

Abstract
Prochlorosins make up a class of secondary metabolites produced by strains of Prochlorococcus, single-cell, planktonic marine cyanobacteria. These polycyclic peptides contain lanthionine and methyllanthionine residues that result in thioether cross-links. In Prochlorococcus MIT9313, a single enzyme, ProcM, catalyzes the posttranslational modification of 29 linear peptide substrates to generate a library of highly diverse cyclic peptides. To investigate the catalytic promiscuity of ProcM, we chose four prochlorosins previously demonstrated to be produced by the organism for detailed structural characterization. Nuclear magnetic resonance studies allowed unambiguous assignment of the ring topologies, demonstrating a high degree of topological diversity. The stereochemistry of the lanthionine and methyllanthionine residues was determined by gas chromatography and mass spectrometry for seven prochlorosins. All methyllanthionines had the (2S,3S,6R) configuration, and the lanthionines had the (2S,6R) configuration, irrespective of the direction of cyclization, ring size, or ring topology. These findings indicate that most, if not all, of the rings in prochlorosins are formed enzymatically by ProcM lanthionine synthetase and not by a nonenzymatic process as previously suggested.
Lantipeptides are ribosomally synthesized and posttranslationally modified peptides, characterized by the thioether amino acids lanthionine (Lan) and methyllanthionine (MeLan).1,2 Lantipeptides with antimicrobial activities are called lantibiotics.3 In lantipeptide-producing organisms, the precursor peptides, termed LanAs, are expressed with an N-terminal leader sequence and a C-terminal core peptide (e.g., Figure 1A). The core peptide is modified by a dehydratase, generating dehydroalanine (Dha) and dehydrobutyrine (Dhb) from Ser and Thr, respectively (Figure 1B). Subsequently, intramolecular additions of the thiols of Cys to the Dha and Dhb residues generate the thioether cross-links (Figure 1B). In class II lantipeptides, these two sequential modifications are conducted by one enzyme, generically called LanM.4 The leader peptide is removed in the final step of lantipeptide biosynthesis to form the mature products.5
Figure 1.

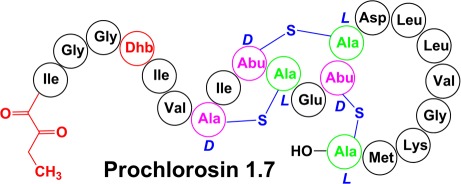
Precursor peptides of the 29 prochlorosins from Prochlorococcus MIT9313 and enzymatic transformations during biosynthesis of lantipeptides. (A) Sequence alignment of the 29 precursor peptides illustrating the high degree of conservation in their leader peptides and the high degree of diversity in their core peptides. The four peptides investigated here are denoted with pink boxes at the left. (B) Dehydration and cyclization reactions during the biosynthesis of lantibiotics and lantipeptides.
Prochlorosins (Pcns) make up a group of lantipeptides produced by Prochlorococcus.6 In Prochlorococcus MIT9313, 29 different peptide substrates are modified by a single promiscuous enzyme, ProcM, forming the characteristic thioether rings.6 The 29 precursor peptides have highly conserved leader peptides but highly diverse core peptides where the posttranslational modifications occur (Figure 1A). This class of lanthionine-containing peptides is of interest not only because of the remarkable promiscuity of the enzyme but also because the organisms carrying these genes are cyanobacteria accounting for as much as half of the chlorophyll in the tropical and subtropical oceans with a lifestyle very different from those of other known lantipeptide-producing organisms.7 They are single-cell organisms living in oligotrophic environments at very dilute concentrations, making it debatable whether prochlorosins serve as antimicrobial defenses like most other known lantipeptides.8
Analysis of the Global Oceanic Survey showed that the genes for production of these lanthionine-containing peptides are widespread.6,9 Their transcription levels respond to nitrogen starvation, suggesting that the prochlorosins are functional.6 However, it remains unclear why the organisms produce this class of secondary metabolites and what their functions are. To address these questions as well as understand the remarkable substrate tolerance of ProcM, it is important to know the chemical structures of prochlorosins, including their ring topologies and the stereochemical configurations of their Lan and MeLan residues.
Preliminary structural characterization of several prochlorosins has been achieved by tandem mass spectrometry (MS).6 The presence of a thioether cross-link prohibits fragmentation of the peptide in the ring, and therefore, the fragmentation pattern can be used for ring topology prediction.4,6 However, tandem MS encounters difficulties in elucidating the ring topology for structures that contain overlapping rings. In such cases, mutagenesis is usually employed to prevent the formation of one or several overlapping rings such that the topology of the remaining rings can be established using tandem MS. However, because ProcM exhibits a very low substrate specificity under laboratory conditions and can generate a variety of ring structures, it is possible that the ring topology of the mutants is not that of the wild-type prochlorosins. Indeed, a single point mutation in ProcA4.3 has been shown to change the original ring pattern.6 Therefore, tandem MS has its limitations for the determination of the structures of prochlorosins, and other complementary techniques are required. In this study, we used nuclear magnetic resonance (NMR) spectroscopy to determine the ring topologies of four prochlorosins (Pcn1.1, Pcn1.7, Pcn3.3, and Pcn4.3) for which the precursor genes have been shown to be transcribed under nitrogen starvation.
