

Abstract
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) with varied clinical presentations and heterogeneous histopathological features. The underlying immunological abnormalities in MS lead to various neurological and autoimmune manifestations. There is strong evidence that MS is, at least in part, an immune-mediated disease. There is less evidence that MS is a classical autoimmune disease, even though many authors state this in the description of the disease. We show the evidence that both supports and refutes the autoimmune hypothesis. In addition, we present an alternate hypothesis based on virus infection to explain the pathogenesis of MS.
1. Introduction
Studies using imaging, serology, pathology and genetics, and patient response to anti-inflammatory treatments indicate that multiple sclerosis (MS) is primarily an inflammatory demyelinating disease of the central nervous system (CNS) with varied clinical presentations and heterogeneous histopathological features. The disease has a peak onset between ages 20 and 40 years [1]; however it may also develop in children and in addition has been reported in individuals aged above 60 years. MS affects women approximately twice as often as men [2–5]. MS results in a plethora of neurological manifestations and is a leading cause of nontraumatic disability among young adults and has great socioeconomic impact in developed countries [6]. Based on the epidemiological studies, approximately 400,000 people have MS in the United States, with 200 new cases added every week. The pathogenesis of MS remains elusive and there were no definitive cause and no effective cure. Therefore, MS can be classified as an episodic demyelinating disease of the central nervous system. Disease pathophysiology is complex and involves genetic susceptibility, environmental factors, and development of a pathologic immune-mediated response leading to focal myelin destruction, axonal loss, and focal inflammatory infiltrates.
The pathophysiology of MS is further fraught with confusion as researchers struggle to classify the disease as either pathological [7] or clinical [8]. Investigators and clinicians who have studied MS agree that the immune system plays a critical role in the development of lesions, especially during the acute early phases of the disease characterized by relapses. Relapses are fundamentally a manifestation of an inflammatory response occurring mostly in the white matter of the nervous system but also within myelin tracts in the gray matter. This results in focal demyelination with relative axonal sparing. The best evidence for inflammation-induced relapses comes from work in MRI, which demonstrates the association of relapses with gadolinium enhancement that is disruption of the blood brain barrier. The main pathologic hallmark of MS is the demyelinated plaque, which has specific histological and immunocytological characteristics depending on the activity of the disease [9–12]. Histologically, an MS plaque is characterized by marked predominance of CD8+ T cells and a relative lack of CD4+ T cells (ratios of 100 : 1 to 50 : 1). In addition, there is a sea of macrophages, which may have a primary role in engulfing myelin debris. Whether they are also primary effectors in the disease process is unknown. Another important immunopathological feature is continuous synthesis of immunoglobulins (oligoclonal IgG's) in cerebrospinal fluid (CSF). The evidence associating antibodies with MS derives from studies such as by Kabat et al., who described increased levels of immunoglobulin (Ig) in the cerebrospinal fluid (CSF) [13]. CSF IgG and oligoclonal bands remain the most predictive immunological test for the diagnosis of MS. All immunoglobulin subtypes have been implicated in MS. The underlying immunological abnormalities lead to presentation of different autoimmune manifestations.
