

SUMMARY
Irs2-mediated insulin/IGF1 signaling in the CNS modulates energy balance and glucose homeostasis; however, the site for Irs2 function is unknown. The hormone leptin mediates energy balance by acting on leptin receptor (LepR-b)-expressing neurons. To determine whether LepR-b neurons mediate the metabolic actions of Irs2 in the brain, we utilized Leprcre together with Irs2L/L to ablate Irs2 expression in LepR-b neurons (LeprΔIrs2). LeprΔIrs2-mice developed obesity, glucose intolerance, and insulin resistance. Leptin action was not altered in young LeprΔIrs2-mice, although insulin-stimulated FoxO1 nuclear exclusion was reduced in LeprΔIrs2-mice. Indeed, deletion of Foxo1 from LepR-b neurons in LeprΔIrs2-mice normalized energy balance, glucose homeostasis, and arcuate nucleus gene expression in LeprΔIrs•2ΔFoxo1-mice. Thus, Irs2 signaling in LepR-b neurons plays a crucial role in metabolic sensing and regulation. While not required for leptin action, Irs2 suppresses FoxO1 signaling in LepR-b neurons to promote energy balance and metabolism.
Introduction
The rates of obesity and type 2 diabetes continue to increase throughout the world, and represent major contributors to morbidity and mortality (White, 2003). It is thus crucial to understand the processes that underlie energy homeostasis, as their dysregulation can promote metabolic disease. Many aspects of metabolism and energy balance are coordinated by the CNS, where specialized sets of neurons integrate information from circulating nutrients and hormones, such as insulin and leptin, to control feeding, energy expenditure, and nutrient homeostasis (Myers, Jr. et al., 2009; Schwartz and Porte, Jr., 2005).
Insulin and insulin-like growth factors (Igf1 and Igf2) act through related tyrosine kinase receptors whose signals converge on their downstream insulin receptor substrate proteins, including Irs1, Irs2 and Irs4 (Kahn and White, 1995). Tyrosine phosphorylated IRS-proteins recruit and activate phosphatidylinositol 3-kinase (PI3K) to promote PDK1→Akt signaling, which mediates the phosphorylation, inactivation and nuclear export of FoxO1 (Matsumoto and Accili, 2005). Of the IRS-proteins, Irs1 plays a prominent role in organismal growth, especially in lean tissues (Tamemoto et al., 1994). By comparison, Irs2 signaling is crucial for the proliferation and survival of pancreatic beta cells (Hennige et al., 2003), fertility (Burks et al., 2000), energy balance and glucose metabolism, life span, and the progression of neurodegenerative disease (Lin et al., 2004; Taguchi et al., 2007; Freude et al., 2009; Sadagurski et al., 2011). Regarding metabolism, disruption of Irs2 in the brain produces an early-onset syndrome of obesity, insulin resistance and glucose intolerance, suggesting that central Irs2 signaling is a crucial mediator of energy balance and metabolic homeostasis (Taguchi et al., 2007).
While the importance of brain Irs2 for metabolic homeostasis is clear, the relevant site of metabolic action has not been revealed. The identity of this site is important in order to avoid potential reproductive or neurodegenerative pathologies. A great deal of attention has been paid to distinct populations of arcuate (ARC) nucleus neurons that express proopiomelanocortin (POMC) or agouti-related peptide (AgRP) (Morton et al., 2006); however, deletion of Irs2 (or the insulin receptor, IR) from these neurons in mice negligibly impacts metabolic homeostasis (Choudhury et al., 2005; Hill et al., 2010). Similarly, disruption of the pathway (which lies downstream of Irs2 and many other growth factors) in POMC and AgRP neurons produces relatively subtle alterations in energy balance and glucose homeostasis (Hill et al., 2008; Belgardt et al., 2008; Cao et al., 2011; Al-Qassab et al., 2009). Thus, the relevant site at which Irs2-signaling contributes to metabolic regulation must be a distinct, or broader, set of neurons.
Leptin is a hormone produced by adipocytes in approximate proportion to triglyceride energy stores, which acts via its receptor, LepR-b, to diminish feeding, promote energy expenditure, and modulate nutrient flux (Myers, Jr. et al., 2009). Brain LepR-b is required for these leptin actions (Myers, Jr. et al., 2009; Cohen et al., 2001; de et al., 2005). While POMC and AgRP neurons are crucial for the control of energy homeostasis and leptin action via POMC and AgRP neurons has been studied extensively (Elmquist et al., 2005), it is clear that direct leptin action on these neurons contributes only modestly to overall leptin action (Myers, Jr. et al., 2009; Balthasar et al., 2004; van de Wall et al., 2008). Indeed, while LepR-b neurons are only found in relatively few areas of the CNS, POMC and AgRP neurons represent only a small percentage of total LepR-b neurons (Myers, Jr. et al., 2009; Patterson et al., 2011; Scott et al., 2009). Furthermore, leptin action via non-POMC, non-AgRP neruons plays a crucial role in energy balance and the control of the hypothalamic melanocortin system (Vong et al., 2011). Overall, the hypothalamic, midbrain and brainstem sites in which LepR-b neurons are located are known to play crucial roles in metabolism and energy balance. In aggregate, LepR-b neurons sense and integrate signals relevant to nutrient homeostasis to control energy balance and metabolism.
Given the crucial overlapping roles for brain leptin and insulin/Igf signaling in the control of metabolism and energy balance, along with the presumed importance of LepR-b neurons for the sensing and modulation of nutrient homeostasis, we hypothesized that LepR-b neurons represent the crucial locus for Irs2 signaling to control metabolism. We therefore deleted Irs2 specifically in LepR-b neurons and examined energy balance and metabolism in these mice, and investigated whether FoxO1 contributes to their phenotype.
Results
Generation of LeprΔIrs2 mice
To inactivate Irs2 specifically in LepR-b neurons, we crossed Leprcre , which drives the expression of cre recombinase in LepR-b cells, onto the Irs2L/L background, in which the Irs2 coding sequence was surrounded by LoxP sites (Figure 1A) (Leshan et al., 2006; Dong et al., 2006; Leshan et al., 2009). Since Leprcre mediates modest cre expression and cre-mediated excision by this allele is more complete in Leprcre/cre homozygous animals, we compared Leprcre/cre•Irs2L/L (LeprΔIrs2) mice to Irs2L/L control littermates. Leprcre does not disrupt LepR-b expression or function, and Leprcre/cre animals never displayed a pathological phenotype (Leshan et al., 2006).
Figure 1.
