

SUMMARY
The cytotoxic activities of five new benzopyranone derivatives containing basic amino side chain are described. Their cytotoxicities against ER (+) MCF-7 and ER (−) MDA-MB-231 human breast cancer cell lines, and Ishikawa human endometrial cell line were determined after 72 h drug exposure employing CellTiter-Glo assay at concentrations ranging from 0.01 – 1.0 × 105 nM. The antiproliferative activities of these compounds were compared to tamoxifen (TAM), 4-hydroxytamoxifen (4-OHT, active metabolite of tamoxifen) and raloxifene (RAL). In vitro results indicated that compounds 9, 10, 12 and 13 were more potent than TAM against the human breast cancer cell lines with IC50 < 20 µM. The in silico structure-activity relationships of these compounds and their binding mode within the estrogen receptor (ER) binding site using AutoDock vina are discussed.
Keywords: Coumarin, estrogen receptors, anticancer activity, antiproliferative agents, cytotoxic activity
INTRODUCTION
Breast cancer is the second leading cause of cancer death in American women behind lung cancer [1]. Postmenopausal women, whose production of ovarian estrogen has ceased with remaining estrogens originating in extra-glandular tissues, accounted for approximately 80% of breast cancer cases [2]. It has been reported that about one-third of postmenopausal breast cancer patients have hormone-dependent tumors, which involve the stimulation of cancer cell proliferation by estrogens [3]. Therapeutic agents, which prevent the biosynthesis (e.g. aromatase inhibitors) and physiological action of estrogen on tumor cells (e.g. Selective Estrogen Receptor Modulators, SERMs), represent a new and very successful approach for the treatment of postmenopausal women with hormone-dependent breast tumors [4]. Currently, one way of blocking the estrogen action on tumor cells is preventing the binding of estrogen to estrogen receptor (ER) by using an antiestrogen compound capable of blocking the effects of 17β-estradiol (E2) [5–7].
Antiestrogens exhibit antitumor effect by displaying an antagonist action at the ER, and are widely used in the treatment of hormone-dependent ER (+) breast cancer [6, 7]. SERMs are antiestrogenic compounds possessing high affinity for ER. They are classified as ER agonists or antagonists based on conformational changes of the receptors, particularly at the helix 12 (H-12) [8, 9]. The nature of the amine-bearing side chain (dialkylamoinoalkoxy group) and its orientation relative to the ligand backbone play an important role in the determination of SERMs’ tissue-selective activity by preventing the proper positioning of H-12 for agonistic activity [10–13]. In this antagonist conformation, the basic amino side chain forces H-12 to move into a position occluding a hydrophobic pocket before coactivators can bind and produce a transcription complex, an interaction that is important in mediating agonist activity [14, 15]. Clinically, SERMs belong to two chemical families of non-steroidal compounds, triphenylethylenes e.g. TAM 1 and benzothiophenes e.g. RAL 2 (Fig. 1) with basic amino side chains [16, 17]. TAM is an antiestrogen drug currently used as adjuvant chemotherapy for the treatment of ER (+) breast cancer, particularly in the post-menopausal women [18–20]. TAM behaves as ER antagonist in the breast tissue and as ER agonist in bone, and has prophylactic use in breast cancer [21]. Although TAM has been very successful in breast cancer treatment, its agonistic effect on the uterus is said to be associated with increased risk of developing endometrial cancer [22]. Thus, alternative and similar effective drugs for the treatment of breast cancer are being sought.
Figure 1.

Structures of Tamoxifen 1, Raloxifene 2, Coumarin-E2 conjugate 3, Coumarin-based benzopyranone, SP500263 4 and 7-methoxy-3-phenyl-4-phenylvinyl benzopyranone derivative 5.