In addition to questions about the ring patterns of prochlorosins, the stereochemical configurations of their Lan and MeLan residues have not been determined. Lan and MeLan residues in lantipeptides and lantibiotics for which the stereochemistry has been determined all exhibit (2S,6R) and (2S,3S,6R) configurations, respectively.10 Because ProcM generates such a diversity of ring topologies, we previously speculated that perhaps only some rings are formed enzymatically, thus preorganizing the partially cyclized peptides for nonenzymatic cyclization to generate the final ring structures.6 Determination of the configuration of Lan and MeLan residues in prochlorosins could potentially serve to assess whether a ring is formed enzymatically or nonenzymatically.
To address this hypothesis, we investigated the stereochemical configuration of Lan and MeLan residues of seven prochlorosins (Pcn1.1, Pcn1.7, Pcn2.8, Pcn2.11, Pcn3.2, Pcn3.3, and Pcn4.3) by gas chromatography and MS (GC–MS) with a chiral stationary phase and compared them with synthetic standards.11−13 We were able to conduct these experiments by producing milligram quantities of the selected prochlorosins by heterologous coexpression of the precursor peptides with ProcM in Escherichia coli.14
Materials and Methods
General Methods
All polymerase chain reactions (PCRs) were conducted on a C1000 thermal cycler (Bio-Rad). DNA sequencing was performed by ACGT, Inc. Preparative high-performance liquid chromatography (HPLC) was performed using a Waters Delta 600 instrument equipped with appropriate columns. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) was conducted on a Voyager-DE-STR (Applied Biosystems). Overexpression and purification of the modified precursor peptides were performed using previously reported procedures.14
Construction of pRSFDuet-1 Derivatives for Coexpression of ProcM and ProcAs
The genes encoding ProcA1.1G–1E and ProcA4.3 were cloned from previously reported vectors6 and inserted into multiple cloning site 1 of the pRSFDuet-1/procM-2 vector14 using the EcoRI and NotI restriction sites. Primer sequences are listed in Table S1 of the Supporting Information. Negative numbers are used for amino acids in the leader peptide counting backward from the putative leader peptide cleavage site. The sequences of the constructed vectors were confirmed by DNA sequencing. The pRSFDuet-1/procM-2, pRSFDuet-1/procM-2-procA3.3G–1K, pRSFDuet-1/procM-2-procA1.7G–1R, pRSFDuet-1/procM-2-procA2.8, pRSFDuet-1/procM-2-procA2.11G–1K, pRSFDuet-1/procM-2-procA3.2, pRSFDuet-1/procM-2-procA4.3G–1R, and pRSFDuet-1/procM-2-procA4.3G–1E vectors were obtained as reported previously.14
Protease Cleavage and Purification of Prochlorosin Core Peptides
Posttranslationally modified ProcA mutants were treated with the commercial protease GluC to remove the leader peptides. For some peptides, additional proteases were used to process the long leader peptides into smaller fragments, which sometimes proved to be advantageous for purification (LysC for ProcA1.1G–1E and ProcA3.3G–1K and trypsin for ProcA4.3). In such instances, the modified core peptide was not proteolyzed even if it contained potential cleavage sites for LysC or trypsin, presumably because the presence of dehydrated amino acids and/or (Me)Lan residues greatly reduced the proteolytic efficiency. The protease cleavage reactions were quenched with 0.5% TFA, and the desired products were confirmed by MALDI-TOF MS and purified by reversed phase HPLC (RP-HPLC) using a Jupiter proteo C12 column (5 μm, 90 Å, 250 mm × 10.0 mm). Solvents for RP-HPLC were solvent A (0.1% TFA in water) and solvent B (0.086% TFA in 80% acetonitrile and 20% water). The desired core peptides eluted from the column between 40 and 60% solvent B, and pure fractions were lyophilized.
NMR Spectroscopy
NMR spectra were acquired at the NMR Facility (School of Chemical Sciences, University of Illinois at Urbana-Champaign) on Varian INOVA 600 or 500 MHz spectrometers equipped with a 5 mm triple-resonance (1H, 13C, 15N) triaxial gradient probe. Lyophilized peptides were dissolved in a 90% H2O/10% D2O mixture to a final concentration of 5–7 mM and a pH of 3.5 to improve solubility. NMR spectra were acquired at 20 °C for 2048 direct and 256 of 400 indirect data points with 16 or 32 scans depending on the peptide concentration. TOCSY15 (mixing time of 0.080 s), gradient double-quantum-filtered COSY (gDQCOSY),16 and NOESY17 spectra were acquired with solvent suppression by presaturation or water gate pulse sequences. The lyophilized peptides were dissolved in 100% D2O, and NOESY spectra were acquired without exchangeable proton signals. NOESY spectra of Pcn1.1 and -3.3 were acquired with a mixing time of 0.20 s, while those of Pcn1.7 and -4.3 were acquired with a mixing time of 0.40 s, which resulted in clearer nuclear Overhauser effect (NOE) signals. Spectra were processed with NMRPipe18 and analyzed in Sparky.19
GC–MS Analysis
The synthesis of Lan and MeLan standards and the preparation of samples for GC–MS analysis were conducted following a reported procedure.12 For Pcn1.1, -1.7, -3.3, and -4.3, the purified modified core peptides were used, whereas for Pcn2.8, -2.11, and -3.2, the ProcM-modified precursor peptides with their leader sequences still attached were used. The derivatized samples were analyzed by GC–MS using an Agilent HP 6890N mass spectrometer equipped with a Varian CP-Chirasil-l-Val fused silica column (25 m × 0.25 mm × 0.15 μm). Samples were dissolved in methanol and introduced into the instrument via splitless injection at a flow rate of 1.7 or 2.0 mL/min in helium gas. The following temperature gradient was used: 160 °C for 5 min, from 160 to 180 °C at a rate of 3 °C/min, and 180 °C for 10 min. The mass spectrometer was operated in simultaneous scan/selected-ion monitoring (SIM) mode, monitoring at the characteristic fragment masses of 365 Da for Lan and 379 Da for MeLan residues.
