

Abstract
The objectives were to review available PK models for saturable FcRn-mediated IgG disposition, and to explore an alternative semimechanistic model. Most available empirical and mechanistic PK models assumed equal IgG concentrations in plasma and endosome in addition to other model-specific assumptions. These might have led to inappropriate parameter estimates and model interpretations. Some physiologically based PK (PBPK) models included FcRn-mediated IgG recycling. The nature of PBPK models requires borrowing parameter values from literature, and subtle differences in the assumptions may render dramatic changes in parameter estimates related to the IgG recycling kinetics. These models might have been unnecessarily complicated to address FcRn saturation and nonlinear IgG PK especially in the IVIG setting. A simple semimechanistic PK model (cutoff model) was developed that assumed a constant endogenous IgG production rate and a saturable FcRn-binding capacity. The FcRn-binding capacity was defined as MAX, and IgG concentrations exceeding MAX in endosome resulted in lysosomal degradation. The model parameters were estimated using simulated data from previously published models. The cutoff model adequately described the rat and mouse IgG PK data simulated from published models and allowed reasonable estimation of endogenous IgG turnover rates.
1. Introduction
Under physiological conditions, IgG has prolonged half-life (t1/2) relative to other plasma proteins, while IgG elimination is faster at much higher concentrations. These observations prompted Brambell et al. to propose a theoretical saturable protective Fc receptor in 1964 [1]. Subsequently, Brambell demonstrated that a neonatal intestinal receptor (FcRn) is responsible for maternal-to-infant IgG transport [2]. Waldmann and Strober later showed that FcRn preferentially binds to IgG Fc at lower pH [3]. The expression of FcRn in neonatal gut is transient [4], while in adults FcRn is expressed primarily in muscle, skin, vascular endothelium at relatively high levels [5–8]. The FcRn-mediated IgG recycling can be described as a 3-step process: (1) passive pinocytosis of IgG into endothelial cells; (2) acidification in endosome by H+ ATPase to approximately pH 6 allowing IgG binding to FcRn; (3) unbound IgG is destined to lysosomal degradation while bound IgG is diverted back to circulation. Fc-FcRn binding in endosome may change FcRn distribution and intracellular trafficking of FcRn-coated endosome vesicles [9]. This directed endosome trafficking mechanism may further explain the high efficiency of IgG recycling.
FcRn is similar to the MHC class I molecule, consisting of a three-domain alpha subunit coupled with β-2 microglobulin. Mutation of the FcRn β-2 microglobulin in mice (β2m −/−) led to approximately 8–15-fold increase in IgG clearance [4, 6, 10]. This more-than-8-fold lower IgG clearance due to FcRn protection implies that an IgG molecule undergoes multiple cycles of FcRn-mediated endosomal recycling before its degradation in lysosome in mice.
Quantitative understanding of IgG recycling has been evolving. The first attempts in the 1960s by Brambell et al. [1] and Waldmann and Strober [3] were empirical by correlating fraction catabolic rate with plasma IgG concentrations. In 1966, Wochner et al. studied IgG disposition in patients with myotonic dystrophy, a hereditary error resulting in plasma deficiency of IgG but not albumin [11]. In 1990, Waldmann and Terru published IgG disposition in two patients with familial hypercatabolic hypoproteinemia [12]. These data provided insightful human kinetic data of the impact of FcRn on IgG disposition. In 2006, Kim et al. developed a mechanistic PK model for FcRn-mediated IgG PK in human [13]. The paper was an elegant review of available quantitative human IgG data in this field; it was also the first attempt to quantitatively assess IgG homeostasis and its disturbance by genetic FcRn diseases. Despite the several identified gaps due to limited available experimental data [14], the proposed model seems to be so far the best mechanistic model in human.
Saturation of FcRn by intravenous administration of high doses of pooled immunoglobulin (IVIG) has been a successful approach to clear pathogenic autoimmune antibodies from circulation, especially in patients with immune thrombocytopenia (ITP) [15]. Several semimechanistic models have been proposed to describe IgG disposition in IVIG setting in experimental animal models [16–19]. These models are discussed in detail in this paper. Quantitative models in clinical IVIG settings have not been published.
In addition, physiologically based PK (PBPK) models have been developed [20–23]. The models included multiple organs/tissues of interest, investigated the kinetics and determinants of IgG tissue distribution, and allowed studying the nonlinear FcRn and IgG recycling kinetics. However, these models are complicated in nature, and interpretations of FcRn-mediated IgG recycling seem very different among the models.
A new semimechanistic PK model is proposed. In this model, the saturable FcRn binding in endosome and subsequent IgG degradation are captured using a simple cutoff approach. The model avoided several questionable assumptions in previous work and adequately described data simulated from a previously published rat and mouse IVIG model.
2. Methods
2.1. Source IgG PK Data for Model Fitting
The rat and mouse semimechanistic IgG PK models [24] developed by Hansen and Balthasar were reconstructed in NONMEM (version 6.2.0; Icon Development Solutions, Ellicott City, MD). Since Hansen's model was derived from [17, 25] and adequately described [24] the experimental data, Hansen's models were used to simulate (reconstruct) the rat and mouse PK data according to the designs of the original experiments. Intersubject variability was fixed to 0. The simulated data were then used as source data to estimate the PK parameters in the new proposed rat and mouse cutoff models.
The rat experimental designs were as follows. In Study 1, an IV bolus dose of a monoclonal antibody 7E3, a platelet-binding antibody, was given to rats at 0.8, 4, or 8 mg/kg. Plasma PK was determined at 1, 3, 6, 12, 24, 48, 72, 96, and 168 hours after dose. In Study 2, rats received a single high dose IVIG at 0.4, 1, or 2 g/kg, followed by a single IV bolus dose of 7E3 at 8 mg/kg. Plasma PK was determined at 1, 3, 6, 12, 24, 48, 96, and 168 hours after dose. The simulated PK profiles using the reconstructed Hansen's model looked identical to the published results [24]. Both 7E3 and endogenous IgG concentration-time profiles were simulated, but only the plasma 7E3 PK data was used to estimate the parameter values of the new models.
