

Abstract
We report a rare case of Hunter syndrome—mucopolysaccharidosis type II (MPS II) with atypical presentation of mild mental retardation, acrocephalic head without corneal clouding, and multiple skin eruptions along with oral, dental, and radiographic findings. It is a rare syndrome with a very low prevalence of 1:100,000 births and as such the clinician should be aware of this syndrome.
Keywords: Hunter syndrome, mucopolysaccharidosis, skin eruptions
INTRODUCTION
Mucopolysaccharidosis (MPS) is a group of autosomal recessive metabolic disorders caused by the absence or malfunctioning of the lysosomal enzymes needed to break down molecules called glycosaminoglycans (GAGs). These are long chains of sugar carbohydrates in each cell that help build bone, cartilage, tendons, corneas, skin, and connective tissues. GAGs (formerly called mucopolysaccharides) are also found in the fluid that lubricates joints. People with MPS either do not produce enough of one of the 11 enzymes required to break down these sugar chains into proteins and simpler molecules, or they produce enzymes that do not work properly. Over time, these GAGs collect in the cells, blood, and connective tissues. This results in permanent, progressive cellular damage which affects the appearance, physical abilities, organ, and system functioning and, in most cases, mental development. Common clinical presentation includes facial dysmorphism, hepatosplenomegaly, joint stiffness and contractures, pulmonary dysfunction, myocardial enlargement, valvular dysfunction, and neurological involvement.[1–5]
We report this case of MPS type II because of its rarity and the atypical features. The purpose of presenting report is to highlight the distinctive manifestation of the Hunter syndrome.
CASE REPORT
An 11-year-old boy was referred to the outpatient department, College of Dental Surgery, Aljabal-Algharby-Zawia University, Zawia, Libya, for routine dental examination. Medical history reported that he had frequent respiratory infections with hepatomegaly. The boy had been followed regularly by the hospital pediatrics unit with the diagnosis of MPS. The boy was the fifth child of healthy non-consanguineous parents. There was no family history of similar symptoms.
Clinically, on physical examination the patient had retarded growth with a short stature for his chronologic age. Bony deformities, including kyphosis, rotated legs, and short stubby hands with a clumsy and stiff gait were evident. The following facial features were observed: oblique palpebral fissures, along with skin eruptions which appeared as red colored rashes on the face, hands, legs, and abdomen [Figure 1]. The patient also had multiple, whitish to red skin-coloured papules and nodules symmetrically distributed on the scapular region, the extensor aspects of the upper arms [Figure 2] and thighs. There was mild mental retardation. On examination, the lips were incompetent and dry. Intraorally, the enamel of permanent teeth was slightly hypoplastic with a clinical appearance of pitted enamel [Figure 3]. There was a carious lower left deciduous first molar and root stumps in relation to lower right second deciduous molar. There was high arched palate, macroglossia, increased salivation, and anterior open bite. The oral hygiene was good.
Figure 1.
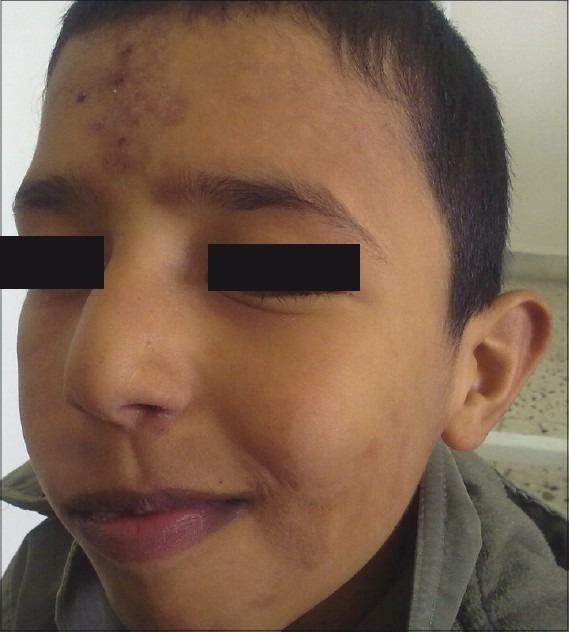
Extra oral picture showing the facial features and skin eruptions on the forehead
Figure 2.