Most recently, our group has been interested in the coumarins (2H-l-benzopyran-2-one) ring system 3 (Fig. 1) as a core pharmacophore of potential therapeutic agents for the treatment of hormone-dependent breast cancer [23]. Coumarin derivatives have been found to be useful in photochemotherapy, antitumor, anti-HIV therapy [24, 25] central nervous system (CNS) stimulants [26], antibacterial [27, 28], anti-inflammatory [29], anti-coagulants [30] and dyes [31]. In breast cancer cells, in particular, numerous coumarins have shown interesting pharmacological activities including binding affinities for ERs, antiproliferative activities, and inhibitory activities against sulfatase and aromatase enzymes. For example, SP500263 4 (Fig. 1), a new category of SERM called a benzopyranone molecule or coumarin-based SERM, produced similar effects as the ideal SERMs like TAM [32, 33]. It binds with high affinity to ERα, functions as potent antiestrogen in in vivo models of breast cancer, and potently inhibits estrogen-dependent MCF-7 proliferation with similar IC50 value to TAM [33, 34]. Compound 4 possessing an amine-bearing side chain attached to coumarin has a different core structural feature with respect to other SERMs currently on the market or in clinical development [34, 35]. Furthermore, recent investigation involving another type of benzopyranone compounds as ER ligands revealed that compound 5 and analogs possess both estrogen agonistic and antagonistic activities, and demonstrate significant in vitro antiproliferative activity with moderate in vivo anti-implantation activity [36].
We herein report the in vitro antiproliferative activity of new benzopyranone derivatives containing basic amino side chain 9–13 (Table 1) against ER (+) MCF-7 and ER (−) MDA-MB-231 human breast cancer cell lines, and Ishikawa human endometrial adenocarcinoma cell line. These cell lines are widely accepted models for assessing potent antiproliferative and antiestrogenic agents including SERMs. An in silico analysis of these antiproliferative agents for their relative binding affinities (RBAs) to the ERs using molecular modeling studies has also been reported here. TAM, 4-OHT and RAL were used as standards for comparison purposes in these studies.
Table 1.
IC50 values (µM) for compounds 9–13 tested against MCF-7 (ER+) and MDA-MB231 (ER−) human breast cancer cell lines, and Ishikawa human endometrial cells.
| Compounds | MCF-7 | IC50 values (µM) MDA-MB231 |
Ishikawa |
|---|---|---|---|
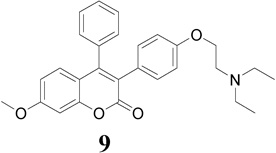 |
10.9 | 13.9 | 40.6 |
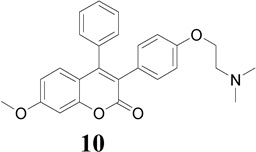 |
16.5 | 12.3 | 18.3 |
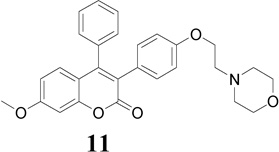 |
48.3 | 33.5 | 41.8 |
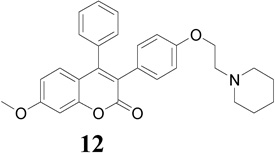 |
10.1 | 17.0 | 26.2 |
 |
10.3 | 12.4 | 15.6 |
| RAL-HCl | 0.66 | 32.8 | 28.0 |
| 4-OHT | 1.2 | 7.8 | 8.3 |
| TAM | 26.2 | 18.7 | 33.0 |
The data represent the average of triplicate determinations at various concentrations.
The IC50 values were determined from the graphs (GraphPad Prism) using mean values of data points at various concentrations.
RESULTS AND DISCUSSION
Cytotoxicity
Compounds 9–13 were evaluated for in vitro antiproliferative activity against ER (+) MCF-7 and ER (−) MDA-MB-231 human breast cancer cell lines and Ishikawa human endometrial cell line at concentration range 0.01 to 1.0 × 105 nM in the presence of 10 nM estradiol (E2) using CellTiter-Glo assay. Cell proliferation involved measuring the light emitted during the bioluminescence reaction of luciferine in the presence of ATP and luciferase. As shown in Table 1 and Fig. 2, compounds 9, 12 and 13 (IC50 = 10.9, 10.1 and 10.3 µM, respectively) demonstrated significant antiproliferative activity against ER (+) MCF-7 human breast cancer cell line in comparison to TAM (IC50 = 26.2 µM), but were less potent than 4-OHT and RAL (IC50 = 1.2 and 0.66 µM, respectively) (Note: IC50 is the concentration of test drug where a 50% reduction is observed in cell growth compared to the untreated control after a 72 h period of exposure to test drug). E2 at physiological concentration (10 nM) was used to do competitive growth inhibitory studies. The results showed that compounds 9, 12 and 13 may act as ER antagonists in the context of human breast cancer cell and compound 12 possessing piperidinylethoxy side chain exhibits the most potent antiproliferative activity in MCF-7 cell line in comparison to TAM (Fig. 2a).
