

Abstract

Ring a ding: The first catalytic enantioselective total synthesis of the meroterpenoid natural product (+)-liphagal is disclosed. The approach showcases a variety of technology including enantioselective enolate alkylation, a photochemical alkyne-alkene [2+2] reaction, microwave-assisted metal catalysis, and an intramolecular aryne capture cyclization reaction. Pivotal to the successful completion of the synthesis was a sequence involving ring expansion from a [6-5-4] tricycle to a [6-7] bicyclic core followed by stereoselective hydrogenation of a sterically occluded tri-substituted olefin to establish the trans homodecalin system found in the natural product.
Keywords: total synthesis, terpenoids, arynes, asymmetric catalysis, palladium, rearrangement
The tetracyclic meroterpenoid natural product (+)-liphagal (1) was isolated in 2006 by Andersen and co-workers from the Caribbean sponge Aka coralliphaga[1] and is one of a number of natural product inhibitors of phosphatidyl inositol 3-kinase (PI3K) including myricetin, quercetin, resveratrol, staurosporine, viridin, and wortmannin (Figure 1).[2] The PI3K family of enzymes participates in the regulation of numerous biological functions and has been directly implicated in the pathogenesis of diabetes and cancer.[3] The inhibitory activity of liphagal is noteworthy due to selective inhibition of PIK3α, a lipid kinase isoform that holds a central role in several cancers.[4] In this context, liphagal was found to have an IC50 of 100 nM against PI3Kα and was 10-fold more potent against isoform α than γ. Additionally, liphagal is cytotoxic toward LoVo (human colon), CaCo (human colon), and MDA-468 (human breast) tumor cell lines with IC50 values of 0.58, 0.67, and 1.58 µM, respectively.[1]
Figure 1.

Natural Product Inhibitors of PI3K.
From a structural perspective, liphagal possesses an unprecedented [6-7-5-6] tetracyclic skeleton and has attracted significant attention from the synthetic organic community. Andersen’s isolation and structure determination was reported concomitantly with the first total synthesis of (±)-1 using a biomimetic strategy.[1,5] Subsequently, the same group determined the absolute stereochemistry,[6] which was corroborated by other laboratories with formal and total syntheses.[7,8,9] Our laboratory became interested in both the potent biological activity and the complex tetracyclic structure of liphagal, highlighted by the chiral quaternary carbon center at C(11). Herein, we report the first catalytic enantioselective total synthesis of (+)-liphagal.
Retrosynthetically, we envisioned simplification of the aromatic ring of liphagal to dimethoxybenzofuran 2, a known precursor to the natural product (Scheme 1).[7] Disconnection of the tetracycle along the benzofuran moiety led back to α-bromoaryl dienone 3. Reduction of the sterically hindered tri-substituted olefin to establish the trans ring fusion was seen as a major challenge. The α-bromoaryl dienone (3) could arise from a ring expansion of strained cyclobutene 4. Excision of the cyclobutene and α-aryl group from ketone 4 revealed chiral cyclopentenone (R)-5. The enantiomeric enone (S)-5[10] was previously prepared from achiral enol carbonate 6[11a] as part of our ongoing research program aimed at the stereoselective synthesis of natural products containing quaternary carbon centers.[11,12] To this end, we have reported a series of palladium-catalyzed enantioselective decarboxylative alkylation reactions that employ the t-Bu-PHOX ligand scaffold in conjunction with allyl enol-carbonates, silyl enol-ethers, and racemic β-ketoesters to produce a wide array of α-quaternary substituted ketones.[11,12,13,14] With this general strategy in mind, we initiated efforts toward a total synthesis of (+)-liphagal.
Scheme 1.

Liphagal Retrosynthesis
The forward synthesis commenced with a palladium-catalyzed decarboxylative alkylation of enol carbonate 6 to furnish tetrasubstituted ketone 7 in 87% yield and 92% ee (Scheme 2).[11,15] This intermediate was elaborated to bicycle 5 following our previously reported two-step sequence.[10] The synthesis continued with exposure of enone 5 to trimethylsilylacetylene under UV irradiation, which promoted a [2+2] photocycloaddition.[16] Exposure of the crude reaction mixture to BF3•OEt2 formed a single silylated cyclobutene product (8a).[17] Subsequent removal of the trimethylsilyl group with TBAF yielded the chromatographically stable and pleasantly fragrant cyclobutene 8b, a compound that contains three contiguous quaternary centers within the strained carbon framework.[18,19] A microwave-assisted palladium-catalyzed α-arylation with 4-bromoveratrole installed the electron rich aromatic moiety, producing aryl ketone 4 as a single diastereomer.[20,21]
Scheme 2.