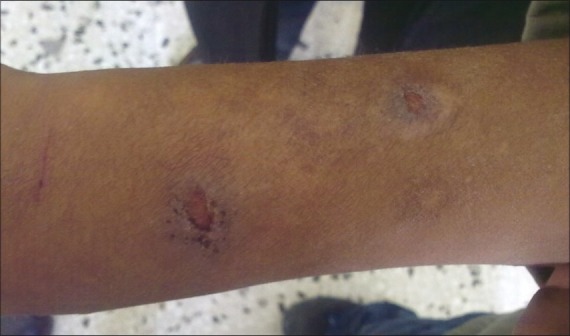
Picture showing the skin lesions on the forehand
Figure 3.
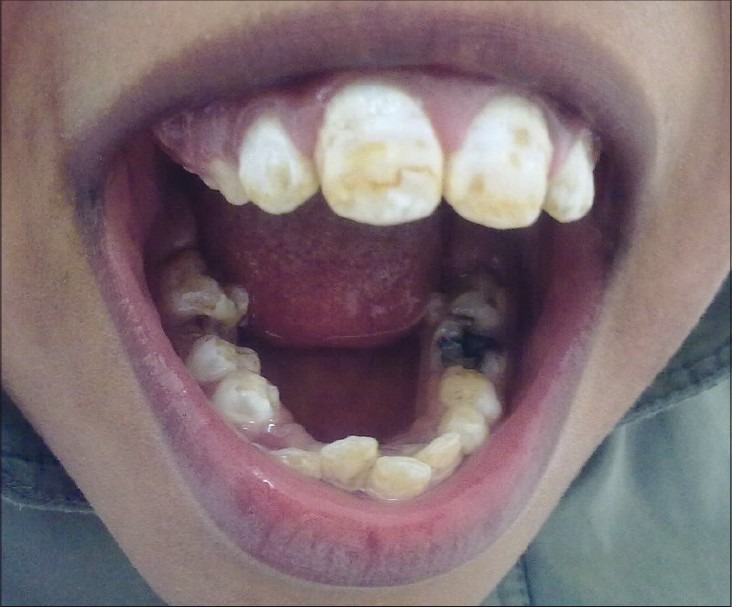
Intraoral picture revealing hypoplastic enamel and carious teeth
IOPA radiograph revealed cyst-like radiolucent lesion in between the mesial root of lower right first permanent molar and root stumps of deciduous second molar [Figure 4]. The involved roots were pushed apart, and there was a well-defined radiolucency with a well-defined radio-opaque border. OPG did not reveal any other periapical radiolucencies [Figure 5]. Aspiration from the cystic lesion revealed a radicular cyst in relation to the root stump of second deciduous molar.
Figure 4.
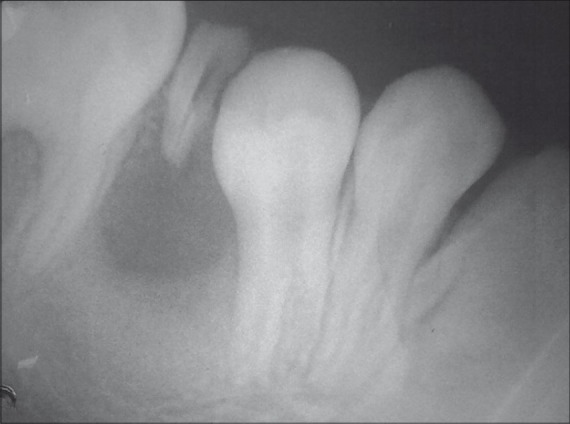
IOPA radiograph revealing a cystic lesion with an associated carious root stump of deciduous second molar
Figure 5.