Figure 2.



In vitro antiproliferative activity of (a) compound 12 against MCF-7 (ER+) cell line, (b) compound 10 against MDA-MB-231 (ER−) cell line and (c) compound 13 against Ishikawa human endometrial cell line; in comparison to TAM, 4-OHT and RAL
The antiproliferative mechanism of TAM in ER (+) MCF-7 breast cancer cells is mainly related to the inhibition of E2 binding to the ER, however ER-independent mechanisms have also been suggested [37, 38]. The ER (−) MDA-MB-231 breast cancer cell line constitutes an original model for identifying the ER-independent mechanisms of TAM antiproliferative effects [39, 40]. Thus, in the present study the antiproliferative activity of compounds 9–13 against ER (−) MDA-MB-231 human breast cancer cell lines were also investigated to shed some light on their mechanism of action. The results showed that compounds 9, 10, 12 and 13 (IC50 = 13.9, 12.3, 17.0 and 12.4 µM, respectively; Table 1) were more potent in this cell line than TAM (IC50 = 18.7 µM) and RAL (IC50 = 32.8 µM), but less potent than 4-OHT (IC50 = 7.8 µM). Furthermore, compound 10 possessing dimethylaminoethoxyl side chain exhibits the most potent antiproliferative activity in comparison to TAM and RAL, suggesting an additional ER-independent mechanism (Fig. 2b).
In the development of antiestrogenic SERM drugs, it is critical that the candidate drug does not cause estrogenic stimulation of the uterus, which could lead to both increase in uterine bleeding and an increased risk of developing uterine cancer. The Ishikawa cell line is an endometrial adenocarcinoma cell line that expresses functional estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ) isofomers [41]. In the present investigation, evaluation of the antiproliferative activity of these compounds on the uterus using the Ishikawa human endometrial cell line revealed that compounds 10 and 13 (IC50 = 18.2 and 15.6 µM, respectively; Table 1) were more potent than TAM (IC50 = 32.9 µM) and RAL (IC50 = 26.1 µM), but less potent than 4-OHT (IC50 = 8.3 µM). Compound 13 possessing pyrrolidinoethoxyl side chain exhibits the most potent antiproliferative activity in the presence of E2 (Fig. 2c).
Molecular Modeling Studies
The X-ray co-crystal structure of the ER-ligand binding domain (LBD) has provided a significant understanding of the ER binding site [42]. In the present study, we examined various binding modes of compounds 9–13 in the receptor of antagonist ERα-4-OHT complex (PDB code 3ERT) and ERβ-RAL complex (PDB code 1QKN) using AutoDock Vina. The best poses (Fig. 3) retrieved through re-docking with AutoDock Vina were almost identical with the original poses of the cognate ligands with the root mean square deviation (rmsd) values between the two poses for both 4-OHT and RAL being <2Å, a criterion often used for the correct bound structure prediction [43]. This led us to conclude that the predicted binding modes from AutoDock Vina can be used reliably as a docking tool in our modeling studies. Then, the optimized structures of compounds 9–13 were docked in the ligand’s binding pocket followed by the determination of the binding affinity.
Figure 3.