2. Is MS an Autoimmune Disease?
From most references gleaned in the literature, MS is boldly stated as an autoimmune disorder. However, the evidence for such a statement is weak and circumstantial. We have updated and revised criteria for determining whether a disease is autoimmune in nature [14]. The main criterion of a given autoimmune disease is that a precise autoantigen be present in all patients with the disease. Despite multiple attempts to identify various proteins, lipids, and gangliosides in myelin as potential MS antigens, none have been proven or confirmed. Secondly, administration of autoantibody or T cells induces autoimmune disease in normal animals. These approaches have been attempted in animal models of MS with contrasting results [15, 16]. A third criterion is the ability to induce lesions by immunizing animals with relevant autoantigen. This had been partially achieved but with problems. The fact that multiple different antigens can induce the disease process in animal models without one specific antigen being superior to the other makes the results ambiguous from the standpoint of identifying the relevant antigen. The fourth criterion is the ability to isolate autoantibody or autoreactive T cells from the lesion or from serum. Many investigators have suggested a higher precursor frequency of T cells, specifically of the CD4 subgroup, in patients with MS when compared to healthy controls, which recognize MBP, proteolipid protein (PLP), MOG, or other such antigens from myelin. Unfortunately, because similar positive results are obtained from normal individuals, this criterion is not satisfied. The fifth criterion is the correlation between the autoantigen or the autoreactive T cells with disease activity. Autoreactive T cells occur with greater frequency in patients experiencing an exacerbation than in patients with progressive disease, which suggests a possible correlation between auto-reactive T cells and disease activity. Even though the precursor frequency of autoreactive T cells may be higher in MS than in normal controls, the presence of autoreactive T cells demonstrated in normal controls makes a definitive conclusion about MS as autoimmune more difficult to accept. The sixth criterion is the presence of other autoimmune disorders or autoantigens associated with the disease. This issue has been addressed by a number of investigators, and there have been occasional case reports demonstrating the presence of MS with other autoimmune diseases, for example, myasthenia gravis [17] and diabetes mellitus [18]. However, population-based cohort studies performed in the Olmsted county, Minnesota, failed to show any association between autoimmune diseases and MS [19]. The only possible increased odds ratio was found with thyroid disease, when both hyperthyroidism and hypothyroidism were combined. In addition, rare cases have been described in patients with both MS and inflammatory bowel disease [17, 18]. There have also been multiple studies looking at the presence of autoantibodies, a characteristic of patients with autoimmune diseases, such as the antibodies seen in Sjögren's syndrome, systemic lupus erythematosus (SLE), or myasthenia gravis, but to date, no evidence indicates that the presence of these antibodies is greater in MS patients than in normal controls. Of interest, this differs greatly from neuromyelitis optica (NMO) [20, 21], where there is clearly an association between the presence of autoantibodies and NMO (discussed later in this paper).
3. Immune Manifestations of MS: Role of Antibodies
There is evidence to suggest that part of the immunopathogenesis of MS is mediated by antibodies. Studies have indicated intrathecal production of antibodies, which occur after clonal expansion manifested by the identification of oligoclonal bands after CSF electrophoresis [22]. Studies at the Mayo Clinic, Austria, and Germany reported on the heterogeneity of MS lesions in CNS tissue and their implications for the pathogenesis of demyelination. A detailed immunohistochemical study was performed in active MS lesions from 83 biopsies and autopsies of MS patients, following, which were identified four different pathologic subtypes of active MS lesions. One of the subtypes, Pattern II, demonstrated the presence of macrophages and T cells but, in addition, a prominent display of antibodies and complement [9]. This data provided evidence that lesion patterns were heterogeneous among patient subgroups but homogeneous in the same patient. Barnett and Prineas studied acute MS lesions and found complement activation, oligodendrocyte apoptosis, and remyelination, findings that overlapped the Mayo/Germany/Austria studies [23]. A recent study identified activated complement (C3d and C9neo) on fragmenting myelin sheaths in the outer actively demyelinating lesions in 20 patients with relapsing MS (58/58 active lesions) [24]. The authors reported the presence of activated complement on disintegrating sheaths in diverse diseases affecting white matter, including viral and autoimmune encephalitis, NMO, and even ischemic infarcts, suggesting that this phenomenon is not limited to MS. In a more recent study, Breij et al. [25] investigated to what extent the four pathological pattern criteria translated to active lesions from patients with established MS. These authors concluded that MS lesions displayed a homogenous profile. The authors were unable to confirm the lesion heterogeneity or interindividual heterogeneity with respect to Ig and complement immuno-reactivity. However, it is possible that all the lesions studied were not active. It is possible that the heterogeneous features reported in active MS lesions that were sampled at varied time-points are the evolution of a single pathophysiological process, rather than discrete immunopathogenic patterns. This may be the case, or the majority of active lesions will present with Pattern II phenotype, as supported by findings of a study by Barnett and Sutton, where the authors described that in 22 patients drawn from a large unselected pool of MS material, 33 actively demyelinating lesions presented Pattern II pathology [26]. The four-pattern system has not been completely independently verified to date because of the lack of available highly comparable pathologic material.