OPG showing the radiolucency in relation to right mandibular deciduous second molar
Analysis of the urine revealed a marked increase in dermatan sulfate and heparan sulfate. An enzyme assay for iduronate sulfatase could not be carried out due to limited availability of such tests. A definitive diagnosis was previously made upon detection of a significant increase of dermatan sulfate in urine and a marked deficiency of l-iduronidase activity in his leukocytes.
Our diagnosis of Hunter syndrome—MPS II was confirmed from his history, clinical examination and biochemical reports.
DISCUSSION
Mucopolysaccharidosis was first described by Charles Hunter, a Canadian physician, who in 1917 described a rare disease found in two brothers.[1,2] Mucopolysaccharidosis is a group of inherited diseases characterized by defective lysosomal enzymes responsible for the degradation of mucopolysaccharides, which are major components of intercellular connective tissue. This leads to an accumulation of incompletely degraded mucopolysaccharides in the lysosomes which affect various body systems through enzymatic activity.[3] The accumulation of GAG within the lysosomes is responsible for the clinical manifestations of this disorder.[4]
Mucopolysaccharidosis type II or Hunter syndrome is rare and is caused by a deficiency of iduronate-2-sulfatase. Hunter syndrome is one of the most common MPS with a prevalence of one in 170,000 male live births. MPS type II is classified into mild (type II, HB) and severe (type II, A) and this classification is based on the length of survival and the presence or absence of central nervous system (CNS) disease.[5] Patients typically appear normal at birth in both types. In the severe form, the clinical features appear between two and four years of age while in the mild form, the clinical features appear in the second decade of life. In the severe form, there is severe mental retardation and loss of skills. Death usually occurs in the first or second decade of life and the main cause of death is obstructive airway disease or cardiac failure.[4] In the milder form, there is mild mental retardation but intelligence is normal, stature is near normal, and clinical features are less obvious and progress very slowly.[5] We feel our patient may have a mild form of the disease.
In patients with neurologic involvement, intelligence is impaired, and death usually occurs in the second decade of life, whereas those patients with minimal or no neurologic involvement may survive into adulthood with normal intellectual development.[6–8] In our case there was mild mental retardation with normal intellectual development. Young et al[6] established that patients with Hunter's syndrome did clearly fall into one of two groups according to the presence or absence of intellectual deterioration. Yatziv et al[9] suggested that the presence or absence of severe mental retardation and the longevity of the affected individuals be distinguishing factors in order to help clinically determine the difference between these two forms.
Patients with MPS II show dental abnormalities including enamel defects, carious teeth, dentigerous cysts, and abscesses.[10] Our patient also had enamel hypoplasia, carious teeth, and radicular cyst.
Diagnosis often can be made through clinical examination and urine tests as excess mucopolysaccharides such as heparin and dermatan sufates are excreted in the urine. Enzymes are also used to provide definitive diagnosis of one of the mucopolysaccharidoses.
Prenatal diagnosis using amniocentesis and chorionic villus sampling can verify if a fetus either carries a copy of the defective gene or is affected with the disorder. Genetic counselling can help parents who have a family history of the mucopolysaccharidoses to determine if they are carrying the mutated gene that causes the disorders.[6]
Enzyme replacement therapy has emerged as a new treatment for mucopolysaccharidosis disorders, including Hunter syndrome. Enzyme replacement therapy using idursulfase (Elaprase), a recombinant human 12S produced in the human cell line, has been recently approved in the United States and the European Union for the management of MPS type II.[5] Weekly intravenous infusion is given over 3 h at a dose of 0.5 mg/kg diluted in saline. Bone marrow transplantation and umbilical cord blood transplantation are definitive treatments for MPS. Apart from these, supportive management is very important. Physical therapy and daily exercise may improve mobility of joints. Blood transfusion, infection, and nutritional management are also important in the management of MPS type II. A mild form is compatible with survival into adulthood, and reproduction is known to have occurred.[11] Six cases of this mild form of Hunter syndrome have been described, and the patients survived to the ages of 65 and 87 in two cases. Three of these affected men had children.[12] The enzyme deficient in this disorder is iduronate sulfatase, as described by Neufeld.[13] With the advent of hematopoietic stem cell transplantation and more recently, there exists a need for early diagnosis, better disease recognition, and management.
