

Abstract
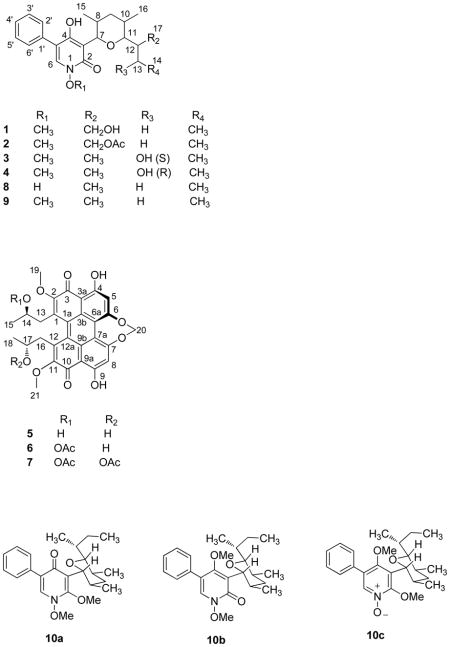
Four new 1,4-dihydroxy-5-phenyl-2-pyridinone alkaloids, 17-hydroxy-N-(O-methyl) septoriamycin A (1), 17-acetoxy-N-(O-methyl)septoriamycin A (2), 13-(S)-hydroxy-N-(O-methyl)septoriamycin A (3) and 13-(R)-hydroxy-N-(O-methyl)septoriamycin A (4), together with the known compounds (+)-cercosporin (5), (+)-14-O-acetylcercosporin (6), and (+)-di-O-acetylcercosporin (7), lumichrome, and brassicasterol were isolated from an ethyl acetate extract of a culture medium of Septoria pistaciarum. Methylation of septoriamycin A (8) with diazomethane yielded three di-O-methyl analogues, two of which existed as mixtures of rotamers. We previously reported antimalarial activity of septoriamycin A. This compound also exhibited significant activity against Leishmania donovani promastigotes. Compounds 5–7 showed moderate in vitro activity against L. donovani promastigotes and chloroquine-sensitive (D6) and -resistant (W2) strains of Plasmodium falciparum, whereas compound 5 was fairly active against methicillin-sensitive and methicillin-resistant strains of Staphylococcus aureus. Compounds 5–7 also displayed moderate phytotoxic activity against both a dicot (lettuce, Lactuca sativa) and a monocot (bentgrass, Agrostis stolonifera), and cytotoxicity against a panel of cell lines.
Phytotoxins from plant pathogenic fungi may act as inhibitors of plant-like metabolic pathways in the apicoplast, a chloroplast-like organelle, essential for the survival of plasmodium species.1 Previously we reported2 the isolation and identification of the 2-pyridinone alkaloid, septoriamycin A3 (8) and three of its derivatives from the causative agent of septoria leaf spot disease in pistachio, Septoria pistaciarum (Ascomycetes), as part of our program to identify antimalarial compounds from plant pathogenic fungi. Although we detected several minor alkaloids of the same class in the original extract, their structural elucidation was not possible due to insufficient quantities. In order to isolate the minor compounds, we recultured the same fungus under the identical conditions at a larger scale. In the ethyl acetate extract of this fermentation culture broth, a series of pigments were detected with 2-pyridinone alkaloidal architecture. The extract showed herbicidal, antimicrobial, antiplasmodial, and antileishmanial activities albeit with low selectivity indices. From this extract, four more new minor septoriamycin A analogues, 17-hydroxy-N-(O-methyl)septoriamycin A (1), 17-acetoxy-N-(O-methyl) septoriamycin A (2), 13-(S)-hydroxy-N-(O-methyl)septoriamycin A (3), and 13-(R)-hydroxy-N-(O-methyl)septoriamycin A (4), in addition to the parent and its previously reported analogues were identified. Septoriamycin A (8), the major 2-pyridinone alkaloid in this extract showed significant antileishmanial activity in addition to its reported antiplasmodial activity. The pigments were identified as three known perylenequinones, (+)-cercosporin (5),4 (+)-14-O-acetylcercosporin (6),4 and (+)-di-O-acetylcercosporin (7).5 These pigments have previously been identified as phytotoxins produced by a number of phytopathogenic Cercospora species and have been linked to their pathogenicity.6 Their biosynthesis appeared to be controlled by numerous environmental and physiological factors and the presence of even small amounts of certain compounds in the medium was found to have a strong stimulatory or inhibitory effect on their production.6,7 Their ability to generate reactive oxygen species in the presence of light has been attributed to their phytotoxic activity.6 Cercosporin and its esters have also been reported to have antibacterial and antifungal8 activities as well as growth inhibitory effects on lettuce4 and tomato seeds.8 In this study, the perylenequinones showed antileishmanial, antiplasmodial, and cytotoxic activities in addition to antibacterial and antifungal activities. Two more known compounds, lumichrome,9 and brassicasterol10 were also isolated and identified.
RESULTS AND DISCUSSION
Fractionation of an EtOAc extract of a culture medium of S. pistaciarum by Sephadex LH-20 gel column chromatography followed by purification using silica gel and RP C18 chromatography afforded four minor 2-pyridinone alkaloids 1–4 in addition to the known septoriamycin A (8) and its three derivatives,2 three known perylenequinones (+)-cercosporin (5), (+)-14-O-acetylcercosporin (6), (+)-di-O-acetyl-cercosporin (7), lumichrome, and brassicasterol.