Results
Coexpression of ProcAs and ProcM in E. coli
To obtain milligram quantities of peptides required for NMR characterization, a heterologous coexpression system was employed using E. coli as a host.14,20−23 Using a pRSFDuet vector, ProcM and His-tagged ProcAs were coexpressed and the modified precursor peptides were purified by immobilized metal affinity chromatography (IMAC) followed by RP-HPLC. Using this procedure, ∼80 mg of modified prochlorosins with their leader peptides still attached could be obtained from 1 L of LB culture. Protease cleavage sites were engineered at the −1 position to remove the leader peptides and produce the mature core peptides. These mutant peptides were all fully processed by ProcM except for ProcA4.3G–1R and ProcA4.3G–1E. As a result, wild-type ProcA4.3 was used for coexpression, and the Glu at position −6 was used as a cleavage site for endoproteinase GluC to generate a core peptide with five residues at its N-terminus that remained from the leader peptide. Unlike ProcA4.3, ProcA1.7 and ProcA3.3 mutated at position −1 (G–1R and G–1K, respectively) were still processed by ProcM to generate the fully modified products. However, for ProcA3.3, the engineered cleavage site was close to a dehydrated amino acid, resulting in a greatly reduced efficiency of removal of the leader peptide by endoproteinase LysC, whereas for ProcA1.7, the selected protease (trypsin) caused cleavage in the core peptide, which greatly reduced the yield. To obtain sufficient amounts of the modified core peptides, the Glu at position −6 was also used for these two peptides to remove most of the leader peptide with GluC endoproteinase. MALDI-TOF mass spectra of the resulting modified core peptides are shown in Figure S1 of the Supporting Information.
NMR Spectral Assignments
To determine the ring topology, the observed NMR resonances were first assigned. A series of one-dimensional (1D) and two-dimensional NMR experiments were performed for this purpose. The amino acid assignments were obtained primarily from TOCSY and gDQCOSY spectra. The TOCSY spectrum of Pcn1.7 is shown in Figure 2 as an example. TOCSY spectra for the other three prochlorosins are shown in Figure S3 (Pcn1.1), Figure S6 (Pcn3.3), and Figure S9 (Pcn4.3) of the Supporting Information. The chemical shifts of amide protons in these peptides in 90% H2O and 10% D2O were between 7 and 10 ppm, most of which showed good dispersion. The number of amide protons was determined by integration of 1D 1H NMR spectra. Each vertical line in each TOCSY spectrum corresponds to the spin system of one amino acid, with the amide proton resonance on the x-axis and the chemical shifts of the side chain proton resonances on the y-axis. The N-terminal residues have no amide resonance and hence were not observed. The amide signals of all amino acids were observed for each of the four prochlorosins. They were assigned to a specific amino acid by taking into account the observed chemical shifts and numbers of resonances of the side chain protons to which the amide protons were correlated in the TOCSY spectra. The amide proton signals of Dhb residues always appeared as singlets in the low field region with chemical shifts of >9.3 ppm, and the β/γ proton signals of Dhb displayed weak correlations to their corresponding amide protons in the TOCSY spectra.
Figure 2.

Water-suppressed TOCSY spectrum identifying all residues of Pcn1.7. Each vertical line indicates a spin system corresponding to an amino acid. A water-suppressed NOESY spectrum (Figure S2 of the Supporting Information) along with the gene sequence was used to assign the residue numbering.
Assignments of each residue to a specific position in the amino acid sequence were made on the basis of the connectivity of dNN(i,i+1) as well as dαN(i,i+1) in the NOESY spectra (Figure S2 for Pcn1.7, Figure S4 for Pcn1.1, Figure S7 for Pcn3.3, and Figure S10 for Pcn4.3), taking into consideration the amino acid sequences deduced from the procA genes.
Ring Topology Assigned by NMR Spectroscopy
In the discussion below, the fragment of the Lan residues originating from Ser will be designated Ala(S) and the fragment that originates from Cys will be designated Cys (Ala and Cys are colored red and green, respectively, in Figure 3A). Similarly, the fragment of MeLan residues that originates from Thr will be designated Abu(S) (for 2-aminobutyric acid), and the fragment originating from Cys will be designated Cys. The chemical shifts for the β protons of putative Ala(S) and Abu(S) residues observed in the TOCSY experiments provided the first evidence of ring formation. Dha/Dhb residues that were converted into thioether cross-links had chemical shifts of their β protons of ∼3 ppm, whereas Dha/Dhb residues that were not involved in ring formation displayed β proton chemical shifts between 6 and 7 ppm.
