

Abstract
Excessive NMDA receptor activation and excitotoxicity underlies pathology in many neuropsychiatric and neurological disorders, including hypoxia/ischemia. Thus, the development of effective therapeutics for these disorders demands a complete understanding of NMDA receptor (NMDAR) activation during excitotoxic insults. The extrasynaptic NMDAR hypothesis posits that synaptic NMDARs are neurotrophic/neuroprotective and extrasynaptic NMDARs are neurotoxic. The extrasynaptic hypothesis is built in part on observed selectivity for extrasynaptic receptors of a neuroprotective use-dependent NMDAR channel blocker, memantine. In rat hippocampal neurons, we found that a neuroprotective concentration of memantine shows little selectivity for extrasynaptic NMDARs when all receptors are tonically activated by exogenous glutamate. This led us to test the extrasynaptic NMDAR hypothesis using metabolic challenge, where the source of excitotoxic glutamate buildup may be largely synaptic. Three independent approaches suggest strongly that synaptic receptors participate prominently in hypoxic excitotoxicity. First, block of glutamate transporters with a nonsubstrate antagonist exacerbated rather than prevented damage, consistent with a primarily synaptic source of glutamate. Second, selective, preblock of synaptic NMDARs with a slowly reversible, use-dependent antagonist protected nearly fully against prolonged hypoxic insult. Third, glutamate pyruvate transaminase, which degrades ambient but not synaptic glutamate, did not protect against hypoxia but protected against exogenous glutamate damage. Together, these results suggest that synaptic NMDARs can mediate excitotoxicity, particularly when the glutamate source is synaptic and when synaptic receptor contributions are rigorously defined. Moreover, the results suggest that in some situations therapeutically targeting extrasynaptic receptors may be inappropriate.
Introduction
Excitotoxicity is an important contributor to many neurodegenerative diseases and neuropsychiatric disorders. Excitotoxicity is initiated in part through excessive NMDA receptor (NMDAR) activation (Rothman and Olney, 1987). However, NMDAR antagonists have deleterious side effects due to the critical role NMDARs play in normal synaptic signaling and plasticity (Olney et al., 1989; Ikonomidou et al., 1999). Thus, there is a need for therapeutics to balance preservation of normal function with neuroprotection. One hypothesis, the extrasynaptic NMDAR toxicity hypothesis (Hardingham et al., 2002; Lipton, 2007; Hardingham and Bading, 2010), is attractive in this regard because extrasynaptic receptors could be therapeutically targeted without compromising normal synaptic function. This hypothesis posits that extrasynaptic NMDARs selectively trigger cell death pathways while synaptic receptors trigger trophic pathways. However, the breadth of the extrasynaptic hypothesis in models of excitotoxicity has not been fully tested.
Accumulating evidence suggests that extrasynaptic NMDARs play a unique role in cell death. Extrasynaptic receptors may become activated when excessive levels of glutamate spill out of the synapse during prolonged depolarization. Another source of extrasynaptic glutamate during depolarizing insults is the reverse operation of glutamate transporters (Jabaudon et al., 2000; Rossi et al., 2000). These factors could conspire to initiate signaling cascades that are uniquely activated by extrasynaptic receptors (Hardingham et al., 2002; Dick and Bading, 2010) and that overwhelm trophic effects of synaptic receptor activation (Hardingham et al., 2002; Soriano et al., 2006; Papadia et al., 2008). Support for the extrasynaptic receptor hypothesis comes from neuroprotective effects of a drug, memantine, which may selectively target extrasynaptic receptors (Léveillé et al., 2008; Okamoto et al., 2009; Xia et al., 2010). However, memantine's selectivity has not been evaluated under conditions of excessive receptor stimulation that may occur during excitotoxic insults. Other studies supporting the extrasynaptic hypothesis have used exogenous excitotoxic agonists, which may be prevented from reaching synaptic receptors by glial glutamate uptake (Sattler et al., 2000; Sinor et al., 2000) and therefore provide a somewhat artificial excitotoxic stimulus. Whether the extrasynaptic hypothesis applies to insults involving endogenous glutamate, such as metabolic challenge, remains uncertain.
Here we specifically characterized memantine's selectivity for receptor populations during excitotoxic paradigms and directly investigated the role synaptic receptors play in hypoxia-induced excitotoxic insults. We demonstrate that memantine blocks synaptic receptors when activated equivalently to extrasynaptic receptors. Furthermore, our findings suggest that memantine's protection may result from blocking excessively activated synaptic receptors. We demonstrate that synaptic receptors appear to account for virtually all the MK-801-sensitive cell death caused by hypoxia. Our findings argue for a tempered view of the extrasynaptic receptor hypothesis.
Materials and Methods
Cell cultures.
Hippocampal cultures were prepared as either mass cultures or microcultures (as indicated in Figure legends) from postnatal day 1–3 rat pups of either sex, which were anesthetized with isoflurane, under protocols consistent with NIH guidelines and approved by the Washington University Animal Studies Committee. Protocols were adapted from earlier descriptions (Huettner and Baughman, 1986; Bekkers et al., 1990; Tong and Jahr, 1994; Mennerick et al., 1995). Hippocampal slices (500 μm thickness) were digested with 1 mg/ml of papain in oxygenated Leibovitz L-15 medium (Invitrogen). Tissue was mechanically triturated in modified Eagle's medium (Invitrogen) containing 5% horse serum, 5% fetal calf serum, 17 mm d-glucose, 400 μm glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells were seeded in modified Eagle's medium at a density of ∼650 cells mm−2 as mass cultures (onto 25 mm cover glasses coated with 5 mg/ml collagen or 0.1 mg/ml poly-d-lysine with 1 mg/ml laminin) or 100 cells/mm2 as “microisland” cultures (onto 35 mm plastic culture dishes coated with collagen microdroplets on a layer of 0.15% agarose). Cultures were incubated at 37°C in a humidified chamber with 5% CO2 and 95% air. Cytosine arabinoside (6.7 μm) was added 3–4 d after plating to inhibit glial proliferation. The following day, half of the culture medium was replaced with Neurobasal medium (Invitrogen) plus B27 supplement (Invitrogen).
Hypoxia exposure.
Mass cultures 14–15 DIV were subjected to hypoxia in a commercially available chamber (Billups-Rothenberg), humidified, and saturated with 95% nitrogen and 5% CO2 at 37°C for the specified amount of time (2.5 h). The gas exchange followed the specifications of the chamber manufacturer (flow of 20 L/min for 4 min to achieve 100% gas exchange). After the insult, we removed cells from the chamber and incubated them under standard culture conditions until the cell death assay (24 h later). Procedures for controls included all media changes and incubation times relevant to the insult conditions. For experiments in which exogenous glutamate exposure was used in place of hypoxia, medium was exchanged with conditioned Neurobasal medium from sibling cultures, 50 μm glutamate was added, and cultures were incubated for 2.5 h at 37°C. The concentration of glutamate was chosen to approximate the death observed during the same period (2.5 h) of hypoxia. The cultures were then returned to their original medium for 24 h until the cell death assay was performed. For experiments in which glutamate pyruvate transaminase (GPT) was used, medium was replaced with conditioned medium prepared with GPT (5 U/ml, Roche) and 2 mm pyruvate (Sigma-Aldrich). The neurons were then exposed to hypoxia or 50 μm glutamate. Following 2.5 h, cultures were returned to their original medium for 24 h until the cell death assay was performed.
Cell death assay.
All cell death studies were performed on mass cultures at 14–15 DIV. Mass cultures at this age contained an astrocyte monolayer, upon which neurons sit. Thus, neurons have strong diffusional contact with the extracellular medium, but processes also exhibit some glial ensheathment (Mennerick et al., 1996). Absolute ratios of astrocyte numbers to neuronal numbers from 19 fields in four platings was 5 ± 1 astrocytes to one neuron, assessed by counts of nuclei stained with Hoechst dye. Although this is a larger astrocyte-to-neuron ratio than found in vivo, diffusional contact in the cultures is higher, thereby limiting transmitter pooling.
We used trypan blue dye to identify compromised neurons 24 h after the insult. Culture medium was removed and replaced with 1 ml of 0.4% trypan blue dissolved in PBS. Cells were incubated in dye at 37°C for 5 min, washed with PBS, then fixed with 4% paraformaldehyde and 0.2% glutaraldehyde at room temperature. Cells were visualized with a 20× objective using both phase-contrast and brightfield microscopy to confirm healthy neuronal profiles (phase-contrast) and verify trypan blue uptake (brightfield). The total numbers of dead and intact neurons were counted and expressed as a percentage of trypan blue-negative cells among total cells. The average of 10 microscope fields for each condition was treated as a single data point for purposes of statistics. Although trypan blue in principle may miss apoptotic cell death, in which membranes remain uncompromised until late in the degeneration process, the total number of viable plus nonviable cells did not differ statistically between control and hypoxia conditions (p = 0.4, n = 6 experiments). Therefore, our assay accounted for all or nearly all cells compromised by the insult. We also cannot exclude that possibility that some trypan blue-positive cells could recover following insult. However, this possibility was minimized by use of the 24 h latent period between insult and assessment. In most cases, trypan-positive cells by this time no longer had discernable plasma membranes under brightfield or phase-contrast optics.
Electrophysiology.