Catalytic enantioselective preparation of synthetic building block (+)-7 and chemical elaboration to (+)-4.
At this stage in our synthesis, Lewis acid-mediated ring expansion by selective cleavage of strained cyclobutane 4 was attempted (Scheme 3). Exposure of tricyclic ketone 4 to BF3•OEt2 at 50 °C, provided the desired cycloheptadienone product 9 in modest yield. Serendipitously, this compound was isolated alongside a crystalline byproduct (i.e., 10), which was suitable for X-ray diffraction analysis and structure determination.[22] Bridged polycyclic ketone 10 is presumably the result of a Cargill rearrangement, which proceeds through two concerted [1, 2]-carbon–carbon bond migrations.[23] More specifically, activation of ketone 4 with BF3 (i.e., 11) promotes carbon bond migration to rupture the cyclobutene and produce an allylic carbocation intermediate (12). The second carbon bond migration forms a [2.2.1] bridged bicyclic core of Lewis acid complex 13. Finally, loss of BF3 reveals the isolated product (10). Importantly, the stereospecific rearrangement mechanism allowed assignment of the relative stereochemistry of cyclobutenes 8 from the unequivocal assignment of bridged bicycle 10. In addition to BF3•OEt2, we discovered that AlCl3 also promotes ring expansion of aryl cyclobutene 4 without formation of Cargill product 10. However, under these reaction conditions we were intrigued to find a new side-product, enone 14, arising from intramolecular 1,6-addition of the electron-rich arene fragment of 9 to the cycloheptadienone system. This result suggests that the arene resides in close proximity to the tri-substituted olefin and also indicated that the aromatic moiety should be deactivated before ring expansion to avoid formation of 14.
Scheme 3.

Unexpected rearrangement and reactivity of strained cyclobutane 4.
With this in mind, we sought to install a functional group handle on the aromatic ring that could be utilized for eventual benzofuran formation and could serve to deactivate the aromatic residue of 9 toward unwanted Friedel–Crafts chemistry. We were impressed to find that chemoselective aromatic bromination occurred in the presence of the strained cyclobutene, furnishing bromoarene 15 (Scheme 4). At this stage, crystallization of the crude product increased the enantiomeric excess to >99%. With the deactivated aromatic ketone in hand, we were pleased to find that treatment of bromide 15 with AlCl3 furnished much improved yields of the corresponding ring expanded product (3). An optimized ring expansion from the [6-5-4] system to the desired [6-7] core (3) was accomplished in the absence of a Lewis acid with microwave heating at 250 °C in o-dichlorobenzene.[24] Chemoselective reduction of dienone 3 with Adams’ catalyst in ethyl acetate furnished ketone 16, leaving the aromatic halide intact.
Scheme 4.

Completion of the total synthesis of Liphagal (+)-1.
With the core carbon framework of liphagal (1) secured, our focus turned to the challenging stereoselective hydrogenation of the tri-substituted olefin to establish the desired [6-7] trans ring fusion.[25] Our strategy to effect this transformation was guided by our previous isolation of 1,6-addition product 14, which provided evidence that hydrogenation to form a trans ring fusion would be sterically demanding (vide supra). To alleviate steric congestion, we proceeded with epimerization of the aryl substituent, forming the β-oriented α-aryl ketone (17). The mass recovery for this equilibration averaged 97% and a 78% overall yield of 17 was obtained after three cycles of equilibration (Keq(av) = 0.76).[25] With the arene substituent further removed from the tri-substituted olefin we planned to rigidify the polycyclic system via formation of the fourth ring. This began with a diastereoselective methylation, affording the desired α-methyl cycloheptenone (18) in 68% yield.[26] Reduction of this hindered ketone with DIBAL produced alcohol 19, a substrate poised for dihydrobenzofuran formation. Initial attempts to form dihydrobenzofuran 21 were unsuccessful[27] and prompted an unconventional strategy to accomplish the desired transformation. Gratifyingly, formation of dihydrobenzofuran 21 was accomplished upon exposure of bromoarene 19 to LDA, proceeding through a putative aryne intermediate (20).[28] This powerful aryne capture cyclization strategy generated the highly congested dihydrobenzofuran product in 83% yield. With tetracycle 21 in hand, we set out to test the key stereoselective hydrogenation of the tri-substituted olefin. To our delight, using catalytic Pd/C in ethanol under 1 atm H2, we isolated 97% yield of saturated homodecalin 22, observing exclusive formation of the [6-7] trans ring fusion.