The mouse study was as follows: an IV bolus dose of 7E3 was given to FcRn wild-type (WT) mice with or without concomitant dosing of 1 g/kg IVIG. PK plasma samples were collected at 1 and 12 hour, and 1, 2, 4, 7, 14, 23, 30, and 60 days after dose. Similarly, an IV bolus dose of 7E3 was given to FcRn knockout (KO) mice with or without concomitant dosing of 1 g/kg IVIG. PK plasma samples were collected at 1 and 12 hour, and 1, 2, 3, and 4 days after dose. Note that the mouse PK sampling time points were estimated based on the published figures [24] and may be slightly different from the original designs.
2.2. Full Mechanistic PK Model
The concept of the model is presented in Figure 1. In the full model scheme (upper panel), IgG disposition involves 3 compartments, that is, a central (plasma) compartment, a peripheral (tissue) compartment, and an endosome compartment nested in the peripheral compartment. As in Hansen's paper, IgG of different sources (i.e., 7E3, IVIG, and endogenous) were assumed to follow the same disposition kinetics [24]. In addition to first-order distribution (Q) between the central and peripheral compartments and first-order elimination (CL) from the central compartment, IgGs in the peripheral compartment also undergo endosome circulation/degradation. This starts with a nonspecific uptake by pinocytosis of first-order kinetics (CLU), and followed by two transient steps governed by transit time τ1 and τ2. During τ1, the endosomal pH decreases to approximately 6 by H+ ATPase in parallel to IgG sorting when IgG molecules compete for FcRn. During τ2, unbound IgGs get degraded. The FcRn-bound IgG is then recycled to circulation. A convenient assumption was made that the endosome vesicle volume does not change from the initial pinocytosis to the final fusion to plasma membrane. This allowed inferring FcRn saturation and fraction of unbound drug (fU) using the total IgG concentration in the peripheral compartment. The total volume of endosome at a given time is determined by CLU and the transit time τ1 and τ2.
Figure 1.

The proposed cutoff PK models. The full physiological model scheme (upper panel) describes physical processes, including IgG distribution in central, peripheral, and endosome compartments, nonspecific elimination from central compartment (CL), and FcRn-mediated endosomal IgG recycling and elimination in the endosome. The endosome space is nested in the peripheral compartment. In the reduced cutoff PK model scheme (lower panel), the endosome compartment is collapsed. The net endosomal IgG clearance is determined by endosomal uptake clearance (CLU) from the peripheral compartment, the FcRn capacity (Max), the total IgG concentration in peripheral compartment (CP,T), and the unbound fraction (fU), where fU = (CP,T − Max)/CP,T if CP,T > Max; fU = 0 if CP,T < Max or in FcRn KO mice.
2.3. Reduced Semimechanistic PK Model (Cutoff Model)
It was reported that the endosome transit time is about 7 min [26, 27]. The whole recycling cycle can be instantaneously compared with the relatively slow elimination phase. For this reason, a simplified model was proposed where the endosome process collapsed into a nonlinear elimination directly from the peripheral compartment, that is, CLU × fU (Figure 1, lower panel).
The cutoff model has 6 compartments (CMTs). All the three sources of IgGs (i.e., 7E3, IVIG, and endogenous IgGs) were incorporated in the model and share the same PK parameters including CL, Q, CLU, the central compartment volume (VC = V1 = V3 = V5), and the peripheral compartment volume (VP = V2 = V4 = V6). In addition, it was assumed that kin is the same regardless of the FcRn status in mice.
A key assumption was made for the fU calculation. A hypothetical FcRn capacity parameter (Max) was proposed, fU = (CP,T − Max)/CP,T, where CP,T is the total IgG concentration in the peripheral compartments (CMTs 2, 4, and 6). When CP,T ≤ Max, fU = 0. In FcRn KO mice (i.e., MAX = 0), fU = 1. It was assumed that 7E3, IVIG, and endogenous IgGs have similar binding affinity for FcRn. In addition, instead of assuming that the microrate constants for endosome uptake (kup) and return rate (kret) are equal as in other semimechanistic models [13, 16–19], a constant intercompartment clearance was assumed where the pinocytosis uptake clearance (CLU) is the sum of endosome degradation clearance (CLU × fU) and the returning clearance (CLU × (1 − fU)).
The baseline endogenous plasma IgG concentrations in FcRn WT animals (C5,WT,0) were fixed to 103 μM and 14.7 μM for rats and mice, respectively [24], where the subscript 5 is the compartment number (Figure 1, lower panel), WT stands for FcRn WT, and 0 represents time 0 (baseline). Since the endosome recycling and elimination are linked to the peripheral compartments (CMTs 2, 4, and 6), C6,WT,0 may be lower than C5,WT,0 if fU > 0. As a result, conditional equations were derived to calculate secondary parameters, including C6,WT,0, the endogenous IgG production rate (kin), and baseline fU (fU,WT,0)
| (1) |
| (2) |
| (3) |
However, when C6,WT,0 ≤ MAX,
| (4) |
| (5) |
| (6) |
Note that (1)–(6) rely on the C5,WT,0 values reported in the literature with normal FcRn expressions. In order to allow the model to predict endogenous IgG levels in FcRn KO mice, baseline endogenous IgG levels need to be defined independent of C5,WT,0
| (7) |
| (8) |
| (9) |
where A5,0 and A6,0 are the endogenous IgG levels at baseline in the central and peripheral compartment, respectively.
In FcRn KO animals, MAX = 0 and the baseline can be simplified to the following:
| (10) |
| (11) |
| (12) |
When A6,0/VP ≤ MAX, there is no IgG elimination from the peripheral compartment, and the baseline condition is defined as follows:
| (13) |
The derivations of the above equations are provided in the appendix.
The PK of 7E3, IVIG, and endogenous IgGs were further defined by differential equations:
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
where CP,T = (A2 + A4 + A6)/VP, and fU = (CP,T − MAX)/CP,T.
