

Abstract
We recently described the in vitro and in vivo properties of an engineered homotrimeric antibody made by fusing the N-terminal trimerization region of collagen XVIII NC1 domain to the C-terminus of a scFv fragment [trimerbody (scFv-NC1)3; 110 kDa]. Here, we demonstrated the utility of the N-terminal trimerization region of collagen XV NC1 domain in the engineering of trivalent antibodies. We constructed several scFv-based trimerbodies containing the human type XV trimerization domain and demonstrated that all the purified trimerbodies were trimeric in solution and exhibited excellent antigen binding capacity. Importantly, type XV trimerbodies demonstrated substantially greater thermal and serum stability and resistance to protease digestion than type XVIII trimerbodies. In summary, the small size, high expression level, solubility and stability of the trimerization domain of type XV collagen make it the ideal choice for engineering homotrimeric antibodies for cancer detection and therapy.
Key words: antibody engineering, multivalent antibody, collagen XVIII, collagen XV, tumor targeting
Introduction
The ideal tumor-targeting molecules are intermediate-sized multimeric antibodies, which allow rapid tissue penetration, high target retention and rapid blood clearance,1–3 provided that they are able to avoid proteolytic degradation in plasma or at the tumor site. Studies on tumor localization indicate that bivalent antibodies such as diabodies (60 kDa),4,5 and minibodies (80 kDa),6 may be best suited for tumor imaging and therapy due to a higher total tumor uptake and better tumor-to-blood ratios than intact IgG molecules.
To maximize tumor targeting capabilities, we developed a new class of trivalent antibody, termed “trimerbody.” The trimerbody (110 kDa) is a multivalent antibody comprising a scFv connected to the collagen XVIII NC1 trimerization domain through a flexible peptide linker.7,8 Trimerbodies exhibited excellent antigen binding capacity and were multivalent, which provides them with a significant increase in functional affinity. Furthermore, fluorescently labeled trimerbodies showed efficient tumor targeting in mice bearing human cancers.8,9
Type XV and XVIII collagens are the only known members of the multiplexin (multiple triple-helix domains and interruptions) collagen family.10 Both collagens are homo-trimers composed of a single α-chain that contains a central highly interrupted collagenous domain flanked at the N- and C-terminus by non-collagenous domains.11–13 The highest level of sequence homology between types XV and XVIII lies in the NC1 domain. The NC1 domains of type XV and XVIII collagens are organized into a N-terminal trimerization domain, a central protease-sensitive hinge region and a compact C-terminal endostatin (collagen XVIII) or restin (collagen XV) domain.14,15 The trimerization regions of both NC1 domains have been crystallized.16,17 Despite having only 32% sequence identity, the type XV trimerization domain structure is remarkably similar to, and displays biochemical properties comparable to, the type XVIII trimerization domain.
Here, we demonstrate the utility of the type XV trimerization domain in the engineering of antibody trimers. We constructed several scFv-based trimerbodies containing the human collagen XV trimerization domain. All the purified type XV trimerbodies were trimeric in solution and exhibited excellent antigen binding capacity, similar to that of type XVIII trimerbodies. Importantly, type XV trimerbodies demonstrated greater stability against thermal denaturation and improved resistance against serum and connective tissue proteases than type XVIII trimerbodies.
Results
Design and expression of recombinant antibodies containing the trimerization region from the human collagen XV NC1 domain.