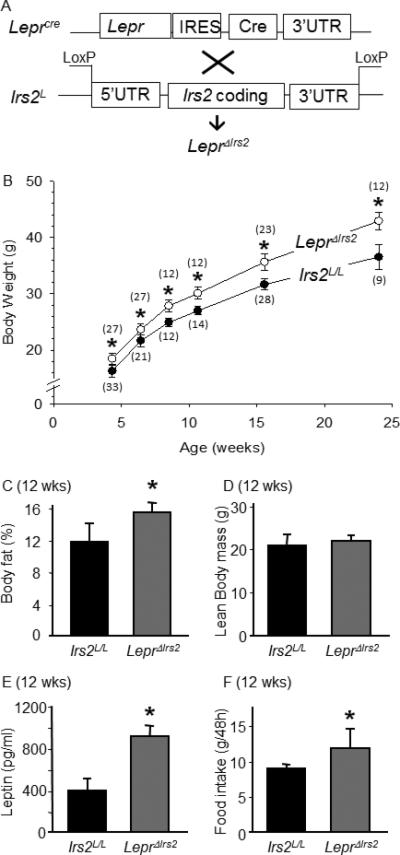
LeprΔIrs2 mice are obese. (A) Schematic representation of our breeding strategy to generate LeprΔIrs2 mice. (B) Average body weights of male LeprΔIrs2 -mice (closed circles) and control Irs2L/L mice (open circles) on regular chow diet was determined in each average age group by generalized linear regression (SPSS, v19). The number of mice in each group is indicated in parentheses (mean ± SD; *, Bonferroni p<0.001). (C) Percent body fat and (D) lean body mass was determined by DEXA using 12-week-old Irs2L/L (n=12) and LeprΔIrs2 (n=12) mice (mean ± SEM; *, p<0.05). (E) Serum leptin levels of 12-week-old male Irs2L/L (n=7) and LeprΔIrs2 (n=7) mice (mean ± SEM; *, p<0.05). (F) Food intake over 48 hours in 12-week-old male Irs2L/L (n=12) and LeprΔIrs2 (n=12) mice fed regular chow diet (mean ± SEM; *, p<0.05). See also Figure S1.
LeprΔIrs2 mice were born at the expected Mendelian ratio, were of normal size and appearance, and were fertile. PCR analysis confirmed the excision of both Irs2L alleles in Lepr-containing brain regions of LeprΔIrs2 mice, and the reduction in Irs2 mRNA and Irs2 protein in the hypothalamus, but not in other tissues or irrelevant brain areas, such as the cortex (Figure S1). Within the hypothalamus, Irs2 expression was substantially but not entirely reduced (Figure S1), consistent with the percentage of total hypothalamic neurons that express LepR-b (Myers, Jr. et al., 2009; Patterson et al., 2011; Scott et al., 2009).
Obesity in LeprΔIrs2 mice
To assess the impact of Irs2 deletion in LepR-b neurons on energy homeostasis, we compared body weight and composition of control and LeprΔIrs2 mice. Male LeprΔIrs2 mice were about 20% heavier than controls between 4-24 weeks of age (Figure 1B). As shown previously for mice null for Irs2 throughout the brain (Lin et al., 2004; Taguchi et al., 2007), 12 week old male LeprΔIrs2 mice displayed more body adiposity but normal lean mass relative to controls (Figures 1C and D). Circulating leptin concentrations were also increased (Figure 1E). However, LeprΔIrs2 mice consumed approximately 20% more chow than controls during a 48-hour interval (Figure 1F), suggesting that hyperphagia contributed to their obesity. Thus, LeprΔIrs2 mice developed hyperphagic obesity from an early age, consistent with an important role for Irs2-mediated insulin/Igf signaling in LepR-b neurons for the control of food intake and energy balance.
To determine the role for Irs2 in LepR-b neurons for the control of energy utilization, we analyzed components of energy expenditure in ad libitum-fed 6-month-old male control and LeprΔIrs2 mice (Figure 2A-D). During the dark and the light phases, O2 consumption, CO2 production and heat generation were reduced significantly in LeprΔIrs2 mice compared to control littermates. Furthermore, ambulatory movement was reduced by more than 50% in LeprΔIrs2 mice. The decreased metabolic rate and heat production of LeprΔIrs2 mice revealed a potential impairment of brown adipose tissue (BAT) function in these animals. Indeed, BAT from young and old LeprΔIrs2 mice contained large, unilocular, lipid-filled vacuoles that resemble those in white adipocytes (Figure 2E; Figure S2). Moreover, the expression of Ucp1 (mitochondrial uncoupling protein 1) and Pgc1α (Ppargc1a, peroxisome proliferator-activated receptor gamma, coactivator 1 alpha) was reduced significantly in the BAT of LeprΔIrs2 mice compared to controls (Figure 2F), suggesting that Irs2 signaling in LepR-b neurons is crucial for the control of BAT function, which presumably contributes to the decreased energy expenditure in these animals. Thus the combination of increased feeding and decreased energy expenditure via multiple mechanisms promotes obesity in LeprΔIrs2 mice, demonstrating the importance of Irs2 signaling in LepR-b neurons for the overall control of energy balance.
Figure 2.
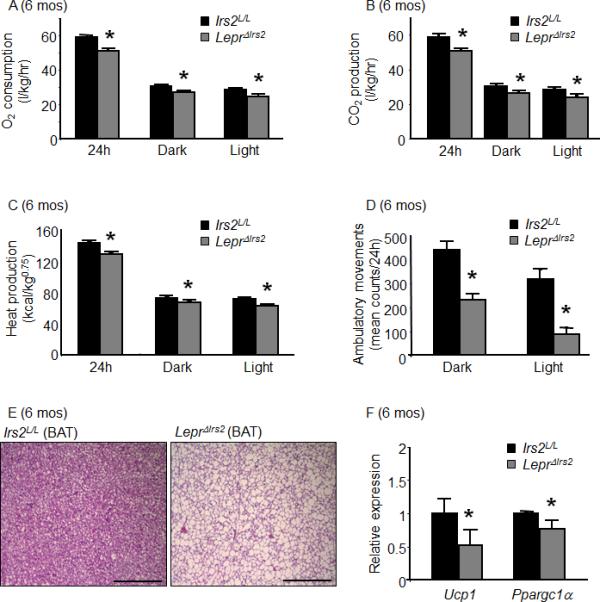
Energy expenditure in LeprΔIrs2 mice. 24 week-old male mice of the indicated genotype were monitored for 72 hours in the CLAMS (n=10/genotype) to assess (A) oxygen consumption (O2, l/kg/h), (B) carbon dioxide production (CO2, l/kg/h), (C) heat production (kcal/kg 0.75) and (D) locomotor activity during the light and dark cycles. Each group was analyzed by generalized linear regression (SPSS, v19), (mean ± SD; *, Bonferroni p<0.001). (E) Representative H&E staining of brown adipose tissue (BAT) of Irs2L/L (left panel) and LeprΔIrs2 mice (right panel) mice aged 24 weeks. Scale bar: 500 μm. (F) Levels of mRNA by semi-quantitative RT-PCR of Ucp1 and Ppargc1α from BAT of 24-week-old Irs2L/L and LeprΔIrs2 mice (n=6). Data are presented as mean ± SEM; *, p < 0.05. See also Figure S2.