2.4. Cutoff Model Parameter Estimation and Simulation
The models were coded in NONMEM (version 6.2.0) with FOCE method. A total of 6 model parameters (i.e., VC, VP, CL, Max, Q, and CLU) were estimated by fitting the model with the simulated rat or mouse 7E3 plasma PK data. A constant coefficient of variation error model was used. Model performance was assessed visually by diagnostic plots. The model was then used to simulate 7E3, IVIG, and endogenous IgG concentrations under the experimental conditions. A sensitivity test was conducted to assess the effects of Max and CLU on the 7E3 PK profile in the presence or absence of IVIG (2 g/kg) using the rat model.
3. Results
The parameter estimates of the reduced models are summarized in Table 1.
Table 1.
Summary of the cutoff PK model parameter estimates in rats and mice based on the PK data simulated by using Hansen's PK models.
| Parameter | Rat | Mouse | ||
|---|---|---|---|---|
| Estimate | %RSE | Estimate | %RSE | |
| V C (mL/kg) | 39.8 | 0.563% | 66.8 | 1.46% |
| Max (μM) | 108 | 0.0756% | 20.1 | 0.557% |
| Q (mL/d/kg) | 81.0 | 0.519% | 72.1 | 3.47% |
| CLU (mL/d/kg) | 2580 | 6.7% | 564 | 29.8% |
| V P (mL/kg) | 29.3 | 0.594% | 65.5 | 7.43% |
| CL (mL/d/kg) | 24.5 | 0.420% | 5.63 | 12.5% |
| C 0 (μM) | 103 (fixed) | — | 14.7 (fixed) | — |
| EPS | 0.000302 | 43.7% | 0.00289 | 68.5% |
In the mouse model, the MAX estimate is for FcRn WT mice; MAX is 0 in FcRn KO mice.
The rat cutoff model adequately described the simulated plasma 7E3 concentrations with or without IVIG using Hansen's model (Figures 2(a) and 2(b)). The estimated VC and VP values were 39.8 and 29.3 mL/kg, respectively, comparable to typical values for IgG reported in the literature [27]. The Cutoff Max was estimated to be 108 μM (16.2 mg/mL). With the endogenous IgG level of 103 μM, rat FcRn should not be saturated at baseline, that is, fU = 0. CLU, CL, and Q were estimated to be 2580, 24.5, and 81.0 mL/d/kg, respectively. The high CLU value was somewhat unexpected, but physiologically it may make sense because a big component of the empirical peripheral compartment is comprised of endothelial cells expressing FcRn. As schematically shown in Figure 1, the nested structure and the fast CLU suggest high intracompartment trafficking rate and endosomal recycling rate. Since FcRn was not saturated at baseline, the zero-order kin was estimated to be 2.52 μmol/d/kg (or 378 μg/d/kg) using (5) instead of (2). All disposition PK parameters were estimated with good precisions (%RSE ≤ 6.7%). However, cautions should be used in interpreting the %RSE values because of the use of simulated data in the model fitting.
Figure 2.

Cutoff model fitting of plasma 7E3 PK data that were simulated using Hansen's models. (a) Rat 7E3 PK data fitting at indicated dose levels without IVIG. (b) Rat 7E3 PK data fitting when 8 mg/kg 7E3 were dosed concomitant with IVIG of indicated dose levels. (c) Mouse 7E3 PK data fitting when 8 mg/kg 7E3 was given to FcRn WT or KO mice, with or without 1 g/kg IVIG.
The plasma and peripheral concentration profiles of 7E3, IVIG, and endogenous IgGs were simulated using the rat cutoff model (Figure 3). Similar to plasma 7E3 PK profiles, the peripheral 7E3 exposures were lower with the increasing IVIG. The IVIG exposures increased at higher dose levels, with the highest plasma IVIG IgG concentration of 336 μM following 2 g/kg IVIG, approximately 3.3 times the endogenous level. This increased the total IgG concentration above MAX and was the driver for the nonlinear 7E3 PK. The endogenous IgG PK was minimally impacted by 7E3 alone (8 mg/kg), but decreased following IVIG treatment in an IVIG-dose-dependent manner, reaching maximum suppression of 13%, 28%, and 42% with 0.4, 1, and 2 g/kg IVIG, respectively. The extent of endogenous IgG suppression was similar to the values estimated using Hansen's model (differences within ±2%) [24].
Figure 3.

Simulated PK profiles using the rat cutoff model. All rats receive 8 mg/kg 7E3, as well as IVIG at indicated dose levels. Plasma and peripheral concentrations of 7E3, IVIG IgG, and endogenous IgG are plotted.
Similarly, the mouse cutoff model adequately described the simulated plasma 7E3 concentrations using Hansen's model (Figure 2(c)), with or without IVIG, and in FcRn WT or KO mice. The estimated VC and VP values were 66.8 and 65.5 mL/kg, respectively, larger than the estimates in the rat cutoff model. The CLU, CL, and Q values were 564, 5.63, and 72.1 mL/d/kg, respectively, similar to the rat estimates. FcRn was not expected to be saturated at the baseline as MAX was estimated to be 20.1 μM, higher than the mouse baseline endogenous IgG concentration of 14.7 μM. Consistent with Hansen's model, the levels of 7E3 were eliminated monoexponentially in FcRn KO mice regardless of IVIG dose as IgGs were no longer protected by FcRn (Figure 2(c)). The endogenous IgG production rate was estimated to be 0.0827 μmol/d/kg. All PK parameters were estimated with reasonable precisions except Q that had a %RSE of 29.8% (Table 1).
The mouse cutoff model was then used to simulate the plasma and peripheral concentration profiles of 7E3, IVIG, and endogenous IgG under the experimental conditions (Figure 4). Similar to plasma 7E3 PK profiles, the peripheral 7E3 exposures were lower with IVIG in FcRn WT mice. The highest plasma IVIG IgG concentration following 1 g/kg IVIG was approximately 100 μM (7.8 times the endogenous level), thus transiently decreased the endogenous IgG level by up to 62%. In FcRn KO mice, IgG is not protected by FcRn (MAX = 0 and fU = 1) while kin remained the same as in FcRn WT mice. As a result, the plasma endogenous IgG levels at the baseline were 14.7 and 1.19 μM in WT and KO mice, respectively.
Figure 4.