We have previously shown that fusion of the N-terminal trimerization region of the murine collagen XVIII NC1 domain to the C-terminus of a scFv antibody confers a trimeric state to the fused antibody (”trimerbody“).7,8 Purified trimerbodies are trimeric in solution, and exhibit excellent antigen binding capacity. Surface plasmon resonance analysis demonstrated that an anti-NIP trimerbody has at least a 100-fold increase in apparent functional affinity compared with its monovalent counterpart.8
In the study reported here, we extended the concept by designing recombinant antibodies using the N-terminal trimerization region of the human collagen XV NC1 domain (from amino acid 1,135 to 1,198, accession number P39059). Starting from the L36 scFv encoding gene,18 a new recombinant trimerbody was generated (Fig. 1). The L36 scFv-based type XV trimerbody (trimerbodyXV) was secreted as soluble functional protein by transfected HEK-293 cells (Fig. 2). Western blot analysis demonstrated that under reducing conditions the trimerbody consists of a single chain type with a mass of 39.7 kDa (Fig. 2A). Typical yields of secreted functional trimerbodyXV after 3 d of transfection ranged between 1–5 µg/ml, similar to that observed after transfecting HEK-293 cells with L36 scFv-based type XVIII trimerbody (trimerbodyXVIII) gene construct. Both type XV and type XVIII trimerbodies were purified from conditioned medium by immobilized metal affinity chromatography, which yielded trimerbodies that were >95% pure by reducing SDS-PAGE (Fig. 2C). The functionality of the purified antibodies was demonstrated by ELISA against plastic immobilized laminin-111. As shown in Figure 2D, antibody titration analysis showed a dose-dependent binding of L36 scFv, L36 scFv-based type XV trimerbody and L36 scFv-based type XVIII trimerbody, with the lowest apparent functional affinity for the monomeric scFv. These result demonstrated that the L36 scFv-based type XV trimerbody recognized its cognate antigen as efficiently as the L36 trimerbody with the trimerization domain from mouse collagen XVIII NC1 domain (trimerbodyXVIII).
Figure 1.

The concept of creating multimeric antibodies using the human collagen XV trimerization domain (collagen XV TD) and scFv fragments. (A) Schematic diagram of the scFv (i) and trimerbody (ii) gene constructs. L, linker peptide. The genes are under the control of the cytomegalovirus promoter/enhancer, and a heterologous signal peptide from the Oncostatin M (onco M) gene is used to direct secretion of recombinant antibodies. Below trimerbody diagram is the sequence of the peptide linker used to join the scFv fragment to the collagen XV TD. Schematic showing arrangement of VH and VL domain in monomeric scFv (B), and arrangement of VH, VL and collagen XV TD in trimeric trimerbody (C).
Figure 2.

The presence of secreted recombinant antibodies (L36 scFv, L36 scFv type XVIII trimerbody and L36 scFv type XV trimerbody) in the supernatant of gene modified HEK-293 cells was demonstrated by western blot analysis (A) and by ELISA (B) against plastic immobilized laminin-111. Migration distances of molecular mass markers are indicated (kDa). The blot was developed with anti-c-myc mAb. Reducing SDS-PAGE of purified type XVIII and type XV trimerbodies (C), and antigen titration ELISA of purified L36 scFv, L36 scFv type XVIII trimerbody and L36 scFv type XV trimerbody against plastic immobilized laminin-111 (D).
Characterization of recombinant trimerbodies.
The oligomerization state of purified type XV trimerbody and type XVIII trimerbody was assessed by analytical gel filtration chromatography, as well as by analytical ultracentrifugation and sedimentation equilibrium gradient. Both trimerbodies eluted from the column as a single peak with estimated masses of 111.4 kDa and 117.6 kDa for the trimerbodyXVIII and the trimerbodyXV, respectively (Fig. 3A and C, respectively). Sedimentation equilibrium experiments could only be fitted to a trimer (not to a monomer or a dimer) (Fig. 3B and D). These results demonstrate the trimeric nature of both antibodies, a feature conferred by the trimerization region from NC1 collagen XVIII and NC1 collagen XV.
Figure 3.

Structural characterization of the purified trimerbodies. Analytical gel filtration chromatograms (A and C) and sedimentation equilibrium gradient (B and D) were performed as described in material and methods, with purified L36 scFv type XVIII trimerbody (A and B), and purified L36 scFv type XV trimerbody (C and D).
To further evaluate type XV trimerization domain as a new platform for engineering multivalent antibodies, we designed scFv-based trimerbodyxv constructs with specificity for the hapten NIP or the tumor-associated antigen CEA. The scFv B1.8 (anti-NIP) and the scFv MFE-23 (anti-CEA) were similarly assembled and expressed as soluble secreted trimerbodyXV in human HEK-293 cells. The expression, functionality and specificity of B1.8 and MFE23 type XV trimerbodies was demonstrated by western blot (Fig. 4A) and ELISA against plastic immobilized BSA, CEA and NIP10-BSA conjugates (Fig. 4B). As shown in Figure 4C, the B1.8 type XV trimerbody displayed better binding to NIP10-BSA conjugates than the monomeric scFv.