The molecular formula of compound 1 was determined as C23H31NO5 by HRESIMS. UV maxima (205.0, 240.9, and 297.0 nm) and IR absorptions (3400, 3207, 1647, and 1555 cm−1) of 1 were consistent with those of a 2-pyridinone moiety.2 The aromatic regions of the 1H NMR and COSY spectra were similar to those observed for 5-phenyl-2-pyridinones previously isolated from this species, and consisted of a one-proton singlet and an A2B2C spin system due to a monosubstituted phenyl group.2 The aliphatic region exhibited two oxymethine and two methyl doublets indicating the presence of a 2,4-dimethyltetrahydropyran moiety and an N-methoxy group. HMBC correlations of H-2′ and H-6′ (δH 7.43) with C-3′ and C-5′ (δC 128.6), C-4′ (δC 127.8), C-1′ (δC 133.3), and C-5 (δC 114.5), and H-6 (δH 7.44) with C-2 (δC 158.0), C-4 (δC 161.9), C-5 (δC 114.5), and C-1′ (δC 133.3) supported the partial structure of the substituted pyridinone ring moiety. HMBC correlations of the H-7 oxymethine doublet (δH 4.73) with C-4 (δC 161.9), C-2 (δC 158.0), C-3 (δC 111.6), C-11 (δC 86.3), C-9 (δC 36.4), C-8 (δC 39.8), and C-15 (δC 17.8) supported the partial structure of the tetrahydropyran moiety. The major difference in the rest of the 1H NMR resonances of compound 1 and the reported2 N-(O-methyl) septoriamycin A (9) is the replacement of the methyl doublet in the chain attached to C-11 of the tetrahydropyran ring by two diastereotopic oxymethylene hydrogens. This indicated that compound 1 was the C-17 oxygenated analogue of 9. The COSY spectrum of compound 1 displayed cross peaks between the C-17 oxymethylene (δH 3.60, 3.49) and C-12 methine proton (δH1.87). In the HMBC spectrum the oxymethylene protons showed cross peaks with C-11 (δC 86.3), C-12 (δC 36.5), and C-13 (δC 22.3), and the H-11 oxymethine doublet at δH 3.36 with C-7 (δC 81.2), C-9 (δC 36.4), C-12 (δC 36.5), C-13 (δC 22.3), C-16 (δC 12.6), and C-17 (δC 63.6) further confirming that compound 1 was 17-hydroxy-N-(O-methyl)septoriamycin A. (Figure 1)
Figure 1.

COSY, HMBC, and ROESY correlations of compound 1.
The HRESIMS data established the molecular formula of compound 2 as C25H33NO6. The 1H and 13C NMR spectra were similar to those of 1 except for the presence of additional resonances [(δH 2.06) δC 21.1 (CH3), δC 171.2 (CO)] originating from an O-acetyl group. HMBC correlations between the C-12 oxymethylene group and the acetyl carbonyl carbon indicated that compound 2 was 17-O-acetyl-N-(O-methyl)septoriamycin A. The remaining HMBC and COSY correlations were identical to those of compound 1. Both 3J7,8 and 3J10,11 values were 10.2 Hz indicating that these protons are trans – diaxially oriented. ROESY correlations of compound 1 (Figure 1) and 2 were identical to those observed for septoriamycin A (8) suggesting that these two compounds had the same relative configurations. Since we have previously assigned the absolute configuration of septoriamycin A on the basis of X-ray diffraction data,2 and all these compounds presumably share a common biosynthetic origin, compounds 1 and 2 also have 7R, 8R, 10S, 11R, and 12R absolute configuration. It is further supported by their dextrorotatory specific rotations.
The HRESIMS data of 3 established its molecular formula as C23H31NO5. Comparison of the NMR spectra of 3 with those of 8 showed that the major difference was the replacement of a methyl triplet (δH 0.89) of the latter by a methyl doublet (δH 1.16) and an oxymethine doublet (δH 3.78) in the former. These changes could be attributable to the substitution of one of the C-13 diastereotopic methylene hydrogens in 8 with a hydroxy group indicating that compound 3 is the 13-hydroxy analogue of N-(O-methyl)septoriamycin A (9). The methyl doublet at δH 0.93 (17-CH3) showed HMBC cross peaks with C-11 (δC 88.9), C-12 (δC 41.4), and C-13 (δC 68.9) and the oxymethine multiplet at δH 3.78 with C-11 (δC 88.9), C-12 (δC 41.4), and C-17 (δC 15.9) confirming the C-13 location of the secondary hydroxy group and, thus, compound 3 was 13-hydroxy-N-(O-methyl)septoriamycin A.
ROESY data and 1H NMR coupling constants of compound 3 showed close correlations to those reported2 for septoriamycin A (8). Our attempts to determine the absolute configuration at C-13 by Mosher analysis were unsuccessful. Treatment of compound 3 with the R- and S-Mosher’s acid chlorides afforded a mixture not worth resolving. Methylation prior to acylation also yielded a mixture of products. The absolute configuration of compound 3 was proposed by a 3J12, 13 - based comparison of compounds 3 and 4 (vide infra).
Compound 4 had the same molecular formula, C23H31NO5, as that of 3 based on HRESIMS data. The 1H NMR spectra of these compounds were similar except for the down-field shift of the oxymethine proton (δH 4.20) and the up-field shift of a methyl doublet (δH 1.00). The COSY and HMBC spectra of compound 4 showed the same correlations as those observed for compound 3 suggesting that both compounds have the same gross structure. Identical coupling constants and ROESY correlations for the protons in the tetrahydrofuran moiety indicated that they had the same relative configuration except at C-13. Since compounds 3, 4, and 8 presumably share a common biosynthetic origin, compounds 3 and 4 also have 7R, 8R, 10S, 11R, 12R absolute configuration. As described earlier for compound 3, our attempts to determine the absolute configuration of the C-13 stereogenic center by Mosher analysis were unsuccessful. Thus, we used a J-based approach relying on 1H NMR coupling constants combined with ROESY correlations, to assign the C-13 absolute configuration of compounds 3 and 4. An observed 3J12, 13 value of 7.2 Hz of 3, suggested a dihedral angle of ca 30° or 150° between H-12 and H-13. Eight rotamers are possible for 3 with these dihedral angles for the S (3a – 3d) and R (3e – 3h) epimers (Figure 2). Observed ROESY correlations between H-12 and H-13, H-13 and CH3-17, and CH3-14 – CH3-17 ruled out all conformers except 3a as the probable most abundant rotamer for compound 3 in solution indicating a 13S absolute configuration. Similarly, an observed 3J12, 13 of 1.0 Hz for 4 supported the fact that the dihedral angle between H-12 and H-13 was 90° as shown in Figure 2 for the 13S (4a – 4b) and R (4c – 4d) epimers. In the ROESY spectrum, H-12 showed correlation with H-13 and CH3-14 and absence of interaction between CH3-14 and CH3 -17 indicating 4a as the dominant rotamer and, hence, 13R as absolute configuration.