Whole-cell recordings were performed at room temperature from neurons cultured for 10–15 d using a Multiclamp 700B amplifier (Molecular Devices). For recordings, cells were transferred to an extracellular solution containing the following (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 10 glucose, 10 HEPES, 0.01 glycine (a saturating concentration for the coagonist site on the NMDA receptor), and 0.001 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), pH 7.25. In some experiments, d-2-amino-5-phosphonovalerate (d-APV, 10–50 μm) was added to block NMDARs as indicated. In some experiments, synaptic NMDARs were preblocked with 10 μm dizocilpine maleate (MK-801). In these experiments, we preblocked in a bath solution with lowered Ca2+ (1 mm), 50 μm bicuculline, no added NBQX, and no added Mg2+, unless otherwise indicated. The tip resistance of patch pipettes was 3–6 MΩ when filled with an internal solution containing the following (in mm): 130 potassium gluconate, 2 NaCl, 0.1 EGTA, and 10 HEPES, pH 7.25, adjusted with KOH. In experiments examining current responses to exogenous agonists, cesium methanesulfonate was used in place of potassium gluconate to block potassium channels. Holding voltage was typically −70 mV. Access resistance for synaptic recordings (8–10 MΩ) was compensated 80–100%. For autaptic responses, cells were stimulated with 1.5 ms pulses to 0 mV from −70 mV to evoke transmitter release. In experiments in which NMDAR EPSCs were examined, AMPAR EPSCs and GABAergic IPSCs were blocked with 1 μm NBQX and 25 μm bicuculline respectively. Drugs were applied with a gravity-driven local perfusion system from a common tip. The estimated solution exchange times were <100 ms (10–90% rise), estimated from junction current rises at the tip of an open patch pipette.
Multielectrode array studies.
Experiments were performed as previously described (Mennerick et al., 2010). Briefly, multielectrode arrays (MEAs) were coated with poly-d-lysine and laminin per the manufacturer's instructions, and dispersed cultures were grown as described above. At 7 DIV and 10 DIV, one-third of the media was removed and replaced with fresh Neurobasal supplemented with B27 and glutamine. Recordings were made with the MEA-60 recording system (MultiChannel Systems) with the headstage in an incubator set at 29°C and equilibrated with 5% CO2 in room air with no additional humidity. The lower temperature was necessary because the electronics in the headstage generate ∼7°C of excess heat. The MEA itself rested on a heating plate inside the headstage to maintain the cultures at 37°C. To allow extended recordings in the dry incubator, cultures were covered with a semipermeable membrane that allows diffusion of oxygen and carbon dioxide but not water (Potter and DeMarse, 2001). Data were amplified 1100× and sampled at 5 kHz. Spikes were detected by threshold crossing of high-pass filtered data. The threshold was set individually for each contact at five SDs above the average RMS noise level. Baseline data were recorded in culture media. We then recorded activity in culture media containing an additional 50 μm glutamate. Finally, we collected another dataset in original growth media. Statistics were performed on 30 min treatment periods.
Data analysis.
Electrophysiology data acquisition and analysis were performed primarily using pClamp 10 software (Molecular Devices). All cell counts and electrophysiological measurements were processed with Microsoft Excel and are presented as mean ± SEM. Statistical significance was determined using a Student's two-tailed t test with a Bonferroni correction for multiple comparisons where appropriate. Data plotting, statistical analysis, and figure preparation were performed with SigmaPlot (Systat Software) and Adobe Photoshop. Decay time constants were measured using standard exponential fitting functions using the variable metric algorithm in pClamp software.
Drugs.
All drugs were obtained from Sigma-Aldrich, except for d-APV, NBQX, and dl-threo-β-benzyloxyaspartic acid (TBOA), which were obtained from Tocris Bioscience, and GPT, from Roche. GPT was supplied as a suspension in 3.2 m ammonium sulfate and was prepared by pelleting out the suspended protein with 32,500 rpm centrifugation, removing the supernatant and dissolving in HEPES-buffered saline (138 mm NaCl, 4 mm KCl, 10 mm HEPES). This procedure resulted in a small volume of residual suspension medium in final working solutions (discussed in Results).
Results
Memantine shows strong synaptic receptor blockade during sustained or repetitive activation
To begin to assess the potential role of synaptic and extrasynaptic receptors in excitotoxic cell death mediated by endogenous glutamate, we first examined effects of the neuroprotective open-channel blocker memantine. Cultures were subjected to 2.5 h of hypoxia and allowed to recover for 24 h to capture delayed cell loss using a trypan blue stain. Under these conditions, we consistently achieved robust cell death of ∼50% (Fig. 1). We found that memantine at concentrations <10 μm conferred only variable and trend-level protection from hypoxic excitotoxicity, but 10 μm memantine clearly protected neurons (Fig. 1). This is consistent with previous reports suggesting that concentrations ≥5 μm memantine are neuroprotective (Chen et al., 1998; Volbracht et al., 2006). We therefore focused additional studies on 10 μm memantine.
Figure 1.

Ten micromolar memantine protects against hypoxic insults. A, Brightfield photomicrographs of a representative experiment demonstrating the protection from different concentrations of memantine during hypoxia. Neurons were exposed to control (95% O2, 5% CO2, top left) or hypoxic (95% N2, 5% CO2, top right and bottom) conditions for 2.5 h in the presence (bottom) or absence (top) of indicated memantine concentrations. Cultures were stained for cell death with trypan blue 24 h postinsult. Trypan-positive nuclei are apparent (arrows). Brightness and contrast of panels were digitally adjusted individually to approximately match background brightness levels. Scale bar: 20 μm. B, Average neuronal survival across the four conditions in A. Asterisks, p < 0.05 by a two-tailed Student's t test. NS, not significant. N = 5 independent experiments in mass cultures.
Memantine is selective for extrasynaptic receptors over synaptic receptors when the presentation of glutamate to synaptic receptors is transient, which is the case under physiological conditions (Lester et al., 1990; Xia et al., 2010). However, the basis for memantine's apparent selectivity remains unclear. Memantine could possess a higher affinity for extrasynaptic versus synaptic receptor populations. Alternatively, as an open-channel blocker, memantine's selectivity could arise as a result of phasic versus sustained patterns of synaptic versus extrasynaptic receptor activation (Johnson and Kotermanski, 2006; Xia et al., 2010). To determine which of these alternatives forms the basis for memantine's selectivity, we developed protocols involving sustained agonist presentation that would equivalently activate synaptic and extrasynaptic receptor populations.
During hypoxia, glutamate may accumulate tonically to activate both synaptic and extrasynaptic receptors. To partly simulate tonic glutamate buildup, we used sustained glutamate (1 μm) application onto NMDAR populations and evaluated antagonism by 10 μm memantine. The concentration of glutamate was chosen to be less than the EC50 concentration of 2.3 μm (Patneau and Mayer, 1990). We used a low concentration of NMDA to probe the availability of receptors before and after conditioning with glutamate plus memantine (Fig. 2A). When NMDA was reapplied following conditioning, currents exhibited an initially reduced response (Fig. 2A, inset, red trace). This initial suppression (Fig. 2A, inset, red trace) is attributable to memantine bound to (trapped in) closed NMDAR channels (Johnson and Kotermanski, 2006; Kotermanski et al., 2009). When all NMDAR populations were activated with glutamate, memantine inhibited currents by ∼80% compared with the baseline NMDA currents (Fig. 2A,D). During the sustained NMDA reapplication, current reemerged gradually as channels were reopened and allowed trapped memantine to dissociate.
Figure 2.