Figure 3.

Illustration of the method used to establish thioether connectivities. (A) Schematic illustration of the proton correlations used to determine ring topologies. (B) Section of the NOESY spectrum (mixing time of 0.40 s) of Pcn1.7 in D2O showing the correlations between β protons. (C) Section of the NOESY spectrum showing the correlations of the α protons with α and β protons across the thioether bridge. (D) Section of the NOESY spectrum showing the correlations of the γ protons with α and β protons across the thioether bridge. In panels B–D, correlations are assigned to rings A, B, and C of Pcn1.7 (see also Figure 4). For enlarged panels B and C, see Figures S18 and S19 of the Supporting Information.
Next, NOESY spectra were acquired in D2O to simplify the spectrum and focus on the NOEs involving the Lan/MeLan protons, which allow assignment of the ring topology24−29 (Figure 3 for Pcn1.7, Figure S5 for Pcn1.1, Figure S8 for Pcn3.3, and Figure S11 for Pcn4.3). A longer NOESY mixing time of 0.40 s was used for Pcn1.7 and -4.3. Intense NOE signals between the β protons of an Ala(S)/Abu(S) and a Cys were taken as evidence of a cross-link between these residues (Figure 3B). These assignments were further supported by correlations of α protons of Ala(S)/Abu(S) with the β protons of the Cys with which it was cross-linked, as well as correlations between α protons of Cys residues and the β protons of Ala(S)/Abu(S) residues (Figure 3C). In the case of MeLan, the γ protons on the methyl groups of Abu(S) residues showed additional correlations with the α and β protons of their partner Cys (Figure 3D). For some prochlorosins (i.e., Pcn1.1, -3.3, and -4.3), these latter correlations were particularly diagnostic to confirm the deduced ring structures because less signal overlap was present in this region of the spectra (Figures S5, S8, and S11 of the Supporting Information). Collectively, this approach allowed assignment of all resonances to individual residues and therefore established the topology of the rings (Figure 4). For Pcn1.1, Pcn1.7, and Pcn3.3, the NMR assignments agreed with the previous tandem MS data.6 For Pcn4.3, the NMR results in this study allowed assignment of the second ring that was not possible by MS.6 The NMR data clearly show that this ring is formed from Thr8 and Cys12, and that Ser9 remained unmodified.
Figure 4.

Ring topology of four selected prochlorosins assigned by NMR spectroscopy in this work. Arrows indicate the start of the putative core peptide. Asterisks indicate prochlorosins containing five more residues at their N-terminus that originate from the leader peptides. Blue residues are the engineered cleavage sites at position −1 in the leader peptides.
We note that the additional amino acids remaining from the leader peptides in prochlorosins 1.7, 3.3, and 4.3 showed fewer correlation signals in the NOESY spectrum, suggesting that the N-terminus is conformationally more flexible than the modified core peptide.
Determination of the Stereochemistry of Lan and MeLan Residues
To determine the stereochemical configurations of the Lan and MeLan residues in the four prochlorosins for which the NMR structures were determined, the peptides were hydrolyzed in 6 M HCl, derivatized to their corresponding pentafluoropropionamide methyl esters, and analyzed by GC–MS with a chiral stationary phase. Lan and MeLan standards with different stereochemical configurations were synthesized and derivatized using previously reported methods.12 A Chirasil-l-Val-coated GC column was used for analysis, and the GC trace was monitored for a characteristic fragment of 365 Da for Lan and 379 Da for MeLan using electron impact mass spectrometry. Figure 5 displays the GC–MS traces for the products from Pcn1.7 as well as the Lan and MeLan standards. As shown in Figure 5A, the derivatized MeLan standards with dl (2S,3S,6R) and ll (2R,3R,6R) configuration were well separated, with retention times of 14.1–14.4 and 14.4–14.7 min, respectively. The derivatized Lan standards with dd (2S,6S), dl (2S,6R), and ll (2R,6R) configurations eluted later than the MeLan standards and were also well separated with retention times of 18.2–18.6, 18.6–19.0, and 19.0–19.4 min, respectively (Figure 5B). Derivatized MeLan originating from Pcn1.7 eluted with a retention time of 14.1–14.4 min (Figure 5C), indicating that the MeLan in Pcn1.7 has a dl configuration. To further confirm this result, the derivatized dl and ll MeLan standards were co-injected with the derivatized MeLan originating from the peptide. As illustrated in Figure 5, the derivatized MeLan fragment from Pcn1.7 coeluted with the dl MeLan standard (Figure 5C), but not with the ll MeLan standard (Figure 5D). MeLan standards with the d-allo-l (2S,3R,6R) and l-allo-l (2R,3S,6R) configuration were not synthesized in this study because the reported elution times of the derivatized MeLan diastereomers on a Chirasil-l-Val-coated GC column increase in the following order: dl < ll < l-allo-l < d-allo-l.12 In this work, no derivatized MeLan residues were observed to elute after the ll standard, ruling out the presence of MeLan residues with an l-allo-l or d-allo-l configuration. The MeLan residues present in the other three prochlorosins were analyzed using the same procedure, and all had the dl configuration (Figure S12 for Pcn1.1, Figure S13 for Pcn3.3, and Figure S14 for Pcn4.3).