Simulated PK profiles using the mouse cutoff model. All mice receive 8 mg/kg 7E3 with or without concomitant 1 g/kg IVIG in either FcRn WT or KO mice. Plasma and peripheral concentrations of 7E3, IVIG IgG, and endogenous IgG are plotted.
Sensitivity test was conducted with the rat model to evaluate the effects of Max and CLU on plasma 7E3. In the absence of IVIG, the plasma 7E3 level increased with increasing Max when Max was less than total IgG level, but remained superimposable as Max was ≥108 μM and fU = 0 (Figure 5). Concomitant 2 g/kg IVIG potentiated the effect of Max, and plasma 7E3 levels increased with the increasing Max throughout the tested Max range. On the other hand, changes in CLU did not show appreciable impact on 7E3 PK profiles (data not shown), indicating that CLU could not be well estimated. This is consistent with the higher %RSE values for CLU in both species (Table 1) and is likely due to lack of informative data about endosomal IgG elimination kinetics.
Figure 5.

Sensitivity tests for Max. The effect of different MAX values on the plasma 7E3 pharmacokinetics was simulated in the presence or absence of IVIG (2 g/kg) using the rat PK model. The tested range was 0.1- to 2-fold the estimated Max (108 μM).
In most published models, the FcRn-IgG binding and FcRn saturation were based on either multiexponent functions [1, 3, 20], Langmuir-type binding isotherm, and quadratic equation solution of unbound concentration [16, 19, 24], or Michaelis-Menten kinetics [13]. In this proposed model, a simplified cutoff concept was introduced to describe the saturable FcRn-mediated kinetics. The relationship between the model estimated fU and total peripheral IgG concentration is plotted in Figure 6. Under the simulated experimental conditions, the fU ranged from 0 to 0.076 in rats, 0 to 0.25 in FcRn WT mice, and remained 1 in FcRn KO mice. For both species (FcRn WT), the cutoff model estimated fU-total peripheral IgG concentration profiles are in parallel with the fU-total plasma IgG concentration profiles estimated using Hansen's models. The difference is largely due to the different model structures and the additional nonspecific linear IgG elimination (CL) in the cutoff models which is absent in Hansen's models.
Figure 6.
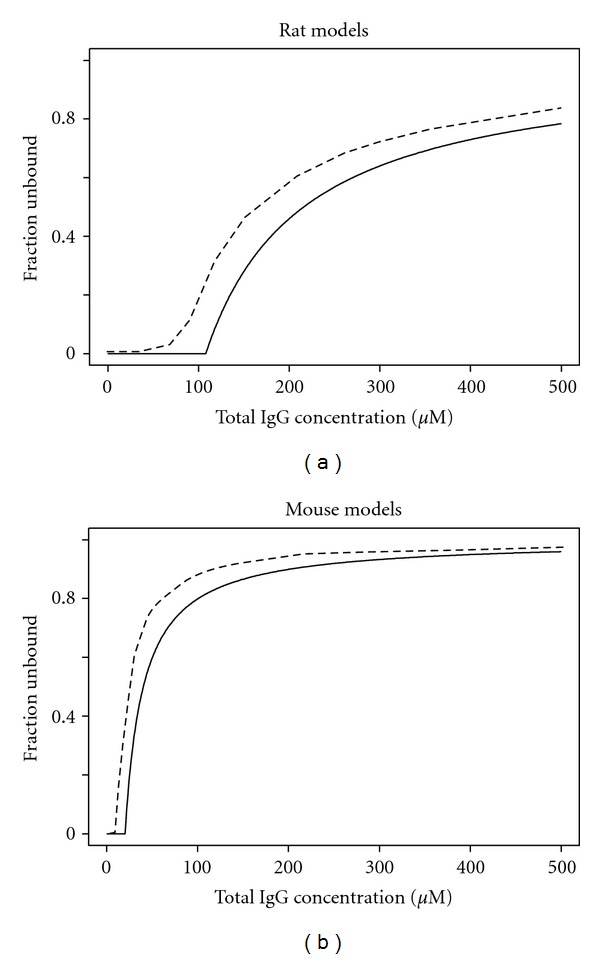
Relationship between fU and total peripheral IgG concentration (cutoff models, solid lines) or total plasma IgG concentration (Hansen's models, dashed lines). The fU values for Hansen's models were calculated according to the published models [4]. The fU values for the cutoff models were based on the model parameters in Table 1.
4. Discussion
4.1. Bleeker's Model [28]
The relationship between fraction clearance rate (FCR) and plasma IgG concentration was described by exponential equations in both human and mice using previously published data. This approach was empirical and did not allow physiological interpretation such as recycling rate and efficiency [13]. A mammillary PK model coupled with the predicted FCR was used for simulation and the results were consistent with IgG t1/2 under physiological conditions and some clinical data. However, it underestimated IVIG effect on autoantibody suppression and overpredicted IgG exposures in FcRn KO model [18].
4.2. Hansen's Model [24]
The model was comprised of the central and endosomal compartments (Figure 7(a)). It considered endogenous IgG, autoantibody (7E3), and IVIG simultaneously. The strengths of the model include simplicity, mechanistic basis, and ability to adapt to different situations. The model adequately described 7E3 PK data in normal and FcRn KO mice, and 7E3 PK data in rats with or without IVIG. The model was also used to infer the total FcRn-binding capacity and endogenous IgG turnover rate. However, the assumption of kup = kret is questionable. At steady-state in endosome, C1 × kup = CE × kret(1 − fU) + CE × kdeg × fU. It implied that V1 = VE when the equation was to be scaled from concentration to amount. This may not be likely as it was estimated by the same group later that endosomal space volume is approximately 5 mL/kg in mice [21], much smaller than the plasma volume of 50 mL/kg [29].
Figure 7.

Published semimechanistic PK models. (a) Hansen's model; (b) Xiao's model; (c) Kim's model. Models schemes are redrawn based on published papers.