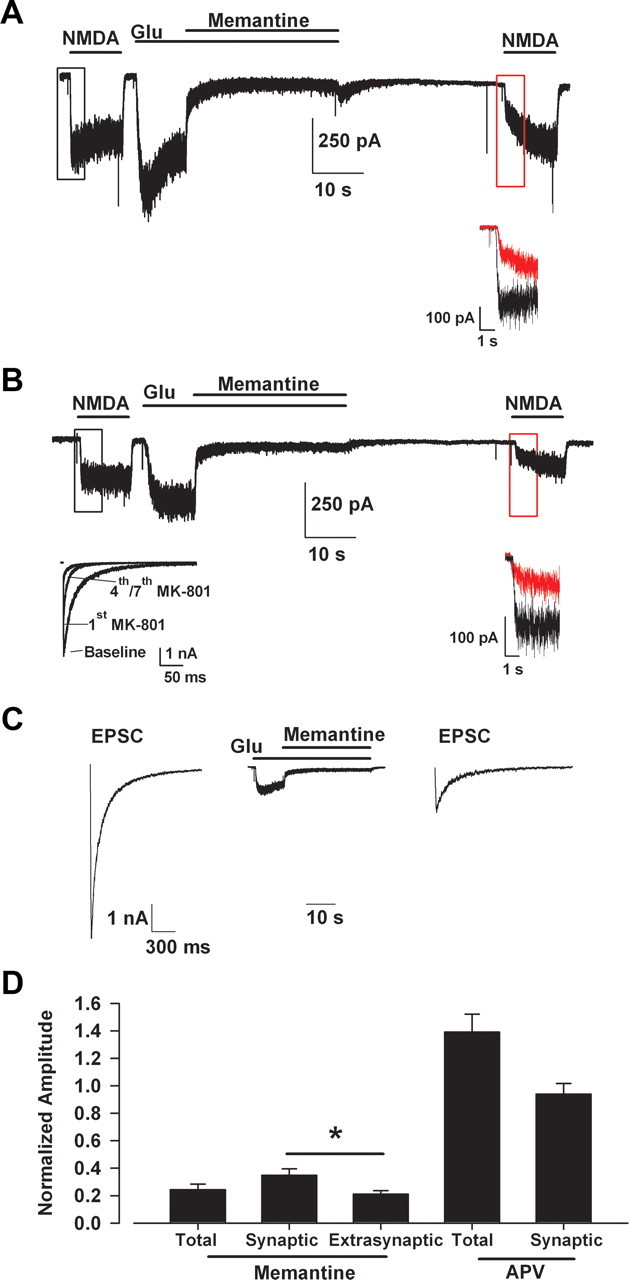
Memantine shows significant trapping block of synaptic NMDARs during tonic activation. A, Trapping protocol performed on all receptors on a synaptically isolated microculture hippocampal neuron: baseline NMDAR current was recorded with exogenous NMDA (10 μm), followed by application of a low concentration of glutamate (Glu, 1 μm) and the coapplication of glutamate and memantine (10 μm). The expected EC50 concentrations of the agonists are 2.3 μm for glutamate and 35 μm for NMDA (Patneau and Mayer, 1990). Note that a saturating glycine concentration (10 μm) was used throughout the experiment to diminish NMDAR desensitization (Lester et al., 1993; Wilcox et al., 1996). Agonist and antagonist were washed away with saline containing 10 μm d-APV, to maintain channels in a nonconducting state (Kotermanski et al., 2009), for 30 s before reapplication of NMDA. Memantine relief from block (untrapping) occurred during the second NMDA application. Inset, Expanded view of region outlined in black and red boxes, demonstrating the trapping apparent with initial NMDA reapplication. The red trace represents the initial current following memantine application attributable to unblocked receptor channels. The black trace represents the corresponding NMDA current before memantine exposure for comparison. B, Memantine inhibition of extrasynaptic receptors. The experiment was similar to A, except that synaptic receptors were blocked by repetitive EPSCs in the presence of 10 μm MK-801 (left lower inset), leaving mainly extrasynaptic receptors to respond to NMDA and glutamate applications. The right lower inset shows the degree of memantine inhibition in the isolated extrasynaptic receptor population. C, Memantine trapping in synaptic receptors. Autaptic NMDA-mediated EPSCs before and after trapping protocol where memantine (10 μm) was coapplied with glutamate (1 μm) to all receptors. Memantine and glutamate were washed for 30 s with d-APV containing saline (trace not shown) before evoking an EPSC. EPSCs were significantly depressed compared with the initial baseline EPSC. D, Average normalized amplitude of test responses following trapping. The initial amplitude, before unblock (e.g., A, B, insets, red trace) was normalized to peak NMDA current (e.g., A, B, insets, black traces) for total and extrasynaptic values in the graphs. Asterisk, p < 0.5, Student's t test. As a comparison, parallel control experiments used 10 μm d-APV in place of memantine (APV bars; traces not shown) in the total receptor (A) and synaptic receptor block (C) protocols. d-APV did not exhibit residual antagonism, and there was no appreciable rundown of responses in these conditions. N = 5–10 cells per condition.
We next enriched responses for extrasynaptic NMDARs by eliciting action potential-dependent EPSCs in the presence of the slowly dissociating open-channel blocker MK-801 (Fig. 2B, left inset). Memantine antagonism at the enriched extrasynaptic NMDAR pool was again assessed by NMDA reapplication and showed similar initial memantine inhibition compared with the total NMDAR pool (Fig. 2A,B,D). To determine whether memantine blocked synaptic receptor channels during sustained exogenous glutamate application, we examined evoked NMDAR EPSCs on neurons before and after the memantine antagonism protocol (Fig. 2C). Memantine inhibition of EPSCs was strong, although slightly weaker than antagonism exhibited at isolated extrasynaptic receptors (Fig. 2D). These results are consistent with the idea that, compared to synaptic receptors, extrasynaptic receptors are slightly more sensitive to memantine (Okamoto et al., 2009; Xia et al., 2010). However, the strong inhibition of synaptic receptors at neuroprotective memantine concentrations suggested that synaptic receptors deserve attention as possible targets of memantine and as mediators of toxicity.
Figure 2 examines effects of memantine on tonically activated NMDAR populations. It is also possible that synaptic receptors are phasically but repeatedly activated during endogenous insults. Although low memantine concentrations (1 μm) have been comprehensively evaluated for effects on repetitive activation of synaptic receptors, neuroprotective concentrations of memantine (10 μm) have not been fully investigated. We found that 10 μm memantine applied continuously while evoking NMDA EPSCs initially produced only ∼30% inhibition. However, over the course of multiple stimuli, inhibition reached ∼80% (n = 5; Fig. 3) and recovered very little with memantine washout (Fig. 3B), suggesting inhibition by memantine bound to closed channels. Steady-state inhibition was likely not achieved in these experiments; with additional stimulation, it is possible that memantine might inhibit EPSCs more completely. Nevertheless, the observations demonstrate that neuroprotective concentrations of memantine strongly antagonize synaptic receptors when receptors are repetitively activated.
Figure 3.
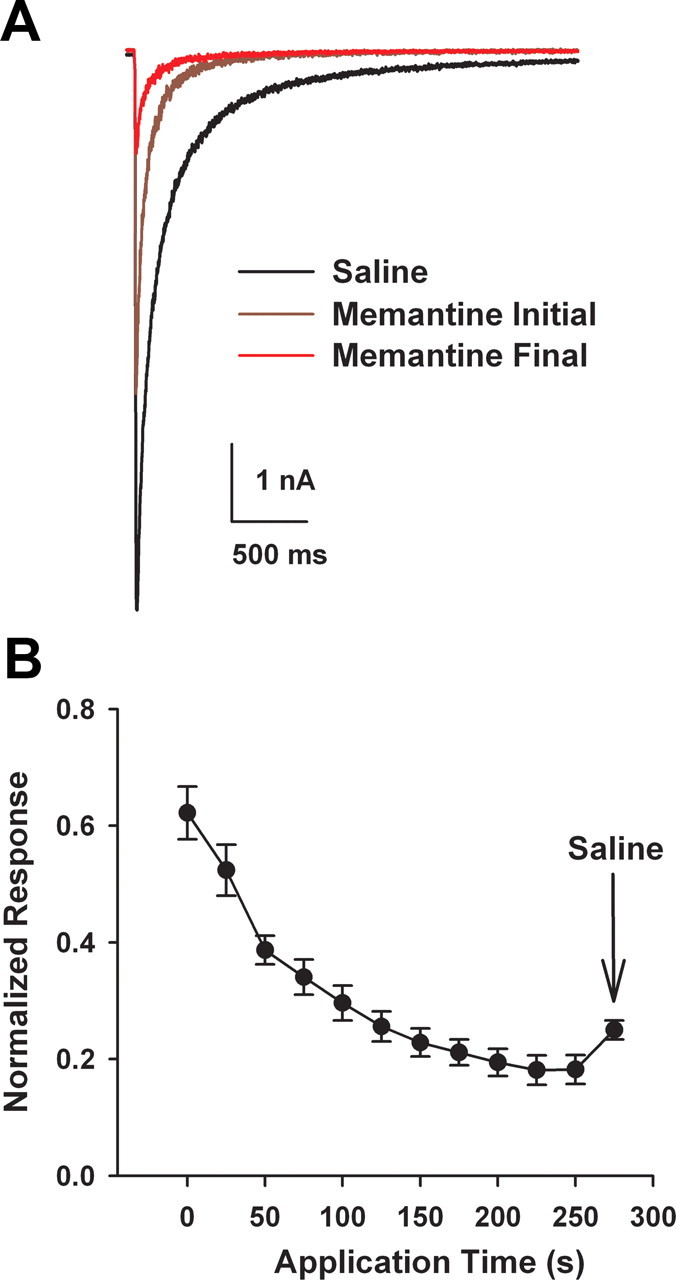
Progressive memantine block of synaptic receptors during repetitive stimulation of a microculture neuron. A, Autaptic baseline NMDA-mediated EPSCs (black), after initial memantine (10 μm) presentation (brown), and after sustained memantine application during repeated stimulation (red). EPSCs were evoked every 30 s, and baseline was stabilized (black) before application of 10 μm memantine. B, Average normalized progressive inhibition summarized from five cells. Arrow designates partial relief from trapping upon memantine washout (saline).
Although the absolute magnitude of memantine's effect were likely exaggerated in these experiments by the absence of Mg2+, which is known to interact with memantine antagonism (Kotermanski and Johnson, 2009), the results confirm that memantine's selectivity arises largely as a result of different dynamics of agonist presentation at synaptic and extrasynaptic receptors. If during hypoxic insult the mode of agonist presentation is synaptic but becomes more sustained or repetitive than under physiological conditions, then it is possible that memantine (and other neuroprotective NMDAR antagonists) act by blocking synaptic receptors. This would contradict recent evidence indicating that extrasynaptic receptors play a privileged role in excitotoxicity.
