

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV, human herpesvirus 8) is etiologically associated with three neoplastic syndromes: Kaposi sarcoma and the uncommon HIV-associated B-cell lymphoproliferative disorders primary effusion lymphoma and multicentric Castleman disease. The incidence of the latter B-cell pathology has been increasing in spite of antiretroviral therapy; its association with lytic virus replication has prompted interest in therapeutic strategies aimed at this phase of the virus life cycle. We designed and expressed a recombinant immunotoxin (2014-PE38) targeting the gpK8.1A viral glycoprotein expressed on the surface of the virion and infected cells. We show that this immunotoxin selectively kills KSHV-infected cells in dose-dependent fashion, resulting in major reductions of infectious virus release. The immunotoxin and ganciclovir, an inhibitor of viral DNA replication, showed marked reciprocal potentiation of antiviral activities. These results suggest that the immunotoxin, alone or in combination, may represent a new approach to treat diseases associated with KSHV lytic replication.
Key words: targeted cytotoxic proteins, human herpesvirus-8, KSHV surface glycoprotein, KSHV lytic infection, multicentric Castleman disease, pseudomonas exotoxin A, ganciclovir, reciprocal drug potentiation
Introduction
Kaposi sarcoma-associated herpes virus (KSHV, human herpes virus 8) is a human oncogenic DNA virus belonging to the gammaherpesviridae family, rhadinoviridae genus.1 Like all herpesviruses2 KSHV displays two distinct transcriptional programs, latency and lytic replication. The KSHV latency phase is associated with restricted gene expression involved in maintenance of the viral genome as a nuclear episome, cell proliferation, immune evasion and suppression of apoptosis; the lytic phase involves temporal expression of genes leading to virus replication and infectious virion release.3
Since its initial identification in 1994 by representational differential analysis of AIDS-related Kaposi sarcoma (KS) tissues,4 KSHV was subsequently implicated in two uncommon, typically human immunodeficiency virus (HIV)-associated, B-cell lymphoproliferative disorders—primary effusion lymphoma (PEL),5 and the plasmablastic variant of multicentric Castleman disease (MCD).6 Its etiological role in all three diseases is now firmly established.7–10 Among these KSHV-associated pathologies, there are marked differences in the patterns of latent vs. lytic phase gene expression as judged by protein expression patterns,11,12 quantitative DNA analysis coupled with computerized immunohistochemistry,13 and serum antibody profiles.14 MCD is distinctive for the prominent involvement of KSHV lytic phase replication. The plasmablastic form involves monotypic (IgM, λ) polyclonal B-cell proliferation accompanied by episodic life-threatening flare-ups of fever, peripheral lymphadenopathy, splenomegaly, severe cytopenia and elevated levels of interleukin-6 (including the virus-encoded homolog) and other inflammatory markers.15–18 Consistent with the involvement of lytic phase replication, high viral loads in peripheral blood mononuclear cells accompany acute episodes,19 and high level KSHV DNA in plasma has been proposed as a predictor of an active attack.20 Oddly while the incidence of HIV-associated KS has declined with access to highly active antiretroviral therapy (HAART),21 HIV-associated MCD incidence has been increasing despite HAART.22
Plasmablastic MCD is a highly aggressive syndrome; left untreated, it is generally fatal within two years, with median survival around 1 y.23 While standardized treatment modalities have not yet been established, several promising approaches are being pursued.16,24,25 Antineoplastic chemotherapy with single or combination agents has yielded clinical remissions, though the responses are typically transient and drug toxicities limit extended use. Successes have been reported with immunotherapy using monoclonal antibodies (mAbs) tocilizumab (Actemra®),26 and siltuximab27 against interlekin-6 or rituximab against the B-cell antigen CD20,18,28–31 though the latter agent has been complicated by aggravation of KS lesions.28,29,32 A recent report described MCD remission in a multiple myeloma patient undergoing treatment with the proteosome inhibitor bortezomib.33 Finally, herpesvirus-directed treatments using inhibitors of the viral DNA polymerase have shown promise,34,35 consistent with the involvement of lytic phase replication in MCD; however, as for chemotherapy, the benefits are transient and long-term use is complicated by dose-limiting toxicities of these agents.
As a potential therapeutic intervention against MCD, we propose specific killing of KSHV-infected cells using targeted cytotoxic proteins (toxic moieties linked to mAbs or ligands) that specifically recognize a KSHV-encoded glycoprotein displayed on the infected cell surface. This concept, which has been vigorously pursued for decades in the oncology field, exploits three variations of the theme of targeted cytotoxic proteins:36 (1) antibody-drug conjugates (ADCs) in which mAbs are chemically coupled to low molecular weight cytotoxic compounds,37,38 (2) radioimmunotherapy whereby radionuclide-conjugated mAbs deliver lethal doses of radiation to the targeted cells,39,40 and (3) immunotoxins wherein the antigen-binding regions of mAbs are linked (chemically or genetically) to the effector domains of protein toxins typically of bacterial or plant origin.41,42 The targeted cytotoxic protein approach is receiving increasing attention for treatment of infectious diseases,43 with pre-clinical antiviral activities demonstrated for each mode, i.e., ADCs,44 radioimmunotherapy45 and immunotoxins.46–50
In the present report, we describe a novel recombinant immunotoxin based on Pseudomonas aeruginosa exotoxin A (PE), in which the normal cell binding moiety is replaced by an antibody fragment against the KSHV gpK8.1A glycoprotein.51 gpK8.1A is a 228 amino acid late lytic glycoprotein52–54 displayed on KSHV virions;55,56 it binds to heparin sulfate on the target cell surface,57,58 thereby facilitating virus entry and consequent spread of infection. Antibodies against gpK8.1A are important serological markers for KSHV disease.59 Antibody profiling for the various KSHV diseases has demonstrated that serum antibodies against gpK8.1A are 5-fold higher in patients with MCD compared with KS and PEL.14 Importantly for the study reported here, the lytic phase of the KSHV infection cycle is associated with expression of gpK8.1A on the surface of at least a fraction of infected cells.53–55,60 This viral glycoprotein therefore appears to be a potential target for an immunotoxin designed to kill lytically infected cells.
Results
Design and production of KSHV gpK8.1A-targeted recombinant immunotoxin.
We designed an immunotoxin targeting gpK8.1A, based on the previously described murine mAb 4C3.55 As shown schematically in Figure 1, the variable heavy and light chain cDNAs from the 4C3 hybridoma were isolated and used to generate a sequence encoding the corresponding single chain variable region fragment; this was linked to the cDNA encoding the PE38 moiety of PE, which lacks the normal N-terminal cell binding domain of the native toxin but contains the effector domains involved in translocation and cytotoxicity (ADP-ribosylation of elongation factor 2). The immunotoxin was expressed in E. coli, the inclusion body fraction was subjected to standard protocols of solubilization, denaturation and refolding, and the protein was purified by sequential ion exchange and size exclusion chromatography61 (see Materials and Methods). This anti-gpK8.1A immunotoxin is herein designated 2014-PE38.
Figure 1.

Schematic of construction of the 2014-PE38 immunotoxin expression plasmid and relative positions of the PCR primers used for the cloning procedure. The VH and VL segments of anti-KSHV K8.1A mAb 4C3 were linked by a 15 amino acid linker (G4S)3 and ligated in frame to PE38.
Target cell system for analyzing immunotoxin activity.