Impaired glucose homeostasis with preserved endocrine function in LeprΔIrs2 mice
Next we investigated glucose homeostasis in LeprΔIrs2 mice. While the blood glucose concentration was indistinguishable between ad libitum-fed control and LeprΔIrs2 mice at 6 weeks of age (151±13.4 and 152±5.5 respectively), these mice displayed substantial hyperinsulinemia at baseline, as well as glucose intolerance and insulin intolerance (Figure 3A, C, E). By 6 months of age, LeprΔIrs2 mice displayed elevated fasting blood glucose concentrations (96±14.8 versus 171.25±20.5) in addition to increased insulin levels and worsened glucose and insulin intolerance at 6 months of age (Figure 3B, D, F). Hyperinsulinemia in the LeprΔIrs2 mice suggested an impairment of insulin sensitivity and glucose handling independent of any changes in islet function. However, while islet area was normal in young mice, islet area in older (1 year) mice increased commensurate with a compensatory increase in insulin production (Figure S3).
Figure 3.
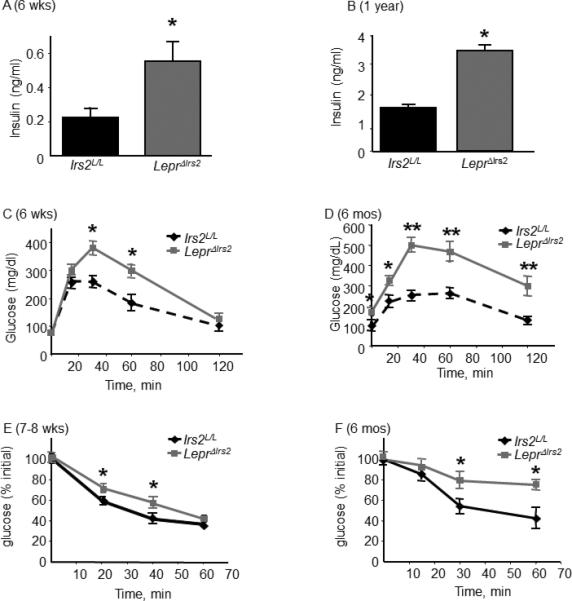
Glucose homeostasis in LeprΔIrs2 mice. (A) Fasting serum insulin levels (n = 10/genotype) for 6 week-old male mice and (B) 1-year-old mice (n=5/genotype) of the indicated genotypes. Glucose tolerance test of (C) 6-week-old male mice or (D) 24-week-old mice of the indicated genotypes (n=10/genotype). (E) Insulin tolerance test of 7-8 weeks old mice (n=9-10/genotype) or (F) 24-week-old mice (n=8-10/genotype). Data are expressed as mean ± SEM. Irs2L/L -mice versus LeprΔIrs2-mice: *, p < 0.05; **, p < 0.01. See also Figures S3, S4 and Table S1.
In contrast to the decreased insulin sensitivity and impaired glycemic control in LeprΔIrs2 mice, other measures of endocrine function were not impaired in these animals (Figure S4). All matings between Irs2L/L and LeprΔIrs2 mice produced normal-sized, healthy litters within 4 weeks (Supplemental Table 1), and LeprΔIrs2 females displayed normal reproductive organs and onset of estrus (Figure S4C and D). Furthermore, serum thyroxine and corticosterone concentrations were indistinguishable between LeprΔIrs2 and control mice (Figure S4A and B). Thus, whereas Irs2 signaling in LepR-b neurons was crucial for the control of energy balance and glucose homeostasis, it did not contribute to the wider control of endocrine function. As the absence of leptin action in LepR-b neurons alters dramatically these endocrine axes along with nutrient homeostasis (Ahima and Flier, 2000), these findings suggest that leptin and insulin/Igf→Irs2 signaling mediate separable functions in LepR-b neurons.
LepR-b signaling and leptin sensitivity in LeprΔIrs2 mice
Given the distinct phenotypes resulting from the absence of leptin action compared to Irs2 signaling in LepR-b neurons, we examined the response to leptin in LeprΔIrs2 mice to determine whether disruption of Irs2 in LepR-b neurons interferes with LepR-b signaling or anorectic leptin action. Since obesity itself impairs each of these aspects of leptin action by a number of mechanisms, we assayed leptin responsiveness in young animals to mitigate against this possibility (Myers, Jr. et al., 2010; Myers, Jr. et al., 2012).
Leptin binding to LepR-b activates an associated Jak2 tyrosine kinase, thereby promoting the phosphorylation of LepR-b and the recruitment and tyrosine phosphorylation of STAT3 (signal transducer and activator of transcription-3) (Robertson et al., 2008). The leptin-stimulated accumulation of pSTAT3 in the brain reflects cell-autonomous LepR-b signaling, which is impaired in states associated with diminished leptin action (Vaisse et al., 1996; Munzberg et al., 2004; Myers, Jr. et al., 2010; Myers, Jr. et al., 2012). Acute leptin treatment promoted similar levels of hypothalamic pSTAT3 in control and LeprΔIrs2 mice (Figure 4A-B). Leptin was similarly effective in control and LeprΔIrs2 mice for the stimulation of cFos-immunoreactivity, a measure of neuronal activation (Figure S5). Thus, Irs2 in LepR-b neurons was not required for acute LepR-b signaling.
Figure 4.
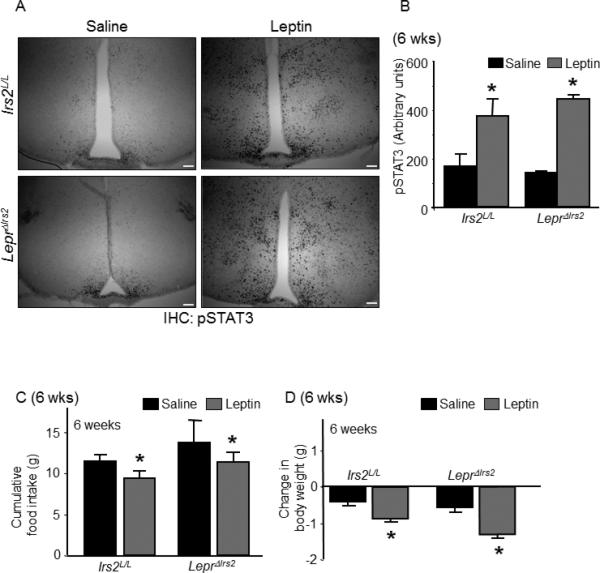
LepR-b signaling and leptin sensitivity in LeprΔIrs2 mice. (A) Immunostaining for pSTAT3 in 6-week-old mice of the indicated genotypes injected intraperitoneally with vehicle or leptin (5 mg/kg, i.p., 2 hours, 3v- 3rd cerebral ventricle). Representative images from the hypothalamus are shown. Scale bar: 100 μm (B) Quantification of pSTAT3 immunoreactivity (n=4/genotype). (C and D) Cumulative food intake and reduction in body weight were measured over a 3-day period of treatment with leptin (5 mg/kg i.p.; BID) or vehicle in 6-week-old male mice of the indicated genotypes. Data are presented as mean ± SEM; *, p < 0.05; **, p < 0.01. See also Figure S5.