The source of hypoxic glutamate is synaptic
In contemplating a role for synaptic receptors in toxicity during energy deprivation, we started by considering the assumption that the source of hypoxic glutamate is synaptic. There is some experimental support for the idea that synaptic release is a major source of glutamate during energetic insults (Rothman, 1983; Kaplan et al., 1989; Monyer et al., 1992; Andrade and Rossi, 2010). The major alternative mechanism of glutamate release is glial reverse glutamate uptake, which may occur under particularly severe conditions of oxygen-glucose deprivation combined with chemical ischemia (Jabaudon et al., 2000; Rossi et al., 2000). Reverse transport could produce excessive tonic glutamate presumably most concentrated at extrasynaptic regions where transporters are primarily located (Danbolt, 2001; Gouix et al., 2009), although synaptic NMDARs could also be recruited since they comprise the majority of cellular receptors (Rosenmund et al., 1995; Harris and Pettit, 2007). We reasoned that if reverse uptake participates strongly in hypoxic glutamate release, blocking the (reverse) activity of transporters during hypoxia would be protective. However, if transporters remove toxic levels of glutamate released synaptically, then blocking transporters would exacerbate the damage. Thus, to test the role of glutamate transporters during hypoxia, we coapplied TBOA, a broad-spectrum, nonsubstrate glutamate transporter inhibitor (Shimamoto et al., 2004), during hypoxia. We found that application of TBOA significantly exacerbated cell death, suggesting that glutamate transporters normally play a protective role in hypoxia (p < 0.05, n = 5; Fig. 4). Although these results do not directly determine the NMDAR populations responsible for cell death, the data are consistent with a synaptic rather than an extrasynaptic source of glutamate.
Figure 4.
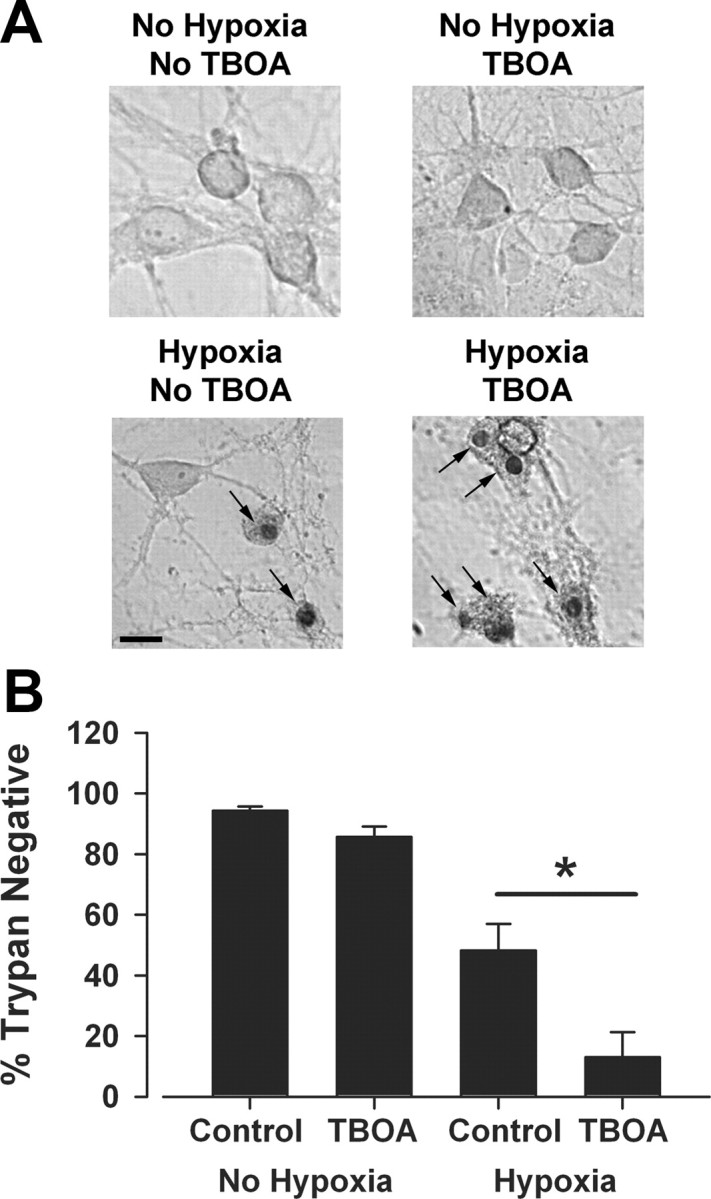
Glutamate transporters protect from hypoxic insults. TBOA (100 μm), a nonsubstrate, broad-spectrum glutamate transporter blocker, was used to diminish reverse or forward contributions of glutamate transporters during an insult. Culture medium was removed and replaced with conditioned medium from sibling cultures containing TBOA. Neurons were then incubated under hypoxic conditions for 2.5 h, and original medium was replaced until assessment of survival 24 h later. A, Representative photomicrographs of sample fields from a mass culture in control (No Hypoxia) or insult (Hypoxia) conditions with and without TBOA, as indicated. Note prominent trypan-positive nuclei, indicated by arrows. Scale bar: 20 μm. B, Survival across conditions summarized from five independent experiments. TBOA significantly exacerbated cell death (asterisk, p < 0.05, two-tailed t test).
To initially test whether milder synaptic receptor activity can be toxic to neurons, we queried whether bicuculline, commonly used to enhance synaptic activity, could kill neurons. Indeed, 5 h treatment of cells with 50 μm bicuculline, 10 μm glycine, 2 mm Ca2+, and no added Mg2+ produced mild but reliable d-APV-sensitive neurotoxicity measured 24 h later (82 ± 3% vs 90 ± 2% cell survival, p < 0.05, n = 6). Although this is a mild effect, it demonstrates that prolonged and excessive synaptic activity may kill neurons, which is contrary to the prevailing idea that any synaptic receptor activation is neuroprotective (Hardingham and Bading, 2010).
Selective synaptic receptor block protects against hypoxia and exogenous insults
The evidence that neuroprotective memantine exhibits strong synaptic receptor (EPSC) block during sustained or repetitive receptor activation, coupled with data suggesting that the glutamate source in hypoxia is synaptic, prompted us to investigate whether synaptic NMDARs may mediate cell death during hypoxia. Hypoxia is an insult that we and others have used as a model of excitotoxic damage (Rothman, 1983; Hogins et al., 2011). Unlike bath application of NMDA or glutamate, other common experimental excitotoxic challenges, hypoxia triggers endogenous glutamate release. In this sense, then, energetic insults like hypoxia are more similar to in vivo insults such as ischemia and stroke (Choi and Rothman, 1990; Lipton, 1999). To test the hypothesis that excessive activation of synaptic NMDARs during hypoxia mediates cell death, we selectively blocked synaptic receptors before hypoxia using the very slowly dissociating, use-dependent antagonist, MK-801. This protocol has been previously used extensively (Hardingham and Bading, 2002; Ivanov et al., 2006; Liu et al., 2007; Stanika et al., 2009; Bordji et al., 2010; Stark and Bazan, 2011; Zhang et al., 2011). First, we exchanged the culture medium for isoosmotic defined recording solution (see Materials and Methods) containing 10 μm MK-801 for 15 min. The preblock bath also contained low (1 mm) Ca2+ concentration, 50 μm bicuculline (to decrease network inhibition), no Mg2+, and 10 μm glycine. This solution exchange prevented background glutamate in the culture medium from activating receptors, thus minimizing the chance of nonspecific MK-801 block. Low Ca2+ concentration ensured a low quantal content for each action potential, minimizing spillover onto extrasynaptic receptors (Mennerick and Zorumski, 1995). Furthermore, we omitted 4-aminopyridine, a potassium-channel blocker commonly used in the preblock protocol (Hardingham et al., 2002; Bordji et al., 2010; Zhang et al., 2011), but which may enhance quantal content and spillover onto extrasynaptic receptors.
This synaptic NMDAR preblock protocol abolished NMDA-mediated EPSCs in mass cultures in which toxicity experiments were performed with relatively little reduction in normal synaptic activity, measured by AMPA receptor-mediated EPSCs (Fig. 5A). AMPA receptor-mediated EPSCs sometimes changed amplitude or frequency following MK-801 preblock (Fig. 5A2), but these changes exhibited no consistent direction, and the overall charge transfer mediated by AMPAR over 30 s was not statistically different from pretreatment levels (Fig. 5C). Preblock with MK-801 preserved ∼30% of bath-applied NMDA current (Fig. 5B,C), consistent with previous studies suggesting that ∼70% of cellular NMDARs are synaptic (Rosenmund et al., 1995; Harris and Pettit, 2007). Following this synaptic preblock protocol, we replaced the original culture medium and subjected the neurons to hypoxia. Preblocking synaptic receptors successfully reduced cell death, evaluated 24 h following insult, from 45 ± 4% to 17 ± 2% (p < 0.05, n = 10) during a 2.5 h hypoxic insult (Fig. 5D).
Figure 5.