Having executed the synthesis of the challenging trans fused system, the completion of (+)-liphagal required three additional transformations: 1) benzofuran construction, 2) aldehyde installation, and 3) demethylation, the final two of which were known from previous syntheses.[1,7] Oxidation of dihydrobenzofuran 22 to benzofuran 2 proved surprisingly difficult, and with DDQ a tendency for over-oxidation was observed.[29] Upon switching to nitrosonium tetrafluoroborate, which oxidizes by hydride abstraction, dehydrogenation occured in 70% yield to give benzofuran 2.[30] Aryl lithiation with n-butyllithium•TMEDA and quenching with anhydrous DMF installed the aldehyde functional group in 23.[7] Finally, this was followed by demethylation using boron triiodide to generate (+)-liphagal (1), which was identical in all respects to data reported in the literature.1
In summary, we have successfully completed the first catalytic enantioselective total synthesis of (+)-liphagal (1) in 15 steps from known compounds (19 steps from commercially available materials). By applying a combination of catalytic enantioselective alkylation (6→7), two-carbon ring expansion via cyclobutene 15, and an intramolecular aryne cyclization (19→20→21) we were able to access the tetracyclic core of the natural product in enantioenriched form. Judicious choice of tetracyclic hydrogenation substrate 21 established the critical trans-[6-7] ring fusion and enabled completion of the total synthesis.
Footnotes
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). The authors wish to thank NIH-NIGMS (R01GM080269-01), the Gordon and Betty Moore Foundation, Abbott, Amgen, Boehringer Ingelheim, and Caltech for generous funding. R.M.M. thanks Eli Lilly for a graduate fellowship. H.K. acknowledges the rejselegat of the Danish Technical University, the Jorcks foundation, and the Otto Mønsteds foundation for financial support. J.L.A. gratefully acknowledges the Amgen Foundation for funding through the Amgen Scholars program. We thank Professor E. N. Jacobsen and Dr. S. J. Zuend for a kind donation of both (R)-t-leucine and their optimal Strecker catalyst.15
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Marion F, Williams DE, Patrick BO, Hollander I, Mallon R, Kim SC, Roll DM, Feldberg L, Van Soest R, Andersen RJ. Org. Lett. 2006;8:321–324. doi: 10.1021/ol052744t. [DOI] [PubMed] [Google Scholar]
- 2.Sundstrom TJ, Anderson AC, Wright DL. Org. Biomol. Chem. 2009;7:840–850. doi: 10.1039/b819067b. [DOI] [PubMed] [Google Scholar]
- 3.a) Cantley LC. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]; b) Engelman JA, Luo J, Cantley LC. Nat. Rev. Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]; c) Ward SG, Finan P. Curr. Opin. Cell Biol. 2003;3:426–434. doi: 10.1016/s1471-4892(03)00078-x. [DOI] [PubMed] [Google Scholar]; d) Ward S, Sotsios Y, Dowden J, Bruce I, Finan P. Chem. Biol. 2003;10:207–213. doi: 10.1016/s1074-5521(03)00048-6. [DOI] [PubMed] [Google Scholar]; e) Sundstrom TJ, Anderson AC, Wright DL. Org. Biomol. Chem. 2009;7:840–850. doi: 10.1039/b819067b. [DOI] [PubMed] [Google Scholar]
- 4.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Science. 2004;304:554. doi: 10.1126/science.1096502. , and references therein. [DOI] [PubMed] [Google Scholar]
- 5.For a recent biomimetic formal synthesis of (±)-liphagal, see: Mehta G, Likhite NS, Kumar CSA. Tetrahedron Lett. 2009;50:5260–5262.
- 6.a) Andersen R, Hollander I, Roll DM, Kim SC, Mallon RG, Williams DE, Marion F. WO2006081659 F. Patent. 2006 Oct 08;; b) Pereira AR, Strangman WK, Marion F, Feldberg L, Roll D, Mallon R, Hollander I, Andersen RJ. J. Med. Chem. 2010;53:8523–8533. doi: 10.1021/jm100531u. [DOI] [PubMed] [Google Scholar]
- 7.George JH, Baldwin JE, Adlington RM. Org. Lett. 2010;12:2394–2397. doi: 10.1021/ol100756z. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez-Manzaneda E, Chahboun R, Alvarez E, Cano MJ, Haidour A, Alvarez-Manzaneda R. Org. Lett. 2010;12:4450–4453. doi: 10.1021/ol101173w. [DOI] [PubMed] [Google Scholar]
- 9.For additional studies directed toward the total synthesis of liphagal, see: Deore V, Lohar MK, Mundada R, Roychowdhury A, Vishwakarma R, Kumar S. Syn. Comm. 2011;41:177–183. Zhang Y, Oblak EZ, Bolstad ESD, Anderson AC, Jasinski JP, Butcher RJ, Wright DL. Tetrahedron Lett. 2010;51:6120–6122.
