

Abstract
NADPH oxidases are a family of enzymes that generate reactive oxygen species (ROS). The NOX1 (NADPH oxidase 1) and NOX2 oxidases are the major sources of ROS in the artery wall in conditions such as hypertension, hypercholesterolaemia, diabetes and ageing, and so they are important contributors to the oxidative stress, endothelial dysfunction and vascular inflammation that underlies arterial remodelling and atherogenesis. In this Review, we advance the concept that compared to the use of conventional antioxidants, inhibiting NOX1 and NOX2 oxidases is a superior approach for combating oxidative stress. We briefly describe some common and emerging putative NADPH oxidase inhibitors. In addition, we highlight the crucial role of the NADPH oxidase regulatory subunit, p47phox, in the activity of vascular NOX1 and NOX2 oxidases, and suggest how a better understanding of its specific molecular interactions may enable the development of novel isoform-selective drugs to prevent or treat cardiovascular diseases.
The primary catalytic function of the NADPH oxidase family of enzymes is the generation of reactive oxygen species (ROS). This property sets them apart from all other ROS-generating enzymes that produce radical species, either as a by-product of their normal catalytic activity or as a result of aberrant functioning in disease. Members of the NADPH oxidase family are expressed in most — if not all — mammalian cell types, in which they catalyse the reduction of molecular oxygen to generate superoxide and/or hydrogen peroxide in various intracellular and extracellular compartments. The ROS generated by NADPH oxidases have crucial roles in various physiological processes, including innate immunity, modulation of redox-dependent signalling cascades, and as cofactors in the production of hormones.
For several decades, it has been recognized that the rare condition known as chronic granulomatous disease (CGD; see BOX 1)1 is caused by an underactive NADPH oxidase system, in which the capacity of phagocytic leukocytes to generate a microbicidal burst of ROS is impaired, leaving the individual susceptible to severe, life-threatening infections by opportunistic microbes. By contrast, it has only recently emerged that excessive ROS production by an overactive NADPH oxidase system, both in phagocytic and non-phagocytic cell types of the artery wall, may set in motion a vicious cycle of radical and non-radical oxidant generation in various cellular compartments, which disrupts redox circuits that are normally controlled by thiol-dependent antioxidant defences2,3. This induces a state of oxidative stress, which is necessary for the initiation and progression of vascular disease that may ultimately lead to heart attacks and strokes.
Box1 | Chronic granulomatous disease.
Chronic granulomatous disease (CGD)1,199–202 is a primary immunodeficiency that affects phagocytes of the innate immune system, and is characterized by a markedly increased susceptibility to severe bacterial and fungal infections.
CGD is caused by any of the >400 mutations that have been identified so far in one of the four genes that encode the subunits of the phagocytic NOX2 subunit-dependent NADPH oxidase complex.
The incidence of CGD is approximately 1 in 200,000 live births. Most (95%) of the mutations that cause CGD lead to complete or partial loss of protein expression, whereas approximately 5% of mutations are loss-of-function mutations that result in normal levels of protein expression — although with impaired function.
Most (more than two-thirds) of CGD cases are X-linked recessive and result from defects in the CYBB gene that encodes the NOX2 subunit. The remaining cases of CGD are autosomal recessive and caused by defects in the CYBA, NCF1 and NCF2 genes, which encode p22phox, p47phox (also known as neutrophil cytosol factor 1) and p67phox (also known as neutrophil cytosol factor 2), respectively. To date, there are no reports of CGD caused by defects in the gene encoding a fifth NADPH oxidase subunit, p40phox. One patient has been identified with a related immunodeficiency resulting from a defect in the gene that encodes the small GTPase RAC2.
Because the disease is often X-linked, female carriers of genes with mutations that lead to CGD may have either one or no normal copies of the affected gene, whereas unaffected female subjects usually have two normal copies of the gene. Importantly, this so-called gene-dosing effect has identified that despite a reduced level of NADPH oxidase activity, there is no CGD pathology in female subjects with only one copy of causative genes.
A recent study in patients with CGD demonstrated that severe illness and poor long-term survival was only evident in individuals whose phagocytic ROS production was more than two orders of magnitude lower than in healthy controls155.
The above two points may be regarded as indirect evidence that it is feasible to reduce — rather than abolish — NADPH oxidase activity without compromising the innate immune system in patients with cardiovascular risk factors and vascular oxidative stress.
However, as most previous studies with NADPH oxidase inhibitors have been performed in experimental animals housed under specific pathogen-free conditions, there is a lack of proof of concept — from long-term experimental studies — of the dose-related effects of chronically administered NADPH oxidase inhibitors (for example, apocynin) on infection rate versus oxidative stress.
The knowledge that ROS are involved in the pathogenesis of vascular disease should facilitate the development of therapies that directly target ROS production to prevent cardiovascular events. However, most current cardiovascular therapies are still merely aimed at either reducing a patient’s risk profile (for example, by lowering blood pressure or reducing cholesterol levels) or alleviating the symptoms of vascular disease (for example, by reducing the cardiac workload to prevent angina attacks). Hence, existing approaches do not yet adequately address the ongoing oxidative processes in the diseased vessel wall, and thus it may be speculated that with current cardiovascular therapies patients remain at an increased risk of a cardiovascular event. It has therefore been disappointing to find that antioxidant drugs (for example, vitamins C and E, and β-carotene) do not appear to have the clinical efficacy to prevent cardiovascular events4, even though these drugs can, high concentrations, chemically remove ROS in in vitro systems and in experimental animal models of cardiovascular disease. However, given that conventional antioxidants display poor reactivity with many endogenous ROS, in retrospect the failure of these clinical trials perhaps not so surprising. Therefore, targeting ROS production by blocking the activity of culprit enzymes — such as NADPH oxidases — is likely to be a far superior approach for combating oxidative stress and preventing the progression of vascular disease than previous attempts aimed at scavenging ROS.
This Review provides an overview of the mechanisms by which ROS can cause vascular disease and the probable reasons why previous attempts to eliminate these species with antioxidants failed. We summarize the evidence that NADPH oxidases are key generators of ROS in the blood vessel wall and other tissues during cardiovascular disease progression. We also discuss why targeting these enzymes in an isoform-selective manner (by exploiting specific molecular features of the different isoforms) is likely to offer therapeutic advantages over conventional antioxidants while having minimal impact on immune cell function by professional phagocytes or on ROS-dependent processes that are essential for normal cell function.
Oxidative stress
A link between risk factors and vascular disease
Vascular disease is a general term that encompasses several pathological states of the arterial wall that may lead to obstructed blood flow and thereby give rise to acute cardiovascular events. Such pathological states include arterial remodelling, atherosclerosis, thrombosis and restenosis. Cardiovascular risk factors — such as hypertension, hypercholesterolaemia, smoking, obesity, diabetes and ageing — almost invariably precede the onset of vascular disease, which is indicative of a causal link. Oxidative stress is associated with all known cardiovascular risk factors, and a large body of evidence indicates that it is implicated in many of the processes that are involved in the formation of atherosclerotic plaques5,6. These observations highlight that oxidative stress is a link between cardiovascular risk factors and vascular disease, and thus a compelling target for disease prevention or therapy.
Oxidative stress is defined as an excess in the levels of of oxidants over antioxidants within a biological system, and the direct consequence of this is a shift in the redox state of the biological compartment (for example, the nucleus, mitochondria, cytosol or extracellular space) towards one that is more oxidizing2,7. ROS — which include radical species such as superoxide anions and hydroxyl radicals, and non-radical oxidants such as hydrogen peroxide — are particularly important effectors of cellular redox status. It is the overproduction of these species that is considered to be a major cause of oxidative stress in the arterial walls of individuals with cardiovascular risk factors, and in organs such as the heart, brain and kidneys following ischaemia–reperfusion injury7,8.
Oxidative stress not only causes direct and irreversible oxidative damage to macromolecules but also disrupts key redox-dependent signalling processes in the arterial wall9. Perhaps the best characterized mechanism by which oxidative stress can promote vascular disease is via disruption of the vasoprotective nitric oxide (NO) signalling pathway10. It has been known for many years that superoxide anions can chemically react with and inactivate NO. This nullifies the anti-inflammatory and vasodilator functions of NO, and promotes the formation of peroxynitrite11,12. Peroxynitrite is a powerful oxidant in its own right, and may thus contribute to oxidative stress through oxidation of small-molecule antioxidants such as glutathione, cysteine and tetrahydro—biopterin13. Reduced bioavailability of tetrahydrobiopterin results in uncoupling of endothelial nitric oxide synthase (eNOS), which causes it to change from being an NO-producing, vasoprotective enzyme to being a superoxide-producing, oxidative stress-causing enzyme14,15. Peroxynitrite can also oxidatively inactivate antioxidant enzymes, such as glutathione reductase, glutaredoxin and superoxide dismutase13, as well as dimethylarginine dimethylaminohydrolase, which metabolizes the endogenous inhibitor of eNOS — asymmetric dimethylarginine16. Finally, oxidative stress can render the receptor for NO dysfunctional, as the NO-binding haem group of soluble guanylyl cyclase, which normally exists in the Fe2+ state, is highly susceptible to oxidation to the Fe3+ state, which is associated with a markedly reduced affinity of the enzyme for NO17.
In addition to inactivation of NO, ROS may directly promote vascular inflammation and remodelling. Indeed, many adverse effects of ROS on the arterial wall are attributable to the oxidation of key signalling proteins, including kinases and phosphatases, and activation of the pro-inflammatory redox-dependent transcription factor NF-κB18; this leads to the expression of adhesion molecules on the endothelium19, and the proliferation and migration of vascular smooth muscle cells (VSMCs)20. ROS may also oxidize matrix metalloproteinases, leading to their activation and the subsequent promotion of vascular remodelling21. Moreover, it was recently suggested that ROS initiate the assembly of multiprotein signalling complexes known as inflammasomes, which activate caspase 1, and thereby lead to the processing and secretion of the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 (REF. 22).
Why haven’t antioxidants worked?
Previous strategies to alleviate oxidative stress in patients with cardiovascular disease aimed to bolster levels of antioxidants — especially vitamin E — within target tissues by increasing antioxidant intake. However, despite evidence from cohort studies of an inverse relationship between dietary or supplemental vitamin E intake and the development of cardiovascular disease23, several randomized clinical intervention trials have failed to show any net benefit, either by arresting progression of atherosclerosis or by preventing the incidence of further cardiovascular events24–26.
However, owing to limitations in the design of the clinical trials and the inability of most conventional antioxidants to neutralize all ROS and reverse oxidative stress in vivo, these observations should not be regarded as evidence against a role for oxidative stress in cardiovascular disease. First, most of these trials were performed in high-risk patients in which vascular disease was presumably quite advanced, and so it is plausible that when antioxidant therapy was commenced, it was too late for it to have a substantial protective effect.
Second, the doses of vitamin E used varied greatly between trials (that is, 30–800 mg) and in no trial was it determined whether the treatment actually affected the antioxidant concentration at the target site of the disease (that is, the blood vessel wall). Indeed, this issue of antioxidant concentration within the vessel wall is extremely important considering the exceptionally high rate constants of the reactions between ROS and crucial endogenous molecules such as NO and certain amino acids and nucleic acids. Reactions between these molecules and ROS must therefore be outcompeted by the therapeutic antioxidant for it to effectively remove ROS from the blood vessel wall and positively affect vascular health. For example, the reaction between superoxide and NO occurs at a rate of approximately 1.9 × 1010 M−1 s−1 (REF. 27), which is six orders of magnitude faster than the reaction between superoxide and vitamin E28. Thus, for vitamin E to prevent this reaction from occurring in a given cellular compartment, its concentration must exceed that of NO by one million-fold. Given that concentrations of NO within the vessel wall are estimated to be in the low nanomolar range29, this would require a vitamin E concentration in the millimolar range, which is unlikely to be achievable by even the highest levels of vitamin E intake.
Third, even if sufficient concentrations of vitamin E were achieved in appropriate cellular compartments within the blood vessel wall, it is likely that vitamin E would be rapidly oxidized in such an oxidizing environment without any prospect of regeneration. Such a situation would not only lead to rapid depletion of the reduced (active) form of the antioxidant but may also cause accumulation of the oxidized form of the parent molecule, which in some instances (including for the vitamin E radical) may exert its own pro-oxidant and pro-atherogenic effects30,31.
Finally, it is noteworthy that although the reaction of certain ROS with antioxidants may lead to a reduction in the concentration of that particular ROS, the product of the reaction may simply be another type of ROS that does not react with the original antioxidant, and may not be further neutralized by an already overwhelmed endogenous antioxidant defence system32.
Therefore, prevention of ROS formation by targeting the enzymes responsible for their generation could be a more effective strategy for ameliorating oxidative stress than scavenging these highly reactive molecules once they are formed. Such an approach requires knowledge of the key enzymes that are responsible for ROS production in vascular disease, including their cellular localization. The following section outlines the evidence for NADPH oxidases as important ROS-generating enzymes in vascular disease.