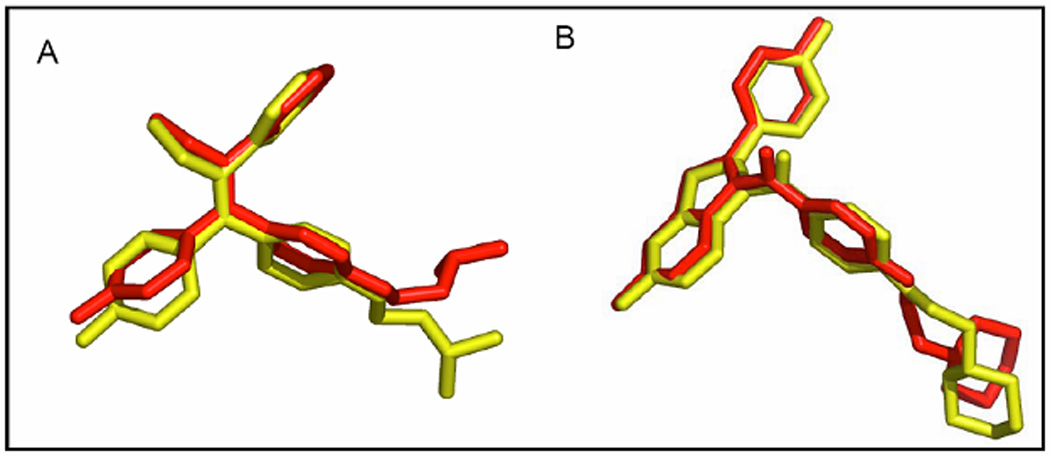
Validation of AutoDock Vina against: A) ERα-4-OHT and B) ERβ-RAL complex.
The triad of Glu 353, Arg 394 and buried water as well as His 524 in the ERs forms the basis of the favorable binding interaction for ER binding pocket. This sort of interaction normally shifts H-12 into a position antiparallel to H-11 (Met 517-Met 528), thereby sealing the ligand in a hydrophobic core and exposing the LXXLL cofactor-binding motif on the receptor surface [44]. The docking results indicated that compounds 9–13 showed a favorable binding affinity toward antagonist ERα (3ERT) binding mode than antagonist ERβ (1QKN) binding mode (Table 2). Furthermore, in the ERα (3ERT) receptor active site compound 9 (Ki = 0.06 µmol) exhibits higher binding affinity and compound 13 (Ki = 0.11 µmol) exhibits comparable binding affinity; in comparison with compounds 10–12 and E2 (Table 2). However, these binding affinities are considerably lower than those of 4-OHT, TAM and RAL.
Table 2.
Binding affinities of the compounds with ERα and ERβ
| ERα (3ERT) | ERβ (1QKN) | |||
|---|---|---|---|---|
| Compound | ΔGbinding (kcal/mol) |
Ki (µmol) |
ΔGbinding (kcal/mol) |
Ki (µmol) |
| 9 | −9.85 | 0.06 | −7.7 | 2.27 |
| 10 | −9.0 | 0.252 | −7.5 | 3.18 |
| 11 | −8.7 | 0.419 | −7.8 | 1.91 |
| 12 | −9 | 0.252 | −7.2 | 5.27 |
| 13 | −9.5 | 0.11 | −7.0 | 7.39 |
| 4-OHT | −11.5 | 0.004 | −9.06 | 0.228 |
| E2 | −9.5 | 0.11 | −8.5 | 0.587 |
| TAM | −10.0 | 0.047 | −8.1 | 1.15 |
| RAL | −11.5 | 0.004 | −10.38 | 0.025 |
The best-docked position of compounds 9 and 13 in the receptor of 3ERT (Fig. 4a) revealed the expected classical ER binding mode. However, a closer inspection of the ER binding pocket of compound 9 revealed that this compound does not exhibit the classical ER binding mode associated with agonists and antagonist compounds i.e. there is no hydrogen bonding interaction between the methoxy group, water, Glu 353, Arg 394 and His 524 (Fig. 4a). This lack of such hydrogen bonding is perhaps largely compensated by hydrophobic interaction. For example, recent investigation of benzochromen-6-one analogs as selective ERβ agonists revealed that the presence of methyl group could allow steric clashes with residues in helix 3 (such as Ala 350), which could prevent hydrogen-bonding interaction with the usual Glu/Arg pair [45]. Based on this result, when we replaced the methoxy group with hydroxyl group in compound 9, hydrogen bonding with Arg 394 and Glu 353 in the receptor of 3ERT (ERα) was observed with the eventually predicted ΔGbinding value of −10.2 kcal/mol (equivalent to 0.033 µM). This finding proved that the presence of the methoxy group was responsible for the lack of the observed hydrogen-bonding interaction with water, Glu 353, Arg 394 and His 524. The aromatic rings and the coumarin moieties are largely accommodated by a hydrophobic cavity lined by residues notably Leu 391, Ala 350, Ile 424, Gly 521, Leu 524, Met 343 and Thr 347 (Fig. 4b). However, the protonated (at physiological pH 7.4) amino nitrogen atom of the basic side chain of compounds (e.g. 9) seems to engage in hydrogen bonding interaction with a buried water molecule that is hydrogen bonded to Thr 347 (Fig. 4b). Kekenes-Huskey and colleagues using computational docking methods have suggested this alternative mode of ER-ligand binding through hydrogen bonding interaction involving the amino nitrogen atom of imidazoles, imidazolines and piperazines with Thr 347 (residue 399 for ERβ) as opposed to His 524 [46]. Most recently, Shankar et al. have also shown such hydrogen bonding interaction involving Thr 347 [47], which was also observed in the present studies.