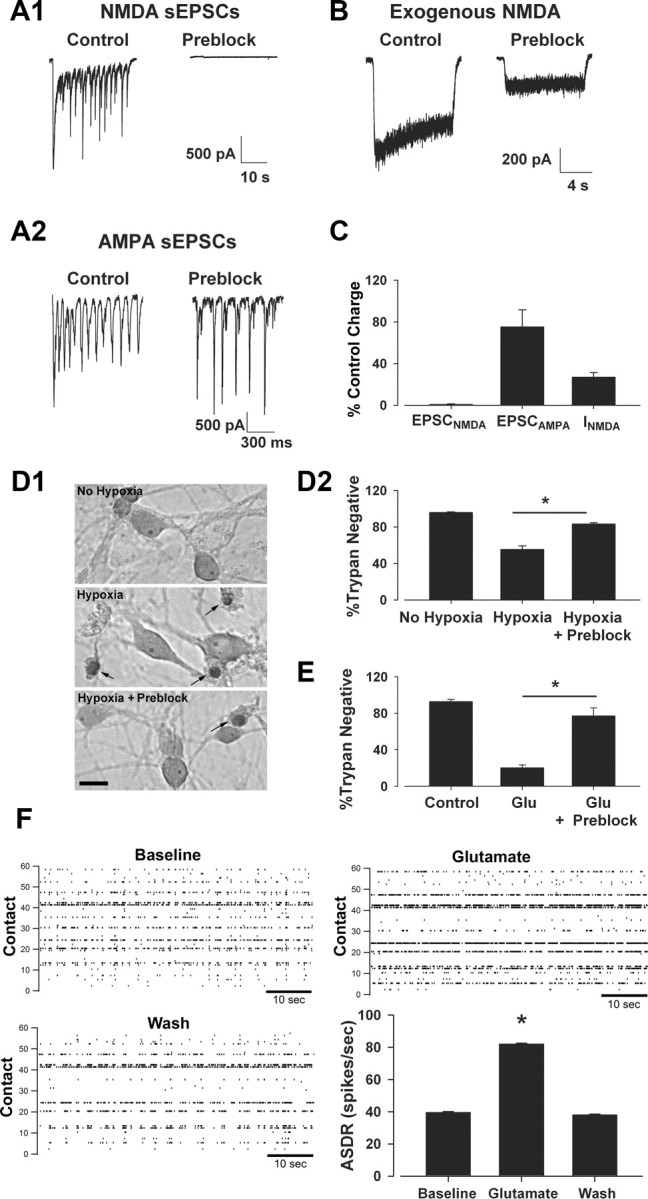
Synaptic NMDAR block significantly protects against hypoxic and exogenous glutamate insults. Mass culture neurons (14–15 DIV) were incubated for 15 min in recording saline containing 1 mm Ca2+, no added Mg2+, bicuculline (50 μm), glycine (10 μm), and MK-801 (10 μm). Neurons were returned to original media before exposure to hypoxia (2.5 h). A1, A2, Bicuculline-induced NMDA spontaneous EPSCs (sEPSCs) isolated with NBQX and recorded in the absence of bath MK-801 were abolished following preblock (A1, right) with little change in AMPA receptor-mediated sEPSCs isolated in the absence of NBQX and presence of d-APV (A2) in the same cell. B, Preblock-reduced exogenous NMDA currents compared with control, untreated sibling cultures, representing block of synaptic receptors and retention of functional extrasynaptic receptors. C, Summary data indicating the remaining percentage across NMDA EPSCs, AMPA EPSCs, and total NMDA current (n = 8–12). The reduction in NMDA-activated current (INMDA) with preblock represents the expected percentage of extrasynaptic receptors estimated previously (Rosenmund et al., 1995). D1, Representative photomicrographs of neurons exposed to 2.5 h hypoxia with or without preblock conditioning. Arrows indicated trypan-positive neurons, labeled 24 h following insult. Conditions are indicated by text labels. Scale bar: 20 μm. D2, Summary data indicating that synaptic NMDAR preblock significantly protected neurons against hypoxia. Asterisk, p < 0.05, two-tailed Student's t test. N = 10 independent experiments. E, Preblock also significantly protected neurons against 2.5 h of 50 μm glutamate (Glu; no receptor antagonists present in the Glu bath). Asterisk, p < 0.05, two-tailed Student's t test. N = 5 experiments. F, MEA recordings from mass cultures in presence of glutamate (50 μm). Raster plots for baseline, glutamate, and wash conditions from a representative experiment are shown with array-wide spike detection rates (ASDR) across conditions. Glutamate significantly increased the firing rate in four of four independent experiments (asterisk, p < 0.05 with ANOVA and pairwise t test). The summary panel reflects 30 min of data in each condition from the single experiment represented in the rasters.
Previous studies investigating the role of synaptic and extrasynaptic receptors in excitotoxic insults typically used exogenous insults in which glutamate or NMDA are bath applied to neurons (Sattler et al., 2000; Hardingham et al., 2001, 2002; Papadia et al., 2008). This insult may activate more extrasynaptic receptors than hypoxic insult. Therefore, we hypothesized that selectively blocking synaptic receptors should not protect against an exogenous insult (Hardingham et al., 2002). We selectively blocked synaptic receptors before insult with 50 μm glutamate, which we found killed approximately as many neurons as hypoxia over 2.5 h. Surprisingly, we found that selective synaptic receptor block significantly protected against exogenous glutamate (Fig. 5E). We hypothesized that this may partly result from increased network activity promoted by exogenous glutamate. This would lead to excessive synaptic receptor activation that could contribute to glutamate-mediated death. To test this, we recorded from a network of dissociated neurons using an MEA before and during treatment with 50 μm glutamate. Exogenous glutamate produced a mild but reliable and reversible increase in network activity in our cultures (p < 0.05 in four of four experiments analyzed with ANOVA and pairwise t test; Fig. 5F). Over the four experiments, the mean increase in array-wide spike rate was 45 ± 22% over baseline. It seems unlikely that this degree of enhanced synaptic activity accounts fully for the neuroprotective effects of synaptic NMDAR preblock (see Discussion). In aggregate, these data suggest that synaptic NMDARs play a critical excitotoxic role across a variety of insult paradigms.
MK-801 synaptic preblock protects strongly against hypoxic damage, but protection is incomplete (Fig. 5D2). Residual cell death could be caused by NMDAR-independent toxicity, rendering even complete MK-801 antagonism ineffective. To test this, we applied 10 μm MK-801 before and during the hypoxic insult, and we continued the MK-801 incubation during the postinsult recovery period (24 h). Despite the constant presence of MK-801, we still found incomplete protection (Fig. 6A). We also directly examined whether total (synaptic and extrasynaptic) NMDAR preblock could confer full protection. This was achieved by coincubation with 50 μm NMDA plus 10 μm MK-801 before hypoxic insult. We found that the total receptor preblock protocol was also incompletely protective against hypoxic damage and exerted protection quantitatively similar to synaptic preblock (Fig. 6B). To verify that MK-801 unblocking is unlikely to underlie the incomplete block, we recorded NMDAR currents immediately after total preblock and immediately after total preblock then 2.5 h hypoxia. Currents immediately following the insult showed only modest recovery (Fig. 6C). Average current amplitudes were −57.5 ± 6.8 pA (recorded immediately following preblock, n = 10), −194.0 ± 29.0 pA (preblocked, recorded posthypoxia, n = 8), and −2146.9 ± 306.7 pA (control, unblocked, n = 5). These observations are consistent with the idea that little relief of block occurs as a result of insult-induced depolarization and channel activation. Finally, the small residual death associated with MK-801 antagonism in Figures 5D, 5E, 6A, and 6B is also similar to the cell death we previously observed with complete block of all NMDA and AMPA-type glutamate receptors during hypoxia (Hogins et al., 2011). This previous evidence together with present evidence is consistent with the idea that nonglutamatergic mechanisms contribute to hypoxic damage in this model (Newell et al., 1995; Besancon et al., 2008), and selective synaptic preblock confers nearly the full measure of possible NMDAR-related protection in this model. Extrasynaptic NMDARs do not appear to be significantly involved in the residual toxicity.
Figure 6.