To examine the anorectic response to exogenous leptin, we administered leptin (5 mg/kg, i.p.) to 6 week old mice twice daily for 3 days. In control animals, this treatment promoted the expected decrease in food intake and weight relative to vehicle-treated animals (Figure 4C-D). Importantly, LeprΔIrs2 mice responded to leptin treatment in a similar manner, demonstrating both decreased food intake and body weight during the treatment. Thus, Irs2 signaling in LepR-b neurons was not required for the ability of leptin to acutely suppress food intake and body weight, suggesting that Irs2 signaling in LepR-b neurons was functionally separable from leptin signaling.
FoxO1 signaling in LeprΔIrs2 mice
Insulin/Igf stimulation leads to the tyrosine phosphorylation of Irs2 and to the activation of the PI3K cascade, which inhibits FoxO1 by promoting its phosphorylation-dependent export from the nucleus (Brunet et al., 2001; Cheng et al., 2010). To establish whether inactivation of Irs2 dysregulates insulin-like signaling in LepR-b neurons, we investigated the distribution of FoxO1 between the nuclear and cytoplasmic compartments in ARC LepR-b neurons of control (Leprcre) and LeprΔIrs2 mice, which were on the cre-inducible ROSA26-EGFP background to reveal LepR-b neurons by the expression of EGFP (LeprEGFP and LeprEGFPΔIrs2 mice, respectively)(Figure 5). Before insulin treatment, about 50% of the ARC LepR-b neurons contained nuclear FoxO1 in fasted control and LeprEGFPΔIrs2 mice. After insulin injection into the lateral ventricle, nuclear FoxO1 was detected in only 30% of the ARC LepR-b neurons of control mice. By contrast, insulin treatment of LeprEGFPΔIrs2 mice did not reduce the percentage of ARC LepR-b neurons containing nuclear FoxO1. These results are consistent with the disruption of the Irs2→PI3K→Akt→FoxO1 pathway, rather than interference with leptin signaling and action, in the LepR-b neurons of LeprΔIrs2 mice.
Figure 5.
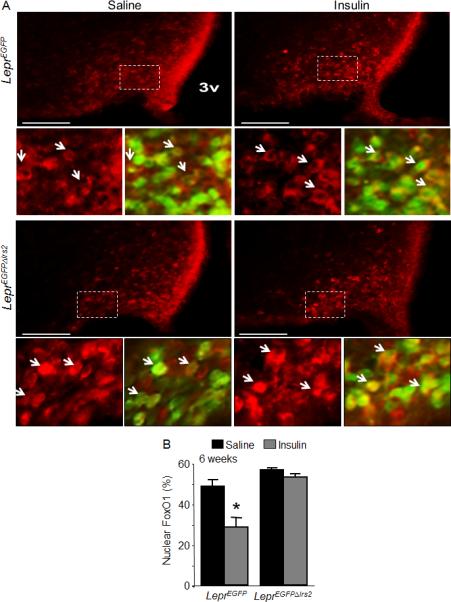
FoxO1 signaling in LeprΔIrs2 –mice. (A) Immunofluorescence for FoxO1 (red) in 6-weeks-old control LeprEGFP and LeprEGFPΔIrs2 mice injected icv with vehicle or insulin (300 mU; 1 hour). Representative images from the hypothalamus of LeprEGFP and LeprEGFPΔIrs2 mice are shown. Scale bar: 100 μm. Lower (small) panels are digital zooms of boxed areas from the top panel, showing FoxO1 (red) alone (left panels) or merged with and EGFP (green) (right panels) in 6-week-old mice of the indicated genotypes. (B) Quantification of LepR-b/EGFP neurons containing nuclear FoxO1 immunoreactivity (n=4/genotype). Data are presented as mean ± SEM; *, p < 0.05.
Restoration of energy homeostasis in LeprΔIrs2 mice by deletion of FoxO1 from LepR-b neurons
Since a major role for Irs2 signaling in other tissues is the suppression of FoxO1 action (Dong et al., 2008), we examined whether FoxO1 mediated the effects of Irs2 in LepR-b neurons. We bred our LeprΔIrs2 mice onto the FoxO1L/L background, in which cre recombinase excises sequences crucial for FoxO1 expression, thereby producing “double-knockout” mice lacking both Irs2 and FoxO1 in LepR-b neurons (LeprΔIrs2ΔFoxO1 mice). PCR analysis confirmed the excision of FoxO1L in Lepr-containing hypothalamic regions of LeprΔIrs2ΔFoxO1 mice, and the reduction of FoxO1 protein in the ARC of these animals (Figure S6).
Whereas LeprΔIrs2 mice showed the expected increase in body weight compared to control littermates, LeprΔIrs2ΔFoxO1 animals displayed normal body weight (Figure 6A). All control genotypes exhibited indistinguishable (normal) body weight (Figure S7). Deletion of FoxO1 from LepR-b neurons also prevented hyperphagia and normalized VO2 and VCO2 in the LeprΔIrs2ΔFoxO1 mice (Figure 6B-D). Moreover, while blood glucose concentrations in 16-week-old LeprΔIrs2 mice were dramatically increased during the glucose tolerance test compared to controls, there was no difference between the controls and LeprΔIrs2ΔFoxO1 mice, and fasting insulin concentrations were normalized in male LeprΔIrs2ΔFoxO1 mice (Figure 6E-F). Overall, these findings indicate that suppression of FoxO1 activity in LepR-b neurons is the principal means by which Irs2 controls energy homeostasis.
Figure 6.
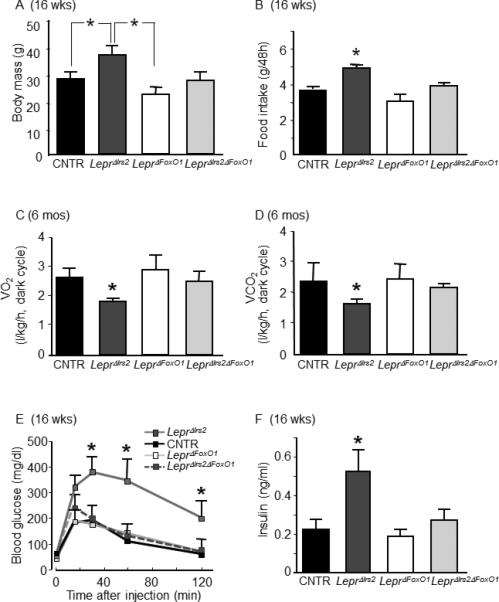
Restoration of energy homeostasis in LeprΔIrs2 mice by deletion of FoxO1 from LepR-b neurons. Parameters of energy balance for 16-week-old chow-fed male mice of the indicated genotypes (n=10/genotype): (A) Body weight, (B) food intake. Parameters of energy balance for 16-week-old chow-fed male mice of the indicated genotypes: (C) oxygen consumption (O2, L•kg-1•h-1), and (D) carbon dioxide production (CO2, L•kg-1•h-1) during the dark cycle. (E) Glucose tolerance test and (F) fasting serum insulin concentrations in 16-week-old mice of the indicated genotypes (n=8-10/genotype). Data are presented as mean ± SEM; *, p < 0.05. See also Figure S6 and S7.