Figure 2.

Rotamer representation of compounds 3 and 4
Methylation of compounds 3 and 4 with diazomethane afforded several products. Treatment of septoriamycin A (8) with diazomethane as a model gave three products, whereas methylation with MeI and Cs2CO3 afforded a single compound which was identified as analogue 9. The products of diazomethane methylation of septoriamycin A were separated by chromatography. All these products had the same molecular formula, C24H33NO4, by HRESIMS, suggesting that they were di-O-methyl derivatives. The 1H NMR spectrum of the least polar compound (10a) was similar to that of 9 except for the presence of an additional O-methyl resonance in the former. The 13C NMR spectrum of this compound showed changes in resonances in the pyridone ring. A down-field shift of C-4 (δC 177) indicated that the carbonyl group now resided at this carbon suggesting 10a was N,2-di-O-methylseptoriamycin A. A hypsochromic shift of the UV absorption (λmax 278.9 nm) when compared to 9 (λmax 295 nm)11 and COSY and HMBC correlations further supported this structure. The 1H and 13C NMR spectra of compound 10b indicated that it existed as a 3:1 mixture of two rotamers about the C-3 – C-7 bond. Their 1H and 13C NMR spectra were similar to those of compound 9 but had an additional methoxy resonance. HMBC correlations showed that the additional methoxy group correlated to C-4 indicating that 10b was N,4-di-(O-methyl)septoriamycin A. At 100 °C in pyridine, duplicated 1H NMR resonances coalesced but were restored to the original ratio when the temperature returned to ambient conditions. Atropisomers of 3-cyclohexyl- or 3-cycloheptyl-N,4-dihydroxy-2-pyridone alkaloids have previously been isolated from several fungal species.11–15 Even though a number of 3-pyrano-N,4-dihydroxy-2-pyridone alkaloids similar to septoriamycin A analogues have been identified16 from fungal sources, none have been reported to exist as rotamers. The introduction of a 4-methoxy group appears to cause restricted rotation about the C-3-C-7 bond giving rise to rotamers. The most polar compound (10c) showed ionic characteristics. 1H and 13C NMR data indicated that the compound also existed as a 3:1 mixture of two diastereomeric rotamers. HMBC correlations showed that the methoxy resonance correlated to C-2 and C-4 suggesting that 10c is the 2,4-di-O-methylpyridinone-N-oxide analogue of septoriamycin A. The presence of the 4-methoxy group appears to cause restricted rotation about the C-3-C-7 bond. At 100 °C in pyridine duplicated 1H NMR resonances coalesced. Even though the original ratio of rotamers reappeared at ambient temperature some decomposition of 10c was also observed.
The known perylenequinones, (+)-cercosporin (5),4 (+)-14-O-acetylcercosporin (6),4,5 and (+)-di-O-acetylcercosporin (7),5 lumichrome,9 and brassicasterol,10 were identified by comparing their spectroscopic data with literature data. The absolute configurations of 5–7 were determined by comparing experimental and reported electronic circular dichroism data.17–18 The helicity of compounds 5–7 was confirmed as M (aS) and the absolute configuration of both C-14 and C-17 as R.19
Compounds 1–4 showed no antimicrobial, antifungal, antiprotozoal, phytotoxic, or cytotoxic activity in vitro. Compounds 5–7 showed moderate in vitro antiplasmodial activity (Table 3) but were cytotoxic to Vero cells. Their low selectivity indices (ratio of cytotoxicity vs. antiplasmodium activity) preclude them as antimalarial drug leads. Even though the plant pathogen S. pistaciarum is host-specific to pistachio, compounds 5–7 showed nonspecific moderate phytotoxic activity towards both bentgrass (A. stolonifera) and lettuce (L. sativa cv. L., Iceberg) in the presence of light (Table 4). General phytotoxcity of phytotoxins from host-specific pathogens is very common. Biosynthesis of cercosporin (5) appeared to be controlled by numerous environmental and physiological factors and their production has been linked to the pathogenicity of fungi.6,7 The possible mechanism of phytotoxic activity of this type of compounds has previously been attributed to their ability to generate reactive oxygen species in the presence of light.6 This suggested that the selective inhibition of the plant-like metabolic pathways in the apicoplast of malaria parasite1 is not responsible for the observed antimalarial activity of compounds 5–7. Septoriamycin A (8), with demonstrated antiplasmodial and antifungal activities,2 exhibited significant antileishmanial activity with an IC50 of 0.11 μM and an IC90 of 0.29 μM (Table 5) and was more potent than the positive controls pentamidine and amphotericin B. Compounds 5–7 also showed significant antileishmanial activity with IC50 values of 1.14 μM, 1.7 μM, and 3.1 μM, respectively (Table 5).
Table 3.