Figure 4.

The functionality and specificity of anti-CEA and anti-NIP scFv type XV trimerbodies was demonstrated by western blot (A), and by ELISA (B and C) against plastic immobilized BSA, NIP10-BSA conjugates and CEA. MFE23 anti-CEA and B1.8 anti-NIP scFvs were used as controls. Antigen titration ELISA of purified B1.8 scFv and B1.8 scFv type XV trimerbody against plastic immobilized NIP10-BSA conjugates (C).
Stability studies.
We examined the thermal stability of the purified scFv-based type XV and type XVIII trimerbodies. Both antibodies showed cooperative thermal denaturations as monitored by the change in circular dichroism signal; however, while the trimerbodyXVIII has a mid-point denaturation temperature of 50°C, the trimerbodyXV is substantially stabilized, with a corresponding value of 67°C (Fig. 5).
Figure 5.
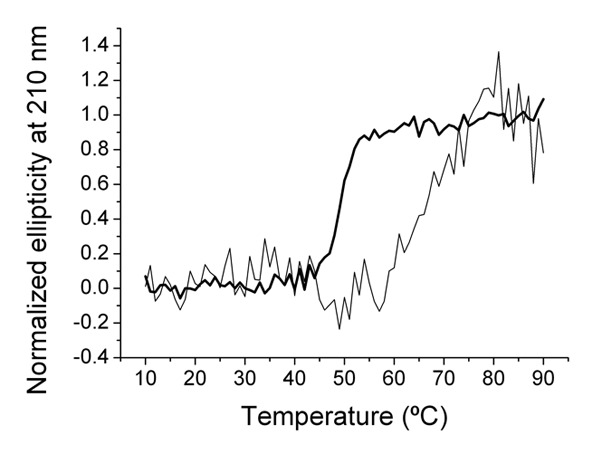
Thermal denaturation curves measured by the change in ellipticity at 210 nm are shown for the type XVIII trimerbody (thick line), and for the type XV trimerbody (thin line).
The stability of engineered antibodies in serum is another key parameter to determine their potential application in vivo. Therefore, we compared the functionality of both type XV and type XVIII trimerbodies after incubation in human or mouse serum at 37°C for prolonged periods of time. As shown in Figure 6, the purified type XV trimerbody had a significantly improved stability over the conventional type XVIII trimerbody [77 and 90% vs. 45 and 59%, in human (Fig. 6A) or mouse (Fig. 6B) serum, respectively].
Figure 6.
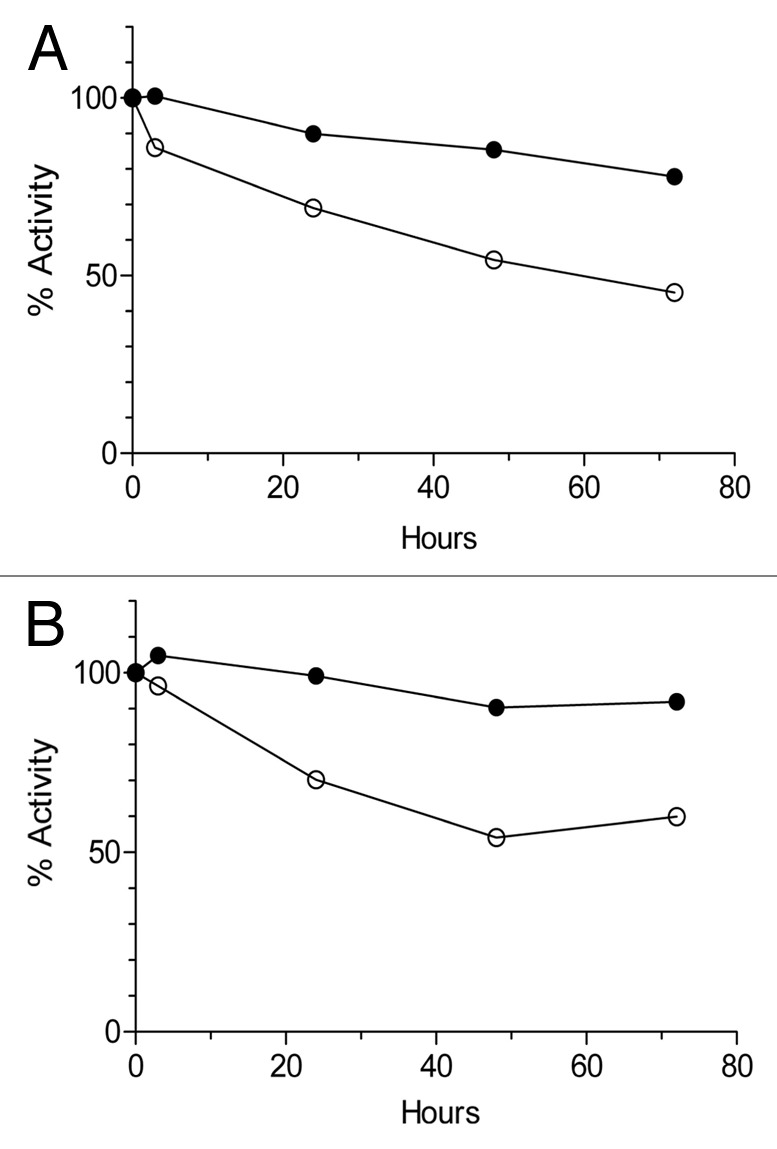
Serum stability of purified L36 scFv type XVIII trimerbody (open circles) and L36 scFvtype XV trimerbody (filled circles), after incubation at 37°C in human (A) or mouse (B) serum, as explained in material and methods. At the indicated times the reaction mixtures were analyzed by ELISA against plastic immobilized laminin-111.
For therapeutic purposes, in addition to serum stability, recombinant antibodies should remain functional in the presence of connective tissue proteases. We therefore compared both type XV and type XVIII trimerbodies in terms of stability and functionality after incubation with two of the proteases present in the tumor microenvironment: cathepsin L and elastase. As shown in Figure 7, type XV trimerbody exhibited greater resistance to protease digestion than the type XVIII trimerbody.
Figure 7.