As a cellular target to test the specific cytotoxic activity of 2014-PE38, we used a Vero cell line derivative (herein referred to as Vero-219) that is chronically infected with a recombinant KSHV (rKSHV.219) that carries two reporter genes, green fluorescent protein (GFP) driven by the constitutive elongation factor 1α strong cellular promoter and red fluorescent protein (RFP) linked to the KSHV lytic PAN promoter; GFP is expressed constitutively, i.e., in both latent and lytic phase, whereas RFP is expressed only upon induction into lytic phase.62 Epifluorescence microscopy confirmed the expected result that without induction only a negligible fraction of Vero-219 cells expressed detectable RFP (Fig. 2A and top parts), whereas in induced cultures most of the cells (77–80% in >10 experiments) were RFP-positive.
Figure 2.

Characterization of Vero-219 cells. (A) The four parts are photomicrographs of Vero-219 cells containing rKSHV.219 showing detection of GFP under both induced (with Back50 + sodium butyrate stimulation, 24 h, lower parts) and uninduced (upper parts) conditions. (B) Flow cytometry analysis of K8.1A on the cell surface of induced Vero-219 cells by anti K8.1A mAb 4C3 (compared with isotype control antibody) at 48 h post-induction. (C) Confocal microscopic analysis of the staining of induced Vero-219 cells at 48 h post induction under un-permeabilized (upper parts) and permeabilized (lower parts) conditions.
We then examined expression of gpK8.1A on Vero-219 cells at 48 h post-induction by flow cytometry, staining with mAb 4C3 (or control IgG1) followed by Alexa Fluor 635-labeled second antibody. As shown in Figure 2B, K8.1 A staining was clearly detected on a small subpopulation of induced cells; this pattern was not observed on uninduced cells (data not shown). The mean fluorescent intensity of the entire population of induced cells was 24.9 with mAb 4C3 compared with 8.0 with control IgG1. Additional perspective was gained by confocal microscopy analysis of the induced Vero-219 cells at 48 h post-induction (Fig. 2C). Beyond confirming the above finding of efficient induction into lytic phase as judged by most GFP-positive cells also expressing RFP, the images verify that gpK8.1A expression is highly variable within the induced population, with a subset of detectably positive cells as judged by surface (unpermeabilized) and intracellular (permeabilized) cell staining, but the majority of cells not detectably stained for gpK8.1A by this method.
Selective immunotoxin killing of induced KSHV-infected Vero-219 cells.
As one test of the specific cytotoxic activity of 2014-PE38, we applied the direct killing assay based on cellular dehydrogenase-mediated reduction of the WST-8 tetrazolium salt to formazan.63 The experiment shown in Figure 3A shows that the immunotoxin killed induced Vero-219 cells in dose-dependent fashion. Consistent with the data above indicating that in induced cultures most GFP positive cells also expressed RFP, most (75–90% in >10 experiments) of the induced cells were susceptible to killing by 2014-PE38. Specificity was verified by the minimal effect of an unrelated immunotoxin (BL22) that targets a B cell antigen not expressed on Vero-derived cells (Fig. 3A), and also by the lack of activity against uninduced Vero-219 cells (Fig. 3B). The killing was mediated by the cytotoxic action of the PE38 moiety, as evidenced by the ineffectiveness of 2014-PE38E553D (Fig. 3A), which contains a single point mutation abolishing the ADP ribosylation activity of the PE38 moiety; this result confirms that the observed killing reflects the enzymatic activity of the immunotoxin and not simply an unrelated toxicity associated with binding of the antibody moiety to the target cells. The IC50 for 2014-PE38 killing of induced Vero-219 cells was 146 ng/ml in Figure 3A, corresponding to 2.3 nM (range in 3 experiments: 139–170 ng/ml, mean 152 ng/ml; corresponding to 2.2–2.6 nM, mean 2.3 nM). These results suggest that despite the above-noted detection of gpK8.1A on only a small subpopulation of induced Vero-219 cells by immunofluorescence staining (Fig. 2B and C), the bulk of the cell population expressed sufficient surface levels of the viral glycoprotein to be sensitized for 2014-PE38-mediated killing.
Figure 3.
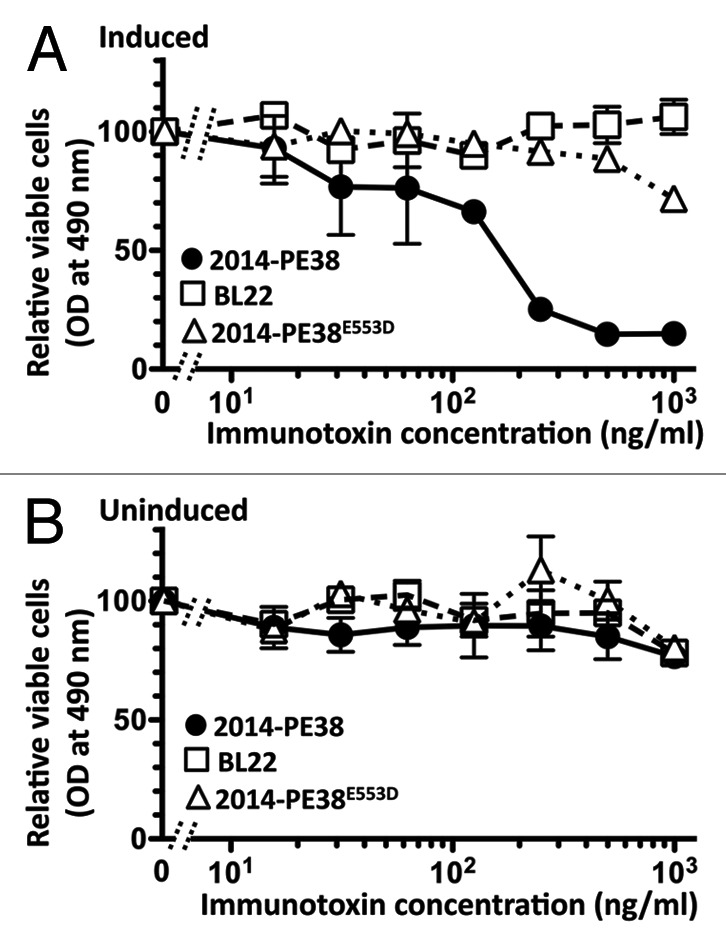
Specific killing of induced Vero-219 cells by 2014-PE38 as measured by the direct cytotoxicity assay. Induced (A) or uninduced (B) cells were treated for 72 h with the indicated concentrations of 2014-PE38 (solid circles) or negative control proteins BL22 (open squares) or 2014-PE38E553 (open triangles). Cell viability was measured by the WST-8 assay. Data points represent mean of duplicate samples; error bars represent standard deviation.
Suppression of infectious KSHV production from Vero-219 cells by immunotoxin-mediated specific cytotoxicity.
As a second test of the selective cytotoxic action of 2014-PE38, we examined the effect of the immunotoxin on infectious KSHV production, by titering culture supernatants of treated Vero-219 producer cells on fresh 293F target cells. The data in Figure 4 demonstrate dose-dependent suppression of infectious virus release. Again specificity was documented by the lack of effect of the unrelated control immunotoxin BL22, and the dependence on immunotoxin-mediated killing of the producer cells was verified by the non-responsiveness to 2014-PE38E553D. The IC50 value of 124 ng/ml, corresponding to 1.9 nM (range in 4 experiments: 111–249 ng/ml, mean 150 ng/ml; corresponding to 1.7–3.8 nM, mean 2.3 nM) was comparable to that noted above in the direct killing assay.