Antiplasmodial Activity of Compounds 5–7 and 10c
| Compound | Chloroquine-sensitive (D6)-clone | Chloroquine-resistant (W2)-clone | Cytotoxicity to Vero cells | ||
|---|---|---|---|---|---|
| IC50 μM | S.I. | IC50 μM | S. I. | IC50 μM | |
| 5 | 1.08 | 4.8 | 1.62 | 3.2 | 5.24 |
| 6 | 2.78 | 1.9 | 3.12 | 1.7 | 5.21 |
| 7 | 2.75 | 1.6 | 1.94 | 2.3 | 4.53 |
| 10c | 6.76 | >1.1 | 6.51 | >1.2 | NC |
| chloroquinea | 0.03 | 0.31 | NC | ||
| artemisinina | 0.02 | 0.01 | NC | ||
Positive controls
NC not cytotoxic
S. I. (selectivity index) = IC50 for cytotoxicity/IC50 for antiplasmodial activity
IC50: concentration causing 50% growth inhibition
Table 4.
Phytotoxic Activity of Compounds 5–7
| Compound | Concentration (mM) | lettuce | bentgrass |
|---|---|---|---|
| 5 | 1.87 | 3 | 2 |
| 6 | 1.73 | 2 | 4 |
| 7 | 1.62 | 3 | 4 |
Concentration (mM) = 1 mg/mL
Ranking based on scale of 0 to 5
0 = no effect
5 = no growth
Table 5.
Antileishmanial Activity of Compounds 5–8
| Compound | IC50 μM | IC90 μM |
|---|---|---|
| 5 | 1.14 | 2.81 |
| 6 | 1.7 | 8.5 |
| 7 | 3.1 | 9.7 |
| 8 | 0.11 | 0.29 |
| pentamidinea | 2.9 | 5.58 |
| amphotericin Ba | 0.18 | 0.38 |
Positive controls
IC50 concentration causing 50% growth inhibition
IC90 concentration causing 90% growth inhibition
Compound 5 was moderately active against both methicillin-sensitive and methicillin-resistant Staph. aureus with MIC values 2.5 and 5.0 Ng/mL (4.7 and 9.4 NM), respectively. In the same assay, the positive control, ciprofloxacin had a MIC value of 0.37 Ng/mL (1.11 NM) against both methicillin-sensitive and methicillin-resistant Staph. aureus.
The cytotoxic potential of compounds 5–7 was further evaluated against a panel of human solid tumor cell lines (SK-MEL, KB, BT-549, SK-OV-3) and pig kidney epithelial cells (LLC-PK11) (Table 6). Moderate cytotoxicity was observed against all the cell lines. The di-O-methyl derivatives of septoriamycin A, 10a, 10b, and 10c were also evaluated for the above activities and compound 10c exhibited weak antiplasmodial activity (Table 3).
Table 6.
Cytotoxic Activity [IC50 (μM)] of Compounds 5–7
| SK-MEL | KB | BT-549 | SK-OV-3 | LLC-PK11 | |
|---|---|---|---|---|---|
| 5 | 3.6 | 7.1 | 10.8 | 3.7 | 3.9 |
| 6 | 3.8 | 8.5 | 8.6 | 4.0 | 10.1 |
| 7 | 4.9 | 8.7 | 8.7 | 4.8 | 9.4 |
| doxorubicina | 1.6 | 2.6 | 2.6 | 1.5 | 1.6 |
Positive control.
IC50 = concentration causing 50% growth inhibition
SK-MEL = human malignant melanoma
KB = human epidermal carcinoma
BT-549 = human breast carcinoma (ductal)
SK-OV-3 = human ovary carcinoma
LLC-PK11 = pig kidney epithelial
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were obtained using a Rudolph Research Analytical Autopol IV automatic polarimeter model 589-546. Melting points were measured with a Uni-melt, Thomas Hoover capillary melting point apparatus. UV and IR spectra were determined on a Varian-50 Bio UV visible spectrophotometer and a Bruker-Tensor-27 infrared spectrophotometer, respectively. NMR spectra were recorded on a Varian-Mercury-plus-400 or Varian Unity-Inova-600 spectrometer using CDCl3 and methanol-d4 unless otherwise stated. MS data were obtained from an Agilent Series 1100 SL equipped with an ESI source (Agilent Technologies, Palo Alto, CA, USA). Column chromatography and preparative TLC were carried out using Merck silica gel 60 (230–400 mesh) and silica gel GF plates (20 × 20 cm, thickness 0.25 mm), respectively. HPLC analysis was conducted on an Hewlett-Packard Agilent 1100 with diode array detector.
Fermentation, Extraction, and Isolation
The plant pathogenic fungus, Septoria pistaciarum Caracc. (ATCC 22201) was obtained from the American Type Culture Collection, Manassas, VA and it was grown as previously reported.2
The oily extract (3.3 g) was chromatographed over Sephadex LH-20 and eluted with 80% MeOH in CHCl3 to give 16 fractions. Fractions 2–11 which showed antiplasmodial activity and were combined and chromatographed over a reversed phase C18 column eluting with a gradient of 10 to 100% MeOH-H2O to yield eight fractions. Subfractions 2, 3, and 4 were combined and separated on Sephadex LH-20 (MeOH) to give compounds 1 (5.0 mg) and 8 (35 mg). Subfraction 5 was chromatographed over a silica gel column using CH2Cl2-hexanes as the eluent to give compound 2 (8.0 mg). Combined fractions 6, 7, and 8 were chromatographed over a silica gel column using CH2Cl2-hexanes as the eluent to give 10 subfractions. Subfraction 2, 3, and 5 were combined and further separated using a C18 reversed phase HPLC column eluting with MeCN:H2O, (1:1), flow rate 3.5 mL/min, to give compounds 3 (3.5 mg) and 4 (8.0 mg). Subfractions 6, 7, and 8 were combined and purified on Sephadex LH-20 (MeOH) to give compounds 5 (14.0 mg), 6 (12.0 mg), and lumichrome (3.0 mg). Subfractions 9 and 10 were combined and chromatographed over a silica gel column using CH2Cl2-hexanes as the eluent to give compound 7 (10.0 mg) and brassicasterol (8.0 mg).