Isoforms of NADPH oxidase in vascular disease
Since their initial identification in endothelial cells and VSMCs33,34, a large and rapidly expanding body of experimental evidence, including studies in knockout mice (TABLE 1), implicates NADPH oxidases in the constitutive cells of the artery wall, as well as in leukocytes and brain cells, as underlying causes of oxidative stress in various cardiovascular disease settings. NADPH oxidases are a family of multisubunit enzyme complexes that are unique in being the only enzymes that have been identified with the primary function of generating superoxide and/or hydrogen peroxide.
Table 1.
Selected studies of NADPH oxidase in models of vascular disease and stroke
| Mouse model | Disease model | Effects | Refs |
|---|---|---|---|
| Hypertension | |||
| Global knockout, Nox1−/− | Ang II | Decreased BP and O2•− levels, no change in medial hypertrophy and increased NO bioavailability | 72 |
| Ang II | Decreased BP and O2•− levels and medial hypertrophy | 71 | |
| Transgenic, NOX1 overexpression in VSMCs | Ang II | Increased BP and O2•− levels and medial hypertrophy | 69 |
| Double transgenic, Nox1−/−/TTRhRen | TTRhRen | No change in BP, decreased O2•− levels and renal fibrosis | 203 |
| Global knockout, Nox2−/− | Ang II | No change in BP, decreased O2•− levels and 3-nitrotyrosine expression | 204 |
| 2K1C | Decreased BP (minimal) and increased NO bioavailability | 56 | |
| Ang II | Increased NO bioavailability in afferent renal arterioles, and decreased constrictor responses to Ang II and adenosine | 205 | |
| Double transgenic, Nox2−/−/TTRhRen | TTRhRen | No change in BP, decreased O2•− levels | 206 |
| Transgenic, NOX2 overexpression in endothelium | Ang II | Increased BP, increased O2•− levels in the endothelium only | 207 |
| Global knockout, p47phox−/− | Ang II | Decreased BP and O2•− levels | 208 |
| DOCA–salt | Decreased BP and O2•− levels | 112 | |
| BMP4 | Decreased BP, increased NO bioavailability | 209 | |
| Adoptive transfer of T lymphocytes from wild-type and p47phox−/− mice to Rag−/− mice | Ang II | p47phox−/− T lymphocytes: decreased BP and O2•− levels, increased NO bioavailability (versus wild-type T lymphocytes) | 115 |
| Atherosclerosis | |||
| Double knockout, Nox2−/−/ApoE−/− | ApoE−/−, HFD | Decreased lesion area in the descending aorta, decreased intimal thickening, decreased O2•− levels and increased NO bioavailability | 81 |
| ApoE−/−, HFD | No change in lesion area in the aortic sinus | 210 | |
| Double knockout, p47phox−/−/ApoE−/− | ApoE−/−, HFD | No change in lesion area in the aortic sinus | 211 |
| ApoE−/−, chow and HFD | No change in lesion area in the aortic sinus, decreased lesion area in the descending aorta | 212 | |
| ApoE−/−, HFD | Decreased lesion area in the descending aorta (p47phox−/− in either vessel-derived or bone marrow-derived cells), decreased oxLDL levels (p47phox−/− in bone marrow-derived cells) and decreased adhesion molecule expression (p47phox−/− in vessel wall) | 213 | |
| Restenosis after arterial injury | |||
| Global knockout, Nox1−/− | Wire-induced injury | Decreased neointimal formation | 76 |
| Transgenic, NOX1 overexpression in VSMCs | Wire-induced injury | No effect on neointimal formation | 76 |
| Adenoviral-mediated overexpression of NOXA1 in VSMCs | Wire-induced injury | Increased neointimal formation, increased O2•− levels | 63 |
| Global knockout, Nox2−/− | Wire-induced injury | Decreased neointimal formation, decreased leukocyte accumulation in the neointima | 214 |
| Cerebral ischaemia | |||
| Global knockout, Nox2−/− | MCAO | Decreased infarct volume, ICAM1 expression and neutrophil infiltration | 54 |
| MCAO | Decreased infarct volume | 55 | |
| MCAO | Decreased infarct volume | 128 | |
| MCAO | Decreased infarct volume (Nox2−/− mice), no change in infarct volume after bone marrow transplant (wild-type bone marrow to Nox2−/− mice, Nox2−/− bone marrow to wild-type mice) | 129 | |
| MCAO | Decreased infarct volume in males only, decreased O2•− production by circulating T cells | 116 | |
| MCAO | Protective effect of apocynin (decreased infarct volume) absent in Nox2−/− mice | 53 | |
| Global knockout, Nox1−/− | MCAO | Increased cortical infarct volume | 131 |
| MCAO | Decreased infarct volume | 132 | |
| Global knockout, Nox4−/− | MCAO | Decreased infarct volume | 130 |
2K1C, two kidneys one-clip; Ang II, angiotensin II; APOE, apolipoprotein E; BMP4,bone morphogenetic protein 4; BP, blood pressure; DOCA, deoxycorticosterone acetate; HFD, high-fat diet; ICAM1, intercellular adhesion molecule 1; MCAO, middle cerebral artery occlusion; NO, nitric oxide; NOX1, NADPH oxidase 1; NOXA1, NOX activator 1; O2•−, superoxide; oxLDL, oxidized low-density lipoprotein; RAG, V(D)J recombination-activating protein; TTRhRen, transgenic mice expressing human renin; VSMC, vascular smooth muscle cell. Table modified from REF. 228.
Seven isoforms of NADPH oxidase have been described in mammals. Each of these isoforms comprises a core catalytic subunit — the so-called NADPH oxidase (NOX) and dual oxidase (DUOX) subunits — and up to five regulatory subunits. These regulatory subunits have important roles in: the maturation and expression of the NOX and DUOX subunits in biological membranes (p22phox, DUOX activator 1 (DUOXA1) and DUOXA2); in enzyme activation (p67phox and NOX activator 1 (NOXA1)); and in spatial organization of the various components of the enzyme complex (p47phox, NOX organizer 1 (NOXO1) and p40phox)35. Some NADPH oxidase isoforms also rely on a small GTPase (RAC1 or RAC2) for their activation. As illustrated in FIG. 1, each NADPH oxidase isoform may be defined by the nature of its core catalytic subunit (NOX1–NOX5, DUOX1 or DUOX2) as well as its suite of regulatory subunits.
Figure 1. Subunit composition of the seven mammalian NADPH oxidase isoforms.

The catalytic core subunits of the enzymes (NADPH oxidase 1 (NOX1)–NOX5, dual oxidase 1 (DUOX1) and DUOX2) are shown in green; NOX and DUOX maturation and stabilization partners (p22phox, DUOX activator 1 (DUOXA1) and DUOXA2) are shown in red; cytosolic organizers (p40phox, NOX organizer 1 (NOXO1) and p47phox) are shown in orange; cytosolic activators (p67phox and NOX activator 1 (NOXA1)) are shown in green; and small GTPases (RAC1 and RAC2) are shown in blue. Also shown (in pink) is polymerase δ-interacting protein 2 (POLDIP2), which is thought to regulate NOX4 activity and link production of reactive oxygen species by this isoform with cytoskeletal organization80. EF hand motifs (yellow circles) are also shown, which bind to Ca2+ and thereby regulate the activity of NOX5, DUOX1 and DUOX2 oxidases. The figure also illustrates the putative additional amino-terminal transmembrane domain and extracellular peroxidase-like region (shown in purple) on DUOX1 and DUOX2. Although NOX3 oxidase activity is enhanced by the expression of organizer and activator proteins, the enzyme displays constitutive activity in their absence.
Mechanism of action of NADPH oxidases
NADPH oxidases catalyse the transfer of electrons from the cytosol across biological membranes and into various intracellular and extracellular compartments. Depending on the biological membrane in which the NADPH oxidase is expressed, such cellular compartments can include the nucleus, endoplasmic reticulum, endosome, phagosome, mitochondria and extracellular space20. Cytosolic NADPH acts as the electron donor for all NADPH oxidase isoforms. When the enzymes are active, electrons are abstracted from NADPH and transferred along an electron transport chain contained within their catalytic subunit. This consists of a single FAD molecule bound to the extended carboxy-terminal tail, and two prosthetic haem groups attached to histidine residues within the transmembrane region of the protein. Oxygen invariably acts as the final electron acceptor. For the NOX1, NOX2 (formerly called gp91phox) and NOX5 oxidases, electron transfer to molecular oxygen results in the release of superoxide from the enzyme. Because superoxide is an anionic molecule, its generation within specific cellular compartments needs to be charge-compensated and, for NOX1 at least, this appears to be achieved by the actions of the chloride–proton antiporter ClC3 (REF. 36). Surprisingly, despite the overall structural similarities among the seven NADPH oxidase family members, NOX4, DUOX1 and DUOX2 oxidases do not appear to generate superoxide as their primary ROS. Instead, the activity of these enzymes results in the direct release of hydrogen peroxide37,38. The biochemistries underlying the direct generation of hydrogen peroxide by NOX4, DUOX1 and DUOX2 oxidases are yet to be fully characterized. However, it has been suggested that the dissociation of superoxide from the catalytic subunit of these enzymes may be delayed, possibly owing to hindrance by the third extracytosolic loop, thus allowing time for the generation of a second molecule of superoxide and its subsequent dismutation prior to its release from the enzyme38,39.
NADPH oxidases in the constitutive cell types of the artery wall
NADPH oxidases are an important source of ROS in blood vessels, and multiple NOX oxidase isoforms are constitutively expressed in each of the predominant cell types of the vascular wall40. Endothelial cells express NOX1 oxidase, NOX2 oxidase, NOX4 oxidase and NOX5 oxidase19,41; VSMCs express NOX1 oxidase, NOX4 oxidase and NOX5 oxidase20,42–44; and adventitial fibroblasts express NOX2 oxidase and NOX4 oxidase45–47 (FIG. 2). One study has also reported that NOX2 oxidase is expressed in VSMCs in human small gluteal arteries48. This pattern of distribution presumably underlies important but still poorly understood physiological signalling and/or protective roles. As vascular NADPH oxidases, especially NOX2 oxidase, are similar in structure to the phagocytic NADPH oxidase (see below and REFS 49,50 for reviews), it is plausible that their localization — particularly in the inner and outer cell layers of the artery wall — normally represents a means for preventing the transfer of pathogens between the bloodstream and surrounding tissues.
Figure 2. Cellular and subcellular expression of NADPH oxidase isoforms in the blood vessel wall.

a | Schematic diagram showing cellular localization (endothelial cells, vascular smooth muscle cells, fibroblasts, macrophages and T cells) of NADPH oxidase isoforms (NOX1 oxidase, NOX2 oxidase, NOX4 oxidase and NOX5 oxidase) through a cross-section of an artery. b | Schematic diagram of a hypothetical cell in which all of the vascular NADPH oxidase isoforms (starting with NOX1 oxidase in the left hand column and finishing with NOX5 oxidase in the right hand column) are expressed in each of their possible subcellular locations. H2O2, hydrogen peroxide; NOXA1, NADPH oxidase activator 1; O2•−, superoxide; p22, p22phox; p40, p40phox; p47, p47phox; p67, p67phox; POLDIP2, polymerase δ-interacting protein 2.
Nevertheless, the expression of the vascular NOX2 subunit and/or its regulatory partners (for example, p47phox or p22phox) is commonly increased by pro-inflammatory stimuli or in the presence of cardiovascular risk factors51. The endothelial injury and reduced NO signalling that occurs in the early stages of vascular disease is largely mediated by excessive levels of NOX2 oxidase-derived superoxide51,52. As the NOX2 subunit can generate considerably large amounts of ROS, and pharmacological inhibition or genetic deletion of the NOX2 or p47phox subunits reduces vascular oxidative stress in several disease models50,53–59 (TABLE 1), there is a strong rationale for therapeutically targeting NOX2 oxidase in the arterial wall for the treatment of vascular disease. Interestingly, in rodents, females appear to have lower levels of vascular NADPH oxidase expression and activity than males or ovariectomized females60, which could contribute to their lower propensity to develop vascular disease before menopause. It is not yet known whether a similar gender difference exists in humans.
The NOX1 subunit is expressed in endothelial cells and in VSMCs, with most information about its regulation and actions obtained from the latter cell type. In VSMCs, the NOX1 subunit is localized to the plasma membrane, caveolae and endosomes, and it forms a complex with the traditional NADPH oxidase subunits (that is, those subunits utilized by the NADPH oxidase complex originally described in professional phagocytes) p22phox, p47phox and RAC1, as well as with the novel activator subunit NOXA1 (REFS 61–63). The expression of the NOX1 subunit is induced in VSMCs by several substances. These include: vasoactive agonists (such as angiotensin II and thrombin); growth factors (such as platelet-derived growth factor (PDGF) and basic fibroblast growth factor); pro-inflammatory cytokines (such as tumour necrosis factor (TNF) and IL-1β); and atherogenic particles (such as oxidized low-density lipoprotein (LDL) and advanced glycation end products) (for a review, see REF. 20). Moreover, studies in animal models of cardiovascular disease — especially of angiotensin II-induced hypertension — implicate this NADPH oxidase isoform as an important contributor to cardiovascular disease pathology.