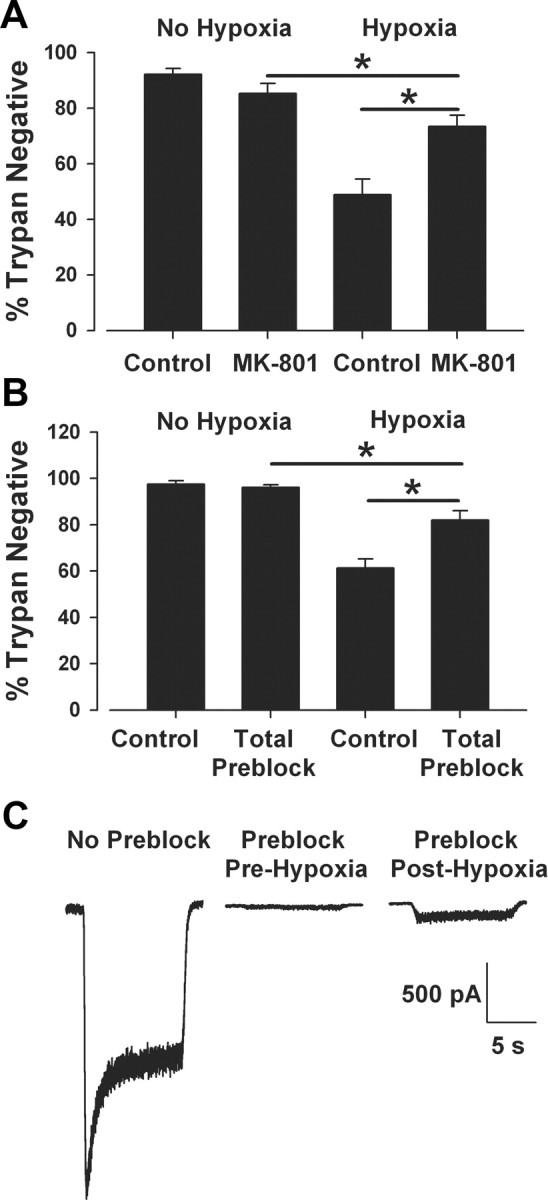
MK-801 partial protection results from non-NMDAR-related toxicity and not from MK-801 unblock. A, Mass culture neurons were exposed to hypoxia or control conditions in the presence or absence (Control) of MK-801 (10 μm) during and after a 2.5 h insult. Despite the continual presence of MK-801, hypoxic protection, although significant, was not complete (hypoxia alone vs hypoxia with MK-801, p < 0.05; MK-801 alone vs hypoxia with MK-801, p < 0.05, two-tailed t test with Bonferroni correction for multiple comparisons, n = 7). B, Total NMDAR preblock protocol: neurons were exposed to NMDA (50 μm) and MK-801 (10 μm) for 15 min in recording solution containing no Mg2+ and 2 mm Ca2+ and then returned to conditioned culture medium before exposure to hypoxic insult. Total receptor block significantly protected against a hypoxic insult, but not completely (hypoxia alone vs hypoxia preblock, p < 0.05; preblock alone vs hypoxia preblock, p < 0.05, t test with Bonferroni correction for multiple comparisons, n = 4). C, To test whether MK-801 escaped the NMDA channel during hypoxic insult to explain partial protection in B, NMDA currents (10 μm) were recorded in unblocked control cultures (No Preblock, n = 5), immediately after the total NMDAR preblock protocol (Preblock, Pre-Hypoxia, n = 10), and then after a preblock protocol, followed by hypoxia (Preblock, Post-Hypoxia, n = 8). Little unblock was evident following 2.5 h hypoxia (representative traces shown; see Results for quantification).
To minimize spillover during our blocking protocol, we reduced bath calcium concentration. This reduction in calcium may have enhanced excitability. (Frankenhaeuser and Hodgkin, 1957). This could counteract the effect of lowering quantal content and result in perisynaptic receptor activation and MK-801 block. To minimize any enhancement in excitability caused by low divalent cation concentration, we omitted bicuculline in our preblock solution and instead added 200 nm of the positive allosteric GABAA receptor modulator allopregnanolone to the low-calcium bath. This protocol significantly reduced the frequency of spontaneous NMDAR-mediated EPSCs but did not abolish activity (p < 0.05, n = 8; Fig. 7A). When MK-801 was added to this more conservative preblock protocol, we still observed significant protection against 2.5 h hypoxia (46.2 ± 6.5% vs 69.7 ± 5.6% cell survival, p < 0.05, n = 5; Fig. 7B). This offers further evidence that synaptic receptors play a vital role in hypoxic excitotoxic death.
Figure 7.
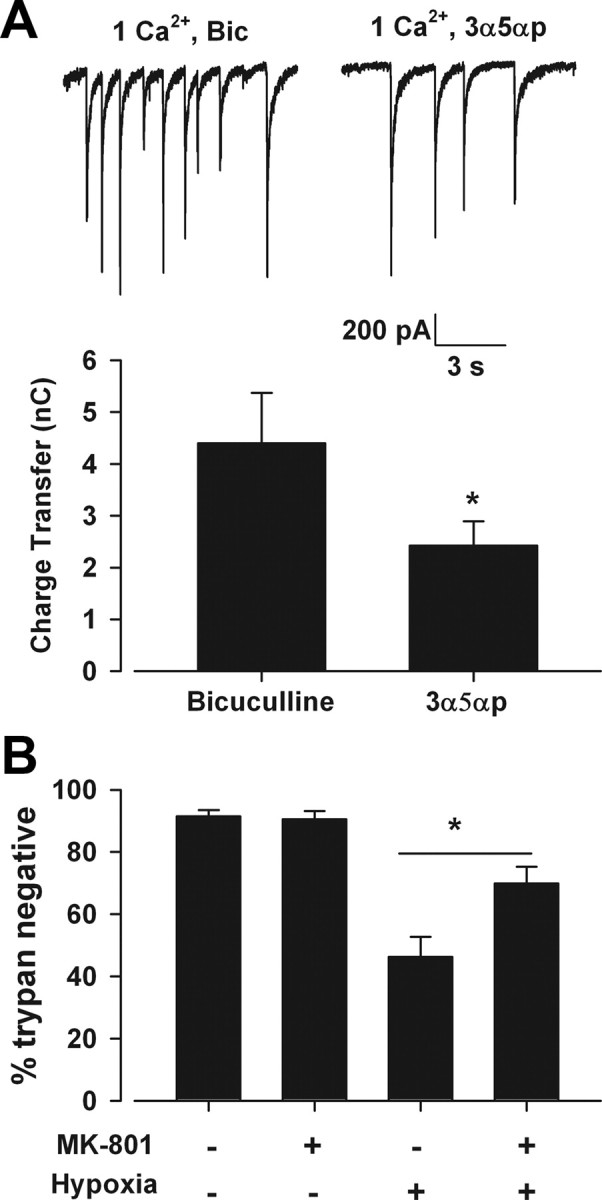
Decreasing synaptic activity during preblock still yields protection. A, The GABAA receptor allosteric modulator allopregnanolone (3α5αp) replaced bicuculline and reduced NMDA-mediated sEPSC frequency in mass cultures. Bar graph shows overall postsynaptic charge transfer from 10 s periods (asterisk, p < 0.05, two-tailed Student's t test, n = 5). B, MK-801 in the 3α5αp preblock solution significantly protected against 2.5 h hypoxic insult (asterisk, p < 0.05, two-tailed Student's t test, n = 5).
Reduction of extrasynaptic glutamate does not protect against hypoxia
As an independent approach to evaluating the role of synaptic receptors in hypoxic damage, we next queried whether selectively reducing contributions of extrasynaptic NMDARs would protect against hypoxic damage. If damage arises mainly through synaptic receptors, then reducing extrasynaptic receptor contributions should offer little protection. If, however, extrasynaptic receptors contribute strongly to damage (Hardingham et al., 2002), perhaps in concert with synaptic receptors, reducing their activation would be protective. To selectively eliminate sustained extrasynaptic NMDAR contributions, we used GPT, which catalyzes pyruvate-dependent glutamate degradation. Catalysis is too slow for GPT to strongly affect the transient, high glutamate concentration in the synaptic cleft, but GPT significantly depresses the activation of extrasynaptic receptors by synaptic overflow (Rossi and Slater, 1993; Rusakov and Kullmann, 1998; Turecek and Trussell, 2000). Consistent with this previous literature, we found that 5 U/ml GPT plus 2 mm pyruvate strongly reduced currents in response to exogenous application of glutamate (1 μm, representing a sub-EC50 concentration), but GPT had a minimal effect on NMDA EPSCs (Fig. 8).
Figure 8.
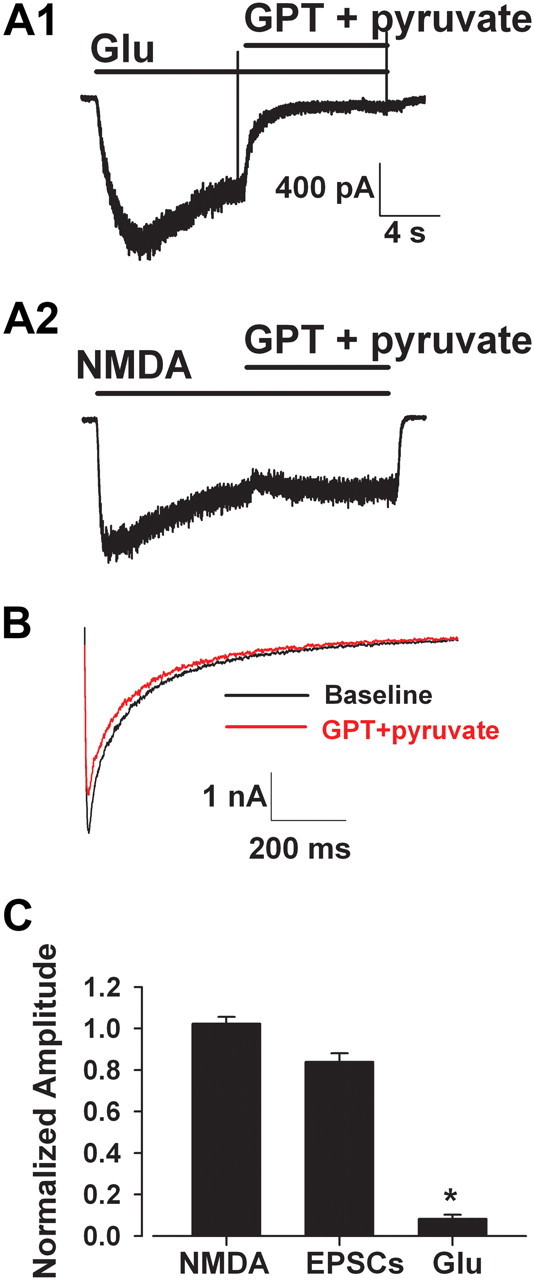
Specificity of glutamate pyruvate transaminase for tonic but not synaptic glutamate. A1, Glutamate pyruvate transaminase (5 U/ml) with 2 mm sodium pyruvate (GPT + pyruvate) significantly depressed current in a microculture neuron in response to exogenous glutamate (Glu, 1 μm). A2, GPT had little effect on exogenous NMDA (10 μm) current in the same cell, suggesting minimal direct effect on NMDAR function. B, GPT with pyruvate minimally depressed NMDA-mediated EPSCs. EPSCs were evoked every 30 s, and baseline was stabilized before application of GPT. C, Summary data indicating the significant selective reduction of tonic glutamate effects over phasic, synaptic effects by enzymatic degradation (8.2 ± 2.1% vs 83.9 ± 4.1%, *p < 0.05, two-tailed t test, n = 8–12).