Figure 4.
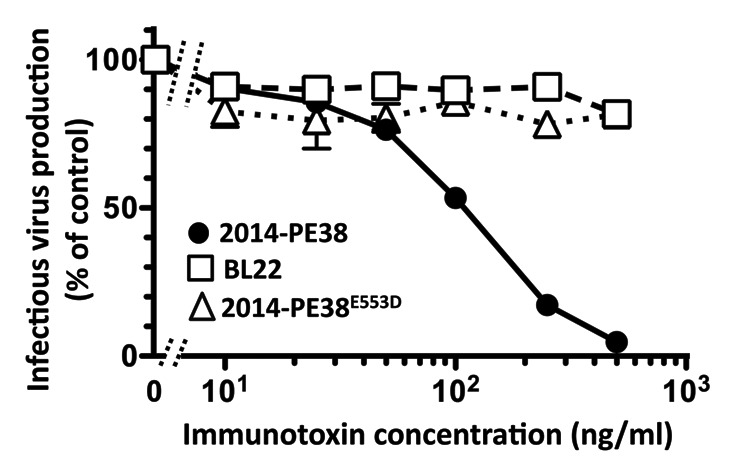
Specific killing of induced Vero-219 cells by 2014-PE38 as measured by the infectious virus release assay. Vero-219 cells were induced by BacK50 plus sodium butyrate and treated for 72 h with the indicated concentrations of 2014-PE38 (solid circles) or the control proteins BL22 (open squares) or 2014-PE38E553 (open triangles). The supernatants were harvested and assayed for infection of fresh 293 cells to measure the infectious KSHV-r129 produced from the activated cells. The amount of infectious virus produced from activated Vero-219 cells with no addition was set as 100%. Each data point represents the mean of duplicate culture wells with error bars as standard deviation.
Complementation of anti-KSHV activity by inhibition of viral DNA replication and targeted killing of infected cells.
Nucleoside analogs that selectively inhibit viral DNA replication are the first line anti-herpesvirus drugs. Originally developed to treat herpes simplex virus infection, they also inhibit KSHV in vitro60,64–66 and have displayed activity against KSHV-associated disease based on reductions in viral oropharyngeal shedding67,68 and clinical relief of MCD symptoms.34,35 At the outset, we considered three alternatives: (1) ganciclovir and the immunotoxin might display additive anti-KSHV activities; (2) they might potentiate one another through their differential mechanisms of inhibiting production of infectious virus during the lytic phase of KSHV replication; (3) ganciclovir might compromise immunotoxin activity by suppressing expression of the gpK8.1A target antigen.60,66 We therefore examined the activities of each agent alone and in combination over wide concentration ranges. In the experiment shown in Figure 5, the individual IC50 values were 249 ng/ml (corresponding to 3.8 nM) for 2014-PE38 and 1.15 µM for ganciclovir. When used in combination, each drug considerably enhanced the potency of the other (i.e., reciprocally reduced the IC50 values). Thus, with ganciclovir at 1 µM (comparable to its intrinsic IC50), the IC50 of the immunotoxin was reduced by more than 5-fold (from 249 ng/ml to 43 ng/ml) (Fig. 5A and inset); conversely with 2014-PE38 at 125 ng/ml (less than its intrinsic IC50), the IC50 of ganciclovir was reduced more than 2-fold (from 1.15 µM to 0.48 µM) (Fig. 2B and inset). Thus there was no evidence for antagonism between these two agents that act by different mechanisms against the lytic phase; instead the opposite effect was observed, i.e., mutual potentiation of the inhibitory activities, beyond simple additivity.
Figure 5.

Mutual potentiation of 2014-PE38 and ganciclovir activities against KSHV infectious virus production from induced Vero-219 cells. Induced Vero-219 cells were treated for 72 h with combinations of 2014-PE38 and ganciclovir over the indicated concentration ranges for each agent. The amount of infectious virus produced in absence of immunotoxin or ganciclovir was set as 100%. Each data point represents the mean of duplicate culture wells with error bars indicating standard deviation. (A) Data are plotted as dose-response curves of 2014-PE38 in presence of the designated concentrations of ganciclovir; (B) Data are plotted as dose-response curves of ganciclovir in presence of the designated concentrations of 2014-PE38. In each part, the insets represent the same data sets, with the values obtained in absence of the agent on the X-axis normalized at 100% for each dose-response curve.
Discussion
The data presented herein demonstrate that 2014-PE38 selectively kills KSHV-infected cells in dose-dependent fashion. The efficacy of this immunotoxin is demonstrated by the potent IC50 values (ranging from 111–249 ng/ml) observed in assays of both direct target cell killing (Fig. 3) and suppression of infectious virus production (Fig. 4); moreover, its activity in the former assay was observed even under conditions of low surface expression of the gpK8.1A antigen on most of the target cells (Fig. 2B and C). This latter phenomenon has been observed with other PE-based immunotoxins; for example the anti-HIV immunotoxins sCD4(178)-PE4069 and 3B3-PE3870 both selectively kill productively HIV-1 infected primary macrophages despite the extremely low surface expression of the corresponding gp120 target antigen on the infected macrophage surface.71 We note that in recent anti-cancer clinical trials of different PE-based recombinant immunotoxins, the mean plasma concentrations achieved at efficacious doses (at or below the maximum tolerated doses) exceeded the IC50 values obtained here for 2014-PE38, e.g., 483 ng/ml for the anti-mesothelin immunotoxin SS1P72 and >700 ng/ml for the anti-CD22 immunotoxin moxetumomab pasudotox.73 That said, the median plasma half-lives of PE-based immunotoxins are relatively short (e.g., ∼7–8 h for SS1P72 and ∼3 h for BL22,74 the earlier version of moxetumomab pasudotox). Thus, it may be critical to enhance the potency of 2014-PE38, e.g., by affinity enhancement of the anti-K8.1A scFv moiety using phage display selection, as has been accomplished in generating the more potent moxetumomab pasudotox from its parent BL22.75
The 2014-PE38 immunotoxin may have particular therapeutic potential for treating MCD, the KSHV-mediated pathology most associated with the lytic phase of the viral life cycle, when gpK8.1A is expressed. This notion is supported by emerging clinical data for MCD treatment testing the effects of inhibitors of herpes virus replication such as ganciclovir and related agents. In one case study, ganciclovir was associated with reduction in the frequency of active MCD episodes,34 although in another report cidofovir was found to be ineffective76 despite its strong anti-KSHV activity in vitro. In a recent pilot study,35 high-dose zidovudine plus valganciclovir produced significant clinical, biochemical and virologic responses in symptomatic MCD patients; the results were interpreted as reflecting mainly (in addition to the possible direct anti-KSHV activity of valganciclovir) the ability of the products of the KSHV genes ORF21 (encoding thymidine kinase) and ORF36 (encoding a phosphotransferase) to activate the conversion of zidovudine and ganciclovir, respectively, into cytotoxic compounds. The efficacy was thus considered to be an example of “virus-activated cytotoxic therapy.” Irrespective of the predominant mechanistic basis for the clinical benefits reported in that study, the present demonstration of selective killing of KSHV infected cells by 2014-PE38 suggests a potential role for this immunotoxin in treating MCD. Moreover, the mutually potentiating effects observed between 2014-PE38 and ganciclovir (Fig. 5) suggest that the combination might be particularly effective, and might achieve clinical benefits at lower doses of each agent, thereby minimizing toxic side effects. We have recently made similar arguments for another KSHV-directed immunotoxin targeting the viral gH glycoprotein.77 Indeed in a murine herpes virus model, complementing activities between an immunotoxin and viral DNA replication inhibitors were observed both in vitro and in vivo.78 Similarly, the robust synergistic activities between an anti-HIV immunotoxin and HAART drugs have been reported. Whereas CD4-PE40 alone or a reverse transcirptase inhibitor alone suppressed but did not eliminate HIV-1 from infected cell cultures,79,80 the combination of the two agents completely “cured” the culture as assessed by eradication of infectious virus and elimination of viral DNA as determined by quantitative polymerase chain reaction analysis.79 Pronounced complementation between anti-HIV immunotoxins and HAART drugs was also observed in an in vivo murine model.81 These results highlight the special advantage of complementing drugs that inhibit the viral replication cycle with immunotoxins that kill already-infected cells.