Control of ARC neuropeptide expression by Irs2 and FoxO1 in LepR-b neurons
To elucidate the mechanisms by which Irs2 and FoxO1 in LepR-b neurons controls energy balance, we employed qPCR to examine the expression of ARC genes involved in the control of energy homeostasis (Figure 7). This analysis revealed the decreased expression of Pomc and increased expression of Agrp in the hypothalamus of LeprΔIrs2, but not LeprΔIrs2ΔFoxO1 mice. While Socs3 expression tended to be slightly reduced in LeprΔIrs2 mice compared to controls, the difference was not statistically significant (0.04±0.009 versus 0.026±0.005, p-value=0.06). Thus, elimination of Irs2 signaling in LepR-b neurons interferes with the normal control of the hypothalamic melanocortin system, and the removal of FoxO1 prevents this dysregulation, suggesting that signaling in LepR-b neurons is crucial for the regulation of this system.
Figure 7.
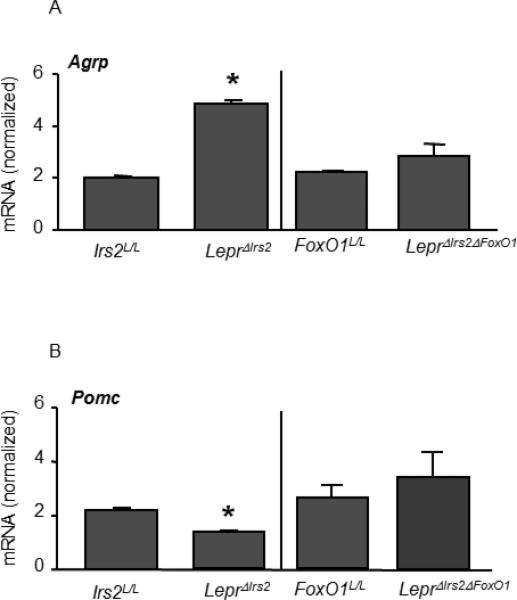
Levels of mRNA by semi-quantitative RT-PCR of Agrp (A) and Pomc (B) from hypothalamus of 16-week-old chow-fed male mice of the indicated genotypes (n=6). Data are presented as mean ± SEM; *, p < 0.05.
Discussion
Our data reveal that Irs2 signaling in LepR-b neurons is crucial for the control of energy balance and glucose homeostasis, since deletion of Irs2 from this restricted subset of CNS neurons dysregulates gene expression within the hypothalamic melanocortin system, increases feeding, decreases energy expenditure, and promotes insulin resistance. Furthermore, the suppression of FoxO1-mediated signaling represents a crucial mechanism for Irs2 action in these neurons.
The manipulation of Irs2 to probe the role for insulin/Igf signaling in neurons has a number of advantages over other approaches. First, given the redundancy and cross-reactivity of insulin and insulin-like growth factors for IR and Igf1R (LeRoith et al., 1994), the deletion of either of these receptors alone in the brain produces relatively modest effects (Bruning et al., 2000; Kappeler et al., 2008), and fails to reveal the full physiologic role for neuronal insulin and Igf signaling in the control of metabolism. Similarly, direct manipulation of the downstream PI3K pathway interferes not only with insulin and Igf signaling, but also dysregulates signals generated by other important growth factor and cytokine receptor pathways, including those of the NGF, PDGF and FGF families, among others (Foukas and Withers, 2011). Thus, in addition to being genetically complex (due to the number of regulatory and catalytic PI3K subunits and downstream effector proteins), manipulation of PI3K and its downstream effectors interferes with the action of multiple receptor systems likely to be important for neuronal growth and differentiation (Cantley, 2002; Brunet et al., 2001). Deletion of Irs2 from LepR-b neurons in LeprΔIrs2 mice reveals the crucial role for insulin/Igf signals specifically in LepR-b neurons for energy balance and metabolism. Moreover, the abrogation of this phenotype by deletion of FoxO1 from these neurons in LeprΔIrs2ΔFoxO1 mice reveals that the inhibition of FoxO1 action represents the main function of Irs2 signaling in LepR-b neurons.
Many reports suggest the importance of brain insulin/Igf signaling in the control of body weight and metabolism (Bruning et al., 2000; Taguchi and White, 2008; Schwartz and Porte, Jr., 2005); however, the site(s) of action at which these signals act to control metabolic homeostasis have remained unclear. While a great deal of research in the control of metabolism by the brain, and especially by insulin action in the brain, has focused upon the POMC and AgRP neurons of the ARC, ablation of IR or Irs2 from these neurons has little effect on body weight or glucose homeostasis (Hill et al., 2010; Plum et al., 2005; Choudhury et al., 2005; Konner et al., 2007). Similarly, deletion of the insulin receptor from SF1 neurons of the VMH promotes modest weight gain on a high fat diet, but does not significantly alter energy balance or insulin sensitivity in chow-fed animals (Klockener et al., 2011). While insulin signaling in TH-expressing catecholaminergic neurons is important for the control of mesolimbic dopamine (DA) signaling, lack of IR signaling in these neurons also fails to change measures of adiposity or glucose homeostasis (Konner et al., 2011). We thus reasoned that the important site of Irs2 signaling in the brain likely represented a more distributed set of neurons involved in the sensing and regulation of metabolic cues.
Leptin is a crucial signal of energy balance and metabolic status, and acts via a distributed set of LepR-b-expressing neurons that represent specialized subpopulations of neurons within areas of the brain known to integrate signals of energy homeostasis (Myers, Jr. et al., 2009; Patterson et al., 2011; Scott et al., 2009). LepR-b neurons include those that express AgRP (as well as subsets of POMC, SF-1 and TH neurons), but also include other metabolically important neurons in the ARC, DMH, and elsewhere. The strong phenotype resulting from Irs2 deletion in LepR-b neurons in LeprΔIrs2 mice thus reveals that the metabolically relevant insulin/Igf signaling in the brain (like leptin signaling) is distributed across multiple groups of specialized neurons that sense and control metabolism. While technical limitations prevent the identification of specific neurons that contain both LepR-b and Irs2, they are largely co-distributed within the ARC, VMH, and DMH, suggesting that LepR-b neurons in these areas, which are known to control energy balance and metabolism, likely mediate important aspects of Irs2 action. Indeed, our findings of decreased ARC Pomc expression and increased Agrp expression in the LeprΔIrs2 mice, but not LeprΔIrs2ΔFoxO1 mice, suggests an important role for Irs2 signaling in LepR-b neurons for the control of hypothalamic melanocortin action. LepR-b neurons in the ARC, VMH, and DMH contribute to the regulation of the hypothalamic melanocortin system, at least in part by indirect action via non-POMC, non-AgRP LepR-b neurons (Vong et al., 2011; Sternson et al., 2005), where Irs2 contributes importantly.