First, NOX1 oxidase expression in the vascular wall is elevated in several in vivo animal models of hypertension64–68. Second, whereas angiotensin II-induced hypertension, vascular superoxide production, endothelial dysfunction and aortic hypertrophy were exacerbated in transgenic mice overexpressing the NOX1 subunit in VSMCs69,70, most of these parameters were blunted in mice that were globally deficient in the NOX1 subunit71,72. Interestingly, mice that were genetically deficient in NOX1 also displayed a marked reduction in aortic expression of type 1 angiotensin II receptors73, which may explain why angiotensin II-dependent but not noradrenaline-dependent hypertension was sensitive to NOX1 oxidase deficiency74. NOX1 oxidase is also implicated in ROS production and in neointimal formation after arterial injury in mice63,75–77, and the demonstration that expression of the NOX1 and/or NOXA1 subunit is elevated in atherosclerotic lesions of apolipoprotein E-deficient (ApoE−/−) mice and humans implies a role for the enzyme in this disease state63,78. In support of this, there is evidence for a greater reduction in the development of atherosclerotic lesions in the aortic arch of Nox1−/−/ApoE−/− double knockout mice compared to ApoE−/− single knockout mice79.
Although there is substantial evidence linking increased levels of NOX1 and NOX2 oxidases to excessive ROS production in the arterial wall, the roles of the other vascular NADPH oxidase isoforms in vascular disease either remain controversial (in the case of NOX4 oxidase) or ill-defined owing to a lack of rigorous interrogation (in the case of NOX5 oxidase). The NOX4 subunit is highly expressed in endothelial cells, VSMCs and adventitial fibroblasts, and owing to its lack of reliance on traditional cytosolic organizer and activator subunits, NOX4 oxidase activity was, until recently80, thought to be regulated primarily through changes in expression of its catalytic subunit. Hence, observations that levels of the NOX4 subunit in vascular cells are either downregulated43,81, unchanged82 or upregulated83 — depending on the pathological stimulus under investigation — provide no clear indication of a homeostatic versus pathophysiological function of the catalytic subunit.
Consistent with this, several reports suggest a role for NOX4 oxidase in promoting endothelial cell survival, proliferation and migration84–87, whereas other studies indicate that NOX4 oxidase mediates effects such as endoplasmic reticulum stress, oxidative DNA damage and apoptotic and necrotic cell death of endothelial cells83,88–90. Likewise, in VSMCs, NOX4 oxidase was initially described as a key regulator of cellular differentiation and quiescence77,91 — which is suggestive of a homeostatic function — but was subsequently shown to contribute to VSMC proliferation, migration and hypertrophy under certain conditions92,93, all of which are important in arterial remodelling and atherogenesis. Finally, even the fact that NOX4 oxidase generates hydrogen peroxide — which is capable of activating pro-inflammatory signalling cascades7 — rather than superoxide37,38,94 raises questions as to whether NOX4 oxidase is likely to have a protective or detrimental role in vascular disease.
Although one study suggested that hydrogen peroxide may impair endothelial NO production via phosphorylation of a crucial tyrosine residue (Tyr657) on eNOS95, other studies have shown that hydrogen peroxide increases eNOS expression and activity96,97 and may itself act as a vasodilator in certain vascular beds98–100. Indeed, this latter action could explain why mice with targeted endothelial overexpression of the NOX4 subunit displayed lower basal blood pressures and improved endothelium-dependent vasodilator function74.
Although the NOX5 subunit was first shown to be expressed in a human endothelial cell line in 2007, its apparent absence from the rodent genome101 has contributed to a relative paucity of additional information since this time about its role in vascular physiology and pathophysiology. Unlike other NADPH oxidases, NOX5 oxidase does not appear to interact with any of the known NADPH oxidase regulatory subunits and is likely to function as a stand-alone protein. NOX5 oxidase also has a unique amino terminal Ca2+-binding domain containing four EF hand motifs, which sets it apart from other NADPH oxidases. This domain allows NOX5 oxidase activity to be regulated by increases in cytosolic Ca2+ concentration102, which could be important in chronic vascular diseases in which cytosolic Ca2+ concentrations in vascular cells are likely to be elevated. Indeed, in human microvascular endothelial cell lines and primary aortic VSMCs, NOX5 oxidase activity was augmented by relevant pro-atherogenic stimuli, including thrombin, PDGF, angiotensin II and endothelin 1, and in each instance NOX5 oxidase activity was suppressed following depletion of intracellular Ca2+ (REFS 41,103). Endothelial cells and coronary arteries from patients with coronary artery disease displayed greater NOX5 subunit expression and Ca2+-dependent ROS production compared to those from healthy subjects104. Importantly, Ca2+-dependent but not Ca2+-independent ROS production was reduced by small interfering RNA against the NOX5 subunit, thus indicating that Ca2+-dependent ROS production is due to NOX5 oxidase activity104. Although these data suggest a strong association between NOX5 oxidase activity and coronary artery disease in humans, evidence for a cause–effect relationship is not yet proven and additional work is needed to characterize its physiological and pathological roles in the arterial wall.
NADPH oxidases in leukocytes
NOX2 oxidase in phagocytic cells was the first member of the NADPH oxidase family to be identified49,105. NOX2 oxidase-expressing leukocytes classically comprise cells of the innate immune system — that is, neutrophils, monocytes and macrophages106,107. Macrophages are the predominant inflammatory cell in atherosclerotic lesions, and are thus a major NADPH oxidase-expressing cell in diseased blood vessels of rodents and primates, including humans108,109 (FIG. 2). Numerous pro-atherogenic stimuli, including Chlamydia pneumoniae110, oxysterols111, angiotensin II112 and oxidized LDL113 activate NOX2 oxidase in macrophages.
In addition, adaptive immune cells (that is, T lymphocytes) express NOX2 oxidase114. NOX2 oxidase activity in circulating T lymphocytes is increased in experimental hypertension115 and stroke116, and in the former condition this contributes to the hypertension and endothelial dysfunction that occurs in association with the infiltration of T lymphocytes into perivascular fat (FIG. 2). NOX2 oxidase-derived superoxide from macrophages is also necessary for the induction of regulatory T cells (which suppress effector T cells) and thus modulation of T cell-mediated inflammation117; however, this occurs through a different and more complex mechanism. Overall, there is strong evidence that excessively high activity of NOX2 oxidase in leukocytes may contribute to vascular diseases through numerous complex mechanisms.
NADPH oxidases in the brain
All of the major cell types that are present in brain tissue (that is, neurons, astrocytes, microglia and vascular endothelial cells) constitutively express NOX1 oxidase, NOX2 oxidase and NOX4 oxidase49, which presumably reflects physiological roles for NOX oxidase-derived ROS in brain function. Expression of NOX5 oxidase in the brain has not yet been demonstrated. Many inflammatory and neurotoxic stimuli can increase NADPH oxidase activity in microglia and/or cause NADPH oxidase-dependent neuronal death, and these effects also appear to occur predominantly via NOX2 oxidase activity49,58,118. Interestingly, oxidative stress in the brain can contribute to systemic hypertension via a mechanism involving activation of T cells119. Recent studies highlight that NOX2 oxidase-derived ROS are generated in the neurons of circumventricular organs in response to circulating angiotensin II, leading to increased sympathetic outflow, which causes hypertension, activation of circulating T cells and increased systemic vascular inflammation118,120.
Cerebral arteries are different from many non-cerebral arteries in that they have higher levels of NADPH oxidase activity121,122. Although — as with most systemic vessels — ROS can increase the contractile tone of cerebral arteries, ROS more typically elicit marked vasorelaxant responses in cerebral vessels52, which tend to increase local blood flow. Interestingly, NOX2 oxidase-derived ROS appear to be important for the normal signalling that occurs in postsynaptic neurons following N-methyl-d-aspartate (NMDA) receptor activation in association with NO-dependent vasodilatation123. Furthermore, mice lacking NOX2 or p47phox subunits have mild deficits in learning and memory124, as has been suggested to occur in patients with CGD125. If NADPH oxidase-derived ROS help to maintain cerebral blood flow and neural function, then great caution will be needed in developing future therapeutic strategies involving inhibition of brain NADPH oxidases in cerebrovascular diseases. Important considerations will include identifying which NADPH oxidases are overactive and in which cells.
Nevertheless, studies using transgenic mice have revealed that excessive levels of NOX2 oxidase-derived superoxide do indeed underlie the oxidative stress and cerebrovascular dysfunction that is associated with angiotensin II-dependent hypertension, hypercholesterolaemia and advanced age118,126,127. Extensive NMDA receptor activation leads to superoxide-induced neuronal death that is absent in p47phox-deficient neurons58 and blocked by the NADPH oxidase inhibitor apocynin (see below), which probably implicates NOX2 oxidase as the source of lethal ROS generation in the brain. Moreover, the accumulation of amyloid-β protein in the brain in Alzheimer’s disease can activate NOX2 oxidase, and this effect accounts for the cerebral oxidative stress, neurovascular dysfunction and behavioural deficits that have been observed in a mouse model of Alzheimer’s disease59. Thus, excessive NOX2 oxidase-derived superoxide levels may represent a link between cerebrovascular dysfunction and cognitive impairment associated with several diseases of advanced age, including Alzheimer’s disease118.
There is evidence from studies on global NOX2 oxidase knockout mice that elevated activity of this NADPH oxidase isoform normally contributes to a worsened outcome following stroke, at least in males53,55,116,128,129, and that the protective effects of apocynin occur via inhibition of NOX2 oxidase53–55. The key cell types involved may include resident brain cells (for example, neurons and microglia) and infiltrating leukocytes116,129. There is evidence that NOX4 oxidase-derived ROS mediate TNF-induced cerebral endothelial apoptosis83 and infarct damage after stroke130. However, there is divided evidence as to whether NOX1 oxidase activity might contribute to stroke damage131,132.
Although NOX4 oxidase and NOX5 oxidase could emerge as validated drug targets from future research, we suggest that the evidence discussed in previous sections points to NOX2 oxidase in several cell types, and to a lesser extent NOX1 oxidase in VSMCs, as the NADPH oxidases that currently represent the most promising therapeutic targets for treating vascular diseases.
Established and novel NADPH oxidase inhibitors
The evidence presented above provides a strong rationale for the use of pharmacological inhibitors of NADPH oxidase to combat oxidative stress and its associated vascular pathologies in cardiovascular diseases. Several compounds are commonly cited in the literature as inhibitors of NADPH oxidase (TABLE 2). Some of these have been the focus of recent comprehensive review articles133–137 and are thus only briefly discussed here. Of these inhibitors, apocynin and diphenyleneiodonium (DPI) are the most widely studied. Although these compounds have proven to be invaluable pharmacological tools in NADPH oxidase research for over two decades, their lack of selectivity for NADPH oxidases over other enzymes and/or their potential to act as pro-oxidants under certain conditions (in the case of apocynin) is likely to limit their clinical utility. The compound 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) is also recognized as an NADPH oxidase inhibitor. However, it has a very low potency (IC50 (half-maximal inhibitory concentration) >1 mM) and off-target effects that include irreversible inhibition of serine proteases, and thus AEBSF has little potential for development into a clinically useful drug. The synthetic peptide Nox2ds-tat (also known as Gp91ds-tat) was designed to penetrate cells and prevent the assembly of the NOX2 oxidase complex (see below). This peptide inhibits NOX2 oxidase activity in vitro and reduces parameters of cardiovascular disease in a mouse model of hypertension57. However, being a peptide it is unlikely to be orally active or display a suitable pharmacokinetic profile to have widespread utility as a clinical drug.
Table 2.
NADPH oxidase inhibitors
| Name | Structure | Best characterized mechanism of action relevant to NADPH oxidase |
Other pharmacological effects | Refs |
|---|---|---|---|---|
| AEBSF | 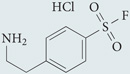 |
Oxidase assembly inhibitor: inhibits association of NOX2 subunit with p47phox. Does not scavenge O2•− generated in cell-free systems | Nonselective serine protease inhibitor | 215 |
| Apocynin | 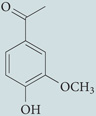 |
Oxidase assembly inhibitor: inhibits association of p47phox with membrane-bound heterodimer | Scavenger of hydrogen peroxide | 216−218 |
| DPI | 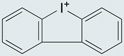 |
Flavoprotein inhibitor: abstracts electrons from FAD and prevents electron flow through the flavocytochrome conduit | Inhibitor of NADH-ubiquinone oxidoreductase, NADH dehydrogenase, xanthine oxidase, cytochrome p450 oxidoreductase, NOS and bacterial nicotine oxidase | 157,158, 219−223 |
| GK-136901 | 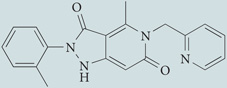 |
Purported NOX1 and NOX4 oxidase inhibitor. Mechanism of action not defined, but structural similarity with NADPH suggests that it may act as a competitive substrate inhibitor of this enzyme | None reported | 148,224 |
| ML171 | 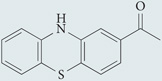 |
A phenothiazine compound with selectivity for NOX1 oxidase (IC50 of 0.25 µM) over other NADPH oxidases (IC50 >3 µM). Does not scavenge ROS generated by xanthine oxidase activity | None reported | 149 |
| Nox2ds-tat | [H]-RKKRRQRRRCSTRIRRQL-NH2 | Oxidase assembly inhibitor: inhibits association of NOX2 subunit with p47phox. Does not scavenge O2•− generated by cell-free systems | None reported | 57,188 |
| Plumbagin | 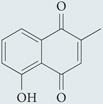 |
Inhibits NAD(P)H-dependent O2•− production in various cell lines that express NOX4 oxidase; mechanism of action unknown | Napthoquinone structure may impart ROS-scavenging effects | 225,226 |
| PR-39 | RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP | Binds to SH3 domains of the p47phox subunit and prevents binding to the p22phox subunit | Not selective for NADPH oxidase as it is likely to inhibit other proteins containing SH3 domains | 184 |
| S17834 | 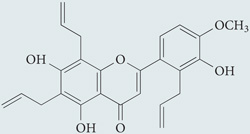 |
Flavonoid derivative proposed to directly inhibit NADPH oxidase activity, although the mechanism of action is undefined | None reported | 140 |
| VAS2870 | 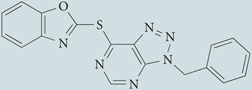 |
Undefined mechanism of action; inhibits NADPH oxidase activity in NOX2 oxidase-containing HL-60 cell line and in vascular endothelial cells containing NOX2 and NOX4 oxidases; does not scavenge O2•− | None reported | 144,145 |
| VAS3947 |  |
A triazolopyrimidine that reduced NADPH oxidase-derived ROS production in several cell lines with low micromolar potency, irrespective of the specific isoforms expressed; showed no inhibitory effects against xanthine oxidase-derived ROS or eNOS activity | None reported | 146 |
eNOS, endothelial nitric oxide synthase; IC50, half-maximal inhibitory concentration; NOX, NADPH oxidase; O2•−, superoxide; ROS, reactive oxygen species; SH3, Src homology 3.