The present findings should be viewed in the context of the general theme of targeted cytotoxic proteins, since major advances have been made in all three modes (i.e., ADCs, radioimmunotherapy, immunotoxins) discussed in the Introduction. In 1999, Ontak, a recombinant fusion protein consisting of IL2 linked to the translocation and cytotoxic domains of diphtheria toxin, became the first targeted cytotoxic protein to receive US Food and Drug Administration (FDA) approval (for treatment of cutaneous T cell lymphoma).82 Impressive clinical results have since been obtained with other immunotoxins,42 including PE-based75 and diphtheria toxin-based83 agents targeting hematologic cancers. The ADC gemtuzumab ozogamicin (Mylotarg®) received FDA approval in 2000 for treatment of acute myeloid leukemia, but was withdrawn due to lack of efficacy and toxicity concerns;84 in subsequent years several new antibody-drug conjugates (e.g., trastuzumab emtansine, inotuzumab ozogamicin) have reached advanced stages of clinical development.37,38,84 Two radioimmunotherapeutic proteins, ibritumomab tiuxetan (Zevalin®) and tositumomab-I131 (Bexxar®) are FDA-approved for treatment of certain types of lymphoma; several other radionuclide-conjugated antibodies are advancing in the clinical pipeline.39,40
In making general comparisons of these approaches, immunotoxins have the disadvantages of relatively short plasma half lives (generally ∼2–4 h) and significant immunogenicity compared with the two alternative approaches employing intact immunoglobulins, which have intrinsically long half lives and can be humanized to reduce immunogenicity. However, marked advances have been achieved in minimizing these drawbacks and enhancing immunotoxin potential.85,86 Efforts to increase the circulation time of these agents include the design of fusion proteins in which the immunotoxins are linked to Fabs87 or to full-length immunoglobulins.88 The immunogenicity problem is being addressed by identifying and eliminating B cell epitopes using site-directed mutagenesis89 or by choosing human-derived proteins as the cytotoxic moieties.90,91 In considering the application of any of these specific cell-killing modalities, treatment of viral infections has the advantage of targeting a viral-encoded antigen, thereby avoiding side-effects frequently encountered in cancer therapy owing to killing of normal cells expressing the targeted human antigen; on the other hand, a single virus-infected cell can release thousands of infectious virions, thereby amplifying the impact of any infected cells that escape killing. Despite these complexities, the specific cytotoxic activity of the 2014-PE38 immunotoxin against K8.1A on KSHV-infected cells, coupled with its mutual potentiating activity when combined with a virus replication inhibitor, support the continued development of targeted cytotoxic protein approaches as components of treatment regimens for KSHV diseases, particularly MCD.
Materials and Methods
Construction and expression of plasmids encoding recombinant Immunotoxins.
Total RNA was extracted from 4C3 hybridoma cells55 by using the Qiagen RNeasy mini kit. Briefly, 5 × 106 cells were disrupted in 350 µl of buffer RLT containing β-mercaptoethanol and homogenized by loading onto a QIAshredder spin column and spinning for 2 min at a maximum speed in a micro centrifuge. 350 µl of 70% ethanol was added and transferred to an RNeasy spin column and centrifuged for 15 sec at 8,000 g. 700 µl of buffer RW1 was then added and spun for 15 sec at 8,000 g. 500 µl of buffer RPE was added and spun for 15 sec at 8,000 g. The RNeasy spin column was placed onto a new collection tube and RNA was eluted in 50 ul RNase-free water by centrifuging for 1 min at 8,000 g.
2.5 µg RNA and isotype-specific VH hinge primer (MG1 hinge 5′-ACC ACA ATC CCT GGG CAC AAT TTT CT-3′) or VL hinge primer (MK-Edge 5′-CTC ATT CTT GTT GAA GCT CTT GAC AAT-3′) were used to set up reaction for cDNA synthesis as described in the SMART RACE cDNA amplification kit (Clonetech, Paulo Alto, CA). The prepared cDNAs were used as templates in the 5′ RACE PCR reactions with 10 pmol of isotype specific primers (MG1-PCR: 5′-AGG GGC CAG TGG ATA GAC AGA TGG GGG TGT-3′, MK-PCR: 5′-GGA TGG TGG GAA GAT GGA TAC AGT TGG TGC AGC-3′). The PCR products were cloned into pCR2.1-TOPO vector using a TOPO TA cloning kit (Invitrogen). At least 20 clones for each chain were sequenced, analyzed and aligned according to the Ig BLAST program using the KABAT database (http://www.ncbi.nlm.nih.gov/igblast/).
The primers used to synthesize VH and VL fragments were designed according to the sequence alignment. The primers used to amplify the heavy chain Fv region were 5′4C3-VH20 (5′-AAA CAT ATG CAG ATC CAG TTG GTG CAG TCT-3′) and 3′4C3-VH20-linker (5′-TCC AGA TCC GCC ACC ACC TGA TCC GCC TCC GCC TGA GGA GAC GGT GAC TGA GGT-3′). The primers used to amplify the light chain Fv region were 5′4C3-VL14-linker (5′-TCA GGT GGT GGC GGA TCT GGA GGT GGC GGA AGC GAC ATT GTG ATG ACA CAG TCT-3′) and 3′4C3-VL14-HindIII (5′-GGA AGC TTT CCG TTT GAT TTC CAG CTT GGT-3′). The PCR products encoding the VH and VL domains of mAb 4C3 connected by a 15 amino acid linker (Gly4Ser)3 were generated by fusion PCR using the purified individual VH and VL PCR fragments through the primers 5′4C3-VH20 and 3′4C3-VL14-HindIII. The PCR product was cloned into pCR2.1 vector, double digested with EcoRV and HindIII and ligated to PE-toxin expression vector pTK21.8,92 previously subjected to sequential single digestion with NdeI (polished with T4 Klenow fragment to generate a blunt end) and HindIII and alkaline-phosphatase treated. The resulting clone was named as 2014-PE38. We also constructed a clone 2014-PE38E553D encoding a negative control protein containing the E553D mutation in the PE38 domain that inactivates its ADP ribosylation activity.93 The corresponding expression plasmids are designated pDC6 and pDC8, respectively. As a control we employed BL22-PE38, directed against CD22 antigen,94 generously provided by Ira Pastan (NCI, NIH).