In addition to early defects in energy balance that result from a combination of increased energy intake and decreased energy expenditure by multiple mechanisms (activity, heat production, etc.), our data reveals early insulin resistance in the LeprΔIrs2 mice that develops concomitantly with weight gain and obesity, suggesting a potential role for obesity in the genesis of this insulin resistance. While it is not clear from our present results which tissue(s) contribute to this diminished insulin action, others have previously suggested that altered hepatic insulin action and glucose handling interferes with glucose homeostasis in animals with defects in hypothalamic signaling (Konner, 2007; Plum et al., 2006). Although Irs2 is essential for pancreatic beta cell proliferation and survival (Hennige et al., 2003; Lin et al., 2004), the metabolic phenotype of the LeprΔIrs2 mice does not include beta cell deficiency: Circulating insulin concentrations are elevated appropriately to compensate for the increased adiposity and insulin resistance, and LeprΔIrs2 mice display increased islet size only at advanced ages.. Indeed, we have failed to observe islet LepR-b expression by the criterion of Leprcre-mediated reporter expression (data not shown). Thus, the previously suggested roles for islet LepR-b expression in physiology likely represent artifacts of cre alleles that mediate recombination in (poorly characterized) groups of brain LepR-b neurons (Covey et al., 2006; Wicksteed et al., 2010). The finding of obesity and insulin resistance in RIPcre•Irs2L/L animals following rescue of beta cell Irs2 suggests that the poorly characterized set of RIPcre–expressing LepR-b neurons may be especially important for the metabolic action of insulin/Igf signaling in the brain (Lin et al., 2004; Wicksteed et al., 2010).
While the crucial set of neurons that mediates the metabolic effects of Irs2 signaling in the brain are by definition those that mediate leptin action (leptin action is mediated by LepR-b and thus by LepR-b neurons), the normal response of young LeprΔIrs2 mice to the anoretic effects of leptin suggests that Irs2 is not required for leptin action. Furthermore, while interference with leptin action in these LepR-b neurons promotes endocrine and reproductive dysfunction, in addition to obesity and insulin resistance (Cohen et al., 2001; van de Wall et al., 2008), deletion of Irs2 and interference with insulin/Igf signaling in LepR-b neurons in LeprΔIrs2 mice does not alter the thyroid, adrenal, or reproductive axes. While leptin can promote some Irs2→PI3K signaling (Niswender et al., 2001), the ability of leptin to stimulate this pathway pales by comparison to that of insulin, suggesting that Irs2 functions primarily to mediate insulin/Igf signaling in leptin-responsive neurons, rather than functioning as a direct mediator of LepR-b signaling. Indeed, while the hypothalamic application of PI3K inhibitors or the genetic blockade of PI3K in some leptin-responsive neurons impairs the acute anorectic response to leptin, these manipulations have little effect on baseline leptin action or energy balance (Morton et al., 2005; Niswender et al., 2003; Niswender et al., 2001). Thus, while Irs2 signaling in LepR-b neurons is crucial for metabolic signaling, this pathway does not appear to play a direct role in leptin action, per se. Hence, while Irs2 and LepR-b signaling act in the same neurons to control a variety of metabolic parameters, the requirement for Irs2 is independent from the cellular action of leptin. Despite continued normal LepR-b signaling in the absence of Irs2, the net tone of the neuron is altered without Irs2 signaling, however.
While the hyperleptinemia of LeprΔIrs2 mice meets one definition of leptin resistance (Myers, Jr. et al., 2010; Myers, Jr. et al., 2012), the unaltered leptin sensitivity in these animals at a young age, prior to exposure to chronic obesity, suggests that the elevated leptin concentration in these animals mainly reflects their increased adipose mass, rather than indicating a primary lesion in leptin action (Myers, Jr. et al., 2010; Myers, Jr. et al., 2012). The finding of hyperleptinemia in leptin sensitive LeprΔIrs2 animals demonstrates the importance of clearly distinguishing between blood leptin levels and leptin action in young animals when assessing leptin resistance or sensitivity, as processes unrelated to cellular leptin signaling or physiologic leptin responsiveness may alter adiposity (Myers, Jr. et al., 2010).
Overall, our findings reveal that LepR-b neurons represent the crucial mediators of brain insulin/Igf signaling in the control of energy balance and glucose homeostasis, revealing the distribution of metabolically important insulin/Igf signaling across a network of specialized neurons that sense and control metabolism. Furthermore, the essential function of insulin/Igf→Irs2 signaling in these neurons is not in the potentiation of leptin signaling, but in the suppression of FoxO1 action.
Experimental Procedures
Animals
Leprcre, Irs2L/L and Leprcre mice on the ROSA26tm2sho background (LeprEGFP) were described previously (Dong et al., 2006; Leshan et al., 2009). Mice were bred in our colony at Children's Hospital Boston, the Harvard School of Public Health, or in the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan. All animals were handled in accordance with all procedures approved by the appropriate Institutional Animal Care and Use Committee (IACUC). Animals were fed breeder chow diet containing 9 kcal %fat (Research diets, Inc).
Metabolic Analysis
Lean and fat body mass were assessed by Dual-Energy X-ray Absorptiometry (DEXA, GE Lunar Corp.) as previously described (Dong et al., 2008). Blood glucose levels were measured on random-fed or overnight-fasted animals in mouse-tail blood using Glucometer Elite (Bayer). Intraperitoneal glucose tolerance test was performed on mice fasted for 16 hours overnight. Animals were then injected intraperitoneally with D-glucose (2 g/kg) and blood glucose levels were measured (Withers et al., 1999). For insulin tolerance tests, mice were fasted for a 4-h period in the light cycle before ip injections of insulin (Humulin R, 0.8 U/kg) diluted in sterile saline. Blood glucose concentrations were measured at indicated time points. Blood insulin and leptin levels were determined on serum from tail vein bleeds using a Rat Insulin ELISA kit and Mouse Leptin ELISA kit (Crystal Chem. Inc.). For food intake measurements mice were singly housed and food intake was measured for 2 consecutive days. For peripheral leptin treatment, mice were treated with either 5 mg/kg recombinant mouse leptin (provided by Dr. A Parlow, National Hormone and Pituitary Program, Torrance, CA) or vehicle, injected i.p. twice per day for 3 consecutive days. Food and body weights were recorded before injection and during the treatment period.