Figure 5.

GC–MS traces (selected ion monitoring, SIM, at 365 Da for Lan and 379 Da for MeLan) of derivatized (Me)Lan standards and co-injections with derivatized Lan/MeLan obtained from Pcn1.7. (A) Derivatized dl-MeLan (—) and ll-MeLan (---) standards. (B) Derivatized dd-Lan (···), dl-Lan (—), and ll-Lan (---) standards. (C) Hydrolyzed and derivatized amino acids from Pcn1.7 (---) and co-injected with the dl-MeLan standard (—). (D) Hydrolyzed and derivatized amino acids from Pcn1.7 (---) and co-injected with the derivatized ll-MeLan standard (—). (E) Hydrolyzed and derivatized amino acids from Pcn1.7 (---) and co-injected with the dd-Lan standard (—). (F) Hydrolyzed and derivatized amino acids from Pcn1.7 (---) and co-injected with the dl-Lan standard (—). (G) Hydrolyzed and derivatized amino acids from Pcn1.7 (---) and co-injected with the ll-Lan standard (—). Lan and MeLan from Pcn1.7 used for overlays were adjusted to 70% intensity for the sake of clarity.
Similarly, derivatized Lan originating from Pcn1.7 coeluted with the Lan standard with the dl configuration but not with either the dd or ll Lan standard (Figure 5E–G). The small shoulders on the derivatized dl-Lan peak arising from Pcn1.7 are believed to be caused by partial epimerization during HCl hydrolysis, which has been reported previously.30 The GC trace for Pcn4.3, the other prochlorosin investigated here that contains Lan, was more complicated. Although the derivatized dl-Lan was still the dominant peak, an additional peak in the GC trace accounted for ∼20% of the total Lan (Figure 6A). The material giving rise to this peak was confirmed to be derivatized ll-Lan by co-injections (Figure S14E of the Supporting Information). This result was in agreement with the NMR data (1D water-suppressed spectrum shown in Figure 6B), which exhibited minor peaks with integration values around 25% of that of the main peaks. These NMR data demonstrate that the ll-Lan was already present in Pcn4.3 before acid hydrolysis and derivatization and was not introduced by epimerization during HCl hydrolysis.
Figure 6.

ll-Lan diastereomer in Pcn4.3 and its relative abundance. (A) GC–MS trace of hydrolyzed and derivatized amino acids of Pcn4.3 monitored for Lan. dd-Lan (18.2–18.6 min), dl-Lan (18.6–19.0 min), and ll-Lan (19.0–19.4 min) eluted from the column, with the relative peak areas indicated. (B) 1D water-suppressed NMR spectrum of Pcn4.3. Minor peaks originate from a different diastereomer of Pcn4.3 containing ll-Lan as shown in panel A. Integrations of two pairs of major and minor peaks are shown to estimate the relative amount of the diastereomer.
In addition to the prochlorosins for which the NMR structures were determined, the stereochemistry of the Lan and MeLan residues was determined for three other ProcA substrates that were posttranslationally modified by ProcM in E. coli (ProcA2.8, ProcA2.11, and ProcA3.2). The corresponding prochlorosins could not be produced in sufficient purity after leader peptide removal for NMR studies, but after hydrolysis and derivatization of the resulting amino acids, GC–MS analysis demonstrated that they contained Lan and MeLan residues with the same configurations as in the other prochlorosins (Figures S15–S17 of the Supporting Information).
Discussion
In this study, we set out to investigate three aspects of prochlorosin biosynthesis: whether tandem MS is reliable for determination of ring topologies, whether sufficient quantities of material can be produced such that NMR spectroscopy can be used when MS data are inconclusive, and whether the stereochemistries of the Lan and MeLan residues are the same as those in other lantipeptides. To address these questions, we first produced larger amounts of prochlorosins by heterologous expression in E. coli because less than 10 μg of impure products was obtained from 20 L of culture of Prochlorococcus MIT9313.6 Although the peptides obtained by proteolysis with commercial proteases contain additional amino acids at their N-termini compared to authentic prochlorosins, for determination of the ring topology and stereochemistry of (Me)Lan residues, these five additional amino acids of the leader peptide are inconsequential.
The NMR data reported here confirm the ring topologies of three prochlorosins that were previously proposed on the basis of tandem mass spectrometry in combination with site-directed mutagenesis. Thus, the concern that a highly promiscuous enzyme like ProcM might process a mutant peptide differently from a wild-type peptide appears not to be corroborated for these compounds. Mass spectrometric approaches used in the previous study could not answer the question of whether Thr8 or Ser9 was involved in the B-ring of Pcn4.3.6 The NMR data in this study show conclusively that it is Thr8 that is dehydrated in wt ProcA4.3 and that it makes a thioether cross-link with Cys12 (Figure 4).
