risser RC. Cortisol secretion and Alzheimer's disease progression. Biol Psychiatry. 1997;42(11):1030–8. doi: 10.1016/s0006-3223(97)00165-0. [DOI] [PubMed] [Google Scholar]
- 68.Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52(2):90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- 69.Yang L, Wang Z, Wang B, Justice NJ, Zheng H. Amyloid precursor protein regulates Cav1. 2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci. 2009;29(50):15660–8. doi: 10.1523/JNEUROSCI.4104-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81(4):525–31. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 71.Zheng H, Koo EH. Biology and Pathophysiology of the Amyloid Precursor Protein. Mol Neurodegener. 2011;6(1):27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Anxiety-related behavior in single mutants and aged animals. (A,B) Animals were tested on the EPM for time on the open arms (A), and time in the closed arms (B). APP/hAβ/PS1 (black bars) animals displayed reduced time on the open arms compared to wildtype animals (white bars). APP single knock-in animals showed similar open arm time as APP/hAβ/PS1 animals (p<0.05 compared to wildtype) whereas PS1 knock-in mutants displayed wildtype levels of anxiety. (C, D) Aged animals were tested for anxiety-related behavior on the EPM. Both wildtype (white bars) and APP/hAβ/PS1 animals displayed decreased open arm time at 12 months of age compared to the same genotype at 3 months of age. At 12 months APP/hAβ/PS1 animals spent significantly less time in the open arms compared to age-matched control animals (p<0.05). (E) CRF levels were measured in the BSTov, PVN, and CeA of wildtype (white bars), APP single knock-in mice (light grey bars), PS1 single knock-in mice (dark grey bars), and APP/hAβ/PS1 mice (black bars). CRF levels are higher in APP single knock-in and APP/hAβ/PS1 mice in the BSTov. In the PVN, CRF levels were higher in APP/hAβ/PS1 mice but not in single knock-in mice. In the CeA, CRF levels were similar in all lines, with APP/hAβ/PS1 having the highest levels of CRF signal.