Proteolytic stability of purified L36 scFv type XVIII trimerbody (open circles) and L36 scFv type XV trimerbody (filled circles), after incubation at 37°C with different amounts of cathepsin L (A) or elastase (B), as explained in material and methods. After incubation the reaction mixtures were analyzed by western blot, using an anti-c-myc mAb, and by ELISA against plastic immobilized laminin-111.
Discussion
In this study, we demonstrated the usefulness of human collagen XV trimerization domain to generate trimeric antibodies. Purified type XV trimerbodies were trimeric in solution, and exhibited excellent antigen binding capacity. The type XV trimerization domain is as efficient as the type XVIII trimerization domain for inducing trimerization of scFv domains located N-terminally. In fact, trimerbodies that shared the scFv domain and the peptide connector, but differed in the trimerization domain possessed similar functional properties. Both types of trimerbodies were isolated in functional active form from conditioned medium of transfected HEK-293 cells and were efficiently purified using standard chromatographic techniques.
Recent crystallographic data for type XV trimerization domain17 demonstrated that the molecular topology and organization of this protein and that of type XVIII trimerization domain16 are comparable. These data indicate that the multiplexin trimerization fold is a robust one, capable of retaining structural integrity with a large sequence variability.
Despite this similarity, stability profiles indicated that the two trimerbodies differed in their resistance to denaturation by heat and proteases. Willuda et al.19 demonstrated that thermal stability of antibody fragments is critical for successfully tumor targeting cells, and in fact type XV trimerbodies showed higher thermal stability than type XVIII trimerbodies. It was encouraging to observe that type XV trimerbodies were significantly more stable in human and mouse serum than type XVIII trimerbodies, as well as in the presence of proteases that are relevant in certain pathological contexts (e.g., cathepsin L and elastase). Cathepsin L is a ubiquitously expressed cysteine protease that localizes primarily to the lysosomes and can be secreted. Expression of cathepsins has been reported to be elevated in multiple cancer types,20 and has been found to be correlated with the metastatic potential of tumor cell lines.21 Furthermore, a local increase in leukocyte elastase is associated with tumor invasiveness in many types of cancer.22 Whereas type XVIII trimerbodies were completely degraded by both proteases, type XV trimerbodies maintained their molecular integrity. Structurally intact type XV trimerbody was detected even at the highest concentrations of both proteases.