- 10.McFadden RM, Stoltz BM. J. Am. Chem. Soc. 2006;128:7738–7739. doi: 10.1021/ja061853f. [DOI] [PubMed] [Google Scholar]
- 11.a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018. Angew. Chem., Int. Ed.2005, 44, 6924–6927. [DOI] [PubMed] [Google Scholar]; c) Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J. Am. Chem. Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]; d) Seto M, Roizen JL, Stoltz BM. Angew. Chem. 2008;120:6979–6982. doi: 10.1002/anie.200801424. Angew. Chem., Int. Ed.2008, 47, 6873–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mohr JT, Krout MR, Stoltz BM. Org. Synth. 2009;86:194–211. doi: 10.15227/orgsyn.086.0194. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew. Chem. 2009;121:6972–6975. doi: 10.1002/anie.200902575. Angew. Chem., Int. Ed.2009, 48, 6840–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Streuff J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2010;2:192–196. doi: 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) McDougal NT, Virgil SC, Stoltz BM. Synlett. 2010:1712–1716. doi: 10.1055/s-0030-1258094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.This methodology has been central to the synthesis of a number of natural products in our laboratory, see: a) (+)-Dichroanone: see reference 10; b) (+)-Elatol: White DE, Stewart IC, Grubbs RH, Stoltz BM. J. Am. Chem. Soc. 2008;130:810–811. doi: 10.1021/ja710294k. c) (−)-Cyanthiwigin F: Enquist JA, Jr, Stoltz BM. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. d) (+)-Carissone: Levine SR, Krout MR, Stoltz BM. Org. Lett. 2009;11:289–292. doi: 10.1021/ol802409h. e) (+)-Cassiol: Petrova KV, Mohr JT, Stoltz BM. Org. Lett. 2009;11:293–295. doi: 10.1021/ol802410t. f) (−)-Laurencenone C and (−)-α-Chamigrene: White DE, Stewart IC, Seashore-Ludlow BA, Grubbs RH, Stoltz BM. Tetrahedron. 2010;66:4668–4686. doi: 10.1016/j.tet.2010.04.128. g) (+)-Hamigeran B: Mukherjee H, McDougal NT, Virgil SC, Stoltz BM. Org. Lett. 2011;13:825–827. doi: 10.1021/ol102669z.
- 13.For related approaches from other laboratories, see: Braun M, Laicher F, Meier T. Angew. Chem. 2000;112:3637–3640. doi: 10.1002/1521-3773(20001002)39:19<3494::aid-anie3494>3.0.co;2-4. Angew. Chem., Int. Ed.2000, 39, 3494–3497. Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. Nakamura M, Hajra A, Endo K, Nakamura E. Angew. Chem. 2005;117:7414–7417. Angew. Chem., Int. Ed.2005, 44, 7248–7251. Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:17180–17181. doi: 10.1021/ja055968f. Burger EC, Barron BR, Tunge JA. Synlett. 2006:2824–2826. Trost BM, Bream RN, Xu J. Angew. Chem. 2006;118:3181–3184. doi: 10.1002/anie.200504421. Angew. Chem., Int. Ed.2006, 45, 3109–3112. Trost BM, Xu J, Reichle M. J. Am. Chem. Soc. 2007;129:282–283. doi: 10.1021/ja067342a. Bélanger É, Cantin K, Messe O, Tremblay M, Paquin J-F. J. Am. Chem. Soc. 2007;129:1034–1035. doi: 10.1021/ja067501q. Schulz SR, Blechert S. Angew. Chem. 2007;119:4040–4044. doi: 10.1002/anie.200604553. Angew. Chem., Int. Ed.2007, 46, 3966–3970. Bélanger É, Houzé C, Guimond N, Cantin K, Paquin J-F. Chem. Commun. 2008:3251–3253. doi: 10.1039/b803097a. Bélanger É, Pouliot M-F, Paquin J-F. Org. Lett. 2009;11:2201–2204. doi: 10.1021/ol9005618. Trost BM, Xu J, Schmidt T. J. Am. Chem. Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. Zhao X, Liu D, Xie F, Zhang W. Tetrahedron. 2009;65:512–517. Braun M, Meier T, Laicher F, Meletis P, Fidan M. Adv. Synth. Catal. 2008;350:303–314. Zheng W-H, Zheng B-H, Zhang Y, Hou X-L. J. Am. Chem. Soc. 2007;129:7718–7719. doi: 10.1021/ja071098l. Yan X-X, Liang C-G, Zhang Y, Hong W, Cao B-X, Dai L-X, Hou X-L. Angew. Chem. 2005;117:6702–6704. doi: 10.1002/anie.200502020. Angew. Chem., Int. Ed.2005, 44, 6544–6546.