Collectively, these results raise the question of why mutants of ProcA1.7 and ProcA3.3 that were made in the previous study6 to deduce their ring topologies by tandem MS were processed correctly by ProcM, as shown by the NMR data in this study, whereas mutants of ProcA4.3 were not. Although we cannot address this question directly, processing of ProcA4.3 by ProcM appears to be less robust in general. First, mutations at the −1 position in ProcA4.3 completely abolished ProcM activity in this study, whereas ProcM fully processed mutants at the −1 position for the other three ProcA substrates. Second, whereas all Lan and MeLan residues in Pcn1.1, Pcn1.7, and Pcn3.3 were formed in high stereochemical purity with the same stereochemistry (dl), the Lan in Pcn4.3 was decidedly less pure and contained ∼20% of the ll diastereomer. As shown in Figure 4, the A-ring of Pcn4.3 contains two Gly residues between the Dha and Cys that form this lanthionine, and perhaps the resulting increased conformational freedom allows enzymatic cyclization that results in protonation of the enolate from the opposite face. Alternatively, the lower stereochemical fidelity of cyclization of this particular Lan structure could be the result of nonenzymatic cyclization as discussed below.
The determination of the ring topologies of four different prochlorosins in this study demonstrates the remarkable substrate flexibility of the ProcM enzyme. Inspection of Figure 4 illustrates that the structures of the four selected prochlorosins do not exhibit any similarity to each other or to any other known lantibiotics/lantipeptides.8 The rings generated by ProcM contain very different numbers of amino acids, from four residues in the A-rings of Pcn1.7 and Pcn4.3 to 11 residues in the A-ring of Pcn3.3. Furthermore, the rings are generated either from a Cys that is located N-terminally to the dehydro amino acid with which it reacts (A-ring of Pcn 1.1 and B-ring of Pcn3.3) or from a Cys located C-terminally of its partner dehydro amino acid (all other rings). Finally, Pcn1.1 and -4.3 have nonoverlapping rings; Pcn1.7 has overlapping rings, and Pcn3.3 contains a ring within a ring. Although many lantipeptide cyclases achieve formation of different sized rings with different topologies, ProcM, and presumably its many orthologs in the world’s oceans, is unique in that it acts on so many different substrates.
It was this unprecedented flexibility that led to a proposal that perhaps the enzyme generates only a subset of the rings and that the partially cyclized intermediates would have a preorganized structure for nonenzymatic cyclization of the remaining rings.6 This proposal was bolstered by the previously observed facile nonenzymatic intramolecular addition of Cys to dehydro amino acids located N-terminal to the Cys residue.31−37 These model studies of nonenzymatic cyclization showed that formation of individual small rings is often stereoselective and results in the same stereochemistry that is observed for enzymatic cyclization. However, for larger rings or multiple-ring structures, nonenzymatic cyclization has been shown to give mixtures of diastereomers and/or constitutional isomers,33,38,39 and cyclization involving a Cys located N-terminally to its partner dehydro amino acid has been shown to be nonstereoselective.35 Therefore, in this investigation, the stereochemistry of the Lan and MeLan structures of prochlorosins was determined to test for nonenzymatic cyclization. The GC–MS data clearly illustrate that the Lan residues in prochlorosins have the (2S,6R) configuration and that the MeLan residues have the (2S,3S,6R) configuration. These configurations are the same as those found for all lantibiotics for which the stereochemistry has been determined.10 As discussed above, the only exception was the Lan in Pcn4.3, which had a 4:1 ratio of dl and ll diastereomers. Although these results do not rule out nonenzymatic cyclization, they also do not provide support for nonenzymatic cyclization, and on the basis of the data in this study on seven prochlorosins with very different ring structures, we favor a model in which ProcM does generate all thioether cross-links. One interesting unique aspect of ProcM and its orthologs is the fact that on the basis of sequence alignments, its catalytic Zn2+ is liganded by three Cys residues rather than the two Cys residues and one His residue that are found for all other LanM and LanC lanthionine cyclases investigated thus far.40−44 The Zn2+ has been proposed to activate the Cys residues of the substrate for nucleophilic attack on the dehydro amino acids.40 Model studies of activation of thiolate nucleophiles by Zn2+ have demonstrated increased reactivity with an increased number of thiolate ligands.45−48 Therefore, ProcM may derive its promiscuity in part from a highly active Zn2+ site with three Cys ligands from ProcM and one Cys ligand from the substrate.
In summary, we report here the NMR structures of four prochlorosins and show for seven prochlorosins that they contain Lan and MeLan residues with the same configuration that is found in other lantibiotics and lantipeptides. These data suggest that most, if not all, of the rings are generated by the ProcM enzyme despite the remarkable structural diversity of its products.
Acknowledgments
We thank Ms. Yanxiang Shi and Ms. Xiao Yang for the preparation of coexpression vectors for ProcA2.8, ProcA4.3G–1R, and ProcA4.3G–1E, Jennifer Rapp and Feng Lin for assistance with acquisition of the NMR spectra, Dr. Alexander V. Ulanov for help with GC–MS, and Prof. Chad Rienstra for helpful discussions.
Glossary
Abbreviations
- Lan
lanthionine
- MeLan
methyllanthionine
- Dha
dehydroalanine
- Dhb
dehydrobutyrine
- Pcn
prochlorosin
- MALDI-TOF MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- IMAC
immobilized metal affinity chromatography.