The response to plasma exchange (PLEX) in acute fulminant MS provides further evidence for the role of immunoglobulin or serum components in the disease. Rodriguez et al. demonstrated conclusively for the first time the detrimental effect of plasma components in inflammatory demyelinating diseases of the CNS; PLEX in acute episodes of fulminant CNS inflammatory demyelination, which did not respond to high-dose methylprednisolone, led to a marked neurologic improvement in 6 patients [27]. These results were confirmed in a randomized, sham-controlled, double-masked study of PLEX without concomitant immunosuppressive treatment in patients with recently acquired, severe neurological deficits resulting from attacks of inflammatory demyelinating disease, who failed to recover after treatment with intravenous corticosteroids [28]. A retrospective study investigated 19 patients treated with PLEX for an attack of fulminant CNS inflammatory demyelinating disease. All patients with pattern II (n = 10), but none with pattern I (n = 3) or pattern III (n = 6), achieved moderate to substantial functional neurological improvement after PLEX (P < 0.0001) [29]. The fact that all cases, which responded to PLEX, had a biopsy demonstrating Igs and complement, whereas none that responded showed this immunologic pattern, provided the strongest proof that the pathologic patterns are unique and have therapeutic significance. Numerous publications during the last few decades have supported the idea that CSF oligoclonal bands correlate to the level of B-cell involvement in MS [30]. In addition, evidence indicates that oligoclonal bands may have a prognostic value. One prospective study of patients with acute isolated demyelinating episode demonstrated intrathecal immunoglobulin synthesis to be a better predictor of MS progression than MRI [31]. Another prospective study showed that presence of CSF oligoclonal bands in early MS generally correlated with a worse outcome [32]. A recent study showed strong correlation between levels of oligoclonal bands (OCBs) and prognosis for MS disability [33].
4. Antigen: Specificity of Antibodies Found in MS
After several years of research, confirmation of the antigen-specificity of antibodies in MS is still lacking. Due to their broad reactivity, IgG in CSF of patients with MS may represent synthesis of “nonsense” antibodies irrelevant to pathogenesis [34–36]. However, other experiments found molecular uniformity and temporal persistence of the Ig response in MS, thus conflicting with the nonsense antibody proposal [37]. It is possible that relevant antigens are limited to the myelin sheath. Studies demonstrated the serological and/or CSF presence of antibodies directed against MBP and/or myelin/oligodendrocyte glycoprotein (MOG) in patients with MS [38]. However, myelin-specific antibodies are not limited to MS. Using an enzyme-linked immunosorbent assay, Karni et al. compared levels and frequencies of anti-MOG antibody between patients with MS, patients with other neurological disorders, and healthy control subjects [39] and found minor differences. In a parallel line of research, some reports suggested lipids or carbohydrates as possible candidate antigens for the humoral immune response [40, 41]. Anti-alpha-glucose-based glycan IgM antibodies have been suggested to be predictors of relapse activity in MS after the first neurological event [42]. Others suggested that serum anti-Glc(alpha1, 4)Glc(alpha) antibodies serve as biomarkers for relapsing-remitting MS [43]. Antibodies to myelin proteins, lipids, and carbohydrates can be extracted from the tissue and sera of some MS patients.