Figure 4.

Best docked position of; a) compounds 9 and 13, and b) compound 9 in the ligand-binding pocket of ERα (PDB 3ERT).
Conclusions
The evaluation of a new group of benzopyranone compounds 9–13 with basic amino side chain as antiproliferative agents have been described. Our studies have shown that addition of basic amino side chains to the coumarin structure widely affect the activity of the molecules, as further demonstrated in this investigation. The in vitro results indicated that compounds 9, 10, 12 and 13 possessing amine groups: piperidine, dialkylamine and pyrrolidine moieties are among the best side chain for antagonist potency against ER (+) MCF-7, ER (−) MDA-MB-231 and Ishikawa human cancer cell lines. The antiproliferative activities of these compounds were compared with TAM, RAL and 4-OHT. Among the synthesized compounds, compound 13 exhibited the highest antiproliferative activity against ER (+) MCF-7 (hormone-dependent) and ER (−) MDA-MB231 (hormone-independent) cancer cell lines as well as Ishikawa human endometrial cell line as evident by lowest IC50 value, in comparison with TAM.
Experimental Section
The Southern Research Institute (SRI) screening procedures [48]
The cytotoxic activity of compounds 9–13 was evaluated at the Southern Research Institute (SRI, Birmingham, Alabama, USA). The compounds were screened against ER (+) MCF-7 and ER (−) MDA-MB-231 human breast cancer cell lines and Ishikawa human endometrical cell line in comparison to TAM, 4-OHT and RAL.
Cytotoxicity Studies
MCF-7 and MDA-MB-231 human breast cancer cell lines were purchased from the NCI. The Ishikawa human endometrial cancer cell line was purchased from Sigma. All three-cell lines were cultured in phenol red-free RPMI-1640 (Hyclone) (500 mL) supplemented with L-glutamine-dipeptide (Hyclone) (5 mL), and 10% fetal bovine serum (Atlanta Biologicals) (50 mL). All three cell lines were maintained in exponential growth phase by sub-culturing twice weekly in 150-cm2 flasks at 37 °C, 95% air with 5% CO2. The media was removed from the flasks, the cells washed with phosphate buffer solution (PBS) (Hyclone), and cells were then detached using 5 mL of TryplExpress solution (Invitrogen) (incubation 5–10 minutes) followed by the addition of growth media. Cells were centrifuged (1,500 rpm) for 5 minutes, and resuspended in growth media at 105 cells per mL. To each well of 96-well microplate, 50 µL of cell suspension was added at final density of 5,000 cells per well. After the cells were allowed to attach and grow overnight, 25 µL of 40 nM estradiol (Sigma) was added to each well, followed by 25 µL of the different drugs treatments (compounds 9–13, and TAM, 4-OHT and RAL) ranging from 400,000 nM to 0.04 nM, resulting in a final concentration of 10 nM estradiol in every well, and drug treatments ranging from 0.01 – 100,000 nM. Incubation lasted for 3 days at 37 °C air with 5% CO2 followed by CellTiter-Glo assay (Promega). All of the compounds were tested in triplicate for each cell lines. The results expressed as IC50 (Inhibitory concentration of 50%) were the averages of three data points for each concentration and were calculated using GraphPad Prism 4.0.