Supporting Information Available
Additional data and experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health Grant GM 58822.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Knerr P. J.; van der Donk W. A. (2012) Discovery, biosynthesis, and engineering of lantipeptides. Annu. Rev. Biochem. DOI: 10.1146/annurev-biochem-060110-113521. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Li B.; Claesen J.; Shi Y.; Bibb M. J.; van der Donk W. A. (2010) Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary insights. PLoS Biol. 8, e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell N.; Entian K.-D.; Schneider U.; Götz F.; Zahner H.; Kellner R.; Jung G. (1988) Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 333, 276–278. [DOI] [PubMed] [Google Scholar]
- Xie L. L.; Miller L. M.; Chatterjee C.; Averin O.; Kelleher N. L.; van der Donk W. A. (2004) Lacticin 481: In vitro reconstitution of lantibiotic synthetase activity. Science 303, 679–681. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; van der Donk W. A. (2010) Follow the leader: The use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 6, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Sher D.; Kelly L.; Shi Y. X.; Huang K.; Knerr P. J.; Joewono I.; Rusch D.; Chisholm S. W.; van der Donk W. A. (2010) Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Z. I.; Zinser E. R.; Coe A.; McNulty N. P.; Woodward E. M. S.; Chisholm S. W. (2006) Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Willey J. M.; van der Donk W. A. (2007) Lantibiotics: Peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501. [DOI] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K.; Mitchell D. A. (2010) Expansion of ribosomally produced natural products: A nitrile hydratase- and Nif11-related precursor family. BMC Biol. 8, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee C.; Paul M.; Xie L. L.; van der Donk W. A. (2005) Biosynthesis and mode of action of lantibiotics. Chem. Rev. 105, 633–683. [DOI] [PubMed] [Google Scholar]
- Küsters E.; Allgaier H.; Jung G.; Bayer E. (1984) Resolution of sulfur-containing amino-acids by chiral phase gas-chromatography. Chromatographia 18, 287–293. [Google Scholar]
- Liu W.; Chan A. S. H.; Liu H. Q.; Cochrane S. A.; Vederas J. C. (2011) Solid supported chemical syntheses of both components of the lantibiotic lacticin 3147. J. Am. Chem. Soc. 133, 14216–14219. [DOI] [PubMed] [Google Scholar]
- Knerr P. J.; Tzekou A.; Ricklin D.; Qu H. C.; Chen H.; van der Donk W. A.; Lambris J. D. (2011) Synthesis and activity of thioether-containing analogues of the complement inhibitor compstatin. ACS Chem. Biol. 6, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. X.; Yang X. A.; Garg N.; van der Donk W. A. (2011) Production of lantipeptides in Escherichia coli. J. Am. Chem. Soc. 133, 2338–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweiler L.; Ernst R. R. (1983) Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528. [Google Scholar]
- Piantini U.; Sørensen O. W.; Ernst R. R. (1982) Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 104, 6800–6801. [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. (1979) Investigation of exchange processes by 2-dimensional NMR spectroscopy. J. Chem. Phys. 71, 4546–4553. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2005) Sparky, University of California, San Francisco. [Google Scholar]
- Ökesli A.; Cooper L. E.; Fogle E. J.; van der Donk W. A. (2011) Nine post-translational modifications during the biosynthesis of cinnamycin. J. Am. Chem. Soc. 133, 13753–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velásquez J. E.; Zhang X. G.; van der Donk W. A. (2011) Biosynthesis of the antimicrobial peptide epilancin 15X and its N-terminal lactate. Chem. Biol. 18, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N.; Tang W.; Goto Y.; van der Donk W. A. (2012) Geobacillins: Lantibiotics from Geobacillus thermodenitrificans. Proc. Natl. Acad. Sci. U.S.A. 109, 5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao J.; Aso Y.; Shioya K.; Nakayama J.; Sonomoto K. (2007) Lantibiotic engineering: Molecular characterization and exploitation of lantibiotic-synthesizing enzymes for peptide engineering. J. Mol. Microbiol. Biotechnol. 13, 235–242. [DOI] [PubMed] [Google Scholar]
- Freund S., Jung G., Gibbons W. A., and Sahl H. G. (1991) NMR and circular dichroism studies on Pep5. In Nisin and Novel Lantibiotics (Jung G., and Sahl H. G., Eds.) pp 103–112, ESCOM, Leiden, The Netherlands. [Google Scholar]
- van de Ven F. J. M.; van den Hooven H. W.; Konings R. N. H.; Hilbers C. W. (1991) NMR studies of lantibiotics. The structure of nisin in aqueous solution. Eur. J. Biochem. 202, 1181–1188. [DOI] [PubMed] [Google Scholar]
- Martin N. I.; Sprules T.; Carpenter M. R.; Cotter P. D.; Hill C.; Ross R. P.; Vederas J. C. (2004) Structural characterization of lacticin 3147, a two-peptide lantibiotic with synergistic activity. Biochemistry 43, 3049–3056. [DOI] [PubMed] [Google Scholar]