5. Immune Manifestations of MS: Role of T Cells
5.1. CD4+ T Cells as Initiators of Disease versus Effectors in Destruction of Myelin
The area of greatest confusion in the MS literature concerns the role of CD4+ T cells in disease pathogenesis. CD4+ cells predominate in experimental autoimmune/allergic encephalomyelitis (EAE) as the effectors that induce disease and destroy myelin. Therefore, due to the influence of the experimental model, many investigators have attempted to show that CD4+ T cells also play a pathogenic role during the evolution of MS. Unfortunately, many findings regarding the role of CD4+ T cells reported have not been reproduced elsewhere [44]. However, there is strong experimental evidence that any immune response must begin through the engagement of the antigen recognized by receptors on CD4+ T cells. In concept, dendritic cells, both outside and/or inside the CNS, take up the exogenous or endogenous antigen and present it to CD4+ T cells. As a result, these CD4+ T cells differentiate into four distinct subtypes depending on the inflammatory milieu (Figure 1). The first is the Th1-type CD4+ T cell, which primarily secretes IFN-γ and TNF-α. The second is a CD4+ T cell frequently called Th2, which secretes primarily TGF-β and IL-10. The third is a CD4+ TREG cell that performs a regulatory function [45]. These T cells express a number of transcription factors including FoxP3 and other molecules [46, 47]. These cells play a major role in downregulating the immune response [48–50]. Finally, there are CD4+ T cells that primarily secrete IL17 called Th17 cells. These Th17 cells induce most of the pathology in EAE. There is evidence of their presence in the MS plaque, where they may preferentially recruit IFN-γ [51]. Data suggest that CD4+ T cells from MS patients use unique human T-cell beta-receptors [52]. In these studies, the investigators used T-cell lines from MS patients as well as healthy controls and showed that these CD4+ T-cell lines reacted against specific human myelin basic proteins, the first being residues 84–102 and the second being residues 143–168. They showed that the CD4+ T-cell receptors being used were primarily of the Vβ17 and Vβ12 family. Vβ12 receptors were used frequently in recognition with the MBP (84–102) peptide, while Vβ17 mostly reacted against MBP (143–168).
Figure 1.
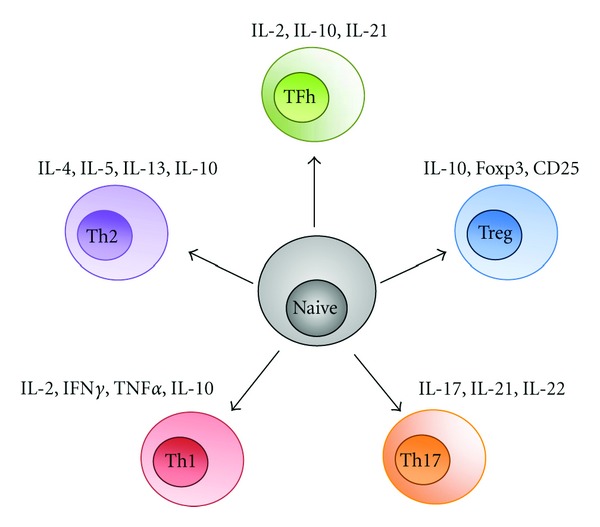
CD4+ T Cells differentiated into subsets. CD4+ T cells can differentiate into different subtypes based on the factors within the inflammatory milieu with which T cells come into contact. TH1+ cells secrete IFN-γ and tumor necrosis factor (TNF) and mediate the pathology in experimental autoimmune encephalomyelitis (EAE). Many of the results previously attributed to TH1 cells are actually mediated by Th17+ cells. Th17+ cells secrete IL17, IL21, and IL22. These cells have been identified in MS lesions, where they may serve as important effectors. As a result of TGF-β stimulation, CD4+ T cells develop into T-regulatory cells. These cells downregulate the immune response and express FOXP3, CD25, and IL10. The mechanism of suppression is by the secretion of factors such as IL10. TH-helper cells provide help to other T cells, such as CD8+ T cells or B cells. Th-helper cells secrete IL2, IL10, and IL21. Th2+ cells downregulate the immune response and are associated with recovery from acute attacks in EAE and, possibly, MS. The cytokines that mediate the downregulation of the immune response are IL4 and IL10, in addition to IL13 and IL5.
The presence of unique T-cell receptor V-β gene usage has generated a series of experimental animal trials as well as early human trials with the goal of deleting specific V-β T cells in MS patients [53]. These experiments have been relatively successful in EAE; however, the approach has been less effective in human patients. Of interest, investigators have also isolated MBP-reactive CD4+ T-cell lines from normal human blood [54]. The fact that these T-cell lines respond to MBP [55], similarly to what is observed in MS patients, has raised major questions as to the specificity of the response of CD4+ T-cell lines to myelin antigen in MS patients [56]. These CD4+ T-cell lines obtained from non-MS patients secreted IL2 similar to that seen with MS patients. All of the T-cell lines isolated from the peripheral blood were of the CD4 phenotype [57]. Investigators have examined peripheral blood lymphocytes from MS patients and other neurological controls in effort to study specific T-cell populations against purified human MBP and other brain antigens [58]. Investigators showed that lymphocytes from MS patients were more likely to react against MBP. Unfortunately, they discerned only minor differences between MS patients and normal controls as to the specificity of the response to any brain tissue antigens. The majority of responses were found in patients with chronic progressive MS, a phase when T cells are least active in the disease. These results also have raised concerns about the specificity of the T-cell immune response to myelin antigens in MS patients given the not easily discernible differences between MS patients and normal individuals. Even those investigators claiming to show a positive, “statistically significant” response show such an overlap in the results between patients and controls that these assays have never been developed as a diagnostic test for MS [59].