Compound 1
amorphous powder: [α]26D +112 (c 0.05, MeOH); UV (MeOH); λmax (log ε) 205.0 (3.56), 240.9 (3.51), 297.0 (2.79) nm; IR (CHCl3 ) νmax 3400, 3207, 2967, 2929, 1647, 1555, 1454, 1049 cm−1; 1H and 13C NMR data (see Table 1); HRESIMS [M − H]+ m/z 400.2118 (calcd for [C23H31NO5 − H]+, 400.2124).
Table 1.
1H, and 13C, NMR Data for Compounds 1–4 in CDCl3/Methanol-d4.
| 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|
| position | δCb | δHa(J in Hz) | δCb | δHa(J in Hz) | δCb | δHa(J in Hz) | δCb | δHa(J in Hz) |
| 2 | 158.0 | 158.0 | 158.0 | 158.2 | ||||
| 3 | 111.6 | 111.4 | 112.0 | 112.0 | ||||
| 4 | 161.9 | 161.8 | 161.6 | 161.7 | ||||
| 5 | 114.5 | 114.3 | 114.5 | 114.8 | ||||
| 6 | 133.0 | 7.43, s | 133.1 | 7.44, s | 132.9 | 7.42, s | 132.8 | 7.43, s |
| 7 | 81.2 | 4.73, d (10.2) | 81.3 | 4.74, d (10.2) | 80.8 | 4.70, d (10.2) | 80.9 | 4.66, d (10.2) |
| 8 | 39.8 | 1.87, m | 36.4 | 1.91, m | 36.2 | 1.91, m | 36.7 | 1.91, m |
| 9 | 36.4 | 1.34, q (12.6) | 36.5 | 1.30, q (12.6) | 42.6 | 1.06, q (12.6) | 42.2 | 1.16, q (12.0) |
| 1.98, brd (13.2) | 2.00, (overlap) | 1.80, dt (13.8, 3.6) | 1.84, dt (13.2, 3.6) | |||||
| 10 | 35.8 | 1.69, m | 36.0 | 1.64, m | 34.1 | 2.03, m | 32.9 | 1.89, m |
| 11 | 86.3 | 3.36, d (10.2) | 86.2 | 3.33, d (10.2) | 88.9 | 3.14, d (10.2) | 88.5 | 3.12, dd (10.2, 2.8) |
| 12 | 36.5 | 1.87, m | 37.0 | 2.00, m | 41.4 | 2.05, m | 38.7 | 1.75, m |
| 13 | 22.3 | 1.02, m, 1.58, m | 22.3 | 1.02, m, 1.55, m | 68.9 | 3.78, m (7.2, 7.2) | 66.0 | 4.20, m (6.6, 1.0) |
| 14 | 12.6 | 0.88, t (7.2) | 12.6 | 0.89, t (7.2) | 23.1 | 1.16, d (6.6) | 22.8 | 1.16, d (6.3) |
| 15 | 17.8 | 0.91, d (6.6) | 17.7 | 0.91, d (7.2) | 17.9 | 0.85, d (6.6) | 17.9 | 0.87, d (6.6) |
| 16 | 17.1 | 0.95, d (6.6) | 17.0 | 0.97, d (7.2) | 17.7 | 0.83, d (6.6) | 17.7 | 0.84, d (6.6) |
| 17 | 63.6 | 3.49, dd (10.6, 3.6) | 65.3 | 3.91, dd (11.2, 5.4) | 15.9 | 0.93, d (7.2) | 10.5 | 1.01, d (7.2) |
| 3.60, dd (10.6, 3.6) | 3.98, dd (11.6, 4.5) | |||||||
| 1′ | 133.3 | 133.4 | 133.4 | 133.7 | ||||
| 2′, 6′ | 129.2 | 7.44, d (7.3) | 129.3 | 7.44, d (7.3) | 129.2 | 7.43, d (7.3) | 129.4 | 7.43, d (7.3) |
| 3′, 5′ | 128.6 | 7.39, t (7.2) | 128.6 | 7.40, t (7.3) | 128.5 | 7.38, t (7.8) | 128.6 | 7.36, d (7.2) |
| 4′ | 127.8 | 7.32, t (7.2) | 127.9 | 7.34, t (7.3) | 127.7 | 7.32, t (7.2) | 127.8 | 7.30, t (7.2) |
| OH | 9.63, s | 9.47, s | 9.63, s | 9.65, s | ||||
| OCH3 | 65.0 | 4.04, s | 65.0 | 4.06, s | 65.0 | 4.04, s | 65.0 | 4.05, s |
| COCH3 | 21.1 | 2.06, s | ||||||
| COCH3 | 171.2 | |||||||
1H NMR spectra recorded at 600 MHz,
13C NMR spectra recorded at 100 MHz
Compound 2
amorphous powder: [α]26D +101 (c 0.05, MeOH); UV (MeOH); UV (MeOH) λmax (log ε) 204.0 (3.44), 240.0 (3.25), 295.9 (2.56) nm; IR (CHCl3 ) νmax 3331, 2967, 2925, 1740, 1651, 1230, 1045 cm−1; 1H and 13C NMR data (see Table 1); HRESIMS [M + H]+ m/z 444.2370 (calcd for [C25H33NO6 + H]+, 444.2386).
Compound 3
amorphous powder: [α]26D −38 (c 0.43, CH3OH); UV (MeOH); λmax (log ε) 208.2 (3.94), 241.9 (3.92), 297.0 (3.19) nm; IR (CHCl3 ) νmax 3405, 3201, 2965, 2929, 1644, 1551, 1455, 1219, 755 cm−1; 1H and 13C NMR data (see Table 1); HRESIMS [M + H + Na]+ m/z 425.2172 (calcd for [C23H31NO5 + H + Na ]+, 425.2178).