Molecular Modeling Studies
Preparation of proteins
Crystal structures of ligand binding domains of human estrogen receptor α (PDB code: 3ERT complexed with 4-hydroxytamoxifen) and β (PDB code: 1QKN, complexed with RAL) were obtained from the protein databank. The water molecules except those within 4.5 Å of the SERM (4-OHT or RAL) binding site were removed and missing hydrogens were added to the protein structures using the Reduce algorithm implemented in the MolProbity server (http://molprobity.biochem.duke.edu/). The latter allows optimization of local H-bonding environment avoiding steric clashes and also necessary correction for misorientation of Asn, Gln and His residues [45]. For further structural refinement, the protein structures together with the associated SERMs were then energy minimized using GROMOS96 force field implemented in Swiss-PdB viewer (Deep View). The associated SERM molecules were later removed from the corresponding energy-minimized protein structures to which the Gasteiger partial atomic charges [49] were added using the AutoDockTool (ADT) [50]. The prepared structures were eventually used as input files for the docking.
Preparation of ligands
The 3D structures of the benzopyranone derivatives containing basic side chain were drawn in ChemBioDraw Ultra 11.0 module of the ChemBioOffice 2008 package. All the structures were then energy-minimized in ChemBio3D ultra 11.0 using MM2 force field. Later, using ADT, the Gasteiger partial atomic charges were added and all flexible torsions were defined for the ligands. The prepared structures were used as input files for the docking.
Docking
Flexible-ligand docking was carried out using AutoDock Vina [51]. Vina uses a sophisticated gradient optimization method in its local optimization procedure and a modified X-score-based scoring function [51]. For this, a grid box of 60 × 60 × 60 number of points (grid spacing of 0.375 Å) was built around the ligand-binding region of the protein chains. Several key residues known to be involved in SERM binding (e.g. Asp 351, Glu 353, Arg 394, Thr 347 and His 524) were held flexible during the docking. From the estimated free energy of ligand binding (ΔGbinding, kcal/mol), the inhibition constants (Ki) was calculated taking into consideration of the dissociation of the enzyme inhibitor complex using basic thermodynamics formula (Arrhenius equation) of ΔG = RT ln Ki. Only the best pose (the one with the lowest ΔGbinding) is considered. The docking was run on windows operating environment implemented on a PC with an Intel dual core processor and the docked structures shown in PyMol.
Acknowledgements
Faculty Research Development Funds (NIH/NCRR grant G12 RR0 3020, NIH/NCRR grant 1 C06 RR12512-01 and NIH/DHHS grant 1 S11 ES011182 01) is gratefully acknowledged for financial support. The authors also gratefully acknowledge Dr. David H. Powell of the Mass Spectrometry Services, Department of Chemistry at University of Florida for assistance with Mass Spectra analysis.
Footnotes
Conflict of Interest: There is no conflict of interest associated with this publication.