These differences may be related to the different protein sequence and localization of type XV and type XVIII collagen. Collagen XVIII is a heparan sulfate proteoglycan23 and collagen XV carries chains of chondroitin/dermatan sulfate alone, or chondroitin/dermatan sulfate together with heparan sulfate in a differential ratio.24 Type XV and XVIII collagens are closely related basement membrane molecules that are broadly expressed and have been localized to a wide variety of basement membranes, including vascular and epidermal basement membranes.25 It is tempting to speculate that the environmentally-induced functional specialization of type XV collagen might be responsible for the dramatic differences observed in stability.
These results have important implications in terms of therapeutic targeting, particularly in oncology. In the hostile microenvironment associated with solid tumors, the function of therapeutic or diagnostic antibodies may be compromised.26 Type XV trimerbodies are remarkably stable, which gives them a substantial advantage in the therapeutic field.
Materials and Methods
Reagents and antibodies.
The monoclonal antibody (mAb) specific for human c-myc used was 9E10 (Abcam, Cambridge, UK). The IRDye 800 (fluorochrome)-conjugated goat anti-mouse IgG polyclonal antibody (Fc specific) was from Rockland Immunochemicals Inc., (Gilbertsville, PA USA). Laminin-111 extracted from the Engelbreth-Holm-Swarm (EHS) mouse tumor was from Becton Dickinson Labware (Bedford, MA USA). Human carcinoembryonic antigen (CEA), pooled human and mouse sera, as well as cathepsin L, and elastase were from Sigma-Aldrich Inc., (St. Louis, MO USA). BSA was conjugated with 4-hydroxy-5-iodo-3-nitrophenyl (NIP) (Sigma-Aldrich) in a molar ratio of 10:1 (NIP10-BSA) as described in reference 27.
Cells and culture conditions. HEK-293 cells (human embryo kidney epithelia; CRL-1573) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) heat-inactivated Fetal Calf Serum (FCS) (all from Invitrogen, Carlsbad, CA) referred as to DMEM complete medium (DCM).
Construction of expression vectors. To construct the pCR3.1-L36-hXVNC1 expression vector, the N-terminal trimerization region from human collagen XV NC1 domain was synthesized by Geneart AG (Regensburg, Germany) and subcloned as NotI-XbaI into the vector pCR3.1-L36,18 containing the L36 (anti-laminin) single-chain Fv (scFv) gene. The MFE23 (anti-human CEA) and the B1.8 (anti-hapten NIP) scFv expression cassettes were subcloned as HindIII-NotI into the vector pCR3.1-L36-hXVNC1, resulting in pCR3.1-MFE23-hXVNC1 and pCR3.1-B1.8-hXVNC1, respectively.
Cell transfections and purification of recombinant antibodies.
HEK-293 cells were transfected with pCR3.1-L36-mXVIIINC1,7 pCR3.1-L36-hXVNC1, pCR3.1-MFE-23-hXVNC1 or pCR3.1-B1.8-hXVNC1 expression vectors using Superfect (QIAGEN GmbH, Hilden, Germany), and selected in DCM supplemented with G418 (500 mg/ml). Supernatants from transient and stably transfected cell populations were analyzed for protein expression by ELISA, SDS-PAGE and western blotting using anti-c-myc mAb. Stably transfected HEK-293 cells were used to collect serum-free conditioned media medium (∼1 L), and loaded onto a HisTrap HP 1 ml column using an ÄKTA Prime plus system (GE Healthcare, Uppsala, Sweden). The purified antibodies were dialyzed against PBS, analyzed by SDS-PAGE under reducing conditions, and stored at −20°C.
ELISA.