According to Hansen's model, it appears that the endosome compartment has dual functions, that is, to explain the saturable elimination of IgG and to serve as a distribution compartment. The PK profiles of 7E3 in the absence of IVIG in the central and the endosome compartments look like typical 2-compartment PK model [24]. In the presence of IVIG, the PK profiles of 7E3 were lowered due to FcRn saturation [24]. While it is theoretically sound and practically convenient to assume that IgG concentration in the early endosome is the same as that in the circulation, dual functions of the endosome compartment had implication on the fU estimation. To illustrate this, two sets of fU values were calculated using the plasma or endosome total IgG concentrations simulated with Hansen's model, and plotted in Figure 8. The significant discrepancies and hysteresis indicate that it may not be appropriate to infer FcRn saturation using plasma total IgG concentrations under the particular model assumptions.
Figure 8.

Comparison of fU values derived from IgG concentrations in endosome and plasma using Hansen's rat model after a single IV dose of 8 mg/kg 7E3 in the presence or absence of IVIG. Significant counter clockwise hysteresis was observed due to IgG distribution delays. In the absence of IVIG, the line visually collapsed into a short line at the ending point of the counter-clockwise hysteresis loop as the lack of significant change in total IgG level kept fU relatively constant.
In addition, fU was first calculated with the total plasma IgG concentration under the assumption that the total concentrations in plasma and endosome were the same. On the other hand, the steady-state endosome IgG concentration was redefined differently based on endosomal mass balance of IgG at steady state. These different definitions and empirical assumptions reflect the lack of informative experimental data of the endosome compartment and casts doubt in the interpretation of the model-derived parameter values. For example, the rat endogenous IgG production rate was estimated to be 62.9 μmol/d/kg in Hansen's model based on steady-state condition [24], while it was reported that the endogenous IgG CL in rats is only 1.9 μmol/d/kg (ranged from 1 to 2.9 μmol/d/kg) following a dose of 131I-labeled rat IgGs [30].
Since the only mechanism of IgG elimination is through the endosome pathway (Figure 6(a)) in Hansen's model, the total FcRn-binding capacity (Rt) had to be smaller than the endogenous levels of IgG in order to describe the PK data (i.e., Rt = 83 μM and 12.2 μM for mice and rats, resp.) [24]. In other words, the model structure predetermined that FcRn had to be saturated at physiological condition. Furthermore, simulations using the mouse model showed an accumulation of IgGs in the endosomal compartment in FcRn KO mice, and the same endogenous IgG concentrations at the baseline between FcRn WT and KO mice (data not shown). These indicate inappropriate model assumptions, and cautions should be used when interpreting the modeling results.
An improved PK/PD model was developed to describe the IVIG effect on the disposition of MWReg30, an anti-platelet antibody, and on MWReg30-induced thrombocytopenia in mice [16]. The PK model was similar to Hansen's model except that the PK data of both MWReg30 and IVIG were available, and the differential equations were written with respect to amounts of MWReg30 and IVIG as opposed to concentrations in Hansen's model. This avoided some of the confusion of distribution volume in Hansen's model. However, the differential equations for endogenous IgGs were still written with respect to concentration. This was probably for model simplification purpose and due to the lack of endogenous IgG PK data. Although this hybrid model offered some improvement, it was still subject to similar concerns.
4.3. Xiao's Model [19]
Another mechanistic FcRn-IgG model was developed by Xiao et al. recently [19] based on total hepcidin and an antihepcidin mAb (Ab 12B9m) concentrations (unbound + complex) in cynomolgus monkeys after single or multiple doses of Ab 12B9m (Figure 7(b)). Higher clearance of Ab 12B9m was observed after multiple weekly IV and SC doses of 300 mg/kg Ab 12B9m and was attributed to FcRn saturation. In addition, target-mediated IgG disposition (TMDD) was incorporated in order to infer the unbound hepcidin and unbound Ab 12B9m concentrations.
The model was a modified version of Hansen's model by inheriting the assumption of Langmuir-type FcRn-IgG interaction and kup = kret. The model had a peripheral compartment, so that the endosomal compartment did not have to bear the dual functions as discussed in Hansen's model. Because it included unbound Ab 12B9m and its complex with hepcidin, endogenous IgG was not considered in the model to minimize the model complexity. As a result, interpretation of FcRn saturation should be viewed with caution. Another difference was that the endosome compartment volume (VE) was included in the model to allow a better sense of mass transfer. However, the endosomal volume VE and the dissociation constant kD for FcRn-IgG binding were estimated as a product rather than separately since they operated in the model only as a product.
4.4. Kim's Model [13]
A mechanism-based PK model for human IgG was developed based on saturable kinetics and data from the literature using easily measurable plasma concentrations (Figure 7(c)). This appeared to be the first paper that addressed FcRn-mediated IgG recycling kinetics quantitatively in human. Note that this model was designed for endogenous IgG only although it may be adapted for therapeutic IgG and/or IVIG. All parameter values, with exception of km, were borrowed from the literature. It was recognized that the plasma concentration was not identical too but as an index for endosomal unbound IgG concentration. In addition, as an improvement from previous models, kup = kret was no longer assumed.
In this model, the rate constants for IgG internalization, catabolism, and recycling were defined as kint, kcat, and krmr, respectively. Because the plasma compartment (V1) and the endosome compartment (VE) were collapsed as the vascular space in the simplified model, the IgG concentrations became the same (CSS). Although the kint retained its physiological meaning to describe the IgG catabolism and trafficking within vascular space, that is, kint = kcat + krmr, according to equations Jrmr = Jmax × CSS/(km + CSS) and Jcat = kcat × Css × V1, the recycling rate Jrmr and the catabolic rate Jcat were directly linked to the vascular space, and no longer rely on kint despite of the sequential events as shown in the model scheme (Figure 7(c)). As a result, kint is not kinetically informed by other processes in the model, and obtaining kint value solely relied on the literature. This might be a problem especially when the values of most parameters were borrowed from literature where different physiological and/or mechanistic assumptions might have been made. Probably because of this, the model did not seem to be able to explain the high fractional catabolic rate of IgG in patients with familial hypercatabolic hypoproteinemia.