The clinical features are limited to short stature, a large head, a short neck, coarse facial features, skin eruptions, and mild mental retardation with normal intelligence, anterior open bite due to enlarged tongue suggested of mucopolysaccharidosis. Frequent upper and lower respiratory tract infections are common and occur secondary to the enlargement of tonsils and adenoids and enlarged tongue,[5,7,8,14–20] This presentation is evident in our patient.
Although the syndrome is uncommon worldwide, a higher incidence has been reported among Jews in Israel.[21] Cutaneous features are peculiar to this syndrome and may be the initial manifestation in the mild form of the disease although patients in both groups may have skin involvement.[22] Firm, skin-colored papules, 2–10 cm in diameter coalesce to form ridges on the scapular and posterior axillary lines. These pebbles are mainly found only in the Hunter Syndrome[22–25] and helps in differentiating it from other MPS. We emphasize that the skin eruption can be the earliest sign of Hunter syndrome, particularly in the mild form presenting with normal development and growth as seen in our case.
CONCLUSION
Mucopolysaccharidosis is a multisystem disorder which presents with a constellation of clinical findings. Based on the clinical representation, it is possible to diagnose a case of MPS. Early detection of the disease and appropriate management through a multidisciplinary approach is recommended to improve the quality of life. A careful and systemic approach is needed to accurately diagnose the exact type as enzymatic studies are not available in most centres.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Wraith JE, Scarpa M, Beck M, Bodamer OA, Meirleir LD, Guffon N, et al. Mucopolysaccharidosis Type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167:267–77. doi: 10.1007/s00431-007-0635-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin R, Beck M, Eng C, Giugliani R, Harmatz P, Mufioz V, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121:377–86. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- 3.Kliegman RM, Behrman RE, Jenson HB, Stanton FB. 18th ed. Vol. 1. Philadelphia: Saunders; 2007. Nelson Textbook of Pediatrics; pp. 620–6. [Google Scholar]
- 4.Tuschl K, Gal A, Paschke E, Kircher S, Bodamer OA. Mucopolysaccharidosis Type II in females: Case report and review of literature. Pediatr Neurol. 2005;32:270–2. doi: 10.1016/j.pediatrneurol.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Shah GS, Mahal T, Sharma S. Atypical clinical presentation of mucopolysaccharidosis type II (Hunter syndrome): A case report. J Med Case Reports. 2010;4:154. doi: 10.1186/1752-1947-4-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter's syndrome. 2. Differences between the mild and severe forms. J Med Genet. 1982;19:408–11. doi: 10.1136/jmg.19.6.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young ID, Harper PS. Mild form of Hunter's syndrome: Clinical delineation based on 31 cases. Arch Dis Child. 1982;57:828–36. doi: 10.1136/adc.57.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hobolth N, Pedersen C. Six cases of a mild form of Hunter syndrome in five generations.Three affected males with progeny. Clin Genet. 1978;20:121. [Google Scholar]