The ability of purified trimerbodies to bind murine laminin-111, human CEA or NIP10-BSA conjugates was studied by ELISA as described in reference 8, 28 and 29.
Analytical ultracentrifugation experiments.
An Optima XL-I analytical ultracentrifuge (Beckman-Coulter, Miami, FL USA) was used to perform the analytical ultracentrifugation experiments. The detection was performed by means of a UV-visible absorbance detection system. Experiments were conducted at 20°C using an AnTi50 eight-hole rotor and Epon-charcoal standard double sector centerpieces (12 mm optical path). Absorbance scans were taken at the appropriate wavelength (240, 280, 297 nm). Sedimentation velocity experiments were performed at 48 krpm using 400 µl samples in PBS buffer. Differential sedimentation coefficient distributions, c(s), were calculated by least squares boundary modeling of sedimentation velocity data using the program SEDFIT.30,31 From this analysis, the experimental sedimentation coefficients were corrected for solvent composition and temperature with the program SEDNTERP to obtain the corresponding standard s values.32 Short column (85 µl) sedimentation equilibrium runs were performed at multiple speeds (9, 11 and 15 krpm). After the equilibrium scans, a high speed centrifugation run (43 krpm) was done to estimate the corresponding baseline offsets. Weight-average buoyant molecular weights of protein and oligonucleotides were determined by fitting a single species model to the experimental data using the HeteroAnalysis program.33 Both analyses gave essentially the same results. The molecular weights of protein and DNA were determined from the experimental buoyant masses using 0.718 and 0.722 as the partial specific volumes of A and B, respectively (calculated from the amino acid composition using the SEDNTERP program32).
Circular dichroism (CD).
Circular dichroism (CD) measurements were performed with a Jasco J-810 spectropolarimeter equipped with Peltier thermal control unit (JASCO). Thermal denaturations were recorded on protein samples at 0.024 or 0.015 mg/mL for trimerbodyXV and trimerbodyXVIII, respectively, in PBS using a 0.2 cm path length stoppered quartz cuvette. Protein unfolding was induced by increasing temperature from 10 to 90°C at a rate of 1°C/min. The ellipticity at 210 nm was monitored every 1°C with a 32 sec response and 4 nm bandwidth and the values were normalized from 0 to 1 (at 5°C) to 1 (at 90°C). Repeated experiments provide an estimate for the error in the mid-point denaturation temperature of trimerbodyXVIII of ±1°C. The denaturation curve of the trimerbodyXV has a smaller signal-to-noise ratio than the curve of trimerbodyXVIII because the total ellipticity change at 210 nm is smaller for this molecule. As a consequence, the error in the mid-point temperature determination is larger for trimerbodyXV, but the shift of the two curves is evident over a range of approximately 20°C, demonstrating a thermal stabilization of trimerbodyXV.
Serum stability.
One microgram of each trimerbody was incubated in 62% human or mouse serum at 37°C for up to 72 h. Samples were removed for analysis 3 h, 24 h, 48 h and 72 h later and frozen until the entire study was completed. As a control, a second set of serum-exposed samples was frozen immediately to represent a zero time point. Aliquots were then subjected to western blot, using an anti-c-myc mAb, and tested for their capability to bind their cognate antigen by ELISA.
Protease stability.
One microgram of each trimerbody was incubated at 37°C for 10 min with 200, 300 and 400 ng cathepsin L in 50 mM sodium acetate [pH 5.5], 2 mM dithiothreitol, 5 mM EDTA; and 50, 100 and 200 ng elastase in 100 mM TRIS-HCl [pH 8.0]. Aliquots were then subjected to western blot, using an anti-c-myc mAb, and tested for their capability to bind laminin-111 by ELISA.
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia e Innovación (BIO2008-03233), the Comunidad Autónoma de Madrid (S-BIO-0236-2006) and the European Union [SUDOE-FEDER (IMMUNONET-SOE1/P1/E014)] to L.A.V.; and by grant CTQ2011-28680 to F.J.B. and by grant PS09/00227 from the Fondo de Investigación Sanitaria to L.S. D.S.M. was supported by a Comunidad Autónoma de Madrid/Fondo Social Europeo training grant (FPI-000531). Angel M. Cuesta was supported by Instituto de Salud Carlos III (CA08/00087).