Energy Expenditure and Locomotor Activity
As previously described (Sadagurski et al., 2010), physical activity and energy expenditure were performed over 72 hour period with a Comprehensive Laboratory Animal Monitoring System (CLAMS, Oxymax Windows 3.0.3; Columbus Instruments, OH, USA). Mice were housed individually at room temperature (22°C) under an alternating 12 hr light/12 hr dark cycle. Heat production was measured and normalized by metabolic body size (WT0.75) to determine the energy expenditure (Xu et al., 2010). For brown adipose tissue histology the hematoxylin and eosin (H&E) staining was performed using paraffin embedded brown adipose tissue sections as previously described (Balthasar, 2004).
RNA extraction and qPCR
Total RNA was extracted from brown adipose tissue or from hypothalamus using Trizol (Gibco BRL) and 1 μg samples were converted to cDNA using the iscript cDNA kit (Bio-Rad Laboratories Inc.). Sample cDNAs were analyzed in triplicate via quantitative RT-PCR for Ucp1 and Ppargc1α in brown adipose tissue and for Pomc and Agrp in hypothalamus with customized primers as previously described (Sadagurski et al., 2010). Actin gene expression was used to normalize RNA content and the relative gene product amounts were reported as mean ±SEM of several animals.
Intracerebral cannulation and insulin administration
As described previously (Leinninger et al., 2009) adult LeprΔIrs2 or control mice were anesthetized using an isoflurane vaporizer and placed in a stereotaxic frame (Kopf Instruments). After exposing the skull and determining coordinates for bregma, a 26g steel guide cannula with stylet (Plastics One) was lowered toward the right lateral ventricle using the following coordinates from bregma: 0.6mm posterior, 1.0mm lateral, 2.1mm ventral. The guide cannula was cemented to the skull using dental acrylic and the skin surrounding the cannula closed with sutures. Post-surgery, mice were singly-housed and received Buprenex analgesia. Animals’ body weights and food intake were monitored daily with additional daily handling that included removal and replacement of the stylet. Two weeks post-surgery, 24 hours before experimental treatment, correct cannula placement was confirmed based on drinking response following angiotensin II injection. On the day of treatment, animals were fasted for 4 hours, followed by administration of insulin (300mU) or an equivalent volume of saline. One hour post-injection, animals were anesthetized and perfused as detailed below.
Perfusion and immunolabeling
Mice were anesthetized with an overdose of intraperitoneal (IP) pentobarbital and transcardially perfused with 10% neutral buffered formalin as previously described (Munzberg et al., 2007). Briefly, brains were post-fixed, dehydrated, then sectioned coronally (30 μm) using a sliding microtome followed by immunohistochemical or immunofluoresent analysis as described. For immunohistochemical labeling, free-floating brain sections were pretreated by sequential incubations in 0.3% H2O2/1% NaOH, 0.3% glycine, 0.03% SDS, followed by blocking in normal donkey serum (NDS) and incubation in primary antibodies (anti-pSTAT3, Cell Signaling; anti-cFos, Calbiochem). Incubation in biotinylated donkey anti-rabbit (Jackson Immunoresearch) preceded avidin-biotin-complex (Vectastain) and development using metal-enhanced DAB (Thermo Scientific). For immunofluorescent detection of rabbit anti-FoxO1 (Cell Signaling) and chicken anti-GFP (Abcam), free-floating brain sections were blocked in NDS, incubated in primary antibodies, followed by AlexaFluor-conjugated secondary antibodies (Invitrogen) Sections were mounted onto Superfrost Plus slides (Fisher Scientific) and coverslipped with ProLong Antifade mounting medium (Invitrogen). Microscopic images were obtained using an Olympus FluoView 500 Laser Scanning Confocal Microscope (Olympus) at the Michigan Diabetes Research and Training Center Microscopy Core. The images were quantified using Imaris (versions 6.4 and 7.0;Bitplane), using the function spot to count nuclei and surface to measure the area or the volume of the different objects.
Statistical analysis
Unless otherwise stated mean values ± SEM were used to make comparisons between 2 groups; significance was determined by a Student's t-test. A p-value less than 0.05 was considered statistically significant. Generalized linear regression (SPSS, v 19) was used to identify significant differences in mice body weight and energy expenditure parameters.
Supplementary Material
HIGHLIGHTS.
Deletion of Irs2 in LEPR-B neurons causes obesity and hyperglycemia
Deletion of Irs2 in LEPR-B neurons impairs energy homeostasis
Irs2 is not required for LEPR-B signaling or leptin action
Irs2 controls energy homeostasis by regulating FoxO1 activity in LEPR-B neurons
Acknowledgments
The authors thank Dr. A. Parlow, National Hormone and Pituitary Program (NHPP) for recombinant mouse leptin. We also thank Dr. K. R. Vella for help with brain microdissections. This project was supported by NIH grants DK38712 and DK55326 (to MFW), DK056731 and DK057768 (to MGM), and by the Ellison Foundation (to MFW and MS). RLL was supported by a grant from the American Heart Association, and CP was supported by T32HL007853.