Expression and purification of immunotoxin.
Immunotoxins were expressed and purified as described previously.61,77 Briefly, the plasmids encoding the 2014-PE38 and 2014-PE38E553D were transformed into Max Efficiency DH5α E. coli BL21 (DE3) cells. Post overnight incubation at 37°C, the colonies were dislodged in 5 ml of super broth media and subsequently added onto a liter of pre-warmed super broth culture containing 20% glucose, 1.68 ml of 1 M MgSO4 and ampicillin (100 µg/ml). Cells were incubated in shaking incubator 220 rpm at 37°C. When the OD600 nm was between 2.0 and 3.0, 500 ml culture was induced with 5 ml of 0.1 M IPTG and incubated at 37°C shaking for 90 min. The bacteria were harvested by centrifuging at 7,500 g at 4°C for 10 min. To prepare inclusion bodies, the bacterial pellet was resuspended in 160 ml of TES buffer (TES: 50 mM TRIS-HCl, pH 8.0, 20 mM EDTA, 100 mM NaCl) followed by treatment with 6.5 ml lysozyme and mixing using a Tissuemizer and incubated at room temperature for 30 min. 20 ml of 25% Triton-X100 was added, mixed with a Tissuemizer and incubated at RT for 30 min. The inclusion bodies were pelleted by centrifugation at 27,000 g for 50 min at 4°C and supernatant was discarded. The inclusion body pellet was washed thrice with TES-25% Triton-X100 and once without the triton-X100. The inclusion body pellet was denatured in 5 ml of denaturing buffer (6 M guanidine HCl, 100 mM TRIS-HCl, pH 8, 2 mM EDTA) using a Tissuemizer. The disulphide bonds in the denatured recombinant immunotoxin were reduced by addition of dithioerythritol powder (concentration 10 mg/ml) and incubation at room temperature overnight. The protein solution was centrifuged at 12,000 g for 10 min to remove the misfolded proteins and other debris in the pellet. Proteins were refolded by brisk stirring the supernatant in 100-fold excess volume of refolding buffer (100 mM TRIS-HCl, 1 mM EDTA, 0.5 M arginine, pH 9.5, chilled to 10°C) containing freshly added oxidized glutathione (551 mg/L). After mixing for 2–3 min, the solution was incubated at 10°C for 36–48 h. The recombinant immunotoxin preparation was finally dialyzed in dialysis buffer (20 mM TRIS-HCl, pH 7.4, 100 mM urea, chilled to 4°C) using dialysis tubing with a molecular weight cutoff of <50 kDa. The correctly folded immunotoxin was then passed through two ion exchange chromatography columns Q Sepharose and MonoQ (Amersham Pharmacia Biotech) and finally through size exclusion chromatography (TSK3000, TOSOH, Tokyo, Japan) to remove the impurities. Protein concentrations of the purified immunotoxins were determined by a protein assay (Bio-Rad) with bovine serum albumin as standard.
Cells and media.
Vero-219 cells that harbor recombinant KSHV-219,62 were maintained in DMEM containing 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin and 5 ng/ml puromycin. 293F cells (derived from human embryonic kidney, Freestyle 293F subclone from Invitrogen) adapted to grow as adherent cultures95 were maintained in DMEM containing 10% FBS, 2 mM L-glutamine, 1% non essential amino acids and 100 U/ml penicillin 100 µg/ml streptomycin.
Flow cytometry analyses.
Vero-219 cells were infected with recombinant baculovirus BacK50 encoding the KSHV RTA trans activator62 (M.O.I. 100) for 3 h at 37°C followed by aspiration of inoculums and addition of fresh media containing 1.25 mM sodium butyrate for overnight incubation. Post 24 h, cells were replenished with fresh media and incubated overnight. The cells were incubated with anti-gpK8.1A mAb 4C3 and IgG1 isotype control at a concentration of 5–10 µg/ml for 1 h at 4°C. The cells were washed with PBS thrice and incubated with secondary anti-mouse antibody conjugated with fluorescent dye Alexa 635 nm (1:500 dilution) for 30 min at 4°C, subsequently washed thrice with PBS, fixed with 2% PFA and run in a FACScalibur flow cytometer.
Confocal microscopy.
12 mm coverslips placed in 24-well culture plates were coated with 0.1% poly l-lysine, incubated overnight in a 37°C tissue culture incubator, washed thoroughly with deionized H2O and seeded with Vero-219 at 100 × 103/well. Vero-219 cells were infected with BacK50 (M.O.I. 100) for 3 h at 37°C followed by aspiration of inoculate and addition of fresh media containing 1.25 mM sodium butyrate for overnight incubation. Post 24 h, cells were replenished with fresh media and incubated overnight. The cells were either left untreated or fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.5% Triton × 100 for 5 min, washed with PBS and stained with anti-gpK8.1A mAb 4C3 and mouse IgG control at a concentration of 5 µg/ml for 1 h at 4°C. Cells were washed thrice with PBS and incubated with secondary anti-mouse antibody conjugated with fluorescent dye 635 nm for 1 h at 4°C. Following three PBS washes, DAPI and mounting media were added and cells were viewed under confocal microscope SP2.
Direct cytotoxicity assay.
Vero-219 were seeded in 96-well plates at 5 × 104 cells/well and incubated overnight at 37°C, 5% CO2. The cells were infected with BacK50 at M.O.I. 100 for 3 h at 37°C, washed with PBS once and replenished with complete medium supplemented with 1.25 mM sodium butyrate and different dilutions of immunotoxin (2-fold serial dilution starting from 2,000 ng/ml to 15.6 ng/ml, 100 µl total volume in each well) and incubated overnight. The supernatant was removed and cells were replenished with fresh media without sodium butyrate containing same immunotoxin dilutions and incubated for an additional 48 h at 37°C, 5% CO2. Ten microliters of the Cell-Counting Kit-8 (CCK-8) solution (Dojindo Inc., Maryland) was added to the cells and incubated for 2 h at 37°C, 5% CO2. The absorbance was measured at 450 nm using a microplate reader.
Virus infectivity assay.
Vero-219 cells were seeded in 24-well plates at 1∼2 × 105 cells/well and incubated overnight. The cells were infected with BacK50 at M.O.I. 100 for 3 h at 37°C, washed with PBS once and replenished with complete medium supplemented with 1.25 mM sodium butyrate and different dilutions of immunotoxin (2-fold serial dilution starting from 2,000 ng/ml to 15.6 ng/ml, 500 µl total volume in each well) or ganciclovir (2-fold serial dilution starting from 4.0 µM to 0.25 µM in each well) and incubated overnight. The supernatants were removed and cells were replenished with fresh media without sodium butyrate containing the same immunotoxin dilutions and incubated for 48 h at 37°C, 5% CO2. Fresh 293F cells were seeded (24 well plates, 2 × 105 cells/well) and spin-inoculated with supernatants from the Vero-219 (1,500 g, 30 min) followed by incubation for 3 h at 37°C, 5% CO2. After 3 h, the supernatants were aspirated and cells were replenished with fresh media and incubated for 48–72 h. The number of infected target cells was determined by flow cytometry; infectivity is expressed as percentage of target cells that scored GFP-positive, which indicated establishment of infection by KSHV.