Abbreviations
- scFv
single-chain Fv
- mAb
monoclonal antibody
- NC1
non-collagenous domain
References
- 1.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 2.Sanz L, Cuesta AM, Compte M, Alvarez-Vallina L. Antibody engineering: facing new challenges in cancer therapy. Acta Pharmacol Sin. 2005;26:641–648. doi: 10.1111/j.1745-7254.2005.00135.x. [DOI] [PubMed] [Google Scholar]
- 3.Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010;28:355–362. doi: 10.1016/j.tibtech.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA. 1993;90:6444–6448. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudson PJ, Kortt AA. High avidity scFv multimers; diabodies and triabodies. J Immunol Methods. 1999;231:177–189. doi: 10.1016/S0022-1759(99)00157-X. [DOI] [PubMed] [Google Scholar]
- 6.Hu S, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JY, et al. Minibody: A novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996;56:3055–3061. [PubMed] [Google Scholar]
- 7.Sánchez-Arévalo Lobo VJ, Cuesta AM, Sanz L, Compte M, García P, Prieto J, et al. Enhanced antiangiogenic therapy with antibody-collagen XVIII NC1 domain fusion proteins engineered to exploit matrix remodeling events. Int J Cancer. 2006;119:455–462. doi: 10.1002/ijc.21851. [DOI] [PubMed] [Google Scholar]
- 8.Cuesta AM, Sánchez-Martín D, Sanz L, Bonet J, Compte M, Kremer L, et al. In vivo tumor targeting and imaging with engineered trivalent antibody fragments containing collagen-derived sequences. PLoS One. 2009;4:5381. doi: 10.1371/journal.pone.0005381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sánchez-Martín D, Cuesta AM, Fogal V, Ruoslahti E, Alvarez-Vallina L. The multicompartmental p32/gClqR as a new target for antibody-based tumor targeting strategies. J Biol Chem. 2011;286:5197–5203. doi: 10.1074/jbc.M110.161927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pihlajaniemi T, Rehn M. Two new collagen subgroups: membrane-associated collagens and types XV and XVII. Prog Nucleic Acid Res Mol Biol. 1995;50:225–262. doi: 10.1016/S0079-6603(08)60816-8. [DOI] [PubMed] [Google Scholar]
- 11.Myers JC, Kivirikko S, Gordon MK, Dion AS, Pihlajaniemi T. Identification of a previously unknown human collagen chain, alpha1(XV), characterized by extensive interruptions in the triple-helical region. Proc Natl Acad Sci USA. 1992;89:10144–10148. doi: 10.1073/pnas.89.21.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehn M, Hintikka E, Pihlajaniemi T. Primary structure of the alpha 1 chain of mouse type XVIII collagen, partial structure of the corresponding gene, and comparison of the alpha1(XVIII) chain with its homologue, the alpha1(XV) collagen chain. J Biol Chem. 1994;269:13929–13935. [PubMed] [Google Scholar]
- 13.Rehn M, Pihlajaniemi T. Alpha1(XVIII), a collagen chain with frequent interruptions in the collagenous sequence, a distinct tissue distribution and homology with type XV collagen. Proc Natl Acad Sci USA. 1994;91:4234–4238. doi: 10.1073/pnas.91.10.4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki T, Fukai N, Mann K, Göhring W, Olsen BR, Timpl R. Structure, function and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. EMBO J. 1998;17:4249–4256. doi: 10.1093/emboj/17.15.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasaki T, Larsson H, Tisi D, Claesson-Welsh L, Hohenester E, Timpl R. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J Mol Biol. 2000;301:1179–1190. doi: 10.1006/jmbi.2000.3996. [DOI] [PubMed] [Google Scholar]