As previously demonstrated in other preparations (Rossi et al., 2002), we found that GPT significantly protected neurons against excitotoxicity induced by 2.5 h of 50 μm glutamate (Fig. 9A). This suggests that the GPT strategy is effective against toxicity mediated by tonic glutamate buildup. However, when tested against hypoxic excitotoxicity, GPT was not protective and instead exacerbated death (Fig. 9B). This exacerbation resulted from residual contaminants in the supplier's enzyme formulation (see Materials and Methods), since equivalent vehicle suspension alone resulted in indistinguishable exacerbation [21 ± 1% survival in hypoxia plus GPT, 24 ± 4% survival with vehicle suspension control in hypoxia (p > 0.05), while hypoxia alone yielded 46 ± 2% survival in six independent, paired experiments on sibling cultures]. As with GPT, the vehicle control did not affect survival in the absence of hypoxia, and the exacerbation was d-APV-insensitive (data not shown). Although the basis of the vehicle effects on hypoxic damage remains unclear, the quantitative similarity of the effect in the presence and absence of GPT suggests that exacerbation did not mask a protective effect of GPT during hypoxia. These results are again consistent with the idea that synaptic receptors can mediate excitotoxicity when the source of glutamate is synaptic.
Figure 9.

Glutamate pyruvate transaminase protects against exogenous glutamate damage but does not protect against hypoxic damage. A, GPT protected against an exogenous insult. Medium was exchanged for conditioned medium from sibling cultures containing 5 U/ml GPT and 2 mm pyruvate (GPT). Glutamate (Glu; 50 μm) was directly added to the enzyme-containing dishes for 2.5 h. Conditioned medium was reintroduced at the end of the insult, and cells were evaluated for cell death 24 h postinsult. GPT significantly protected against exogenous glutamate (asterisk, p < 0.05, two-tailed t test, n = 5). B, GPT did not protect against hypoxia. Culture medium from the mass culture was exchanged for conditioned medium from sibling dishes containing 5 U/ml GPT and 2 mm pyruvate, and neurons were exposed to hypoxia. Original medium was replaced at the end of the insult, and neurons were stained for cell death using trypan blue 24 h postinsult. GPT did not protect. The exacerbation was linked to residual suspension from the supplier's formulation (asterisk, p < 0.05, two-tailed t test; n = 14). See Results.
Discussion
The extrasynaptic NMDAR excitotoxicity hypothesis proposes an attractive dichotomy between trophic synaptic receptor activation and toxic extrasynaptic receptor activation (Hardingham et al., 2002; Papadia et al., 2008). The extrasynaptic hypothesis is built in part on neuroprotection by the use-dependent NMDAR channel blocker memantine, which has been suggested to be selective for extrasynaptic receptors (Lipton, 2007; Léveillé et al., 2008; Xia et al., 2010). Here we demonstrated that a neuroprotective concentration of memantine shows only weak selectivity for extrasynaptic NMDARs when the pattern of synaptic and extrasynaptic receptor activation (channel opening) is identical. The extrasynaptic receptor hypothesis also posits that synaptic receptor blockade should not exhibit neuroprotection during excitotoxic insults and may in fact exacerbate damage. In contrast, we demonstrated that synaptic NMDAR blockade is neuroprotective, especially when the excitotoxic insult is mediated by synaptic glutamate release. Our results contradict the strong form of the extrasynaptic NMDAR excitotoxicity hypothesis and suggest that at least some excitotoxic insults act mainly through overactivation of synaptic NMDARs.
Our data suggest that memantine possesses similar actions at extrasynaptic and synaptic receptors. Although extrasynaptic receptors may be enriched in NR2B subunits (Groc et al., 2006), offering a potential substrate for selectivity, memantine exhibits no clear subunit selectivity (Blanpied et al., 1997). A previous study of memantine found evidence for selectivity at extrasynaptic receptors, but this study did not directly test the contribution of transient versus sustained receptor activation in memantine's apparent selectivity (Xia et al., 2010). Our results suggest that the pattern of receptor activation is the most important determinant of memantine's apparent selectivity, consistent with memantine's open-channel block mechanism (Chen and Lipton, 1997). Transient receptor activation during brief transmitter presence limits the amount of memantine channel block during synaptic activation, especially at low memantine concentrations. By contrast, sustained activation of receptors facilitates memantine inhibition. Previous studies assessed memantine's selectivity using a low concentration (1 μm) (Okamoto et al., 2009; Xia et al., 2010), whereas we focused on a 10-fold higher concentration, which we and others have documented to be neuroprotective (Fig. 1) (Chen et al., 1998; Volbracht et al., 2006). Our results demonstrate that synaptic receptors (EPSCs) can be strongly blocked by this concentration if they are activated by either sustained or repetitive agonist presentation. Either pattern or both patterns could apply to excitotoxic insults, such as hypoxia. It should be noted that our measurements (Figs. 2, 3) were performed at −70 mV in the absence of extracellular Mg2+. It is likely that under pathophysiological conditions, such as hypoxia, the absolute level of memantine block for both extrasynaptic and synaptic receptors will be reduced by physiological Mg2+ and by hypoxic depolarization (Kotermanski and Johnson, 2009; Xia et al., 2010).
We also provide evidence that synaptic receptors can play a critical role in mediating excitotoxic death during a moderate hypoxic challenge. Blocking synaptic NMDARs protected against hypoxia. Key to interpreting this result is the selectivity of our strategy for preblocking synaptic receptors. For our MK-801 preblocking protocol, we followed protocols established in the literature (Hardingham et al., 2002; Papadia et al., 2008). We also defined the synaptic receptor population conservatively by preblocking in bath solutions designed to limit glutamate spillover during synaptic activity, and we ensured minimal background glutamate levels by preblocking in fresh, defined bath solution. Most importantly, we directly verified the selectivity of our preblock strategy (Fig. 5). Finally, we complemented MK-801 preblock experiments with tests of the effect of enzymatic degradation of glutamate. GPT failed to protect against hypoxic damage but significantly reduced damage to exogenous glutamate. This result suggests strongly that tonic buildup at extrasynaptic receptors is not a major source of toxic glutamate under conditions of our hypoxia experiments. A recent study has also shown that tonic NMDAR activity may not play a critical role in excitotoxic insults, particularly in certain neuronal populations vulnerable to excitotoxic insult (Povysheva and Johnson, 2012).
We cannot exclude the possibility that under some conditions extrasynaptic NMDARs may play a more pronounced role in excitotoxicity than we observed here. Insults of altered severity may promote a larger toxic role for extrasynaptic receptors. For instance, reverse glutamate transport has been shown to play a prominent role in glutamate release under very extreme conditions of metabolic poisoning plus oxygen/glucose deprivation (Rossi et al., 2000). Because neuronal and glial transporters are mainly excluded from synapses (Danbolt, 2001), release by reverse glutamate transport may promote a more prominent role for extrasynaptic receptors in toxicity than our studies indicate. Additional conditions fostering an extrasynaptic receptor role could include cell types with a large percentage of extrasynaptic receptors or networks with weak synaptic connectivity. Finally, we note that previous studies have rarely rigorously verified the degree of synaptic receptor block in the oft-used MK-801 preblock protocol. This leaves open the possibility that significant numbers of synaptic receptors remained unblocked in these studies and may have contributed to toxicity attributed to extrasynaptic receptors.
A related issue concerns the definition of synaptic receptors. Our studies, like previous work, used an operational definition of synaptic NMDARs: receptors blocked by MK-801 when synaptically activated. Although our estimates of synaptic and extrasynaptic receptor percentages agree with previous estimates, it is likely too simplistic to dichotomize receptor populations. The literature's operational definition can be affected by factors that, for example, promote transmitter spillover and variably recruit perisynaptic receptors. There is also growing realization that complex trafficking pathways can dynamically affect exchange between these two populations of surface receptors (Gladding and Raymond, 2011).