- 9.Yatziv S, Erickson RP, Epstein CJ. Mild and severe Hunter syndrome (MPS II) within the same sibships. Clin Genet. 1977;11:319–26. doi: 10.1111/j.1399-0004.1977.tb01323.x. [DOI] [PubMed] [Google Scholar]
- 10.Lustmann J, Bimstein E, Yatziv S. Dentigerous cysts and radiolucent lesions of the jaw associated with Hunter's syndrome. J Oral Surg. 1975;33:679–85. [PubMed] [Google Scholar]
- 11.Ben Simon-Schiff E, Bach G, Zlotogora J, Abeliovich D. Combined enzymatic and linkage analysis for heterozygote detection in Hunter syndrome: Identification of an apparent case of germinal mosaicism. Am J Med Genet. 1993;47:837–42. doi: 10.1002/ajmg.1320470608. [DOI] [PubMed] [Google Scholar]
- 12.Berg K, Danes BS, Bearn AG. The linkage relation of the loci for the Xm serum system and the X-linked form of Hurler's syndrome (Hunter syndrome) Am J Hum Genet. 1968;20:398–401. [PMC free article] [PubMed] [Google Scholar]
- 13.Neufeld EF, Liebaers I, Epstein C J, Yatziv S, Milunsky A, Migeon B R. The Hunter syndrome in females: is there an autosomal recessive form of iduronate sulfatase deficiency? Am J Hum Genet. 1977 Sep;29(5):455–461. [PMC free article] [PubMed] [Google Scholar]
- 14.Dorfman A, Vaughan V, McKaRy J, Jr, Behrman RE. Nelson Textbook of Pediatrics. 11th ed. Philadelphia: WB Saunders Co; 1979. Mucopolysaccharidoses; pp. 1845–8. [Google Scholar]
- 15.Gorlin RJ, Cohen M, Jr, Levin LS. Syndromes of the Head and Neck. 3rd ed. New York: Oxford University Press; 1990. Mucopolysaccharidosis II; pp. 106–8. [Google Scholar]
- 16.Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917;10:104–16. doi: 10.1177/003591571701001833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardner DG. The oral manifestations of Hurler's syndrome. Oral Surg. 1971;32:46–57. doi: 10.1016/0030-4220(71)90249-0. [DOI] [PubMed] [Google Scholar]
- 18.Liu KL. The oral signs of Hurler-Hunter syndrome: Report of four cases. ASDCJ Dent Child. 1980;47:122–7. [PubMed] [Google Scholar]
- 19.Angel TD, Tim C, Gerald F. Hunter's syndrome and oral manifestations: A review. Pediatr Dent. 1995;17:98–100. [PubMed] [Google Scholar]
- 20.Ogunbiyi A, Adeyinka AO, Ogah SO, Baiyeroju AM. Hunter syndrome: Case report and review literature. West Afr J Med. 2006;25:169–72. doi: 10.4314/wajm.v25i2.28272. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Simon-Schiff E, Zlotogora J, Abeliovich D, Zeigler M, Bach G. Hunter syndrome among Jews in Israel. Biomed Pharmacother. 1994;48:381–4. doi: 10.1016/0753-3322(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 22.Demitsu T, Kakurai M, Okubo Y, Shibayama C, Kikuchi Y, Mori Y, et al. Skin eruption as the presenting sign of Hunter Syndrome IIB. Clin Exp Dermatol. 1999;24:179–82. doi: 10.1046/j.1365-2230.1999.00448.x. [DOI] [PubMed] [Google Scholar]
- 23.Prystowsky SD, Maumenee IH, Freeman RG, Herndon JH, Jr, Harrod MJ. A cutaneous marker in the Hunter Syndrome. Arch Dermatol. 1977;113:602–5. [PubMed] [Google Scholar]
- 24.Cole HN, Jr, Irving RC, Lund HZ, Mercer RD, Schneider RW. Gargoylism with cutaneous manifestations. AMA Arch Derm Syphilol. 1952;66:371–83. doi: 10.1001/archderm.1952.01530280075012. [DOI] [PubMed] [Google Scholar]
- 25.Thappa DM, Singh A, Jaishankar TJ, Rao R, Ratnakar C. Pebbling of the skin: A marker of Hunters syndrome. Pediatric Dermatol. 1998;15:370–3. doi: 10.1046/j.1525-1470.1998.1998015370.x. [DOI] [PubMed] [Google Scholar]