- Shenkarev Z. O.; Finkina E. I.; Nurmukhamedova E. K.; Balandin S. V.; Mineev K. S.; Nadezhdin K. D.; Yakimenko Z. A.; Tagaev A. A.; Temirov Y. V.; Arseniev A. S.; Ovchinnikova T. V. (2010) Isolation, structure elucidation, and synergistic antibacterial activity of a novel two-component lantibiotic lichenicidin from Bacillus licheniformis VK21. Biochemistry 49, 6462–6472. [DOI] [PubMed] [Google Scholar]
- Smith L.; Novak J.; Rocca J.; McClung S.; Hillman J. D.; Edison A. S. (2000) Covalent structure of mutacin 1140 and a novel method for the rapid identification of lantibiotics. Eur. J. Biochem. 267, 6810–6816. [DOI] [PubMed] [Google Scholar]
- He Z.; Yuan C.; Zhang L.; Yousef A. E. (2008) N-terminal acetylation in paenibacillin, a novel lantibiotic. FEBS Lett. 582, 2787–2792. [DOI] [PubMed] [Google Scholar]
- Kido Y.; Hamakado T.; Yoshida T.; Anno M.; Motoki Y.; Wakamiya T.; Shiba T. (1983) Isolation and characterization of ancovenin, a new inhibitor of angiotensin I converting enzyme, produced by actinomycetes. J. Antibiot. 36, 1295–1299. [DOI] [PubMed] [Google Scholar]
- Polinsky A.; Cooney M. G.; Toy-Palmer A.; Ösapay G.; Goodman M. (1992) Synthesis and conformational properties of the lanthionine-bridged opioid peptide [d-AlaL2,AlaL2]enkephalin as determined by NMR and computer simulations. J. Med. Chem. 35, 4185–4191. [DOI] [PubMed] [Google Scholar]
- Toogood P. L. (1993) Model Studies of Lantibiotic Biogenesis. Tetrahedron Lett. 34, 7833–7836. [Google Scholar]
- Burrage S.; Raynham T.; Williams G.; Essex J. W.; Allen C.; Cardno M.; Swali V.; Bradley M. (2000) Biomimetic synthesis of lantibiotics. Chem.—Eur. J. 6, 1455–1466. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; Zhu Y.; van der Donk W. A. (2000) Facile chemoselective synthesis of dehydroalanine-containing peptides. Org. Lett. 2, 3603–3606. [DOI] [PubMed] [Google Scholar]
- Zhou H.; van der Donk W. A. (2002) Biomimetic stereoselective formation of methyllanthionine. Org. Lett. 4, 1335–1338. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Gieselman M.; Zhou H.; Averin O.; van der Donk W. A. (2003) Biomimetic studies on the mechanism of stereoselective lanthionine formation. Org. Biomol. Chem. 1, 3304–3315. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Ni W.; van der Donk W. A. (2007) On the regioselectivity of thioether formation by lacticin 481 synthetase. Org. Lett. 9, 3343–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. T.; Gieselman M. D.; Zhou H.; Averin O.; van der Donk W. A. (2003) Biomimetic studies on the mechanism of stereoselective lanthionine formation. Org. Biomol. Chem. 1, 3304–3315. [DOI] [PubMed] [Google Scholar]
- Zhang X.; van der Donk W. A. (2007) On the substrate specificity of dehydration by lacticin 481 synthetase. J. Am. Chem. Soc. 129, 2212–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeley N. M.; Paul M.; Stasser J. P.; Blackburn N.; van der Donk W. A. (2003) SpaC and NisC, the cyclases involved in subtilin and nisin biosynthesis, are zinc proteins. Biochemistry 42, 13613–13624. [DOI] [PubMed] [Google Scholar]
- Li B.; Yu J. P.; Brunzelle J. S.; Moll G. N.; van der Donk W. A.; Nair S. K. (2006) Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science 311, 1464–1467. [DOI] [PubMed] [Google Scholar]
- Helfrich M.; Entian K. D.; Stein T. (2007) Structure-function relationships of the lanthionine cyclase SpaC involved in biosynthesis of the Bacillus subtilis peptide antibiotic subtilin. Biochemistry 46, 3224–3233. [DOI] [PubMed] [Google Scholar]
- Li B.; van der Donk W. A. (2007) Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin. J. Biol. Chem. 282, 21169–21175. [DOI] [PubMed] [Google Scholar]
- Paul M.; Patton G. C.; van der Donk W. A. (2007) Mutants of the zinc ligands of lacticin 481 synthetase retain dehydration activity but have impaired cyclization activity. Biochemistry 46, 6268–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilker J. J.; Lippard S. J. (1997) Alkyl transfer to metal thiolates: Kinetics, active species identification, and relevance to the DNA methyl phosphotriester repair center of Escherichia coli Ada. Inorg. Chem. 36, 969–978. [DOI] [PubMed] [Google Scholar]
- Warthen C. R.; Hammes B. S.; Carrano C. J.; Crans D. C. (2001) Methylation of neutral pseudotetrahedral zinc thiolate complexes: Model reactions for alkyl group transfer to sulfur by zinc-containing enzymes. J. Biol. Inorg. Chem. 6, 82–90. [DOI] [PubMed] [Google Scholar]
- Brand U.; Rombach M.; Seebacher J.; Vahrenkamp H. (2001) Functional modeling of cobalamine-independent methionine synthase with pyrazolylborate-zinc-thiolate complexes. Inorg. Chem. 40, 6151–6157. [DOI] [PubMed] [Google Scholar]
- Chiou S. J.; Riordan C. G.; Rheingold A. L. (2003) Synthetic modeling of zinc thiolates: Quantitative assessment of hydrogen bonding in modulating sulfur alkylation rates. Proc. Natl. Acad. Sci. U.S.A. 100, 3695–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.