Numerous studies over the past decade have purported to have identified novel NADPH oxidase inhibitors. However, before any compound can be considered to be a selective NADPH oxidase inhibitor, it must be proven to inhibit ROS production in both NADPH oxidase-expressing cells and in cell-free NADPH oxidase preparations without blocking ROS production from other enzymatic and non-enzymatic sources (for a detailed description of these criteria, see REF. 136). For compounds such as plumbagin and fulvene-5, some or most of these criteria have not yet been established, thus it remains to be determined whether they are true NADPH oxidase inhibitors138,139 as opposed to ROS scavengers or inhibitors of activation mechanisms upstream of NADPH oxidase activity. By contrast, S17834 blocks NADPH oxidase activity in intact endothelial cells and in isolated endothelial cell membranes without inhibiting ROS generated in a cell-free preparation consisting of hypoxanthine and xanthine oxidase140. Furthermore, S17834 suppressed TNF-induced leukocyte adhesion, reduced expression of plasminogen activator inhibitor 1, augmented NO production in endothelial monolayers and displayed atheroprotective properties when it was administered chronically to ApoE- or LDL receptor-deficient mice140–143.
Triazolopyrimidines, including VAS2870 and VAS3947, have also emerged as promising inhibitors of NADPH oxidase activity. These compounds inhibited NADPH oxidase-derived ROS in several cell lines expressing NADPH oxidases and in primary endothelial and VSMC cultures, but had no effect on ROS generated by xanthine oxidase or on eNOS activity144– 146. VAS2870 inhibited elevated ROS production and restored endothelium-dependent relaxations in aortas from spontaneously hypertensive rats82 and limited phenylephrine-induced contractions in rat tail arteries 147. Most recently, intrathecal administration of VAS2870 to mice following experimental stroke was shown to reduce brain infarct volume and improve neurological outcome130. In this study, Nox4−/− mice were similarly protected against brain infarction and neurological deficits following stroke, and administration of VAS2870 to these animals conferred no further benefits130.
Pyrazolopyridine derivatives such as GK-136901, which are structurally related to VAS2870 and VAS3947, were recently identified as potent inhibitors of NOX1 oxidase- and NOX4 oxidase-dependent ROS generation from disrupted cell membrane preparations, with Ki values in the mid-nanomolar range148. These compounds were approximately tenfold less active against NOX2 oxidase-dependent ROS production (Ki >1 µM), and had no detectable inhibitory effects against ROS generated in vitro by xanthine oxidase148. GK-136901 was also highly drug-like in terms of its ease of synthesis, high oral bioavailability, lack of off-target effects against a panel of 135 enzymes (including eNOS), and its in vivo pharmacokinetic and safety profiles148.
ML171 (also known as 2-acetylphenothiazine) was recently identified as a specific NOX1 oxidase inhibitor at nanomolar concentrations, with only marginal activity on other cellular ROS-producing sources — including xanthine oxidase and the other NADPH oxidases149. ML171 appears to target the NOX1 catalytic subunit but not its cytosolic regulators (the NOXO1, NOXA1 or RAC1 subunits) and it blocks the NOX1 oxidase-dependent ROS-mediated formation of extracellular matrix-degrading invadopodia in colon cancer cells149. Finally, a hydrogen peroxide assay using human embryonic kidney cells overexpressing the NOX4 subunit identified several NOX4 oxidase inhibitors from compounds that contained one of five core structures (oxalyl hydrazides, flavonoids, oxindoles, benzoquinolines or benzothiophenes) 150. From these data the authors constructed a pharmacophore model of a NOX4 oxidase inhibitor that comprised two hydrogen bond donors and two aromatic rings150. Further work is needed to determine the isoform selectivity of compounds based on this pharmacophore model as well as their specificity for inhibition of ROS production by NADPH oxidase versus other sources.
All of the emerging NADPH oxidase inhibitor compounds discussed above were either identified directly by high-throughput screening of compound libraries, or they are derivatives of compounds that were initially identified in such screens. Although many of these compounds appear to hold promise as NADPH oxidase inhibitor drugs, it is interesting to note that none of them displays selectivity for NOX2 oxidase over other NADPH oxidases; on the contrary, some compounds were selective for multiple isoforms other than the NOX2 oxidase.
Based on the evidence presented in previous sections of this Review, we conclude that the NOX1 and NOX2 oxidases currently appear to be the major contributors to vascular oxidative stress in cardiovascular disease, and hence are the most logical therapeutic targets. Therefore, given that other NADPH oxidases are known to have important roles in processes such as postural balance (NOX3 oxidase)151,152, vasodilatation and cell survival (NOX4 oxidase)99,100,122,153, and thyroid hormone production (DUOX1 and DUOX2 oxidases)154, one could argue that in cardiovascular disease therapy, inhibitors displaying a dual selectivity for the NOX1 and NOX2 isoforms over other members of the NADPH oxidase family will probably offer the greatest benefits in terms of therapeutic efficacy versus side-effect profiles.
Nevertheless, the potential implications of inhibiting any important extravascular effects of NOX1 and NOX2 oxidases must be considered. This is of particular relevance for NOX2 oxidase, given its crucial role in the innate immune response to invading pathogens, and the possibility that any attempts to pharmacologically inhibit NOX2 oxidase will severely compromise a patient’s immunological function — analogous to the rare, severe genetic condition CGD (BOX 1). It is therefore reassuring to note that a recent study involving patients with CGD demonstrated that severe illness and poor long-term survival were only evident in individuals whose phagocytic ROS production was more than two orders of magnitude lower than healthy controls155.
Findings from clinical and animal studies show that NADPH oxidase activity in the vascular wall increases by no more than fivefold during cardiovascular disease. Therefore, we suggest that an optimal therapeutic intervention dually targeting vascular NOX1 and NOX2 oxidases would only need to reduce NADPH oxidase activity by less than one order of magnitude. Hence, by titrating the dose of an NADPH oxidase inhibitor, we predict that it will be feasible to partially inhibit the target NADPH oxidase in the vascular wall to alleviate oxidative stress without affecting long-term immune function. In the following section we outline a rational approach that we believe has the potential to identify new target sites for novel, dual inhibitors of vascular NOX1 oxidase and NOX2 oxidase.
A rational drug design approach
Towards the discovery of dual NOX1 and NOX2 oxidase inhibitors
We contend that the goal of selectively targeting the isoforms of NADPH oxidase that are responsible for the pathophysiology of vascular disease could be achieved via a directed drug discovery approach, starting with an extensive knowledge of the subunit–subunit interactions that are common to the NOX1 and NOX2 oxidase complexes, but not used by the other NADPH oxidase family members.
Because protein–protein interactions often involve binding pockets that are too large to be occupied by small-molecule inhibitors, it will be essential to resolve the three-dimensional structures of each of these interacting domains to identify which protein–protein interactions could represent feasible targets for future drugs.
One could postulate that because the catalytic NOX subunit is what primarily distinguishes each NADPH oxidase isoform, drugs targeting the NOX1 and NOX2 subunits should have the highest likelihood of achieving isoform selectivity. However, it must be noted that despite their diverse amino acid sequences, the functional domains on the different NOX catalytic subunits — which are responsible for binding to NADPH, FAD and haem prosthetic groups — are highly conserved156 and provide little scope for selective pharmacological targeting. A good example to illustrate this is DPI, which forms a covalent adduct with FAD or on a site within the NOX protein that is adjacent to this moiety, thereby preventing electron flow through the catalytic subunit157,158. As a result of this mechanism of action, DPI shows no selectivity towards any isoform of NADPH oxidase and also fails to discriminate between NADPH oxidases and other FAD-containing enzymes — including NOS, xanthine oxidase and cytochrome P450 (REF. 50).
This is not to say that other, hitherto undefined sites on NOX subunits do not have the potential to be exploited by selective inhibitors. However, as NOX subunits are integral membrane proteins50, resolution of their three-dimensional structure to identify druggable pockets is likely to represent a major technical challenge. Therefore, we suggest that a feasible strategy with a high potential to yield clinically relevant, isoform-selective lead compounds would be to pharmacologically target the cytosolic regulatory subunits that are used exclusively by the vascular NOX1 and NOX2 oxidases.
The subunit composition of each of the seven isoforms of NADPH oxidase is summarized in TABLE 3. Several of the regulatory subunits used by NOX1 and NOX2 oxidases (including p22phox, NOXA1, NOXO1 and RAC1) are also used to varying degrees by other isoforms of NADPH oxidase (including the NOX3 and NOX4 oxidases). Therefore, agents that target these subunits are unlikely to represent specific inhibitors of NOX1 and NOX2 oxidases. By contrast, p47phox is the only subunit that is used specifically by NOX2 oxidase and by the NOX1 oxidase expressed in VSMCs; that is, interactions between p47phox and both of these two isoforms are reasonably selective in the cell types that are involved in the generation of NADPH oxidase-derived ROS in vascular disease. Moreover, p47phox is a soluble protein50 for which some aspects of its three-dimensional structure have already been resolved by nuclear magnetic resonance (NMR) and X-ray crystallography, and thus further studies to explore its three-dimensional structure for potential druggable sites are highly feasible.
Table 3.
Comparison of subunit utilization of the seven NADPH oxidases
| NOX1 | NOX2 | NOX3 | NOX4 | NOX5 | DUOX1 | DUOX2 | |
|---|---|---|---|---|---|---|---|
| p22phox | + | + | + | + | − | − | − |
| DUOXA1 | − | − | − | − | − | + | + |
| DUOXA2 | − | − | − | − | − | + | + |
| p67phox | − | + | − | − | − | − | − |
| NOXA1 | + | − | +* | − | − | − | − |
| p47phox | +‡ | + | − | − | − | − | − |
| NOXO1 | +§ | − | +* | − | − | − | − |
| p40phox | − | + | − | − | − | − | − |
| POLDIP2 | ND | ND | ND | + | ND | ND | ND |
| RAC1 | + | + | + | − | − | − | − |
| RAC2 | − | + | − | − | − | − | − |
DUOXA1, dual oxidase activator 1; NOXA1, NADPH oxidase activator 1; NOXO1, NOX organizer 1; ND, not determined; POLDIP2, polymerase δ-interacting protein 2.
NOX3 is constitutively active when it forms a complex with p22phox; however, its activity is further elevated by NOXA1 and NOXO1 subunits (REF. 227).
Expressed with NOX1 in vascular smooth muscle cells only.
Not detectable in vascular smooth muscle cells.
p47phox as a target for current and future NADPH oxidase isoform-selective inhibitors
The p47phox subunit acts as the ‘organizer’ protein for the NOX2 oxidase complex, as it is the subunit that is responsible for chaperoning the activator subunit, p67phox, to the membrane-bound catalytic domain159. p47phox also serves a similar organizer function for the NOX1 oxidase complex that is expressed in VSMCs, by facilitating the interaction of the cytosolic NOXA1 subunit with the catalytic domain of NOX1 oxidase62,160 (FIG. 1). To achieve these functions, p47phox contains distinct functional domains that allow it to interact simultaneously with the p67phox or NOXA1 subunits, p22phox, and the NOX1 or NOX2 catalytic subunits as well as with phospholipids in the biological membrane to which the whole complex is anchored (FIG. 3). At least one of these domains on p47phox has already been exploited as a target for the novel, rationally-designed NADPH oxidase inhibitor Nox2ds-tat57. We predict that further examination of the physicochemical and structural properties of this functional domain and the remaining functional domains on p47phox will yield new opportunities for the development of highly selective dual NOX1 and NOX2 oxidase inhibitors that could be suitable for the treatment of cardiovascular disease. In the following section we look more closely at the interactions of p47phox with other subunits of the NOX1 and NOX2 oxidase complexes and, where possible, discuss how this information might be translated into the design of novel inhibitor drugs.