Recent work has focused on Th17+ cells in MS [60, 61]. Investigators looked at evidence implicating IFN-γ producing hybrid T cells (so-called Th1 cells as well as IL17+ lymphocytes (Th17+ cells)) in MS. They compared this to animals with EAE and demonstrated expansion of Th17 lymphocytes from the blood of healthy controls as well as from patients with relapsing MS. In response to IL23, which is known to expand the Th17 phenotype, they showed simultaneous expressions of IFN-γ and IL17. They noted that patients with relapsing-remitting MS had increased production of IFN-γ by Th17 cells. The same findings were also present in the experimental model. Both these data sets support the hypothesis that Th17 cells play a role in the pathology of MS and EAE. However, the presence of Th17 cells does not automatically prove that they play a role in pathogenesis [62]. No data is available that deletion of Th17 cells improves MS or that elevated Th17 cells in lesions correlate with disability. This is in contrast to the work done on CD8+ T cells (discussed hereinafter), which reveals a strong correlation between CD8+ T cells, perforin, and other molecules as secreted by CD8+ T cells with disease disability.
5.2. CD8+ T Cells: Primary Mediator of Effector Function in the MS Plaque
Pathological studies demonstrate that the CD8+ T cell is the most common T cell observed in the MS plaque. Conventional perception is that CD8+ T cells have two major functions: cytotoxicity and suppression. In MS, because of the strong bias of the experimental autoimmune encephalomyelitis (EAE) models, the CD8+ T cell has been primarily thought to play a suppressive role. In EAE, the CD4+ T cell, through its Th1 and Th17 function, mediates the disease and induces the inflammatory response, neurological deficits, paralysis, and histological findings. In EAE, CD8+ T cells are associated with recovery of neurologic function and have been shown to have suppressive properties. In contrast, in the MS plaque, the CD8+ T cells appear to play a much more aggressive role rather than just suppressing the inflammatory response. CD8+ T cells interact with major histocompatibility (MHC) class I antigens to induce their response. In normal CNS, class I MHC is observed only in vascular cells and rare meningeal cells. However, in the midst of an inflammatory process such as MS, class I MHC is observed in astrocytes, oligodendrocytes, and neurons and even rarely on axons [63]. In addition, the CD8+ T cells correlate with axonal injury, and there is strong evidence in vitro that CD8+ T cells play a major role in transecting axons [64, 65]. Relapses of MS are associated with increased CD8+ T cell cytotoxicity in the CSF [66]. A number of clinical trials with monoclonal antibodies, specifically against CD4+ T cells [67], failed to show any therapeutic benefit in MS as opposed to broader spectrum antibodies (alemtuzumab CD52), which are able to deplete all T cells [68], including CD8+ T cells. It is also important to emphasize that CD8+ T cells may play a major role in a number of proven autoimmune disease including SLE, diabetes mellitus, Crohn's disease, Graves' disease, and autoimmune Addison's disease [63].