References
- 1.Boyle P. Breast. 2005;14:429–438. doi: 10.1016/j.breast.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Woo LL, Purohit A, Malini B, Reed MJ, Potter BV. Chem. Biol. 2000;7:773–791. doi: 10.1016/s1074-5521(00)00023-5. [DOI] [PubMed] [Google Scholar]
- 3.Henderson IC, Canellos GP. N. Engl. J. Med. 1980;302:17–30. doi: 10.1056/NEJM198001033020104. [DOI] [PubMed] [Google Scholar]
- 4.Hamelers IH, Van Schaik RF, Sussenbach JS, Steenbergh PH. Cancer Cell. Int. 2003;3:1–10. doi: 10.1186/1475-2867-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Early Breast Cancer Trialists' Collaborative Group. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 6.Coombes RC, Gibson L, Hall E, Emson M, Bliss J. J. Steroid. Biochem. Mol. Biol. 2003;86:309–311. doi: 10.1016/s0960-0760(03)00372-8. [DOI] [PubMed] [Google Scholar]
- 7.Johnston S. Br. J. Cancer. 2004;90(Suppl.1):S15–S18. doi: 10.1038/sj.bjc.6601632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taras TL, Wurz GT, DeGregorio MW. J. Steroid Biochem. Mol. Biol. 2001;77:271–279. doi: 10.1016/s0960-0760(01)00066-8. [DOI] [PubMed] [Google Scholar]
- 9.Draper MW. Eur. J. Cancer. 2002;38(Suppl. 6):S35. doi: 10.1016/s0959-8049(02)00278-2. [DOI] [PubMed] [Google Scholar]
- 10.Wallace OB, Bryant HU, Shetler PK, Adrian MD, Geiser AG. Bioorg. Med. Chem. Lett. 2004;14:5103–5106. doi: 10.1016/j.bmcl.2004.07.072. [DOI] [PubMed] [Google Scholar]
- 11.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Embo. J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 13.Kim YW, Mobley JA, Brueggemeier RW. Bioorg. Med. Chem. Lett. 2003;13:1475–1478. doi: 10.1016/s0960-894x(03)00132-x. [DOI] [PubMed] [Google Scholar]
- 14.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis JS, Jordan VC. Mutat. Res. 2005;591:247–263. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 16.Muchmore DB. Oncologist. 2000;5:388–392. doi: 10.1634/theoncologist.5-5-388. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein SR, Siddhanti S, Ciaccia AV, Plouffe L., Jr. Hum. Reprod. Update. 2000;6:212–224. doi: 10.1093/humupd/6.3.212. [DOI] [PubMed] [Google Scholar]
- 19.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 19.Jordan VC. Cancer. 1992;70:977–982. [PubMed] [Google Scholar]
- 20.Seeger H, Huober J, Wallwiener D, Mueck AO. Horm. Metab. Res. 2004;36:277–280. doi: 10.1055/s-2004-814480. [DOI] [PubMed] [Google Scholar]
- 21.Phillips DH, Venitt S. Lancet. 1993;341:1485–1486. doi: 10.1016/0140-6736(93)90934-9. [DOI] [PubMed] [Google Scholar]
- 22.Jordan VC. Nat Rev Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 23.Musa MA, Cooperwood JS, Khan OFM. Letters in Drug Design & Discovery. 2009;6:133–138. doi: 10.2174/157018009787582624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nofal ZM, El-Zahar MI, AbdEl-Karim SS. Molecules. 2000;5:99–113. [Google Scholar]
- 25.Xie L, Takeuchi Y, Cosentino LM, Lee KH. Bioorg. Med. Chem. Lett. 1998;8:2151–2156. doi: 10.1016/s0960-894x(98)00367-9. [DOI] [PubMed] [Google Scholar]
- 26.Moffet RS. J. Med. Chem. 1964;7:446–449. doi: 10.1021/jm00334a010. [DOI] [PubMed] [Google Scholar]
- 27.Al-Haiza MA, Mostafa MS, El-Kady MY. Molecules. 2003;8:275–286. [Google Scholar]
- 28.Musiciki B, Periers AM, Laurin P, Ferroud D, Benedetti Y, Lachaud S, Chatreaux F, Haesslein JL, LLtis A, Pierre C, Khider J, Tessol N, Airault M, Demassey J, Dupuis-Hamelin C, Lassaigne P, Bonnefoy A, Vicat P, Klich M. Bioorg. Med. Chem. Lett. 2000;10:1695–1699. doi: 10.1016/s0960-894x(00)00304-8. [DOI] [PubMed] [Google Scholar]