Abbreviations
- CNS
central nervous system
- Lepr
leptin receptor
- Irs2
insulin receptor substrate 2
- FoxO1
forkhead box-containing protein of the O subfamily-1
- JAK2
Jak family tyrosine kinase
- ERK
extracellular signal-regulated kinase
- Socs3
suppressor of cytokine signaling-3
- Stat3
signal transducer and activator of transcription 3
- POMC
proopiomelanocortin
- PI3K
phosphatidylinositol 3-kinase
- ARC
arcuate nucleus of the hypothalamus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413–437. doi: 10.1146/annurev.physiol.62.1.413. [DOI] [PubMed] [Google Scholar]
- Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI, Selman C, Piipari K, Clements M, Lingard S, Chandarana K, Bell JD, Barsh GS, Smith AJ, Batterham RL, Ashford ML, Vanhaesebroeck B, Withers DJ. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 2009;10:343–354. doi: 10.1016/j.cmet.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. 2004. [DOI] [PubMed]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr., Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B, Klockener T, Alessi D, Kloppenburg P, Bruning JC. PDK1 Deficiency in POMC-Expressing Cells Reveals FOXO1-Dependent and -Independent Pathways in Control of Energy Homeostasis and Stress Response. Cell Metab. 2008;7:291–301. doi: 10.1016/j.cmet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Burks DJ, de Mora JF, Schubert M, Withers DJ, Myers MG, Jr., Towery HH, Altamuro SL, Flint CL, White MF. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000;407:377–382. doi: 10.1038/35030105. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Cao Y, Nakata M, Okamoto S, Takano E, Yada T, Minokoshi Y, Hirata Y, Nakajima K, Iskandar K, Hayashi Y, Ogawa W, Barsh GS, Hosoda H, Kangawa K, Itoh H, Noda T, Kasuga M, Nakae J. PDK1-Foxo1 in agouti-related peptide neurons regulates energy homeostasis by modulating food intake and energy expenditure. PLoS. One. 2011;6:e18324. doi: 10.1371/journal.pone.0018324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab. 2010;21:589–598. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury AI, Heffron H, Smith MA, Al-Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, Bedford DC, Hisadome K, Diakonov I, Moosajee V, Bell JD, Speakman JR, Batterham RL, Barsh GS, Ashford ML, Withers DJ. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J. Clin. Invest. 2005;115:940–950. doi: 10.1172/JCI24445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey SD, Wideman RD, McDonald C, Unniappan S, Huynh F, Asadi A, Speck M, Webber T, Chua SC, Kieffer TJ. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006;4:291–302. doi: 10.1016/j.cmet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- de LC, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LepR-b transgenes. J Clin Invest. 2005;115:3484–3493. doi: 10.1172/JCI24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J. Clin. Invest. 2006;116:101–114. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, White MF. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J. Comp Neurol. 2005;493:63–71. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- Foukas LC, Withers DJ. Phosphoinositide signalling pathways in metabolic regulation. Curr. Top. Microbiol. Immunol. 2011;346:115–141. doi: 10.1007/82_2010_59. [DOI] [PubMed] [Google Scholar]
- Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr. Alzheimer Res. 2009;6:213–223. doi: 10.2174/156720509788486527. [DOI] [PubMed] [Google Scholar]
- Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, Dow MA, Leshan R, Zakaria M, Mossa-Basha M, White MF. Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J. Clin. Invest. 2003;112:1521–1532. doi: 10.1172/JCI18581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, Lauzon D, Lee CE, Coppari R, Richardson JA, Zigman JM, Chua S, Scherer PE, Lowell BB, Bruning JC, Elmquist JK. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn CR, White MF. Molecular mechanism of insulin action. In Endocrinology. In: DeGroot LJ, editor. W.B. Saunders Company; Philadelphia: 1995. pp. 1373–1387. et al. [Google Scholar]
- Kappeler L, De Magalhaes Filho CM, Dupont J, Leneuve P, Cervera P, Perin L, Loudes C, Blaise A, Klein R, Epelbaum J, Le BY, Holzenberger M. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008;6:e254. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockener T, Hess S, Belgardt BF, Paeger L, Verhagen LA, Husch A, Sohn JW, Hampel B, Dhillon H, Zigman JM, Lowell BB, Williams KW, Elmquist JK, Horvath TL, Kloppenburg P, Bruning JC. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci. 2011;14:911–918. doi: 10.1038/nn.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konner AC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. 2007. [DOI] [PubMed]
- Konner AC, Hess S, Tovar S, Mesaros A, Sanchez-Lasheras C, Evers N, Verhagen LA, Bronneke HS, Kleinridders A, Hampel B, Kloppenburg P, Bruning JC. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011;13:720–728. doi: 10.1016/j.cmet.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Bruning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Munzberg H, Myers MG., Jr. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoith D, Sampson PC, Roberts CT., Jr. How does the mitogenic insulin-like growth factor I receptor differ from the metabolic insulin receptor? Horm. Res. 1994;41(Suppl 2):74–78. doi: 10.1159/000183964. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Bjornholm M, Munzberg H, Myers MG., Jr. Leptin receptor signaling and action in the central nervous system. Obesity. (Silver. Spring) 2006;14(Suppl 5):208S–212S. doi: 10.1038/oby.2006.310. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Louis GW, Jo YH, Rhodes CJ, Munzberg H, Myers MG., Jr. Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. doi: 10.1523/JNEUROSCI.0155-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Taguchi A, Park S, Kushner JA, Li F, Li Y, White MF. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. Journal of Clinical Investigation. 2004;114:908–916. doi: 10.1172/JCI22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Accili D. All roads lead to FoxO. Cell Metab. 2005;1:215–216. doi: 10.1016/j.cmet.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005;2:411–420. doi: 10.1016/j.cmet.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Jobst EE, Bates SH, Jones J, Villanueva E, Leshan R, Bjornholm M, Elmquist J, Sleeman M, Cowley MA, Myers MG., Jr. Appropriate inhibition of orexigenic hypothalamic arcuate nucleus neurons independently of leptin receptor/STAT3 signaling. J. Neurosci. 2007;27:69–74. doi: 10.1523/JNEUROSCI.3168-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr., Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, Tschop MH, Yanovski JA. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012;15:150–156. doi: 10.1016/j.cmet.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr., Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr., Munzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr., Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–231. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr., Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- Patterson CM, Leshan RL, Jones JC, Myers MG., Jr. Molecular mapping of mouse brain regions innervated by leptin receptor-expressing cells. Brain Res. 2011;1378:18–28. doi: 10.1016/j.brainres.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 2006;116:1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Schubert M, Bruning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005;16:59–65. doi: 10.1016/j.tem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Robertson SA, Leinninger GM, Myers MG., Jr. Molecular and neural mediators of leptin action. Physiol Behav. 2008;94:637–642. doi: 10.1016/j.physbeh.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Cheng Z, Rozzo A, Palazzolo I, Kelley GR, Dong X, Krainc D, White MF. IRS2 increases mitochondrial dysfunction and oxidative stress in a mouse model of Huntington disease. J Clin Invest. 2011;121:4070–4081. doi: 10.1172/JCI46305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Norquay L, Farhang J, D'Aquino K, Copps K, White MF. Human IL6 enhances leptin action in mice. Diabetologia. 2010;53:525–535. doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Porte D., Jr. Diabetes, Obesity, and the Brain. Science. 2005;307:375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH --> arcuate nucleus microcircuits and their reorganization by fasting. Nat. Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 Signaling Coordinates Life Span and Nutrient Homeostasis. Science. 2007;317:369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 2008;70:191–212. doi: 10.1146/annurev.physiol.70.113006.100533. [DOI] [PubMed] [Google Scholar]
- Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, Sekihara H, Yoshioka S, Horikoshi H, Furuta Y, Ikawa Y, Kasuga M, Yazaki Y, Aizawa S. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr., Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, Elmquist J, Lowell BB, Barsh GS, de LC, Myers MG, Jr., Schwartz GJ, Chua SC., Jr. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, Elghazi L, Roe MW, Labosky PA, Myers MG, Jr., Gannon M, Powers AC, Dempsey PJ. Conditional gene targeting in mouse pancreatic beta-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59:3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 1999;23:32–40. doi: 10.1038/12631. [DOI] [PubMed] [Google Scholar]
- Xu Y, Hill JW, Fukuda M, Gautron L, Sohn JW, Kim KW, Lee CE, Choi MJ, Lauzon DA, Dhillon H, Lowell BB, Zigman JM, Zhao JJ, Elmquist JK. PI3K signaling in the ventromedial hypothalamic nucleus is required for normal energy homeostasis. Cell Metab. 2010;12:88–95. doi: 10.1016/j.cmet.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.