Compound 4
amorphous powder: [α]26D +130 (c 0.2, MeOH); UV (MeOH); λmax (log ε) 207.1 (3.01), 241.0 (2.95), 295.1 (2.10) nm; IR (CHCl3 ) νmax 3422, 3159, 2926, 1641, 1541, 1456 cm−1; 1H and 13C NMR data (see Table 1); HRESIMS [M + H + Na]+ m/z 425.2174 (calcd for [C23H31NO5 + H + Na ]+, 425.2178).
Methylation of Septoriamycin A (8) with MeI
A mixture of MeI (4 mL), Cs2CO3 (10.0 mg), and compound 8 (10.0 mg) in acetone was stirred at room temperature for 5 h. The reaction mixture was filtered, and the solvent was evaporated. The product was dissolved in CH2Cl2 and passed through a plug of florisil to give N-(O-methyl)septoriamycin A (9).
Methylation of Septoriamycin A (8) with Diazomethane
A solution of 8 (60.0 mg) in MeOH was treated with excess diazomethane in Et2O at 0 °C for 2 h. The solvent was evaporated and the mixture was separated by PTLC (40% EtOAc in hexanes) to yield compounds 10a (8 mg), 10b, and 10c. Compounds 10b and 10c were further purified by HPLC using a reversed phase Luna C18 column (1×25 cm) with MeOH: H2O (92:8) as the mobile phase at a flow rate of 4 mL/min, to give compounds 10b (5 mg) and 10c (6.0 mg).
Compound 10a
amorphous powder: [α]26D +14 (c 0.5, MeOH); UV (MeOH); λmax (log ε) 205.0 (3.75), 232.0 (3.68), 278.9 (3.41) nm; IR (CHCl3 ) νmax 2959, 2929, 2873, 1627, 1547, 1469, 1046 cm−1; 1H and 13C NMR data (see Table 2); HRESIMS [M + Na]+ m/z 422.2307 (calcd for [C24H33NO4 + Na]+, 422.2307).
Table 2.
1H and 13C NMR Data Methylated Compounds 10a, 10b, and 10c in CDCl3/Methanol-d4.
| 10a | 10b | 10c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| position | δCb | δHa (J in Hz) | δCb | δHa(J in Hz) | δCb | δHa(J in Hz) | ||||
| 2 | 155.0 | 159.2 | 157.2 | 158.0 | 156.0 | |||||
| 3 | 119.5 | 124.3 | 122.8 | 125.6 | 125.3 | |||||
| 4 | 177.0 | 164.9 | 164.8 | 155.7 | 157.0 | |||||
| 5 | 134.6 | 117.2 | 115.9 | 128.5 | 128.5 | |||||
| 6 | 132.7 | 7.51, s | 133.4 | 134.0 | 7.44, s | 7.49, s | 139.6 | 139.7 | 8.15, s | 8.14, s |
| 7 | 78.4 | 4.63, d (10.2) | 79.4 | 79.5 | 4.56, d (10.2) | 4.21, d (10.2) | 79.4 | 80.7 | 4.33, d (10.4) | 4.33, d (10.4) |
| 8 | 36.3 | 2.0, m | 33.8 | 33.8 | 2.3, m | 2.3, m | 32.6 | 32.9 | 2.44, m | 2.52, m |
| 9 | 43.4 | 1.78, m | 43.2 | 43.1 | 1.79, m | 1.84, m | 43.1 | 43.1 | 1.86, m | 1.86, m |
| 1.02, m | 1.04, m | 1.06, m | 0.96, m | 0.96, m | ||||||
| 10 | 33.7 | 1.69, m | 32.3 | 31.9 | 1.72, m | 1.72, m | 32.4 | 32.5 | 1.88, m | 1.88, m |
| 11 | 89.2 | 2.92, d (12.0) | 89.0 | 88.9 | 2.97, d (12.0) | 2.87, d (12.0) | 89.8 | 90.0 | 2.92, d (10.0) | 2.94, d (10.0) |
| 12 | 34.2 | 1.54, m | 36.1 | 36.7 | 1.57, m | 1.57, m | 36.0 | 36.0 | 1.60, m | 1.60, m |
| 13 | 22.8 | 1.54, m | 22.6 | 21.9 | 1.53, m | 1.53, m | 22.5 | 22.5 | 1.5, m | 1.5, m |
| 0.82, m | 0.85, m | 0.85, m | 1.1, m | 1.1, m | ||||||
| 14 | 13.0 | 0.84, t (7.2) | 12.7 | 12.4 | 0.84, t (6.6) | 0.84, t (6.6) | 12.9 | 12.8 | 0.87, t (6.6) | 0.87, t (6.6) |
| 15 | 17.6 | 0.68, d (6.6) | 17.5 | 17.8 | 0.80, d (6.6) | 0.80, d (6.6) | 17.6 | 17.6 | 0.81, d (6.6) | 0.81, d (6.6) |
| 16 | 17.7 | 0.77, d (6.6) | 17.3 | 17.6 | 0.75, d (6.6) | 0.68, d (6.6) | 17.7 | 17.8 | 0.64, d (6.4) | 0.67, d (6.6) |
| 17 | 17.0 | 0.89, d (6.6) | 16.8 | 17.0 | 0.91, d (6.6) | 0.91, d (6.6) | 17.2 | 17.2 | 0.90, d (6.6) | 0.90, d (6.6) |
| 1′ | 126.6 | 133.8 | 133.8 | 133.0 | 133.0 | |||||
| 2′, 6′ | 128.9 | 7.56, d (7.2) | 128.6 | 128.5 | 7.43, d (7.2) | 7.43, d (7.2) | 129.0 | 128.9 | 7.48, d (6.8) | 7.48, d (7.5) |
| 3′, 5′ | 128.3 | 7.33, t (7.2) | 128.4 | 128.5 | 7.39, t (7.2) | 7.39, t (7.2) | 128.9 | 128.7 | 7.42, t (6.8) | 7.42, d (7.5) |
| 4′ | 127.8 | 7.25, t (7.2) | 127.6 | 127.5 | 7.34, t (7.2) | 7.34, t (7.2) | 129.8 | 129.8 | 7.39, t (7.3) | 7.39, t (7.3) |
| OCH3 | 66.8 | 4.01, s | 64.5 | 64.5 | 4.08, s | 4.08, s | 61.7 | 62.1 | 3.36, s | 3.37, s |
| OCH3 | 64.4 | 4.0, s | 61.5 | 61.2 | 4.01, s | 3.34, s | 60.6 | 60.4 | 4.16, s | 4.14, s |
1H NMR spectra recorded at 600 MHz,
13C NMR spectra recorded at 100 MHz
Compound 10b
amorphous powder: UV (MeOH) λmax (log ε) 204.0 (3.39), 235.0 (3.18), 308.0 (2.65) nm; IR (CHCl3) νmax 2958, 2929, 1740, 1656, 1524, 1457 cm−1; 1H and 13C NMR data (see Table 2); HRESIMS [M +H] + m/z 400.2472 (calcd for [C24H33NO4 + H]+, 400.2487).