Acknowledgments
We thank Jeffrey Vieira (University of Washington, Seattle WA) for donating the recombinant KSHV-infected stable cell lines as well as Joseph Newland and George Katsafanas (NIAID, NIH) for technical support as well as John Weldon, Tapan Bera and Ira Pastan (NCI, NIH) for providing control PE-based immunotoxins and technical advice. We are grateful to Bertrand Saunier and Yingyun Cai (NIAID, NIH) for sharing experimental insights. The excellent technical assistance of Virgilio Bundoc (NIAID, NIH) is acknowledged. This work was funded in part by the Intramural Program of the NIH, NIAID.
Abbreviations
- KSHV
Kaposi sarcoma associated herpes virus
- KS
Kaposi sarcoma
- MCD
multicentric Castleman disease
- HAART
highly active antiretroviral therapy
- mAb
monoclonal antibody
- PE
Pseudomonas aeruginosa exotoxin A
- GCV
ganciclovir
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author's Contributions
D.C. was involved in design of experiments, acquisition, analysis and interpretation of data, drafting of the manuscript and final approval of the completed version. B.C. was involved in interpretation of data, reviewing/revising the manuscript and final approval of the completed version. E.A.B. was involved in conception and design of the study, analysis and interpretation of data, drafting of the manuscript and final approval of the completed version.
Database submission
The nucleotide sequences of the VH and VL regions of the 4C3 hybridoma will be submitted to GenBank under accession number JQ434470.
References
- 1.Ganem D. Kaposi's sarcoma-associated herpesvirus. In: Knipe DM, Howley PM, editors. Fields Virology. Vol. 5. Philadelphia, PA: Lippincott Williams and Wilkins; 2007. pp. 2847–2888. [Google Scholar]
- 2.Pellett PE, Roizman B. The family Herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth Edition. Philadelphia: Lipincott Williams & Wilkens; 2007. pp. 2479–2499. [Google Scholar]
- 3.Wen KW, Damania B. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 2010;289:140–150. doi: 10.1016/j.canlet.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 5.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 6.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- 7.Moore PS, Chang Y. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol. 2003;57:609–639. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan RJ, Pantanowitz L, Casper C, Stebbing J, Dezube BJ. HIV/AIDS: epidemiology, pathophysiology and treatment of Kaposi sarcoma-associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma and multicentric Castleman disease. Clin Infect Dis. 2008;47:1209–1215. doi: 10.1086/592298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gantt S, Casper C. Human herpesvirus 8-associated neoplasms: the roles of viral replication and antiviral treatment. Curr Opin Infect Dis. 2011;24:295–301. doi: 10.1097/QCO.0b013e3283486d04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cesarman E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011;305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katano H, Sato Y, Kurata T, Mori S, Sata T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma and multicentric Castleman's disease. Virology. 2000;269:335–344. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 12.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, et al. Differential viral protein expression in Kaposi's sarcoma-associated herpesvirus-infected diseases: Kaposi's sarcoma, primary effusion lymphoma and multicentric Castleman's disease. Am J Pathol. 2000;156:743–749. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asahi-Ozaki Y, Sato Y, Kanno T, Sata T, Katano H. Quantitative analysis of Kaposi sarcoma-associated herpesvirus (KSHV) in KSHV-associated diseases. J Infect Dis. 2006;193:773–782. doi: 10.1086/500560. [DOI] [PubMed] [Google Scholar]
- 14.Burbelo PD, Issa AT, Ching KH, Wyvill KM, Little RF, Iadarola MJ, et al. Distinct profiles of antibodies to Kaposi sarcoma-associated herpesvirus antigens in patients with Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. J Infect Dis. 2010;201:1919–1922. doi: 10.1086/652869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stebbing J, Pantanowitz L, Dayyani F, Sullivan RJ, Bower M, Dezube BJ. HIV-associated multicentric Castleman's disease. Am J Hematol. 2008;83:498–503. doi: 10.1002/ajh.21137. [DOI] [PubMed] [Google Scholar]
- 16.Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4:16–21. doi: 10.1097/COH.0b013e328319bca9. [DOI] [PubMed] [Google Scholar]
- 17.Cronin DMP, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16:236–246. doi: 10.1097/PAP.0b013e3181a9d4d3. [DOI] [PubMed] [Google Scholar]
- 18.Bower M, Newsom-Davis T, Naresh K, Merchant S, Lee B, Gazzard B, et al. Clinical features and outcome in HIV-associated multicentric Castleman's disease. J Clin Oncol. 2011;29:2481–2486. doi: 10.1200/JCO.2010.34.1909. [DOI] [PubMed] [Google Scholar]
- 19.Oksenhendler E, Carcelain G, Aoki Y, Boulanger E, Maillard A, Clauvel JP, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10 and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood. 2000;96:2069–2073. [PubMed] [Google Scholar]
- 20.Stebbing J, Adams C, Sanitt A, Mletzko S, Nelson M, Gazzard B, et al. Plasma HHV8 DNA predicts relapse in individuals with HIV-associated multicentric Castleman disease. Blood. 2011;118:271–275. doi: 10.1182/blood-2011-02-335620. [DOI] [PubMed] [Google Scholar]
- 21.Dittmer DP, Vahrson W, Staudt M, Hilscher C, Fakhari FD. Kaposi's sarcoma in the era of HAART-an update on mechanisms, diagnostics and treatment. AIDS Rev. 2005;7:56–61. [PubMed] [Google Scholar]
- 22.Powles T, Stebbing J, Bazeos A, Hatzimichael E, Mandalia S, Nelson M, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman's disease. Ann Oncol. 2009;20:775–779. doi: 10.1093/annonc/mdn697. [DOI] [PubMed] [Google Scholar]
- 23.Mylona EE, Baraboutis IG, Lekakis LJ, Georgiou O, Papastamopoulos V, Skoutelis A. Multicentric Castleman's disease in HIV infection: a systematic review of the literature. AIDS Rev. 2008;10:25–35. [PubMed] [Google Scholar]
- 24.Bower M. How I treat HIV-associated multicentric Castleman disease. Blood. 2010;116:4415–4421. doi: 10.1182/blood-2010-07-290213. [DOI] [PubMed] [Google Scholar]
- 25.El-Osta HE, Kurzrock R. Castleman's disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16:497–511. doi: 10.1634/theoncologist.2010-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuyama M, Suzuki T, Tsuboi H, Ito S, Mamura M, Goto D, et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman's disease. Intern Med. 2007;46:771–774. doi: 10.2169/internalmedicine.46.6262. [DOI] [PubMed] [Google Scholar]
- 27.van Rhee F, Fayad L, Voorhees P, Furman R, Lonial S, Borghaei H, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman's disease. J Clin Oncol. 2010;28:3701–3708. doi: 10.1200/JCO.2009.27.2377. [DOI] [PubMed] [Google Scholar]
- 28.Gérard L, Bérezné A, Galicier L, Meignin V, Obadia M, De Castro N, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25:3350–3356. doi: 10.1200/JCO.2007.10.6732. [DOI] [PubMed] [Google Scholar]