In addition, as the author pointed out, the estimated rate of pinocytosis (kup) was smaller in magnitude than the real unidirectional uptake rate, so the daily fractional uptake rate (18%), the fractional recycling rate (10.6%), and the fraction catabolic rate (7.4%) should be interpreted with cautions. Indeed, the inferred efficiency of FcRn-mediated IgG recycling (~60%) was lower than what can be inferred from the IgG PK data from two reported cases of familial hypercatabolic hypoproteinemia with non-functional FcRn due to a mutant β2m gene. In these patients with normal IgG synthesis rates, IgG survival was short. The IgG fractional catabolic rates increased fivefold to 31% and 36% of the intravenous pool per day (normal 6.7 ± 2%/d) [12]. This approximate 5-fold increase in FCR suggested that under normal physiological conditions, an average IgG molecule is degraded after approximately 5 times endothelial pinocytosis, or approximately 80% efficiency of FcRn-mediated IgG recycling.
Another difference between Kim's model and previously discussed models was the empirical Emax model for FcRn-IgG binding. It was equivalent to early regression models by Brambell and Waldmann [1, 3]. km was defined as the serum IgG concentration at which a half maximal IgG recycling was achieved and was estimated to be 21 mg/mL. Choosing the total serum IgG concentration for FcRn-binding estimation might have been a convenient approximation; however, cautions should be used when comparing results from models with different structures.
4.5. Physiological Models
Several physiologically based PK (PBPK) models were developed for anticarcinoembryonic antigen (CEA) antibodies [20, 22] or 7E3 [21] in mice. The models included organs/tissues of interest, lymphatic circulation, FcRn-IgG binding, and IgG-CEA binding (if applicable). Most physiological parameters regarding plasma flow rate, vascular, interstitial, and total tissue volume were available from the literature. Lymphatic circulation rate was either assumed [21] or estimated [20, 22]. Endosome update and return rates were either fixed to be the same value [22] or estimated [20, 21]. In general, the models described/predicted the anti-CEA antibody PK reasonably well. However, depending on the model assumptions, the FcRn or endosome-related parameter values might be very different. For example, Ferl's estimations of kint and krec were 1.38 day−1 and 0.5 day−1, respectively [20], while in Garg's model, the values were 1.96 day−1 and 20.4 day−1, respectively [21, 24]. In a recent revision of the model, target-mediated disposition was included in addition to the FcRn-IgG binding, where kint and krec were assumed to be the same and estimated to be 0.715 day−1 [22]. In addition, the percentage and mechanism of IgG elimination in each organ were interpreted differently. According to Ferl's model, approximately 40% of IgG eliminated was attributed to linear elimination kinetics in liver and other organs, while in Garg's model, skin, muscle, liver, and gut were the major organs responsible for 33, 24, 16, and 12% of total IgG elimination, respectively.
It is well recognized that the PBPK models have the potential to better describe the physiological processes, and thus may allow more meaningful interpretations. On the other hand, it should be noted that these models usually have complicated structures and heavily depend on available data from the literature. The complexity as an effort to best describe the physiological processes came with the need for more sophisticated modeling assumptions. Multiplicity of assumptions and potential error prorogation may lead to liabilities to model misspecification and result in misinterpretation. The routinely performed “model validation” using a small experimental data set might not be sufficient for model testing and assumption justification, as the inherent model flexibility may sometimes allow apparent good data fitting even when some inappropriate assumptions were made.
4.6. The Proposed Semimechanistic Cutoff Model
It has been reported that some IgGs with higher affinity for FcRn showed prolonged t1/2 [31, 32]. However, affinity for FcRn may not be the only major contributor to IgG catabolic rate [33]. Physiological PK models have been proposed, including the one in Figure 1(a), to depict the saturable FcRn-mediated IgG kinetics based on known physiology. However, in a strict sense, quantitative physiological models have not been published. The reason might be twofold. First, there is insufficient understanding of the intracellular events and difficulty in scaling data from in vitro to in vivo (e.g., volume, rate, and transit time). In addition, there is insufficient or uninformative in vivo PK data to develop and differentiate such PK models. The simulated data using Hansen's model was easily described by the new cutoff model. The model parameters were estimated fairly precisely (Table 1). However, other models with different structural assumptions might be able to describe the data equally well. To better test a model, additional PK data are required, for example, PK of total IgG.
It is fair to say that none of the available models (including the cutoff model) adequately mimicked the physiology of FcRn-mediated IgG recycling, and more sophisticated physiological models cannot be developed simply because the lack of informative experimental data in the endosome compartment. For example, the rate of pinocytosis at a whole body level is difficult to measure experimentally. In addition, although the mean transit time of endosome was inferred to be about 7 min [26, 27], it remains largely unknown how endosome is destined to lysosome kinetically and how IgG degradation changes as a function of the mean transit. For example, it was reported that FcRn-IgG binding in endosome triggers more efficient intracellular trafficking and transcytosis [9], but the quantitative impact on the endosomal recycling is not clear. And the dynamics of pH lowering in early endosome is not easy to describe mathematically. Furthermore, it was realized that FcRn saturation estimated based on plasma IgG levels may not approximate the real situation in endosome as discussed for Hansen's model. The technical difficulties in estimating the parameters governing the FcRn-mediated IgG recycling process prevent developing models that truly resembles the physiology.
Taken together, it might be practically justified to utilize simple models of FcRn-mediated IgG disposition such as the cutoff model, especially in the IVIG setting, and leave more sophisticated models for later optimization when cumulative understanding of this physiological process allows so.
The proposed cutoff model has only 6 parameters (Table 1). The distribution volume parameters VC and VP were clearly defined. The collapsed IgG recycling was based on the relative short endosome mean transit time, and the assumption that FcRn-IgG dissociation did not happen, or more appropriately, only happened to a constant extent. It allows estimation of nonspecific linear CL and nonlinear endosomal elimination. At baseline level in both rats and FcRn WT mice, the linear CL accounted for 100% of endogenous IgG elimination. The apparent nonspecific linear CL at baseline should be further discussed. Mathematically, CL accounted for the linear portion of IgG elimination, but it did not necessarily imply endosome-unrelated processes. For example, a fraction of IgG in endosome could end up in the lysosome degradation pathway even when FcRn is not saturated. This is possible considering variability in the biological processes such as the endosome acidification dynamics, transit time of endosome intracellular trafficking, and conversion to lysosome, as well as proteolysis kinetics. At a whole-body level, the above could have led to an apparent nonspecific linear. It is also important to realize that although several organs and tissues have been identified to express FcRn, the expression levels may be different. It has been reported that the dispersed pinocytotic activities of virtually all cells capture and process all soluble plasma proteins at a rate of ≈2x per day with equivalent degradative rates unless they are protected by specific mechanisms [4]. This high pinocytosis rate not only explains the nonspecific linear CL, but also is consistent with the high CLU values in both species estimated by the cutoff models (Table 1).