Compound 10c
amorphous powder: UV (MeOH) λmax (log ε) 207.0 (2.58), 240.9 (2.47), 307.1 (1.70) nm; IR (CHCl3) νmax 2900, 2850, 1575, 1450 cm−1; 1H and 13C NMR data (see Table 2); HRESIMS [M + Na] + m/z 422.2279 (calcd for [C24H33NO4 + Na]+, 422.2307).
Compound 5
Red crystals: mp 239 °C (lit.17 240–241.5 °C); UV (MeOH) λmax (log ε) 210.0 (3.71), 221.0 (3.72), 267.0 ( 3.55), 470.0 (3.42), 563.0 (2.98) nm; IR (CHCl3) νmax 3396, 2924, 1616, 1267 cm−1 1H and 13C NMR and CD data were consistent with those reported.4,17,19
Compound 6
Red crystals: mp 135 °C (lit.5 134 °C); UV (MeOH) λmax (log ε) 222.0 (3.9), 269.0( 3.74), 471.0 (3.62), 563.0 (3.2) nm; IR (CHCl3) νmax 3338, 1735, 1616, 1266 cm−1 1H and 13C NMR and CD data data were consistent with those reported.4,5
Compound 7
Red amorphous powder: UV (MeOH) λmax (log ε) 222.0 (3.9), 270.0 ( 3.84), 470.1 (4.0), 563.3 (3.26) nm; IR (CHCl3) νmax 2921, 1736, 1617, 1211 cm−1 1H and 13C NMR and CD data were consistent with those reported.5
Antiplasmodial Assay
The antiplasmodial activity was determined against D6 (chloroquine sensitive) and W2 (chloroquine resistant) strains of Plasmodium falciparum in an in vitro assay as described earlier.20 Artemisinin and chloroquine were included as the drug controls and IC50 values were computed from the dose response curves using Microsoft Excel software.
Phytotoxicity Assay
The bioassay for phytotoxicity was carried out according to the procedure described by Dayan et. al.21 using bentgrass (Agrostis stolonifera) and lettuce (Lactuca sativa cv. L., Iceberg), in 24-well plates. Test compounds (1 mg each) were dissolved in 100 μL of acetone and a 20 μL aliquot of each solution was pipetted onto the filter paper and dried for 30 min by airflow in a sterile biohazard hood. Water (200 μL) was added after placing the dried and sample impregnated filter paper in the well. The solvent controls were treated identically, using the solvent described above. Phytotoxicity was ranked visually. The ranking of phytotoxic activity was based on a scale of 0 to 5 with 0 showing no effect and 5 no growth.
Antileishmanial Assay
The in vitro antileishmanial activity of the compounds was carried out on a culture of Leishmania donovani promastigotes as described earlier.22 Pentamidine and amphotericin B were used as standard antileishmanial agents. The IC50 values for each compound were computed from the growth inhibition curve using Microsoft Excel software.
Antimicrobial Assay
All organisms were obtained from the American Type Culture Collection (Manassas, VA) and included the fungi Candida albicans ATCC 90028, Candida glabrata ATCC 90030, Candida krusei ATCC 6258, Cryptococcus neoformans ATCC 90113, and Aspergillus fumigatus ATCC 90906, and the bacteria Staphylococcus aureus ATCC 29213, methicillin-resistant Staphylococcus aureus ATCC 33591 (MRSA), Escherichia coli ATCC 35218, Pseudomonas aeruginosa ATCC 27853, and Mycobacterium intracellulare ATCC 23068. Susceptibility testing was performed using a modified version of the CLSI methods23,24 as described by Samoylenko et. al.25 The drug controls ciprofloxacin (ICN Biomedicals, Ohio) for bacteria and amphotericin B (ICN Biomedicals, Ohio) for fungi were included in each assay.
Cytotoxicity Assay for Mammalian Cells
In vitro cytotoxicity was determined against a panel of mammalian cells that included kidney fibroblast (Vero), kidney epithelial (LLC-PK11), malignant melanoma (SK-MEL), oral epidermal carcinoma (KB), breast ductal carcinoma (BT-549), and ovary carcinoma (SK-OV-3) cells as described earlier.26 The number of viable cells was determined by using Neutral Red dye and IC50 values were obtained from dose response curves. Doxorubicin was used as a positive control.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R21 A1061431-01 and R01 AI 27094) and, in part, by the United States Department of Agriculture, ARS, Specific Cooperative Agreement No. 58-6408-2-009 and US DoD CDMRP Investigator Initiated Grant Award W81XWH-09-2-0093. We thank Dr. B. Avula and Mr. F. T. Wiggers, NCNPR, University of Mississippi, for recording the MS and 1H NMR spectra (600 MHz), and Ms. Marsha Wright, Mr. John Trott, Mr. Surendra Jain, and Mr. Robert Johnson for biological testing.