- 29.Bower M, Powles T, Williams S, Davis TN, Atkins M, Montoto S, et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med. 2007;147:836–839. doi: 10.7326/0003-4819-147-12-200712180-00003. [DOI] [PubMed] [Google Scholar]
- 30.Bestawros A, Michel R, Séguin C, Routy JP. Multicentric Castleman's disease treated with combination chemotherapy and rituximab in four HIV-positive men: a case series. Am J Hematol. 2008;83:508–511. doi: 10.1002/ajh.21108. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt SM, Raible A, Kortüm F, Mayer F, Riessen R, Adam P, et al. Successful treatment of multicentric Castleman's disease with combined immunochemo-therapy in an AIDS patient with multiorgan failure. Leukemia. 2008;22:1782–1785. doi: 10.1038/leu.2008.54. [DOI] [PubMed] [Google Scholar]
- 32.Law AB, Ryan G, Lade S, Prince HM. Development of Kaposi's sarcoma after complete remission of multicentric Castlemans disease with rituximab therapy in a HHV8-positive, HIV-negative patient. Int J Hematol. 2010;91:347–348. doi: 10.1007/s12185-010-0497-9. [DOI] [PubMed] [Google Scholar]
- 33.Yuan ZG, Dun XY, Li YH, Hou J. Treatment of multicentric Castleman's Disease accompanying multiple myeloma with bortezomib: a case report. J Hematol Oncol. 2009;2:19. doi: 10.1186/1756-8722-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103:1632–1634. doi: 10.1182/blood-2003-05-1721. [DOI] [PubMed] [Google Scholar]
- 35.Uldrick TS, Polizzotto MN, Aleman K, O'Mahony D, Wyvill KM, Wang V, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117:6977–6986. doi: 10.1182/blood-2010-11-317610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pasquetto MV, Vecchia L, Covini D, Digilio R, Scotti C. Targeted drug delivery using immunoconjugates: principles and applications. J Immunother. 2011;34:611–628. doi: 10.1097/CJI.0b013e318234ecf5. [DOI] [PubMed] [Google Scholar]
- 37.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 38.Teicher BA, Chari RVJ. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17:6389–6397. doi: 10.1158/1078-0432.CCR-11-1417. [DOI] [PubMed] [Google Scholar]
- 39.Sharkey RM, Goldenberg DM. Cancer radioimmunotherapy. Immunotherapy. 2011;3:349–370. doi: 10.2217/imt.10.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steiner M, Neri D. Antibody-radionuclide conjugates for cancer therapy: historical considerations and new trends. Clin Cancer Res. 2011;17:6406–6416. doi: 10.1158/1078-0432.CCR-11-0483. [DOI] [PubMed] [Google Scholar]
- 41.Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58:221–237. doi: 10.1146/annurev.med.58.070605.115320. [DOI] [PubMed] [Google Scholar]
- 42.Choudhary S, Mathew M, Verma RS. Therapeutic potential of anticancer immunotoxins. Drug Discov Today. 2011;16:495–503. doi: 10.1016/j.drudis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 43.Berger EA. Targeted cytotoxic therapy: adapting a rapidly progressing anticancer paradigm for depletion of persistent HIV-infected cell reservoirs. Curr Opin HIV AIDS. 2011;6:80–85. doi: 10.1097/COH.0b013e3283412515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansson S, Goldenberg DM, Griffiths GL, Wahren B, Hinkula J. Elimination of HIV-1 infection by treatment with a doxorubicin-conjugated anti-envelope antibody. AIDS. 2006;20:1911–1915. doi: 10.1097/01.aids.0000247111.58961.60. [DOI] [PubMed] [Google Scholar]
- 45.Dadachova E, Casadevall A. Radioimmunotherapy of infectious diseases. Semin Nucl Med. 2009;39:146–153. doi: 10.1053/j.semnuclmed.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnett BB, Burns NJ, 3rd, Park KJ, Dawson MI, Kende M, Sidwell RW. Antiviral immunotoxins: antibody-mediated delivery of gelonin inhibits Pichinde virus replication in vitro. Antiviral Res. 1991;15:125–138. doi: 10.1016/0166-3542(91)90030-U. [DOI] [PubMed] [Google Scholar]
- 47.Barnett BB, Smee DF, Malek SM, Sidwell RW. Selective cytotoxicity towards cytomegalovirus-infected cells by immunotoxins consisting of gelonin linked to anti-cytomegalovirus antibody. Antiviral Res. 1995;28:93–100. doi: 10.1016/0166-3542(95)00034-J. [DOI] [PubMed] [Google Scholar]
- 48.Barnett BB, Smee DF, Malek SM, Sidwell RW. Selective cytotoxicity of ricin A chain immunotoxins towards murine cytomegalovirus-infected cells. Antimicrob Agents Chemother. 1996;40:470–472. doi: 10.1128/aac.40.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berger EA, Pastan I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog. 2010;6:1000803. doi: 10.1371/journal.ppat.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mareeva T, Wanjalla C, Schnell MJ, Sykulev Y. A novel composite immunotoxin that suppresses rabies virus production by the infected cells. J Immunol Methods. 2010;353:78–86. doi: 10.1016/j.jim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandran B. Early events in Kaposi's sarcoma-associated herpesvirus infection of target cells. J Virol. 2010;84:2188–2199. doi: 10.1128/JVI.01334-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandran B, Bloomer C, Chan SR, Zhu LJ, Goldstein E, Horvat R. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology. 1998;249:140–149. doi: 10.1006/viro.1998.9316. [DOI] [PubMed] [Google Scholar]
- 53.Li MT, MacKey J, Czajak SC, Desrosiers RC, Lackner AA, Jung JU. Identification and characterization of Kaposi's sarcoma-associated herpesvirus K8.1 virion glycoprotein. J Virol. 1999;73:1341–1349. doi: 10.1128/jvi.73.2.1341-1349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu LJ, Renne R, Ganem D, Forghani B. Human herpesvirus 8 glycoprotein K8.1: expression, post-translational modification and localization analyzed by monoclonal antibody. J Clin Virol. 2000;17:127–136. doi: 10.1016/S1386-6532(00)00085-8. [DOI] [PubMed] [Google Scholar]
- 55.Zhu LJ, Puri V, Chandran B. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology. 1999;262:237–249. doi: 10.1006/viro.1999.9900. [DOI] [PubMed] [Google Scholar]
- 56.Zhu FX, Chong JM, Wu LJ, Yuan Y. Virion proteins of Kaposi's sarcoma-associated herpesvirus. J Virol. 2005;79:800–811. doi: 10.1128/JVI.79.2.800-11.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang FZ, Akula SM, Pramod NP, Zeng L, Chandran B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J Virol. 2001;75:7517–7527. doi: 10.1128/JVI.75.16.7517-27.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Birkmann A, Mahr K, Ensser A, Yağuboğlu S, Titgemeyer F, Fleckenstein B, et al. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J Virol. 2001;75:11583–11593. doi: 10.1128/JVI.75.23.11583-93.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Engels EA, Whitby D, Goebel PB, Stossel A, Waters D, Pintus A, et al. Identifying human herpesvirus 8 infection: performance characteristics of serologic assays. J Acquir Immune Defic Syndr. 2000;23:346–354. doi: 10.1097/00126334-200004010-00011. [DOI] [PubMed] [Google Scholar]
- 60.Zoeteweij JP, Eyes ST, Orenstein JM, Kawamura T, Wu LJ, Chandran B, et al. Identification and rapid quantification of early- and late-lytic human herpesvirus 8 infection in single cells by flow cytometric analysis: characterization of antiherpesvirus agents. J Virol. 1999;73:5894–5902. doi: 10.1128/jvi.73.7.5894-5902.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. In: Lo BKC, editor. Meth Molec Biol. Totowa, NJ: Humana Press Inc; 2003. pp. 503–518. [DOI] [PubMed] [Google Scholar]