- 16.Boudko SP, Sasaki T, Engel J, Lerch TF, Nix J, Chapman MS, et al. Crystal structure of human collagen XVIII trimerization domain: A novel collagen trimerization Fold. J Mol Biol. 2009;392:787–802. doi: 10.1016/j.jmb.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wirz JA, Boudko SP, Lerch TF, Chapman MS, Bächinger HP. Crystal structure of the human collagen XV trimerization domain: a potent trimerizing unit common to multiplexin collagens. Matrix Biol. 2011;30:9–15. doi: 10.1016/j.matbio.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanz L, Kristensen P, Blanco B, Facteau S, Russell SJ, Winter G, et al. Single-chain antibody-based gene therapy: inhibition of tumor growth by in situ production of phage-derived human antibody fragments blocking functionally active sites of cell-associated matrices. Gene Ther. 2002;9:1049–1053. doi: 10.1038/sj.gt.3301725. [DOI] [PubMed] [Google Scholar]
- 19.Willuda J, Honegger A, Waibel R, Schubiger PA, Stahel R, Zangemeister-Wittke U, et al. High thermal stability is essential for tumor targeting of antibody fragments: engineering of a humanized anti-epithelial glycoprotein-2 (epithelial cell adhesion molecule) single-chain Fv fragment. Cancer Res. 1999;59:5758–5767. [PubMed] [Google Scholar]
- 20.Kane SE, Gottesman MM. The role of cathepsin L in malignant transformation. Semin Cancer Biol. 1990;1:127–136. [PubMed] [Google Scholar]
- 21.Denhardt DT, Greenberg AH, Egan SE, Hamilton RT, Wright JA. Cysteine proteinase cathepsin L expression correlates closely with the metastatic potential of H-ras-transformed murine fibroblasts. Oncogene. 1987;2:55–59. [PubMed] [Google Scholar]
- 22.Yamashita J, Tashiro K, Yoneda S, Kawahara K, Shirakusa T. Local increase in polymorphonuclear leukocyte elastase is associated with tumor invasiveness in non-small cell lung cancer. Chest. 1996;109:1328–1334. doi: 10.1378/chest.109.5.1328. [DOI] [PubMed] [Google Scholar]
- 23.Halfter W, Dong S, Schurer B, Cole GJ. Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem. 1998;273:25404–25412. doi: 10.1074/jbc.273.39.25404. [DOI] [PubMed] [Google Scholar]
- 24.Amenta PS, Scivoletti NA, Newman MD, Sciancalepore JP, Li D, Myers JC. Proteoglycan-collagen XV in human tissues is seen linking banded collagen fibers subjacent to the basement membrane. J Histochem Cytochem. 2005;53:165–176. doi: 10.1369/jhc.4A6376.2005. [DOI] [PubMed] [Google Scholar]
- 25.Ackley BD, Crew JR, Elamaa H, Pihlajaniemi T, Kuo CJ, Kramer JM. The NC1/endostatin domain of Caenorhabditis elegans type XVIII collagen affects cell migration and axon guidance. J Cell Biol. 2001;152:1219–1232. doi: 10.1083/jcb.152.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brezski RJ, Vafa O, Petrone D, Tam SH, Powers G, Ryan MH, et al. Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc Natl Acad Sci USA. 2009;106:17864–17869. doi: 10.1073/pnas.0904174106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez-Vallina L, Hawkins RE. Antigen-specific targeting of CD28-mediated T cell co-stimulation using chimeric single-chain antibody variable fragment-CD28 receptors. Eur J Immunol. 1996;26:2304–2309. doi: 10.1002/eji.1830261006. [DOI] [PubMed] [Google Scholar]
- 28.Sanz L, Kristensen P, Russell SJ, Ramirez García JR, Alvarez-Vallina L. Generation and characterization of recombinant human antibodies specific for native laminin epitopes: potential application in cancer therapy. Cancer Immunol Immunother. 2001;50:557–565. doi: 10.1007/s00262-001-0235-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanz L, García-Bermejo L, Blanco FJ, Kristensen P, Feijóo M, Suárez E, et al. A novel cell binding site in the coiled-coil domain of laminin involved in capillary morphogenesis. EMBO J. 2003;22:1508–1517. doi: 10.1093/emboj/cdg150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laue T. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Ultracentrifugation in biochemistry and polymer science. Cambridge: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 33.Cole JL. Analysis of heterogeneous interactions. Methods Enzymol. 2004;384:212–232. doi: 10.1016/S0076-6879(04)84013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]