Footnotes
Supporting Information: NMR spectra of compounds 1–4, 10a, 10b, and 10c. This material is available free of charge via the Internet at http://pubs.acs.org
REFERNCES AND NOTES
- 1.Bajsa J, Singh K, Nanayakkara D, Duke SO, Rimando AM, Evidente A, Tekwani BL. Biol Pharm Bull. 2007;30:1740–1744. doi: 10.1248/bpb.30.1740. [DOI] [PubMed] [Google Scholar]
- 2.Kumarihamy M, Fronczek FR, Ferreira D, Jacob M, Khan SI, Nanayakkara NPD. J Nat Prod. 2010;73:1250–1253. doi: 10.1021/np1000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.We did not assign a trivial name for compound 8. This compound was recently synthesized by Fotiadou and Zografos and named septoriamycin A. Fotiadou AD, Zografos AL. Org Lett. 2011;13:4592–4595. doi: 10.1021/ol2017802.
- 4.Tabuchi H, Tajimi A, Ichihara A. Biosci Biotech Biochem. 1994;58:1956–1959. [Google Scholar]
- 5.Assante G, Locci R, Camarda L, Merlini L, Nasini G. Phytochemistry. 1997;16:243–247. [Google Scholar]
- 6.Daub ME, Ehrenshaft M. Annu Rev Phytopathol. 2000;38:461–490. doi: 10.1146/annurev.phyto.38.1.461. [DOI] [PubMed] [Google Scholar]
- 7.You BJ, Lee MH, Chung KR. Can J Microbiol. 2008;54:259–269. doi: 10.1139/w08-002. [DOI] [PubMed] [Google Scholar]
- 8.Lynch FJ, Geoghegan MJ. Trans Br Mycol Soc. 1979;72:31–37. [Google Scholar]
- 9.Ding ZG, Zhao JY, Yang PW, Li MG, Huang R, Cuio XL, Wen ML. Magn Reson Chem. 2008;47:366–370. doi: 10.1002/mrc.2393. [DOI] [PubMed] [Google Scholar]
- 10.Lee JW, Lee DY, Cho JG, Baek NI, Lee YH. J Appl Biol Chem. 2010;53:207–211. [Google Scholar]
- 11.Cai P, Smith D, Cunningham B, Brown-Shimer S, Katz B, Pearce C, Venables D, Houck D. J Nat Prod. 1999;62:397–399. doi: 10.1021/np980450t. [DOI] [PubMed] [Google Scholar]
- 12.Teshima Y, Shin-ya K, Shimazu A, Furihata K, Chul HS, Furihata K, Hayakawa Y, Nagai K, Seto H. J Antibiot. 1991;44:685–687. doi: 10.7164/antibiotics.44.685. [DOI] [PubMed] [Google Scholar]
- 13.Isaka M, Tanticharoen M, Kongsaeree P, Thebtaranonth Y. J Org Chem. 2001;66:4803–4808. doi: 10.1021/jo0100906. [DOI] [PubMed] [Google Scholar]
- 14.Wagenaar MM, Gibson DM, Clardy J Org Lett. 2002;4:671–673. doi: 10.1021/ol016737q. [DOI] [PubMed] [Google Scholar]
- 15.De Silva ED, Geiermann AS, Mitova MI, Kuegler P, Blunt JW, Cole ALJ, Munro MHG. J Nat Prod. 2009;72:477–479. doi: 10.1021/np800627f. [DOI] [PubMed] [Google Scholar]
- 16.Henning JJ, Gademann K. Nat Prod Rep. 2010;27:1168–1185. doi: 10.1039/b911516c. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki S, Ogawa Tadatomo. Agr Biol Chem. 1972;36:1707–1718. [Google Scholar]
- 18.Morgan BJ, Dey S, Johnson SW, Kozlowski MC. J Am Chem Soc. 2009;131:9413–9425. doi: 10.1021/ja902324j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan BJ, Mulrooney CA, Kozlowski MC. J Org Chem. 2010;75:44–56. doi: 10.1021/jo9013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bharate SB, Khan SI, Yunus NAM, Chauthe SK, Jacob MR, Tekwani BL, Khan IA, Singh IP. Bioorg Med Chem. 2007;15:87–96. doi: 10.1016/j.bmc.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Dayan FE, Romagni JG, Duke SO. J Chem Ecol. 2000;26:2079–2094. [Google Scholar]
- 22.Machumi F, Samoylenko V, Yenesew A, Derese S, Midiwo JO, Wiggers FT, Jacob MR, Tekwani BL, Khan SI, Walker LA, Muhammad I. Nat Prod Commun. 2010;5:853–858. [PMC free article] [PubMed] [Google Scholar]
- 23.NCCLS. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically M7-A5. National Committee on Clinical Laboratory Standards. 2000;20(2) [Google Scholar]
- 24.NCCLS. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Conidium-Forming Filamentous Fungi; Proposed Standard, M38-P. National Committee on Clinical Laboratory Standards. 1998;18(13) [Google Scholar]
- 25.Samoylenko V, Jacob MR, Khan SI, Zhao J, Tekwani BL, Midiwo JO, Walker LA, Muhammad I. Nat Prod Commun. 2009;4:791–796. [PMC free article] [PubMed] [Google Scholar]
- 26.Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Lipids. 2004;39:167–172. doi: 10.1007/s11745-004-1215-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.