Figure 3. Schematic diagram showing p47phox as the central organizer of the vascular NOX1 and NOX2 oxidases.

a | This figure shows the conformation of p47phox in resting cells in which the protein forms a complex with other cytosolic regulatory subunits via an interaction between its carboxy-terminal proline-rich region (PRR) and an Src homology 3 (SH3) domain on one of two potential activator proteins, p67phox or NADPH oxidase activator 1 (NOXA1) subunit (site 1). In the resting state, p47phox adopts a closed conformation in which its tandem-repeat SH3 and phox homology (PX) domains are ensconced and unable to interact with the membrane components. This conformation is largely achieved via intramolecular interactions of both the polybasic autoinhibitory region (AIR) and the PX domain with the tandem-repeat SH3 domain (site 2). b | Phosphorylation of sulfhydryl groups (SH) on crucial cysteine residues within the AIR destabilizes these intramolecular interactions and causes p47phox to unfold. This exposes the tandem-repeat SH3 domain of p47phox, thereby allowing it to associate with a PRR on the amino terminus of membrane-bound p22phox (site 3), and the PX domain of p47phox, which interacts with membrane phospholipids including phosphatidylinositol-3,4-biphosphate (PtdIns(3,4)P2) and phosphatidic acid (PA) (site 4). The association of the cytosolic complex with the membrane subunits is further stabilized by direct protein–protein interactions between p47phox and the NOX2 subunit (site 5). It is unclear whether p47phox undergoes similar protein–protein interactions with the NOX1 subunit. Also shown are likely target sites for conventional NADPH oxidase inhibitors. PKC, protein kinase C.
The p47phox–p67phox interaction
p47phox interacts directly with an Src homology 3 (SH3) domain on p67phox via a PxxP binding motif contained within a proline-rich region (PRR) near its C terminus (amino acids 360–369)161–165. Additional contacts with the SH3 domain of p67phox are made by a domain on p47phox that lies distal to the PRR on its C terminus (amino acids 368–390), and these contacts are important for increasing the affinity of the protein–protein interaction (KA ~20 nM) and for conferring binding partner specificity166,167.
The interaction between the p47phox PRR and the p67phox SH3 domain is important, both for the constitutive association of p47phox and p67phox in the cytosol of resting cells and for the p47phox-dependent recruitment of p67phox to the membrane159. Hence, disruption of this interaction represents a possible strategy for inhibiting NADPH oxidase activation. It is unclear whether any of the current NADPH oxidase inhibitor drugs act by interfering with the interaction between the p47phox PRR and the p67phox SH3 domain. However, a series of synthetic peptides based on the PRR of p47phox — but not on PRRs of other proteins — were effective at preventing the binding of p47phox to p67phox, thus providing proof of principle that this interaction may represent a future target for selective NOX2 oxidase inhibitors162.
As mentioned above, the activation of NADPH oxidase in VSMCs relies on the association of the NOXA1 subunit with the membrane-bound NOX1-p22phox flavocytochrome62. Nonetheless, p47phox is still likely to act as the organizer subunit for this enzyme complex62. The association of p47phox with the NOXA1 subunit is likely to occur via mechanisms that are similar to those involved in the association of p47phox with p67phox. Examination of the C terminus of the NOXA1 subunit reveals it to be highly homologous to that of p67phox, with respect to the presence of the PxxP binding motif-containing SH3-binding domain, and also in the domain further towards the C-terminal end, which is responsible for increasing the binding affinity and specificity of p67phox for p47phox168,169. Moreover, substitution of Arg436 for Trp436 within this C-terminal region results in a complete loss of the interaction between the NOXA1 and p47phox subunits169. As such, pharmacological strategies that inhibit the interaction of the p47phox PRR and the p67phox SH3 domain would also be expected to block the interaction between the p47phox and NOXA1 subunits, and would thereby inhibit the production of ROS by NOX1 oxidase in VSMCs.
Intramolecular interactions of the p47phox autoinhibitory region with its SH3 and Phox homology domains
In resting cells, p47phox is folded in on itself such that the bis-SH3 and Phox homology (PX) domains of the protein are ensconced159. This closed configuration of p47phox is largely due to intramolecular interactions between its polybasic autoinhibitory region (AIR) and bis-SH3 domain170–172, and also between its bis-SH3 domain and PX domains173,174. Activation of p47phox occurs when serine residues within the AIR are phosphorylated by protein kinase C (PKC) or phosphoinositide 3-kinase (PI3K)175–178. This destabilizes the interaction of the AIR with the bis-SH3 domain, allowing p47phox to adopt an open conformation whereby its bis-SH3 and PX domains are free to interact with the p22phox PRR and membrane phospholipids, respectively159.
Based on the above model, one would predict that preventing phosphorylation of the AIR could lock p47phox in its closed state and thereby inhibit its interaction with the membrane components of the NADPH oxidase enzyme. One strategy for achieving this could be the use of compounds that act upstream of p47phox by inhibiting the activity of PKC and other protein kinases that are responsible for phosphorylating the AIR. A more selective approach may involve direct chemical modification of the sulfhydryl side-groups belonging to the crucial serine residues present within the AIR. Indeed, this may be the mechanism by which apocynin achieves its widely reported inhibitory effects on NADPH oxidase179.
The p47phox–p22phox intermolecular interaction
A key interaction in the assembly of the active NADPH oxidase complex occurs between the p47phox bis-SH3 domain — which is exposed following phosphorylation of the AIR — and a PRR present on the carboxy terminal of p22phox (amino acids 151–160)180,181. As is common for many SH3–PRR interactions, the affinity and specificity of the p22phox PRR for the p47phox bis-SH3 domain are enhanced by a series of amino acids that are located just proximal to the PRR of p22phox (amino acids 161–164)182. As proof of its importance to NADPH oxidase activation (and thus its potential as a target site for future drugs), a short synthetic peptide spanning amino acids 159–162 of p22phox was shown to bind to the p47phox bis-SH3 site and inhibit its association with p22phox183. Moreover, PR39 — an endogenous antimicrobial peptide produced by neutrophils — was shown to inhibit the translocation of p47phox to the membrane via a direct interaction with the bis-SH3 domain of the protein184.
NMR solution structures of the p47phox bis-SH3 domain in complex with peptide fragments of either the p22phox PRR185,172 or the p47phox AIR170 have revealed that, in its uninhibited state, the p47phox bis-SH3 domain forms a groove along the surface of the protein. Although the lowest energy structure for this bis-SH3 domain is likely to be too shallow to represent a viable docking site for small-molecule ligands, preliminary data of the relative position and orientation of individual amino acids within this three-dimensional space reveal a single tryptophan residue (Trp193) that is exposed to the solvent (N. Hall, A. Christopoulos and P. Sexton, personal communication). This residue is engaged in minimal interactions with surrounding residues and is thus likely to be highly dynamic in solution. Molecular modelling of feasible alternative p47phox conformations reveals that rotation of Trp193 opens up a deeper groove on the surface of the protein, which is within the size range of known druggable binding pockets (FIG. 4). Although it remains to be determined whether virtual ligand screening with this modified p47phox bis-SH3 structure will identify small molecules with in vitro and/or in vivo NADPH oxidase inhibitory activity, the above discussion nonetheless highlights that potentially specific molecular targets for drugs may arise as more detailed molecular structural information becomes available.
Figure 4. The p47phox tandem-repeat SH3 domain as a potential drug target.

Nuclear magnetic resonance solution structure of the p22phox proline-rich region (PRR)-binding domain formed by the tandem repeat Src homology (SH3) domain of p47phox before (a) and after (b) molecular modelling to rotate a putative dynamic tryptophan residue (Trp193) within its core. Note that rotation of Trp193 results in two smaller pockets (red and orange) combining to form a single large pocket that is potentially druggable.
The p47phox–NOX2 subunit intermolecular interaction
Phage display mapping indicates that p47phox directly interacts with the NOX2 subunit186. Three putative p47phox-interacting domains have been identified on the NOX2 subunit, including one on the first predicted intracellular loop (amino acids 86–93), and two additional domains on the cytosolic C-terminal tail (amino acids 450–457 and 554–564)186. In the absence of detailed three-dimensional structural information for the NOX2 subunit, it is not possible to determine whether these multiple sites lie adjacent to each other in the folded protein to form a single interaction domain or whether they contact p47phox at separate sites. Interestingly, studies looking for regions on p47phox that interact with the NOX2 subunit have so far identified only one site situated within the AIR (amino acids 323–342)187,188, which may indicate a single point of contact for the NOX2 subunit on p47phox.
Short synthetic peptides spanning any one of the three p47phox-interacting domains on the NOX2 subunit acted as competitive inhibitors of NADPH oxidase activation in both cell-free systems and in electropermeabilized neutrophils186,189–191. Elegant follow-up to this work showed that the addition of a human immunodeficiency virus transactivator of transcription (tat) peptide sequence to one of these NOX2 subunit-docking sequences (Nox2ds) resulted in a compound that was effective at inhibiting NOX2 oxidase activity in intact RAW264.7 cells (an immortalized mouse macrophage cell line) and mouse aortas, and at preventing oxidative stress and its vascular sequelae in animal models of hypertension57.
An alternative strategy involving the adenoviral-mediated expression of either Nox2ds or a dominant negative p67phox construct in adventitial fibroblasts of the rat carotid artery in vivo reduced angioplasty-induced ROS production and neointimal proliferation192,193. However, it is unclear where on p47phox the Nox2ds-tat binds. It is also unclear whether the site occupied by Nox2ds-tat is involved in the association of p47phox with the NOX1 subunit in VSMCs and, consequently, whether Nox2ds-tat will be effective at inhibiting ROS production in this cell type. Identification of this site, as well as resolution of its three-dimensional structure, should provide important insight as to whether it may represent a future target for small-molecule dual NOX1 and NOX2 oxidase inhibitors.
The p47phox PX–membrane phospholipid interaction
The PX domain is a conserved sequence of approximately 120 amino acids that is present in p47phox and at least 100 other proteins, including p40phox and NOXO1 subunits194–196. As previously mentioned, the p47phox PX domain contains a PRR that is important for maintaining p47phox in its inactive state in resting cells. Detailed mutagenesis studies and X-ray crystallography of the p47phox PX domain have revealed that it also contains two highly basic pockets that bind to membrane phosphoinositides following unfolding of the protein197,198. Importantly, these binding pockets are structurally distinct from the one that is present on the p40phox PX domain, thus imparting selectivity towards different phospholipids198. The first pocket is relatively large (and therefore probably not an appropriate target for small-molecule drugs) and selectively binds to phosphatidylinositol-3,4-bisphosphate (PtdIns(3,4)P2) with a Kd of ~38 nM. The second pocket is much smaller and has strong affinities towards anionic phospholipids such as phosphatidic acid or phosphatidylserine198. It will be interesting to see whether virtual ligand screening of this pocket yields novel selective dual NOX1 and NOX2 oxidase inhibitors.
Conclusions and perspectives
ROS are compelling targets for the prevention and treatment of vascular diseases. Compounds that inhibit the production of ROS should offer considerable advantages over conventional antioxidants, for which clinical efficacy is likely to have been limited owing to poor bioavailability at the site of disease and paradoxical pro-oxidant effects. The NADPH oxidase family of enzymes, particularly those that contain NOX1 or NOX2 catalytic subunits, are important sources of ROS production in the arterial wall, leukocytes and the brain during vascular disease states. These isoforms of NADPH oxidase rely on their organizer subunit, p47phox, for full activity, thus highlighting this protein as a promising molecular target for therapy. However, although p47phox is important for optimal NADPH oxidase function, the enzyme can be activated (albeit less effectively) in its absence. Given the importance of retaining at least some NADPH oxidase activity for normal immune cell function, p47phox is therefore likely to represent a clinically safer target than other subunits that are absolutely required for enzyme activity. Although several studies have demonstrated protective effects of reputed p47phox inhibitors — including apocynin and Nox2ds-tat — in animal models of hypertension, atherosclerosis and stroke, the clinical translation of such findings is only likely to be achieved with the development of inhibitors with improved efficacy, specificity and pharmacokinetic profiles. Further insight into the molecular structure of p47phox and its interactions with other NADPH oxidase subunits and membrane phospholipids should facilitate the development of the next generation of NADPH oxidase inhibitors.
Acknowledgements
The authors are grateful to R. Brandes, F. Faraci, C. Iadecola and P. Pagano for their expert advice during the preparation of this manuscript, and to J. Rivera for compiling the information presented in table 1. We also acknowledge the National Health and Medical Research Council of Australia for Fellowship and Project Grant support (IDs 606472, 1006017, 545942, 570861, 606488 and 1010984), the Heart Foundation of Australia for Grant-in-aid support (G 09M 4398 and G 10M 5218), the Cancer Council of Victoria for a Project Grant (606674), and the US National Institutes of Health for grant support (HL38206, HL058863, HL092120 and HL095070).