In rats, the estimated production rate of endogenous IgGs in rats was 2.52 μmol/d/kg using the cutoff model, slightly lower than the measured value of 1.9 μmol/d/kg (ranged 1 to 2.9 μmol/d/kg) [30]. In FcRn WT mice, the endogenous production rate was estimated to be 0.0827 μmol/d/kg, lower than previously reported value 0.132 μmol/d/day (assumed 50 mL/kg plasma) [4, 34]. Apparently, there is room for further model improvement and more precise parameter estimation (e.g., CLU) with emerging experimental data and better understanding of the physiology. However, it is important to note there has been a big improvement from in the previous model [24], where the kin estimates were 62.9 and 1.21 μmol/d/kg in rats and FcRn WT mice, respectively.
Different plasma endogenous IgG levels were reported between rats (103 μM) and mice (14.7 μM) at the baseline [24]. According to the cutoff model results, FcRn was almost saturated in rats (MAX = 108 μM) at the baseline but not in FcRn WT mice (MAX = 20.1 μM). In addition, rats seemed to have higher CL and CLU than mice according to the cutoff models (Table 1). These differences translated to an estimated 30-fold faster kin in rats than in mice versus approximately a 14-fold difference reported in the literature [4, 30, 34]. Further investigation would help understand the between-species difference in endogenous IgG turnover.
According to the cutoff model, FcRn should not be saturated at baseline, and the baseline total IgG clearance was determined solely by the nonspecific linear CL. The total body IgG clearance started to increase when the total peripheral IgG concentration exceeded MAX. In both rats and FcRn WT mice, the 7E3 plasma exposure decreased with the concomitant IVIG (Figures 2(b) and 2(c)). This is different from Hansen's model which predicted FcRn saturation at the baseline [24]. In addition, the mouse cutoff model is able to predict the endogenous IgG concentrations for FcRn WT and KO mice. The estimated baseline endogenous IgG concentrations in plasma were estimated to be 14.7 and 1.19 μM for FcRn WT and KO mice, respectively. This 12-fold decrease in the baseline IgG level reflects accelerated IgG catabolism in the absence of FcRn, consistent with reported values of 8–15-fold [4, 6, 10]. On the other hand, Hansen's model infers that the baseline endogenous IgG level remains the same in FcRn WT and KO mice [24].
In conclusion, A simple cutoff model was proposed for IgG PK modeling with saturable FcRn-mediated IgG disposition in rats and mice.
Acknowledgment
The author is an employee of Genentech Inc. and has no conflict of interests to declare.
Appendix
Detailed Derivation of Equations
(1) Derivation of secondary PK parameters with the baseline plasma endogenous IgG concentration in rats and FcRn WT mice.
Based on mass balance of endogenous IgGs between CMT5 and CMT6, C5,WT,0 × Q = C6,WT,0 × Q + C6,WT,0 × CLU × fU, where fU = (C6,WT,0 − MAX)/C6,WT,0.
So C5,WT,0 × Q = C6,WT,0 × Q + CLU × (C6,WT,0 − MAX),
If C6,WT,0 > MAX,
then C6,WT,0 = (C5,WT,0 × Q + CLU × MAX)/(Q + CLU) and fU,WT,0 = (C6,WT,0 − MAX)/C6,WT,0
If C6,WT,0 ≤ MAX, then C6,WT,0 = C5,WT,0 and fU,WT,0 = 0.
For endogenous IgGs at baseline, production rate = elimination rate,
| (A.1) |
If C6,WT,0 > MAX, kin = C5,WT,0 × CL + CLU × (C6,WT,0 − MAX);
If C6,WT,0 ≤ MAX, kin = C5,WT,0 × CL.
(2) Derivation of baseline endogenous IgG levels. The key is to allow model prediction of baseline plasma and peripheral IgG levels in both FcRn WT and KO animals.
With mass balance at baseline,
| (A.2) |
Solve the above equations for A5,0/VC and A6,0/VP,
| (A.3) |
If A6,0/VP > MAX,
then A6,0/VP = (kin × Q + CLU × CL × MAX + CLU × Q × MAX)/(Q × CL + CLU × CL + CLU × Q),
and A5,0/VC = (kin − CLU × (A6,0/VP − MAX))/CL.
If A6,0/VP ≤ MAX,
then A6,0/VP = kin × Q/(Q × CL) = kin/CL,
and A5,0/VC = kin/CL.