- 29.Fylaktakidou KC, Hadipavlou-Litina DJ, Litinas KE, Nicolaides DN. Current Pharmaceutical Design. 2004;10:3813–3833. doi: 10.2174/1381612043382710. [DOI] [PubMed] [Google Scholar]
- 30.Jung J, Kin J, Park O. Synth. Commun. 1999;29:3587–3595. [Google Scholar]
- 31.Wang ZS, Hara K, Dan-oh Y, Kasada C, Shinpo A, Suga S, Arakawa H, Sugihara H. J. Phys. Chem. B. 2005;109:3907–3914. doi: 10.1021/jp044851v. [DOI] [PubMed] [Google Scholar]
- 32.Kung Sutherland MS, Lipps SG, Patnaik N, Gayo-Fung LM, Khammungkune S, Xie W, Brady HA, Barbosa MS, Anderson DW, Stein B. Cytokine. 2003;23:1–14. doi: 10.1016/s1043-4666(03)00179-0. [DOI] [PubMed] [Google Scholar]
- 33.Brady H, Desai S, Gayo-Fung LM, Khammungkhune S, McKie JA, O'Leary E, Pascasio L, Sutherland MK, Anderson DW, Bhagwat SS, Stein B. Cancer Res. 2002;62:1439–1442. [PubMed] [Google Scholar]
- 34.Brady H, Doubleday M, Gayo-Fung LM, Hickman M, Khammungkhune S, Kois A, Lipps S, Pierce S, Richard N, Shevlin G, Sutherland MK, Anderson DW, Bhagwat SS, Stein B. Mol. Pharmacol. 2002;61:562–568. doi: 10.1124/mol.61.3.562. [DOI] [PubMed] [Google Scholar]
- 35.McKie JA, Bhagwat SS, Brady H, Doubleday M, Gayo L, Hickman M, Jalluri RK, Khammungkhune S, Kois A, Mortensen D, Richard N, Sapienza J, Shevlin G, Stein B, Sutherland M. Bioorg. Med. Chem. Lett. 2004;14:3407–3410. doi: 10.1016/j.bmcl.2004.04.081. [DOI] [PubMed] [Google Scholar]
- 36.Gupta A, Sharma R, Balapure AK, Keshri G, Singh MM, Ray S. Letters in Drug Design & Discovery. 2009;6:46–50. [Google Scholar]
- 37.Coletta AA, Benson JR, Baunm M. Breast Cancer Res. Treat. 1994;31:5–9. doi: 10.1007/BF00689672. [DOI] [PubMed] [Google Scholar]
- 38.Butta A, MacLennan K, Flanders KC, Sacks NP, Smith I, McKinna A, Dowsett M, Wakefield LM, Sporn MB, Baum M, et al. Cancer Res. 1992;52:4261–4264. [PubMed] [Google Scholar]
- 39.Charlier C, Chariot A, Antoine N, Merville MP, Gielen J, Castronovo V. Biochem. Pharmacol. 1995;49:351–358. doi: 10.1016/0006-2952(94)00492-5. [DOI] [PubMed] [Google Scholar]
- 40.Obrero M, Yu DV, Shapiro DJ. Journal of Biological Chemistry. 2002;277:45695–45703. doi: 10.1074/jbc.M208092200. [DOI] [PubMed] [Google Scholar]
- 41.Holinka CF, Hata H, Kuramoto H, Gurpide E. Cancer Res. 1986;46:2771–2774. [PubMed] [Google Scholar]
- 42.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structure. 2001;9:145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 43.Bursulaya BD, Totrov M, Abagyan R, Brooks CL., 3rd J. Comput. Aided Mol. Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 44.Pike AC. Best Pract. Res. Clin. Endocrinol. Metab. 2006;20:1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 45.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Nucleic Acids Res. 2007;35(Web Server issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kekenes-Huskey PM, Muegge I, von Rauch M, Gust R, Knapp EW. Bioorg. Med. Chem. 2004;12:6527–6537. doi: 10.1016/j.bmc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 47.Shankar R, Chakravarti B, Singh US, Ansari MI, Deshpande S, Dwivedi SK, Bid HK, Konwar R, Kharkwal G, Chandra V, Dwivedi A, Hajela K. Bioorg. Med. Chem. 2009;17:3847–3856. doi: 10.1016/j.bmc.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 48.Crouch SP, Kozlowski R, Slater KJ, Fletcher J. J. Immunol. Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-u. [DOI] [PubMed] [Google Scholar]
- 49.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 50.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trott O, Olson AJ. J. Comput. Chem. 2009;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]