Glossary
- Innate immunity
The first line of defense against invading microorgansims. This is mediated by a combination of anatomical barriers (such as skin and internal epithelial surfaces), physiological barriers (such as raised body temperature), secretory molecules (such as digestive enzymes, complement and interferons) and certain kinds of leukocytes (such as macrophages, neutrophils, mast cells and so on).
- Microbicidal burst
The rapid release of reactive oxygen species such as superoxide and hydrogen peroxide by white blood cells (for example, neutrophils and macrophages) to eliminate invading microorganisms from the body.
- Oxidative stress
An imbalance between the production and inactivation of reactive oxygen species within a biological system, which often results in redox-sensitive modification of macromolecules — including proteins, nucleotides and lipids — and leads to cellular toxicity.
- Professional phagocytes
Cells that are able to engulf (phagocytose) particles such as invading pathogens, dead host cells and cell debris with high efficiency. Such cells include monocytes, macrophages, neutrophils, dendritic cells and mast cells.
- Acute cardiovascular events
Cardiovascular end points — such as stroke and heart attacks — that result from the sudden cessation of blood flow through arteries owing to a stenosis or a thromboembolus.
- Arterial remodelling
A term used to describe any persistent change in the cross-sectional area of a blood vessel. Remodelling may be outward, in which the vessel expands to accommodate a stenosis, or inward, as a result of medial hypertrophy in hypertension, intimal thickening owing to atherosclerosis, transplant vasculopathy or restenosis after balloon angioplasty.
- Thrombosis
The formation of a blood clot within a blood vessel.
- Restenosis
The reoccurrence of a stenosis leading to restricted blood flow, occurring at a site on the artery that received angioplasty treatment to clear an initial stenosis.
- Atherosclerotic plaque
An accumulation of inflammatory cells — mainly macrophages — and cell debris containing lipids, cholesterol, calcium and fibrous connective tissue, which leads to the enlargement of the artery wall.
- Redox state
The reduction or oxidation state of a molecule, which is governed by the number of electrons gained or transferred in a chemical reaction.
- Adhesion molecules
Integral membrane proteins including integrins, selectins and certain immunoglobulins that mediate the binding of one cell to other cells or extracellular matrix proteins.
- Metalloproteinases
A family of protease enzymes possessing a metal centre (most often Zn2+) as the key functional group in their catalytic site.
- Inflammasomes
Multiprotein signalling platforms that are activated in response to pathogens or host-derived stress signals that trigger the maturation of pro-inflammatory cytokines such as interleukin-1β and interleukin-18.
- Cohort studies
An analytical study that follows the same group of people over a long period of time and is often used to assess risk of disease.
- Endothelial dysfunction
A shift in the activation state of the endothelium towards reduced production of vasodilator substances, such as nitric oxide, and increased production of pro-inflammatory and prothrombotic mediators.
- EF hand motifs
A protein structural domain that is involved in binding Ca2+, and is characterized by a helix–loop–helix topology that has a conformation similar in shape to the spread thumb and forefinger of the hand.
- Binding pockets
Regions on a protein that other smaller molecules, termed ligands, can dock onto in a highly specific fashion through the formation of chemical bonds. Such ligands can include endogenous factors such as hormones or neurotransmitters, or exogenous substances such as drugs.
- X-ray crystallography
A method of determining the three-dimensional arrangement of atoms within a crystalline substance based on the diffraction pattern generated following bombardment of the crystal with a beam of X-rays.
- Phox homology (PX) domain
A conserved protein structural domain that is involved in the targeting of proteins to cellular membranes by binding to phosphoinositides.
- Phage display mapping
A method for the study of protein–protein interactions in which proteins of interest are immobilized and exposed to peptide-presenting bacteriophages. Bacteriophages that bind to the protein are used to infect a bacterial host from which the phagemid is collected and the relevant DNA is sequenced and used to predict the primary sequence of the bound peptide.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Sobey, Drummond and Selemidis — The Vascular Biology and Immunopharmacology Group homepage: http://www.med.monash.edu.au/pharmacology/research/vbig.html
Griendling laboratory homepage: http://www.medicine.emory.edu/cardio/labs/kkg/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Dinauer MC. Disorders of neutrophil function: an overview. Methods Mol. Biol. 2007;412:489–504. doi: 10.1007/978-1-59745-467-4_30. [DOI] [PubMed] [Google Scholar]
- 2.Jones DP. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 4.Morris CD, Carson S. Routine vitamin supplementation to prevent cardiovascular disease: a summary of the evidence for the U. S. Preventive Services Task Force. Ann. Intern. Med. 2003;139:56–70. doi: 10.7326/0003-4819-139-1-200307010-00014. [DOI] [PubMed] [Google Scholar]
- 5.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell. Mol. Med. 2010;14:70–78. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J. Thromb. Haemost. 2005;3:254–267. doi: 10.1111/j.1538-7836.2004.01085.x. [DOI] [PubMed] [Google Scholar]
- 7.Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 8.Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol. Med. 2008;14:495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 10.Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 11.Blough NV, Zafiriou OC. Reaction of superoxide with nitric oxide to form peroxonitrite in alkaline aqueous solution. Inorg. Chem. 1985;24:3502–3504. [Google Scholar]
- 12.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 13.Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Rev. Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 14.Harrison DG, Chen W, Dikalov S, Li L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv. Pharmacol. 2010;60:107–132. doi: 10.1016/B978-0-12-385061-4.00005-2. [DOI] [PubMed] [Google Scholar]
- 15.Laursen JB, et al. Endothelial regulation of vasomotion in ApoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 16.Wadham C, Mangoni AA. Dimethylarginine dimethylaminohydrolase regulation: a novel therapeutic target in cardiovascular disease. Expert Opin. Drug Metab. Toxicol. 2009;5:303–319. doi: 10.1517/17425250902785172. [DOI] [PubMed] [Google Scholar]
- 17.Stasch JP, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anrather J, Racchumi G, Iadecola C. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 19.Dworakowski R, Alom-Ruiz SP, Shah AM. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol. Rep. 2008;60:21–28. [PubMed] [Google Scholar]
- 20.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinon F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 23.Lonn EM, Yusuf S. Is there a role for antioxidant vitamins in the prevention of cardiovascular diseases? An update on epidemiological and clinical trials data. Can. J. Cardiol. 1997;13:957–965. [PubMed] [Google Scholar]
- 24.Bleys J, Miller ER, Pastor-Barriuso R, Appel LJ, Guallar E. Vitamin-mineral supplementation and the progression of atherosclerosis: a meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2006;84:880–887. doi: 10.1093/ajcn/84.4.880. [DOI] [PubMed] [Google Scholar]
- 25.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361:2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 26.Schiffrin EL. Antioxidants in hypertension and cardiovascular disease. Mol. Interv. 2010;10:354–362. doi: 10.1124/mi.10.6.4. [DOI] [PubMed] [Google Scholar]
- 27.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 28.Gotoh N, Niki E. Rates of interactions of superoxide with vitamin E, vitamin C and related compounds as measured by chemiluminescence. Biochim. Biophys. Acta. 1992;1115:201–207. doi: 10.1016/0304-4165(92)90054-x. [DOI] [PubMed] [Google Scholar]
- 29.Simonsen U, Wadsworth RM, Buus NH, Mulvany MJ. In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. J. Physiol. 1999;516:271–282. doi: 10.1111/j.1469-7793.1999.271aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witting PK, Willhite CA, Davies MJ, Stocker R. Lipid oxidation in human low-density lipoprotein induced by metmyoglobin/H2O2: involvement of α-tocopheroxyl and phosphatidylcholine alkoxyl radicals. Chem. Res. Toxicol. 1999;12:1173–1181. doi: 10.1021/tx9900472. [DOI] [PubMed] [Google Scholar]
- 31.Witting PK, Upston JM, Stocker R. Role of α-tocopheroxyl radical in the initiation of lipid peroxidation in human low-density lipoprotein exposed to horse radish peroxidase. Biochemistry. 1997;36:1251–1258. doi: 10.1021/bi962493j. [DOI] [PubMed] [Google Scholar]
- 32.Stocker R. Lipoprotein oxidation: mechanistic aspects, methodological approaches and clinical relevance. Curr. Opin. Lipidol. 1995;5:422–433. [PubMed] [Google Scholar]
- 33. Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am. J. Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568.. This was the first paper to demonstrate that endothelial cells express a functional NAD(P)H oxidase system.
- 34. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141.. This was the first paper to demonstrate that VSMCs express a functional NAD(P)H oxidase system.
- 35.Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller FJ, Jr, et al. Cytokine activation of nuclear factor κB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 2007;101:663–671. doi: 10.1161/CIRCRESAHA.107.151076. [DOI] [PubMed] [Google Scholar]
- 37.Serrander L, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dikalov SI, et al. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takac I, et al. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Csanyi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med. 2009;47:1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.BelAiba RS, et al. NOX5 variants are functionally active in endothelial cells. Free Radic. Biol. Med. 2007;42:446–459. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 42.Lassegue B, et al. Novel gp91(phox) homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 43.Ellmark SH, Dusting GJ, Fui MN, Guzzo-Pernell N, Drummond GR. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc. Res. 2005;65:495–504. doi: 10.1016/j.cardiores.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Moe KT, et al. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-α in human aortic smooth muscle and embryonic kidney cells. J. Cell. Mol. Med. 2006;10:231–239. doi: 10.1111/j.1582-4934.2006.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamseddine AH, Miller FJ., Jr Gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2003;285:H2284–H2289. doi: 10.1152/ajpheart.00459.2003. [DOI] [PubMed] [Google Scholar]
- 46.Pagano PJ, et al. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc. Natl Acad. Sci. USA. 1997;94:14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haurani MJ, Pagano PJ. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc. Res. 2007;75:679–689. doi: 10.1016/j.cardiores.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Touyz RM, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ. Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 49.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 50.Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008;120:254–291. doi: 10.1016/j.pharmthera.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Brandes RP, Schroder K. Composition and functions of vascular nicotinamide adenine dinucleotide phosphate oxidases. Trends Cardiovasc. Med. 2008;18:15–19. doi: 10.1016/j.tcm.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Miller AA, Drummond GR, Sobey CG. Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol. Ther. 2006;111:928–948. doi: 10.1016/j.pharmthera.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 53.Jackman KA, et al. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br. J. Pharmacol. 2009;156:680–688. doi: 10.1111/j.1476-5381.2008.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia–reperfusion. J. Cereb. Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kahles T, et al. NADPH oxidase plays a central role in blood–brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 56.Jung O, et al. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 57. Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ. Res. 2001;89:408–414. doi: 10.1161/hh1701.096037.. This was the first demonstration that a rational drug design approach can yield effective inhibitors of NADPH oxidase that prevent vascular oxidative stress and reduce systolic blood pressure in an in vivo animal model of hypertension.
- 58.Brennan AM, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nature Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park L, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl Acad. Sci. USA. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller AA, De Silva TM, Jackman KA, Sobey CG. Effect of gender and sex hormones on vascular oxidative stress. Clin. Exp. Pharmacol. Physiol. 2007;34:1037–1043. doi: 10.1111/j.1440-1681.2007.04732.x. [DOI] [PubMed] [Google Scholar]
- 61.Ambasta RK, et al. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 62. Ambasta RK, Schreiber JG, Janiszewski M, Busse R, Brandes RP. Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic. Biol. Med. 2006;41:193–201. doi: 10.1016/j.freeradbiomed.2005.12.035.. This study provides evidence that the NOX1 oxidase expressed in VSMCs may be unique from that expressed in other tissues in its utility of the classical organizer subunit p47phox.
- 63.Niu XL, et al. Nox activator 1: a potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation. 2010;121:549–559. doi: 10.1161/CIRCULATIONAHA.109.908319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akasaki T, et al. Increased expression of gp91phox homologues of NAD(P)H oxidase in the aortic media during chronic hypertension: involvement of the reninangiotensin system. Hypertens. Res. 2006;29:813–820. doi: 10.1291/hypres.29.813. [DOI] [PubMed] [Google Scholar]
- 65.Oelze M, et al. NADPH oxidase accounts for enhanced superoxide production and impaired endothelium-dependent smooth muscle relaxation in BKβ1−/− mice. Arterioscler. Thromb. Vasc. Biol. 2006;26:1753–1759. doi: 10.1161/01.ATV.0000231511.26860.50. [DOI] [PubMed] [Google Scholar]
- 66.Wang P, et al. Contribution of different Nox homologues to cardiac remodeling in two-kidney two-clip renovascular hypertensive rats: effect of valsartan. Pharmacol. Res. 2007;55:408–417. doi: 10.1016/j.phrs.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 67.Nakano D, et al. Dietary sesamin suppresses aortic NADPH oxidase in DOCA salt hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2008;35:324–326. doi: 10.1111/j.1440-1681.2007.04817.x. [DOI] [PubMed] [Google Scholar]
- 68.Nakamura T, et al. Beneficial effects of pioglitazone on hypertensive cardiovascular injury are enhanced by combination with candesartan. Hypertension. 2008;51:296–301. doi: 10.1161/HYPERTENSIONAHA.107.099044. [DOI] [PubMed] [Google Scholar]
- 69.Dikalova A, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 70.Dikalova AE, et al. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H673–H679. doi: 10.1152/ajpheart.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gavazzi G, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580:497–504. doi: 10.1016/j.febslet.2005.12.049. [DOI] [PubMed] [Google Scholar]
- 72. Matsuno K, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709.. These two studies (references 71 and 72) emanating from different groups and published within 12 months of each other used novel strains of NOX1-deficient mice to provide the first definitive evidence that NOX1 oxidase is an important contributor to angiotensin II-dependent hypertension and its associated vascular pathologies.