- 14.For recent reviews on the allylic alkylation of ketone enolates, see: Braun M, Meier T. Angew. Chem. 2006;118:7106–7109. Angew. Chem., Int. Ed.2006, 45, 6952–6955. You S-L, Dai L-X. Angew. Chem. 2006;118:5372–5374. Angew. Chem., Int. Ed.2006, 45, 5246–5248. Braun M, Meier T. Synlett. 2006:661–676. Kazmaier U. Curr. Org. Chem. 2003;7:317–328. Trost BM, Jiang C. Synthesis. 2006:369–396. Mohr JT, Stoltz BM. Chem. Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183.
- 15.The synthesis of (R)-t-BuPHOX was greatly enabled by the ready availability of (R)-t-leucine via the catalytic enantioselective Strecker synthesis reported by Jacobsen and co-workers, see: Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968–970. doi: 10.1038/nature08484.
- 16.Miyaoka H, Nagaoka H, Okamura T, Yamada Y. Chem. Pharm. Bull. 1989;37:2882–2883. [Google Scholar]
-
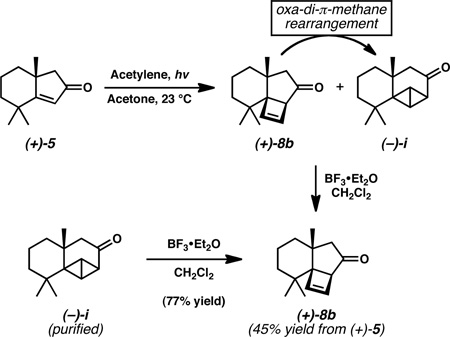
17.Photocycloaddition of enone 5 with acetylene produced a mixture of cyclobutane 8b and bicyclobutane (−)-i, a secondary photoadduct presumably resulting from oxa-di-π-methane rearrangement of 8b (for an excellent review see:
Hixson SS, Mariano PS, Zimmerman HE. Chem. Rev. 1973;73:531–551.
). Exposure of the mixture to BF3•OEt2 resulted in conversion of i to 8b. A related bicyclobutane rearrangement to a cyclobutene mediated by AlBr3 has been reported:
Razin VV, Mostova MI, D’yakonov IA. Zh. Organish. Khim. 1968;4:535.
Although a more complex mixture is believed to result in the case of the trimethylsilylacetylene photochemical reaction, the 3-step process in Scheme 2 is higher yielding (cf. 45% versus 68% yield).
- 18.See Supporting Information for experimental details.
- 19.At this stage in the synthesis it was not possible to determine the relative stereochemistry of tricycle 8.
- 20.The α-arylation conditions were adapted from: Kawatsura M, Hartwig JF. J. Am. Chem. Soc. 1999;121:1473–1478.
- 21.For a review on microwave assisted organic synthesis, including metal catalysis, see: Kappe CO. Angew. Chem. 2004;116:6408–6443. Angew. Chem., Int. Ed.2004, 43, 6250–6284.
- 22.Crystallographic data have been deposited at the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK and copies can be obtained on request, free of charge, by quoting the publication citation and the deposition number 634511.
- 23.Cargill RL, Jackson TE, Peet NP, Pond DM. Acc. Chem. Res. 1974;7:106–113. [Google Scholar]
- 24.For the thermal rearrangement of a cyclobutene stabilized through vinylogy with an electron-withdrawing substituent, see: Wakabayashi S, Sugihara Y, Hayakawa Y, Murata I. J. Chem. Soc., Perkin Trans. I. 1990:1489–1490.
- 25.Hydrogenation of dienes 9 and 3 or alkenes 16–19 did not result in saturation of the tri-substituted olefin or afforded the undesired cis ring fusion.
- 26.Aryl ketone 17 was recovered in 18% yield without epimerization.
- 27.Initial attempts used CuI, TMEDA/H2O: Carril M, SanMartin R, Tellitu I, Domínguez E. Org. Lett. 2006;8:1467–1470. doi: 10.1021/ol060274c.
- 28.For the intramolecular trapping of aryne with a pendant alcohol, see: Birkett MA, Knight DW, Little PB, Mitchell MB. Tetrahedron. 2000;56:1013–1023. , and references therein
- 29.Based on 1H NMR of the crude reaction mixture.
- 30.Olah GA, Salem G, Staral JS, Ho T-L. J. Org. Chem. 1978;43:173–175. [Google Scholar]