In FcRn KO mice, A6,0/VP and A5,0/VC do not need to be separately coded. Instead, an NONMEM input data entry has FCRN = 0 for KO mice or FCRN = 1 for WT mice. MAX is defined as MAX0 × FCRN, where MAX0 is the MAX in FcRn WT mice. So MAX = 0 for KO mice, and A6,0/VP and A5,0/VC equations can be simplified as
| (A.4) |
| (A.5) |
References
- 1.Brambell FWR, Hemmings WA, Morris IG. A theoretical model of γ-globulin catabolism. Nature. 1964;203(4952):1352–1355. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- 2.Brambell FW. The transmission of immunity from mother to young and the catabolism of immunoglobulins. The Lancet. 1966;2(7473):1087–1093. doi: 10.1016/s0140-6736(66)92190-8. [DOI] [PubMed] [Google Scholar]
- 3.Waldmann TA, Strober W. Metabolism of immunoglobulins. Progress in Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- 4.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the β2-microglobulin-containing neonatal intestinal transport receptor. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(11):5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borvak J, Richardson J, Medesan C, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. International Immunology. 1998;10(9):1289–1298. doi: 10.1093/intimm/10.9.1289. [DOI] [PubMed] [Google Scholar]
- 6.Ghetie V, Hubbard JG, Kim JK, Tsen MF, Lee Y, Ward ES. Abnormally short serum half-lives of IgG in β2-microglobulin-deficient mice. European Journal of Immunology. 1996;26(3):690–696. doi: 10.1002/eji.1830260327. [DOI] [PubMed] [Google Scholar]
- 7.Campbell RM, Cuthbertson DP, Matthews CM, McFarlane AS. Behaviour of 14C- and 131I-labelled plasma proteins in the rat. The International Journal of Applied Radiation and Isotopes. 1956;1(1-2):66–84. doi: 10.1016/0020-708x(56)90020-5. [DOI] [PubMed] [Google Scholar]
- 8.McFarlane AS. The behavior of I 131-labeled plasma proteins in vivo. Annals of the New York Academy of Sciences. 1957;70(1):19–25. doi: 10.1111/j.1749-6632.1957.tb35374.x. [DOI] [PubMed] [Google Scholar]
- 9.Ramalingam TS, Detmer SA, Martin WL, Bjorkman PJ. IgG transcytosis and recycling by FcRn expressed in MDCK cells reveals ligand-induced redistribution. The EMBO Journal. 2002;21(4):590–601. doi: 10.1093/emboj/21.4.590. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack β2-microglobulin: possible protective role of FcRn. Immunology. 1996;89(4):573–578. doi: 10.1046/j.1365-2567.1996.d01-775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wochner RD, Drews G, Strober W, Waldmann TA. Accelerated breakdown of immunoglobulin G (IgG) in myotonic dystrophy: a hereditary error of immunoglobulin catabolism. The Journal of Clinical Investigation. 1966;45(3):321–329. doi: 10.1172/JCI105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waldmann TA, Terru WD. Familial hypercatabolic hypoproteinemia. A disorder of endogenous catabolism of albumin and immunoglobulin. The Journal of Clinical Investigation. 1990;86(6):2093–2098. doi: 10.1172/JCI114947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clinical Immunology. 2007;122(2):146–155. doi: 10.1016/j.clim.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurbaxani B. Mathematical modeling as accounting: predicting the fate of serum proteins and therapeutic monoclonal antibodies. Clinical Immunology. 2007;122(2):121–124. doi: 10.1016/j.clim.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Imbach P, Barandun S, d'Apuzzo V, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. The Lancet. 1981;1(8232):1228–1231. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- 16.Deng R, Balthasar JP. Pharmacokinetic/pharmacodynamic modeling of IVIG effects in a murine model of immune thrombocytopenia. Journal of Pharmaceutical Sciences. 2007;96(6):1625–1637. doi: 10.1002/jps.20828. [DOI] [PubMed] [Google Scholar]
- 17.Hansen RJ, Balthasar JP. Pharmacokinetics, pharmacodynamics, and platelet binding of an anti-glycoprotein IIb/IIIa monoclonal antibody (7E3) in the rat: a quantitative rat model of immune thrombocytopenic purpura. Journal of Pharmacology and Experimental Therapeutics. 2001;298(1):165–171. [PubMed] [Google Scholar]
- 18.Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Human Immunology. 2005;66(4):403–410. doi: 10.1016/j.humimm.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 19.Xiao JJ, Krzyzanski W, Wang YM, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS Journal. 2010;12(4):646–657. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferl GZ, Wu AM, DiStefano JJ. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn) Annals of Biomedical Engineering. 2005;33(11):1640–1652. doi: 10.1007/s10439-005-7410-3. [DOI] [PubMed] [Google Scholar]
- 21.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. Journal of Pharmacokinetics and Pharmacodynamics. 2007;34(5):687–709. doi: 10.1007/s10928-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 22.Urva SR, Yang VC, Balthasar JP. Physiologically based pharmacokinetic model for T84.66: a monoclonal anti-CEA antibody. Journal of Pharmaceutical Sciences. 2010;99(3):1582–1600. doi: 10.1002/jps.21918. [DOI] [PubMed] [Google Scholar]
- 23.Davda JP, Jain M, Batra SK, Gwilt PR, Robinson DH. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. International Immunopharmacology. 2008;8(3):401–413. doi: 10.1016/j.intimp.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen RJ, Balthasar JP. Pharmacokinetic/pharmacodynamic modeling of the effects of intravenous immunoglobulin on the disposition of antiplatelet antibodies in a rat model of immune thrombocytopenia. Journal of Pharmaceutical Sciences. 2003;92(6):1206–1215. doi: 10.1002/jps.10364. [DOI] [PubMed] [Google Scholar]
- 25.Hansen RJ, Balthasar JP. Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood. 2002;100(6):2087–2093. [PubMed] [Google Scholar]
- 26.Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. Journal of Cell Biology. 1983;97(2):508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. Journal of Immunology. 2004;172(4):2021–2029. doi: 10.4049/jimmunol.172.4.2021. [DOI] [PubMed] [Google Scholar]
- 28.Bleeker WK, Teeling JL, Erik Hack C. Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood. 2001;98(10):3136–3142. doi: 10.1182/blood.v98.10.3136. [DOI] [PubMed] [Google Scholar]
- 29.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharmaceutical Research. 1993;10(7):1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 30.Cure SF, Cremer NE. Decreased rate of synthesis of immunoglobulin (IgG) in rats infected with moloney leukemia virus. Journal of Immunology. 1969;102(6):1345–1353. [PubMed] [Google Scholar]
- 31.Ghetie V, Popov S, Borvak J, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nature Biotechnology. 1997;15(7):637–640. doi: 10.1038/nbt0797-637. [DOI] [PubMed] [Google Scholar]
- 32.Hinton PR, Johlfs MG, Xiong JM, et al. Engineered human IgG antibodies with longer serum half-lives in primates. The Journal of Biological Chemistry. 2004;279(8):6213–6216. doi: 10.1074/jbc.C300470200. [DOI] [PubMed] [Google Scholar]
- 33.Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Molecular Immunology. 2006;43(9):1462–1473. doi: 10.1016/j.molimm.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 34.Junghans RP. IgG biosynthesis: no ‘immunoregulatory feedback’. Blood. 1997;90(10):3815–3818. [PubMed] [Google Scholar]