- 73.Basset O, et al. NADPH oxidase 1 deficiency alters caveolin phosphorylation and angiotensin II-receptor localization in vascular smooth muscle. Antioxid. Redox Signal. 2009;11:2371–2384. doi: 10.1089/ars.2009.2584. [DOI] [PubMed] [Google Scholar]
- 74.Gavazzi G, et al. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension. 2007;50:189–196. doi: 10.1161/HYPERTENSIONAHA.107.089706. [DOI] [PubMed] [Google Scholar]
- 75.Chu X, et al. A critical role for chloride channel-3 (CIC-3) in smooth muscle cell activation and neointima formation. Arterioscler. Thromb. Vasc. Biol. 2011;31:345–351. doi: 10.1161/ATVBAHA.110.217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee MY, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szocs K, et al. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc. Biol. 2002;22:21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 78.Bengtsson SH, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin. Exp. Pharmacol. Physiol. 2003;30:849–854. doi: 10.1046/j.1440-1681.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- 79.Sheehan AL, Takapoo M, Banfi B, Miller FJJ. Role for Nox1 NADPH oxidase in atherosclerosis. Circulation. 2007;116:II_244. doi: 10.1016/j.atherosclerosis.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lyle AN, et al. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Judkins CP, et al. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H24–H32. doi: 10.1152/ajpheart.00799.2009.. This was the first study to directly implicate a specific isoform of NADPH oxidase, namely NOX2 oxidase, in the pathogenesis of atherosclerosis.
- 82.Wind S, et al. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56:490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 83.Basuroy S, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-α in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2009;296:C422–C432. doi: 10.1152/ajpcell.00381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Basuroy S, Tcheranova D, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase-derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-α-induced apoptosis. Am. J. Physiol. Cell Physiol. 2011;300:C256–C265. doi: 10.1152/ajpcell.00272.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Datla SR, et al. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007;27:2319–2324. doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 86.Petry A, et al. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid. Redox Signal. 2006;8:1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 87.Peshavariya H, et al. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch. Pharmacol. 2009;380:193–204. doi: 10.1007/s00210-009-0413-0. [DOI] [PubMed] [Google Scholar]
- 88.Pedruzzi E, et al. NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell Biol. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gordillo G, Fang H, Park H, Roy S. Nox-4-dependent nuclear H2O2 drives DNA oxidation resulting in 8-OHdG as urinary biomarker and hemangioendothelioma formation. Antioxid. Redox Signal. 2010;12:933–943. doi: 10.1089/ars.2009.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu RF, Ma Z, Liu Z, Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell Biol. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clempus RE, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007;27:42–48. doi: 10.1161/01.ATV.0000251500.94478.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sturrock A, et al. Nox4 mediates TGF-β1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L1543–L1555. doi: 10.1152/ajplung.00430.2006. [DOI] [PubMed] [Google Scholar]
- 93.Ismail S, et al. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takac I, et al. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Loot AE, Schreiber JG, Fisslthaler B, Fleming I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009;206:2889–2896. doi: 10.1084/jem.20090449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 2000;86:347–354. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- 97.Thomas SR, Chen K, Keaney JF., Jr Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J. Biol. Chem. 2002;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- 98.Matoba T, Shimokawa H. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J. Pharmacol. Sci. 2003;92:1–6. doi: 10.1254/jphs.92.1. [DOI] [PubMed] [Google Scholar]
- 99.Paravicini TM, Chrissobolis S, Drummond GR, Sobey CG. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35:584–589. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- 100.Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J. Cereb. Blood Flow Metab. 2006;26:836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- 101.Lambeth JD, Kawahara T, Diebold B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Banfi B, et al. A Ca2+-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200.. This study led to the discovery of the Ca2+-dependent NADPH oxidase NOX5.
- 103.Jay DB, et al. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic. Biol. Med. 2008;45:329–335. doi: 10.1016/j.freeradbiomed.2008.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guzik TJ, et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008;52:1803–1809. doi: 10.1016/j.jacc.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature Rev. Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 106.Segal AW. How neutrophils kill microbes. Annu. Rev. Immunol. 2005;23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid. Redox Signal. 2006;8:1549–1561. doi: 10.1089/ars.2006.8.1549. [DOI] [PubMed] [Google Scholar]
- 108.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kalinina N, et al. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2002;22:2037–2043. doi: 10.1161/01.atv.0000040222.02255.0f. [DOI] [PubMed] [Google Scholar]
- 110.Azenabor AA, Yang S, Job G, Adedokun OO. Elicitation of reactive oxygen species in Chlamydia pneumoniae-stimulated macrophages: a Ca2+-dependent process involving simultaneous activation of NADPH oxidase and cytochrome oxidase genes. Med. Microbiol. Immunol. 2005;194:91–103. doi: 10.1007/s00430-004-0223-4. [DOI] [PubMed] [Google Scholar]
- 111.Rosenblat M, Aviram M. Oxysterol-induced activation of macrophage NADPH-oxidase enhances cell-mediated oxidation of LDL in the atherosclerotic apolipoprotein E deficient mouse: inhibitory role for vitamin E. Atherosclerosis. 2002;160:69–80. doi: 10.1016/s0021-9150(01)00563-9. [DOI] [PubMed] [Google Scholar]
- 112.Harrison DG, Cai H, Landmesser U, Griendling KK. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2003;4:51–61. doi: 10.3317/jraas.2003.014. [DOI] [PubMed] [Google Scholar]
- 113.Bae YS, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res. 2009;104:210–218. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nature Immunol. 2004;5:818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 115. Guzik TJ, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657.. This was a seminal study showing that T cell-derived NOX2 oxidase activity is critical for the development of hypertension and endothelial dysfunction following chronic angiotensin II infusion in mice.
- 116.Brait VH, et al. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J. Cereb. Blood Flow Metab. 2010;30:1306–1317. doi: 10.1038/jcbfm.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kraaij MD, et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl Acad. Sci. USA. 2010;107:17686–17691. doi: 10.1073/pnas.1012016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke. 2009;40:S40–S44. doi: 10.1161/STROKEAHA.108.533638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marvar PJ, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ. Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harrison DG, Vinh A, Lob H, Madhur MS. Role of the adaptive immune system in hypertension. Curr. Opin. Pharmacol. 2010;10:203–207. doi: 10.1016/j.coph.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miller AA, et al. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: role of Nox2. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H220–H225. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- 122.Miller AA, Drummond GR, Schmidt HH, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 123.Girouard H, et al. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kishida KT, et al. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pao M, et al. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics. 2004;45:230–234. doi: 10.1176/appi.psy.45.3.230. [DOI] [PubMed] [Google Scholar]
- 126.Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J. Appl. Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 127.Miller AA, et al. Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke. 2010;41:784–789. doi: 10.1161/STROKEAHA.109.575365. [DOI] [PubMed] [Google Scholar]
- 128.Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J. Cereb. Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- 129. Walder CE, et al. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252.. This was the first demonstration that the absence of NOX2 oxidase in genetically modified mice confers protection against cerebral infarct development after experimental stroke.
- 130.Kleinschnitz C, et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8:e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jackman KA, Miller AA, Drummond GR, Sobey CG. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 2009;1286:215–220. doi: 10.1016/j.brainres.2009.06.056. [DOI] [PubMed] [Google Scholar]
- 132.Kahles T, et al. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol. Dis. 2010;40:185–192. doi: 10.1016/j.nbd.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 133.Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov. Today. 2006;11:524–533. doi: 10.1016/j.drudis.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 134.Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin. Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 135.Cave A. Selective targeting of NADPH oxidase for cardiovascular protection. Curr. Opin. Pharmacol. 2009;9:208–213. doi: 10.1016/j.coph.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 136.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 137.Wingler K, et al. NOX 1, 2, 4, 5: Counting out oxidative stress. Br. J. Pharmacol. 2011 Feb 16; doi: 10.1111/j.1476-5381.2011.01249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bhandarkar SS, et al. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Invest. 2009;119:2359–2365. doi: 10.1172/JCI33877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ding Y, et al. Inhibition of Nox-4 activity by plumbagin, a plant-derived bioactive naphthoquinone. J. Pharm. Pharmacol. 2005;57:111–116. doi: 10.1211/0022357055119. [DOI] [PubMed] [Google Scholar]
- 140.Cayatte AJ, et al. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets NADPH oxidase. Arterioscler. Thromb. Vasc. Biol. 2001;21:1577–1584. doi: 10.1161/hq1001.096723. [DOI] [PubMed] [Google Scholar]
- 141.Jaulmes A, et al. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thromb. Res. 2009;124:439–446. doi: 10.1016/j.thromres.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 142.Kalinowski L, Dobrucki IT, Malinski T. Race-specific differences in endothelial function: predisposition of African Americans to vascular diseases. Circulation. 2004;109:2511–2517. doi: 10.1161/01.CIR.0000129087.81352.7A. [DOI] [PubMed] [Google Scholar]
- 143.Zang M, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- 144.Stielow C, et al. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem. Biophys. Res. Commun. 2006;344:200–205. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 145.ten Freyhaus H, et al. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc. Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 146.Wind S, et al. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tsai MH, Jiang MJ. Reactive oxygen species are involved in regulating α1-adrenoceptor-activated vascular smooth muscle contraction. J. Biomed. Sci. 2010;17:67. doi: 10.1186/1423-0127-17-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Laleu B, et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 149.Gianni D, et al. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 2010;5:981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Borbely G, et al. Small-molecule inhibitors of NADPH oxidase 4. J. Med. Chem. 2010;53:6758–6762. doi: 10.1021/jm1004368. [DOI] [PubMed] [Google Scholar]
- 151.Banfi B, et al. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 152.Paffenholz R, et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004;18:486–491. doi: 10.1101/gad.1172504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cheranov SY, Jaggar JH. TNF-α dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am. J. Physiol. Cell Physiol. 2006;290:C964–C971. doi: 10.1152/ajpcell.00499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ris-Stalpers C. Physiology and pathophysiology of the DUOXes. Antioxid. Redox Signal. 2006;8:1563–1572. doi: 10.1089/ars.2006.8.1563. [DOI] [PubMed] [Google Scholar]
- 155. Kuhns DB, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097.. This study demonstrates that severe illness and poor long-term survival in patients with CGD is only evident in individuals whose phagocytic ROS production is more than two orders of magnitude lower than in healthy controls. This observation provides comfort that therapeutic targeting of NOX2 oxidase activity to alleviate vascular oxidative stress is unlikely to compromise systemic immune function.
- 156. Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8.. This was a study identifying three novel NOX homologues, two of which (NOX4 and NOX5) are likely to have important roles in vascular physiology and pathophysiology.
- 157.O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem. J. 1993;290:41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.O’Donnell VB, Smith GC, Jones OT. Involvement of phenyl radicals in iodonium inhibition of flavoenzymes. Mol. Pharmacol. 1994;46:778–785. [PubMed] [Google Scholar]
- 159.El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009;41:217–225. doi: 10.3858/emm.2009.41.4.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lassegue B, Griendling KK. Nox is playing with a full deck in vascular smooth muscle, a commentary on “Noxa1 is a central component of the smooth muscle NADPH oxidase in mice”. Free Radic. Biol. Med. 2006;41:185–187. doi: 10.1016/j.freeradbiomed.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 161.Finan P, Koga H, Zvelebil MJ, Waterfield MD, Kellie S. The C-terminal SH3 domain of p67phox binds its natural ligand in a reverse orientation. J. Mol. Biol. 1996;261:173–180. doi: 10.1006/jmbi.1996.0450. [DOI] [PubMed] [Google Scholar]
- 162.Finan P, et al. An SH3 domain and proline-rich sequence mediate an interaction between two components of the phagocyte NADPH oxidase complex. J. Biol. Chem. 1994;269:13752–13755. [PubMed] [Google Scholar]
- 163.Fuchs A, Dagher MC, Faure J, Vignais PV. Topological organization of the cytosolic activating complex of the superoxide-generating NADPH-oxidase. Pinpointing the sites of interaction between p47phoz, p67phox and p40phox using the two-hybrid system. Biochim. Biophys. Acta. 1996;1312:39–47. doi: 10.1016/0167-4889(96)00020-1. [DOI] [PubMed] [Google Scholar]
- 164.Hata K, Takeshige K, Sumimoto H. Roles for proline-rich regions of p47phox and p67phox in the phagocyte NADPH oxidase activation in vitro. Biochem. Biophys. Res. Commun. 1997;241:226–231. doi: 10.1006/bbrc.1997.7807. [DOI] [PubMed] [Google Scholar]
- 165.Wilson L, Butcher C, Finan P, Kellie S. SH3 domain-mediated interactions involving the phox components of the NADPH oxidase. Inflamm. Res. 1997;46:265–271. doi: 10.1007/s000110050185. [DOI] [PubMed] [Google Scholar]
- 166.Kami K, Takeya R, Sumimoto H, Kohda D. Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67phox, Grb2 and Pex13p. EMBO J. 2002;21:4268–4276. doi: 10.1093/emboj/cdf428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mizuki K, et al. A region C-terminal to the proline-rich core of p47phox regulates activation of the phagocyte NADPH oxidase by interacting with the C-terminal SH3 domain of p67phox. Arch. Biochem. Biophys. 2005;444:185–194. doi: 10.1016/j.abb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 168.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 169.Takeya R, et al. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J. Biol. Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 170.Yuzawa S, et al. Solution structure of the tandem Src homology 3 domains of p47phox in an autoinhibited form. J. Biol. Chem. 2004;279:29752–29760. doi: 10.1074/jbc.M401457200. [DOI] [PubMed] [Google Scholar]
- 171.Yuzawa S, et al. A molecular mechanism for autoinhibition of the tandem SH3 domains of p47phox, the regulatory subunit of the phagocyte NADPH oxidase. Genes Cells. 2004;9:443–456. doi: 10.1111/j.1356-9597.2004.00733.x. [DOI] [PubMed] [Google Scholar]
- 172. Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/s0092-8674(03)00314-3.. This study reported two crystal structures of the core of p47phox, both in its auto-inhibited form and in its activated form, bound to its cognate target peptide from the cytosolic portion of p22phox.