We were surprised that toxicity in response to 50 μm glutamate was also strongly reduced by synaptic NMDAR block (Fig. 5E). This protection could be explained in at least two ways. First, synaptic receptors constitute the majority of NMDARs (Rosenmund et al., 1995; Harris and Pettit, 2007). Thus, synaptic receptor block may have protected by simply blocking a large number of receptors. A second possibility is that exogenous agonist, perhaps acting preferentially at extrasynaptic receptors (Sinor et al., 2000; Hardingham et al., 2002), evoked secondary synaptic glutamate release from the network, contributing to the observed death (Monyer et al., 1992). By this explanation, extrasynaptic receptors act indirectly, as a conduit to trigger synaptic NMDAR toxicity. MEA data showed a significant increase in overall network activity during prolonged glutamate exposure, consistent with the idea that synaptic activity is increased by exogenous glutamate. However, toxicity from network disinhibition with bicuculline was milder and treatment took longer than that for glutamate exposure. Thus, while repetitive synaptic activity can be toxic, the effects of exogenous glutamate are unlikely to be explained by the increase in network synaptic activity alone. Rather, direct activation of synaptic receptors by glutamate probably mediates most of the effect of synaptic receptor preblock protection. Regardless of whether synaptic NMDAR activation stems directly from exogenous glutamate or indirectly from synaptic glutamate, our data offer strong evidence that synaptic receptors can be neurotoxic in multiple circumstances, including energy deprivation and insults induced by exogenous agonist.
Our results are in broad agreement with a previous paper that selectively disrupted synaptic NMDAR function with actin depolymerization and observed neuroprotection from glutamate released during oxygen/glucose deprivation (Sattler et al., 2000). However, manipulations of actin polymerization can also have presynaptic effects that could confound interpretations (Morales et al., 2000; Yao et al., 2006; Andrade and Rossi, 2010). Our approach avoided this potential complication. The previous study showed that toxicity to exogenous toxin was not prevented by actin depolymerization, apparently implicating extrasynaptic receptors (Sattler et al., 2000). This is in apparent conflict with our studies in which synaptic receptor blockade prevented exogenous excitotoxicity (Fig. 5E). This discrepancy is likely explained by the fact that actin depolymerization did not disrupt the total number of surface receptors; rather, it relocated synaptic receptors to nonsynaptic membrane. Therefore, our approach reduced overall toxic Ca2+ load in response to exogenous glutamate, whereas the approach by Sattler et al. (2000) did not.
Excitotoxicity is a complex phenomenon mediated in part by overactivation of NMDARs. The extrasynaptic NMDAR hypothesis of excitotoxicity has been one of the most exciting proposals in the glutamate toxicity field in recent years, in part because of the clinical implications. However, our observations substantially undermine two arms of the extrasynaptic NMDAR hypothesis. First, we find that memantine at neuroprotective concentrations significantly blocks synaptic receptors during sustained or repetitive stimulation as likely experienced during excitoxicity-triggering insults. Second, we demonstrate that synaptic receptor activation, proposed to be uniquely protective, is toxic during at least some endogenous and exogenous excitotoxic insults. Our findings point toward a need to consider synaptic receptor contributions when developing therapeutics for disorders with excitotoxic components.
Footnotes
This work was supported by National Institute of Health Grants GM08151 (C.M.W), DA07261 (J.H.), MH78823 (S.M.), and NS52823 (S.M). We thank Ann Benz and Amanda Taylor for assistance with cultures and laboratory members for advice and discussion.
References
- Andrade AL, Rossi DJ. Simulated ischaemia induces Ca2+-independent glutamatergic vesicle release through actin filament depolymerization in area CA1 of the hippocampus. J Physiol. 2010;588:1499–1514. doi: 10.1113/jphysiol.2010.187609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci U S A. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J Neurosci. 2010;30:15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM, Lipton SA, Jensen FE. Neuroprotective concentrations of the N-methyl–aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121–1132. doi: 10.1016/s0306-4522(98)00163-8. [DOI] [PubMed] [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dick O, Bading H. Synaptic activity and nuclear calcium signaling protect hippocampal neurons from death signal-associated nuclear translocation of FoxO3a induced by extrasynaptic N-methyl-D-aspartate receptors. J Biol Chem. 2010;285:19354–19361. doi: 10.1074/jbc.M110.127654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–320. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Gouix E, Léveillé F, Nicole O, Melon C, Had-Aissouni L, Buisson A. Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Mol Cell Neurosci. 2009;40:463–473. doi: 10.1016/j.mcn.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A–2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta. 2002;1600:148–153. doi: 10.1016/s1570-9639(02)00455-7. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogins J, Crawford DC, Jiang X, Mennerick S. Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol Dis. 2011;43:516–525. doi: 10.1016/j.nbd.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Baughman RW. Primary culture of identified neurons from the visual cortex of postnatal rats. J Neurosci. 1986;6:3044–3060. doi: 10.1523/JNEUROSCI.06-10-03044.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabaudon D, Scanziani M, Gähwiler BH, Gerber U. Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci U S A. 2000;97:5610–5615. doi: 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2006;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kaplan TM, Lasner TM, Nadler JV, Crain BJ. Lesions of excitatory pathways reduce hippocampal cell death after transient forebrain ischemia in the gerbil. Acta Neuropathologica. 1989;78:283–290. doi: 10.1007/BF00687758. [DOI] [PubMed] [Google Scholar]
- Kotermanski SE, Johnson JW. Mg2+ imparts NMDA receptor subtype selectivity to the Alzheimer's drug memantine. J Neurosci. 2009;29:2774–2779. doi: 10.1523/JNEUROSCI.3703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotermanski SE, Wood JT, Johnson JW. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J Physiol. 2009;587:4589–4604. doi: 10.1113/jphysiol.2009.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- Lester RA, Tong G, Jahr CE. Interactions between the glycine and glutamate binding sites of the NMDA receptor. J Neurosci. 1993;13:1088–1096. doi: 10.1523/JNEUROSCI.13-03-01088.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008;22:4258–4271. doi: 10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Presynaptic influence on the time course of fast excitatory synaptic currents in cultured hippocampal cells. J Neurosci. 1995;15:3178–3192. doi: 10.1523/JNEUROSCI.15-04-03178.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Benz A, Zorumski CF. Components of glial responses to exogenous and synaptic glutamate in rat hippocampal microcultures. J Neurosci. 1996;16:55–64. doi: 10.1523/JNEUROSCI.16-01-00055.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Chisari M, Shu HJ, Taylor A, Vasek M, Eisenman LN, Zorumski CF. Diverse voltage-sensitive dyes modulate GABAA receptor function. J Neurosci. 2010;30:2871–2879. doi: 10.1523/JNEUROSCI.5607-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Giffard RG, Hartley DM, Dugan LL, Goldberg MP, Choi DW. Oxygen or glucose deprivation-induced neuronal injury in cortical cell cultures is reduced by tetanus toxin. Neuron. 1992;8:967–973. doi: 10.1016/0896-6273(92)90211-u. [DOI] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron. 2000;27:539–550. doi: 10.1016/s0896-6273(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Newell DW, Barth A, Papermaster V, Malouf AT. Glutamate and nonglutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J Neurosci. 1995;15:7702–7711. doi: 10.1523/JNEUROSCI.15-11-07702.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SM, DeMarse TB. A new approach to neural cell culture for long-term studies. J Neurosci Methods. 2001;110:17–24. doi: 10.1016/s0165-0270(01)00412-5. [DOI] [PubMed] [Google Scholar]
- Povysheva NV, Johnson JW. Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol. 2012;107:2232–2243. doi: 10.1152/jn.01017.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL. Synaptic NMDA receptor channels have a low open probability. J Neurosci. 1995;15:2788–2795. doi: 10.1523/JNEUROSCI.15-04-02788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Slater NT. The developmental onset of NMDA receptor-channel activity during neuronal migration. Neuropharmacology. 1993;32:1239–1248. doi: 10.1016/0028-3908(93)90018-x. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Rossi P, Sola E, Taglietti V, Borchardt T, Steigerwald F, Utvik JK, Ottersen OP, Köhr G, D'Angelo E. NMDA receptor 2 (NR2) C-terminal control of NR open probability regulates synaptic transmission and plasticity at a cerebellar synapse. J Neurosci. 2002;22:9687–9697. doi: 10.1523/JNEUROSCI.22-22-09687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536–537. doi: 10.1126/science.6836300. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Excitotoxity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel L-threo-β-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Sinor JD, Du S, Venneti S, Blitzblau RC, Leszkiewicz DN, Rosenberg PA, Aizenman E. NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci. 2000;20:8831–8837. doi: 10.1523/JNEUROSCI.20-23-08831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc Natl Acad Sci U S A. 2009;106:9854–9859. doi: 10.1073/pnas.0903546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark DT, Bazan NG. Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J Neurosci. 2011;31:13710–13721. doi: 10.1523/JNEUROSCI.3544-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–2063. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, van Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Fitzsimonds RM, Johnson B, Dichter MA. Glycine regulation of synaptic NMDA receptors in hippocampal neurons. J Neurophysiol. 1996;76:3415–3424. doi: 10.1152/jn.1996.76.5.3415. [DOI] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Qi J, Chen G. Actin-dependent activation of presynaptic silent synapses contributes to long-term synaptic plasticity in developing hippocampal neurons. J Neurosci. 2006;26:8137–8147. doi: 10.1523/JNEUROSCI.1183-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Buchthal B, Lau D, Hayer S, Dick O, Schwaninger M, Veltkamp R, Zou M, Weiss U, Bading H. A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J Neurosci. 2011;31:4978–4990. doi: 10.1523/JNEUROSCI.2672-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]