- 62.Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 63.Tominaga H, Ishiyama M, Ohseto F, Sasamoto K, Hamamoto T, Suzuki K, et al. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal Commun. 1999;36:47–50. doi: 10.1039/a809656b. [DOI] [Google Scholar]
- 64.Kedes DH, Ganem D. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest. 1997;99:2082–2086. doi: 10.1172/JCI119380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Medveczky MM, Horvath E, Lund T, Medveczky PG. In vitro antiviral drug sensitivity of the Kaposi's sarcoma-associated herpesvirus. AIDS. 1997;11:1327–1332. doi: 10.1097/00002030-199711000-00006. [DOI] [PubMed] [Google Scholar]
- 66.Lu M, Suen J, Frias C, Pfeiffer R, Tsai MH, Chuang E, et al. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J Virol. 2004;78:13637–13652. doi: 10.1128/JVI.78.24.13637-52.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Casper C, Krantz EM, Corey L, Kuntz SR, Wang J, Selke S, et al. Valganciclovir for suppression of human herpesvirus-8 replication: a randomized, double-blind, placebo-controlled, crossover trial. J Infect Dis. 2008;198:23–30. doi: 10.1086/588820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cattamanchi A, Saracino M, Selke S, Huang ML, Magaret A, Celum C, et al. Treatment with valacyclovir, famciclovir or antiretrovirals reduces human herpesvirus-8 replication in HIV-1 seropositive men. J Med Virol. 2011;83:1696–1703. doi: 10.1002/jmv.22194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashorn P, Englund G, Martin MA, Moss B, Berger EA. Anti-HIV activity of CD4-Pseudomonas exotoxin on infected primary human lymphocytes and monocyte/macrophages. J Infect Dis. 1991;163:703–709. doi: 10.1093/infdis/163.4.703. [DOI] [PubMed] [Google Scholar]
- 70.Kennedy PE, Bera TK, Wang QC, Gallo M, Wagner W, Lewis MG, et al. Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages and negligible hepatotoxicity in macaques. J Leukoc Biol. 2006;80:1175–1182. doi: 10.1189/jlb.0306139. [DOI] [PubMed] [Google Scholar]
- 71.Potts BJ, Maury W, Martin MA. Replication of HIV-1 in primary monocyte cultures. Virology. 1990;175:465–476. doi: 10.1016/0042-6822(90)90431-P. [DOI] [PubMed] [Google Scholar]
- 72.Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 73.Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Phase I/II trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. doi: 10.1200/JCO.2011.38.1756. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJP, Wilson WH, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23:6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 75.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17:6398–6405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berezne A, Agbalika F, Oksenhendler E. Failure of cidofovir in HIV-associated multicentric Castleman disease. Blood. 2004;103:4368–4369. doi: 10.1182/blood-2004-01-0158. [DOI] [PubMed] [Google Scholar]
- 77.Cai Y, Berger EA. An immunotoxin targeting the gH glycoprotein of KSHV for selective killing of cells in the lytic phase of infection. Antiviral Res. 2011;90:143–150. doi: 10.1016/j.antiviral.2011.03.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smee DF, Sidwell RW, Barnett BB. Combination of antiviral immunotoxin and ganciclovir or cidofovir for the treatment of murine cytomegalovirus infections. Antiviral Res. 1996;32:165–171. doi: 10.1016/S0166-3542(95)00986-8. [DOI] [PubMed] [Google Scholar]
- 79.Ashorn P, Moss B, Weinstein JN, Chaudhary VK, FitzGerald DJ, Pastan I, et al. Elimination of infectious human immunodeficiency virus from human T-cell cultures by synergistic action of CD4-Pseudomonas exotoxin and reverse transcriptase inhibitors. Proc Natl Acad Sci USA. 1990;87:8889–8893. doi: 10.1073/pnas.87.22.8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsubota H, Winkler G, Meade HM, Jakubowski A, Thomas DW, Letvin NL. CD4-Pseudomonas exotoxin conjugates delay but do not fully inhibit human immunodeficiency virus replication in lymphocytes in vitro. J Clin Invest. 1990;86:1684–1689. doi: 10.1172/JCI114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goldstein H, Pettoello-Mantovani M, Bera TK, Pastan IH, Berger EA. Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-SCID-Hu mice. J Infect Dis. 2000;181:921–926. doi: 10.1086/315351. [DOI] [PubMed] [Google Scholar]
- 82.Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther. 2009;9:1445–1451. doi: 10.1517/14712590903348135. [DOI] [PubMed] [Google Scholar]
- 83.Woo JH, Frankel A, Neville DM. Cytotoxic therapy with diphtheria toxin fusion proteins. Drugs Future. 2010;35:823–831. doi: 10.1358/dof.2010.035.010.1534010. [DOI] [Google Scholar]
- 84.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17:6417–6427. doi: 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- 85.Hetzel C, Bachran C, Tur MK, Fuchs H, Stöcker M. Improved immunotoxins with novel functional elements. Curr Pharm Des. 2009;15:2700–2711. doi: 10.2174/138161209788923930. [DOI] [PubMed] [Google Scholar]
- 86.Weldon JE, Pastan I. A guide to taming a toxin—recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278:4683–4700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bera TK, Pastan I. Comparison of recombinant immunotoxins against LeY antigen expressing tumor cells: influence of affinity, size and stability. Bioconjug Chem. 1998;9:736–743. doi: 10.1021/bc980028o. [DOI] [PubMed] [Google Scholar]
- 88.Hakim R, Benhar I. “Inclonals”: IgGs and IgG-enzyme fusion proteins produced in an E. coli expression-refolding system. MAbs. 2009;1:281–287. doi: 10.4161/mabs.1.3.8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Onda M, Beers R, Xiang LM, Lee B, Weldon JE, Kreitman RJ, et al. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc Natl Acad Sci USA. 2011;108:5742–5747. doi: 10.1073/pnas.1102746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mathew M, Verma RS. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009;100:1359–1365. doi: 10.1111/j.1349-7006.2009.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lorberboum-Galski H. Human toxin-based recombinant immunotoxins/chimeric proteins as a drug delivery system for targeted treatment of human diseases. Expert Opin Drug Deliv. 2011;8:605–621. doi: 10.1517/17425247.2011.566269. [DOI] [PubMed] [Google Scholar]
- 92.Bera TK, Kennedy PE, Berger EA, Barbas CFI, 3rd, Pastan I. Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med. 1998;4:384–391. [PMC free article] [PubMed] [Google Scholar]
- 93.Douglas CM, Collier RJ. Exotoxin A of Pseudomonas aeruginosa: substitution of glutamic acid 553 with aspartic acid drastically reduces toxicity and enzymatic activity. J Bacteriol. 1987;169:4967–4971. doi: 10.1128/jb.169.11.4967-4971.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- 95.Dube D, Schornberg KL, Shoemaker CJ, Delos SE, Stantchev TS, Clouse KA, et al. Cell adhesion-dependent membrane trafficking of a binding partner for the ebolavirus glycoprotein is a determinant of viral entry. Proc Natl Acad Sci USA. 2010;107:16637–16642. doi: 10.1073/pnas.1008509107. [DOI] [PMC free article] [PubMed] [Google Scholar]