- 173.Hiroaki H, Ago T, Ito T, Sumimoto H, Kohda D. Solution structure of the PX domain, a target of the SH3 domain. Nature Struct. Biol. 2001;8:526–530. doi: 10.1038/88591. [DOI] [PubMed] [Google Scholar]
- 174.Marcoux J, et al. p47phox molecular activation for assembly of the neutrophil NADPH oxidase complex. J. Biol. Chem. 2010;285:28980–28990. doi: 10.1074/jbc.M110.139824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ago T, et al. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc. Natl Acad. Sci. USA. 2003;100:4474–4479. doi: 10.1073/pnas.0735712100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ago T, Nunoi H, Ito T, Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47phox. Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47phox, thereby activating the oxidase. J. Biol. Chem. 1999;274:33644–33653. doi: 10.1074/jbc.274.47.33644. [DOI] [PubMed] [Google Scholar]
- 177.Hoyal CR, et al. Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase. Proc. Natl Acad. Sci. USA. 2003;100:5130–5135. doi: 10.1073/pnas.1031526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Huang J, Kleinberg ME. Activation of the phagocyte NADPH oxidase protein p47phox. Phosphorylation controls SH3 domain-dependent binding to p22phox. J. Biol. Chem. 1999;274:19731–19737. doi: 10.1074/jbc.274.28.19731. [DOI] [PubMed] [Google Scholar]
- 179.Ximenes VF, Kanegae MP, Rissato SR, Galhiane MS. The oxidation of apocynin catalyzed by myeloperoxidase: proposal for NADPH oxidase inhibition. Arch. Biochem. Biophys. 2007;457:134–141. doi: 10.1016/j.abb.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 180. Sumimoto H, et al. Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc. Natl Acad. Sci. USA. 1994;91:5345–5349. doi: 10.1073/pnas.91.12.5345.. This paper was one of the first reports on the crucial roles of the interactions between SH3 domains and PRR for the association of p47phox with both p22phox in the membrane and p67phox in the cytosol.
- 181.Watanabe Y, et al. Molecular dynamics study on the ligand recognition by tandem SH3 domains of p47phox, regulating NADPH oxidase activity. Comput. Biol. Chem. 2006;30:303–312. doi: 10.1016/j.compbiolchem.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 182.Nobuhisa I, et al. Activation of the superoxide-producing phagocyte NADPH oxidase requires co-operation between the tandem SH3 domains of p47phox in recognition of a polyproline type II helix and an adjacent α-helix of p22phox. Biochem. J. 2006;396:183–192. doi: 10.1042/BJ20051899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Leto TL, Adams AG, de Mendez I. Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc. Natl Acad. Sci. USA. 1994;91:10650–10654. doi: 10.1073/pnas.91.22.10650.. This paper was also one of the first reports on the crucial roles of interactions between SH3 domains and PRRs for the association of p47phox with both p22phox in the membrane and p67phox in the cytosol.
- 184.Shi J, Ross CR, Leto TL, Blecha F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47phox. Proc. Natl Acad. Sci. USA. 1996;93:6014–6018. doi: 10.1073/pnas.93.12.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ogura K, et al. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006;281:3660–3668. doi: 10.1074/jbc.M505193200.. This study shows the three-dimensional NMR structure of one of the sites on p47phox that is crucial to activation of NADPH oxidase. In this article we have provided a working example of how this information, in conjunction with molecular modelling, virtual library screening and in vitro validation studies, may be used as a platform for the discovery of novel lead compounds.
- 186.DeLeo FR, et al. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc. Natl Acad. Sci. USA. 1995;92:7110–7114. doi: 10.1073/pnas.92.15.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.De Leo FR, Ulman KV, Davis AR, Jutila KL, Quinn MT. Assembly of the human neutrophil NADPH oxidase involves binding of p67phox and flavocytochrome b to a common functional domain in p47phox. J. Biol. Chem. 1996;271:17013–17020. doi: 10.1074/jbc.271.29.17013. [DOI] [PubMed] [Google Scholar]
- 188. DeLeo FR, et al. A domain of p47phox that interacts with human neutrophil flavocytochrome b558. J. Biol. Chem. 1995;270:26246–26251. doi: 10.1074/jbc.270.44.26246.. The previous two studies used phage display mapping to demonstrate that p47phox interacts directly with the NOX2 catalytic subunit in the assembled NADPH oxidase complex. Peptide mimetics of these sites of interaction were shown to be effective inhibitors of NADPH oxidase activity.
- 189.Rotrosen D, et al. Evidence for a functional cytoplasmic domain of phagocyte oxidase cytochrome b558. J. Biol. Chem. 1990;265:8745–8750. [PubMed] [Google Scholar]
- 190.Park MY, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Synthetic peptides corresponding to various hydrophilic regions of the large subunit of cytochrome b558 inhibit superoxide generation in a cell-free system from neutrophils. Biochem. Biophys. Res. Commun. 1997;234:531–536. doi: 10.1006/bbrc.1997.6672. [DOI] [PubMed] [Google Scholar]
- 191.DeLeo FR, Jutila MA, Quinn MT. Characterization of peptide diffusion into electropermeabilized neutrophils. J. Immunol. Methods. 1996;198:35–49. doi: 10.1016/0022-1759(96)00144-5. [DOI] [PubMed] [Google Scholar]
- 192.Dourron HM, et al. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- 193.Weaver M, et al. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am. J. Physiol. Heart Circ. Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 194.Cheng G, Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 2004;279:4737–4742. doi: 10.1074/jbc.M305968200. [DOI] [PubMed] [Google Scholar]
- 195.Cheng G, Lambeth JD. Alternative mRNA splice forms of NOXO1: differential tissue expression and regulation of Nox1 and Nox3. Gene. 2005;356:118–126. doi: 10.1016/j.gene.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 196.Ponting CP. Novel domains in NADPH oxidase subunits, sorting nexins, and PtdIns 3-kinases: binding partners of SH3 domains? Protein Sci. 1996;5:2353–2357. doi: 10.1002/pro.5560051122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kanai F, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nature Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 198.Karathanassis D, et al. Binding of the PX domain of p47(phox) to phosphatidylinositol 3,4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002;21:5057–5068. doi: 10.1093/emboj/cdf519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Goldblatt D, Thrasher AJ. Chronic granulomatous disease. Clin. Exp. Immunol. 2000;122:1–9. doi: 10.1046/j.1365-2249.2000.01314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Holland SM. Chronic granulomatous disease. Clin. Rev. Allergy Immunol. 2010;38:3–10. doi: 10.1007/s12016-009-8136-z. [DOI] [PubMed] [Google Scholar]
- 201.Winkelstein JA, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 202.Towbin AJ, Chaves I. Chronic granulomatous disease. Pediatr. Radiol. 2010;40:657–668. doi: 10.1007/s00247-009-1503-3. [DOI] [PubMed] [Google Scholar]
- 203.Yogi A, et al. Renal redox-sensitive signaling, but not blood pressure, is attenuated by Nox1 knockout in angiotensin II-dependent chronic hypertension. Hypertension. 2008;51:500–506. doi: 10.1161/HYPERTENSIONAHA.107.103192. [DOI] [PubMed] [Google Scholar]
- 204.Wang HD, et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res. 2001;88:947–953. doi: 10.1161/hh0901.089987. [DOI] [PubMed] [Google Scholar]
- 205.Carlstrom M, et al. Role of NOX2 in the regulation of afferent arteriole responsiveness. Am. J. Physiol. Regul. Integr Comp. Physiol. 2009;296:R72–R79. doi: 10.1152/ajpregu.90718.2008. [DOI] [PubMed] [Google Scholar]
- 206.Touyz RM, et al. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension. 2005;45:530–537. doi: 10.1161/01.HYP.0000158845.49943.5e. [DOI] [PubMed] [Google Scholar]
- 207.Bendall JK, et al. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007;100:1016–1025. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- 208.Landmesser U, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Miriyala S, et al. Bone morphogenic protein-4 induces hypertension in mice: role of noggin, vascular NADPH oxidases, and impaired vasorelaxation. Circulation. 2006;113:2818–2825. doi: 10.1161/CIRCULATIONAHA.106.611822. [DOI] [PubMed] [Google Scholar]
- 210.Kirk EA, et al. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2000;20:1529–1535. doi: 10.1161/01.atv.20.6.1529. [DOI] [PubMed] [Google Scholar]
- 211.Hsich E, et al. Vascular effects following homozygous disruption of p47phox: an essential component of NADPH oxidase. Circulation. 2000;101:1234–1236. doi: 10.1161/01.cir.101.11.1234. [DOI] [PubMed] [Google Scholar]
- 212. Barry-Lane PA, et al. p47phox is required for atherosclerotic lesion progression in ApoE−/− mice. J. Clin. Invest. 2001;108:1513–1522. doi: 10.1172/JCI11927.. This was the first study to provide definitive evidence that NADPH oxidase activity contributes to the development of atherosclerotic plaques.
- 213.Vendrov AE, et al. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler. Thromb. Vasc. Biol. 2007;27:2714–2721. doi: 10.1161/ATVBAHA.107.152629. [DOI] [PubMed] [Google Scholar]
- 214.Chen Z, et al. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc. Natl Acad. Sci. USA. 2004;101:13014–13019. doi: 10.1073/pnas.0405389101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J. Biol. Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 216.Simons JM, et al. Immunodulatory compounds from Picrorhiza kurroa: isolation and characterization of two anti-complementary polymeric fractions from an aqueous root extract. J. Ethnopharmacol. 1989;26:169–182. doi: 10.1016/0378-8741(89)90064-0. [DOI] [PubMed] [Google Scholar]
- 217.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am. J. Respir. Cell. Mol. Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 218.‘T Hart BA, Simons JM, Knaan-Shanzer S, Bakker NP, Labadie RP. Antiarthritic activity of the newly developed neutrophil oxidative burst antagonist apocynin. Free Radic. Biol. Med. 1990;9:127–131. doi: 10.1016/0891-5849(90)90115-y. [DOI] [PubMed] [Google Scholar]
- 219.Stuehr DJ, et al. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991;5:98–103. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- 220.Gatley SJ, Sherratt HS. The effects of diphenyleneiodonium and of 2,4-dichlorodiphenyleneiodonium on mitochondrial reactions. Mechanism of the inhibition of oxygen uptake as a consequence of the catalysis of the chloride/hydroxyl-ion exchange. Biochem. J. 1976;158:317–326. doi: 10.1042/bj1580317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cross AR, Parkinson JF, Jones OT. The superoxide-generating oxidase of leucocytes. NADPH-dependent reduction of flavin and cytochrome b in solubilized preparations. Biochem. J. 1984;223:337–344. doi: 10.1042/bj2230337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem. J. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Brandsch R, Bichler V. Studies in vitro on the flavinylation of 6-hydroxy-d-nicotine oxidase. Eur. J. Biochem. 1986;160:285–289. doi: 10.1111/j.1432-1033.1986.tb09969.x. [DOI] [PubMed] [Google Scholar]
- 224.Sedeek M, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2010;299:F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 225.Rossary A, Arab K, Steghens JP. Polyunsaturated fatty acids modulate NOX 4 anion superoxide production in human fibroblasts. Biochem. J. 2007;406:77–83. doi: 10.1042/BJ20061009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fan C, Katsuyama M, Nishinaka T, Yabe-Nishimura C. Transactivation of the EGF receptor and a PI3 kinase-ATF-1 pathway is involved in the upregulation of NOX1, a catalytic subunit of NADPH oxidase. FEBS Lett. 2005;579:1301–1305. doi: 10.1016/j.febslet.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 227.Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J. Biol. Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- 228.Rivera J, Sobey CG, Walduck AK, Drummond GR. Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox Rep. 2010;15:50–63. doi: 10.1179/174329210X12650506623401. [DOI] [PMC free article] [PubMed] [Google Scholar]