Finally, CD8+ T cells show oligoclonal expansion in MS brains, blood, and CSF that have not been reported with CD4+ T cells [69–71]. Some of the cytotoxic T cells react against autoantigens such as myelin-basic protein [72]. If these cells traveled randomly in the CNS, then presumably their CD3 junction region length would show a normal Gaussian distribution. In contrast, there is skewing of the CDR3 junction regions in MS, suggesting a selective infiltration or expansion of CD8+ T cell clones into the CNS [71]. Moreover, the T-cell receptors of the CD8+ T cells demonstrate distinct CD8+ T-cell clones with conserved specificity implying recognition of a similar antigen that results in their proliferation in the CNS. Much effort has focused on the potential role of IL17 in the MS plaque, and because of the bias of experiments in EAE, it has been proposed that this comes from the CD4+ T cells. However, there is evidence that IL17 is also made by CD8+ T cells [73]. Defining IL17+ CD8+ T cells opens up new avenues for future research and new targets from the standpoint of immunotherapy. CD8+ T cells secrete a number of molecules including granzymes and perforin. Strong evidence suggests that perforin contributes to axonal injury in the MS plaque (Figure 2). The presence of perforin correlates with neurologic disability and with the presence of “black holes” on MRI. Therefore, CD8+ T cells play a critical role during the acute inflammatory phase of the disease as well as during the neurodegenerative phase. CD8+ T cells account for axonal damage in MS as well as long-term neurological deficits. There is evidence that CD8+ T cells play a major role in the secondary progressive phase of the disease by the secretion of lymphotoxin [74]. In studies of cytokine secretion in patients with secondary progressive MS and normal controls, investigators found clear evidence of anti-CD3-stimulated CD8+ T cells in the patients with secondary progressive MS. These cells secreted lymphotoxin and other cytokines, which play a critical role in the evolution of the progressive phase of the disease. This provides strong evidence that the CD8+ T cell plays a role in the neurodegenerative aspect of the progressive phase of the disease as well as in the early acute phase.
Figure 2.
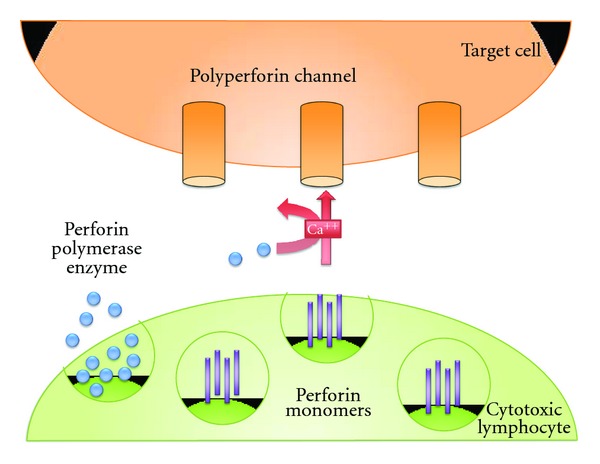
Perforin is the primary mediator of injury by CD8+ T cells. Perforin is the primary molecule known to mediate injury by CD8+ T cells. Perforin mediates axonal transection in multiple sclerosis (MS) and correlates with neurological disability. Cytotoxic T cells secrete perforin in the form of granules along with granzymes. This release activates calcium, which results in “poly-perforin” channels on the target cells. This results in holes in the membrane of the target cells, causing leakage of intracellular material, which results in cell death.
It is also important to emphasize a possible regulatory role for CD8+ T cells in MS. Investigators have identified CD8+/CD25+ Foxp3+ as regulatory T cells in MS patients [75]. In these studies, they examined the peripheral blood, CSF, and CD8+ T cell clones from patients with MS exacerbations, patients with remissions, healthy individuals, and patients with other inflammatory neurological diseases. The inhibition of CD4+ self-reactive T-cell proliferation by CD8+ regulatory cells was mediated by IL10 and transforming growth factor beta (TGF-β). Any attempt to delete CD8+ T cells from the MS lesion could potentially worsen the disease by eliminating regulatory cells. Therefore, caution must be taken in any effort to manipulate the CD8+ population.
6. Autoimmunity-Based Evidence for NMO Pathogenesis
Hinson et al. [76–78] discovered the occurrence of anti-AQP4 IgG in patients with NMO. It was further demonstrated that AQP4-reactive antibodies appear in the pathologic lesions [79] and that levels of AQP4 antibody and disease activity were correlated [80]. NMO is associated with other autoimmune disorders [20]. In addition, autoantibodies against other common autoimmune diseases, such as Sjogren's syndrome and systemic lupus erythematosus, appear in the serum of NMO patients [21] but not in the serum of MS patients [19]. Given the autoimmune hypothesis associated with NMO, we hypothesized that PLEX, a conventional method to remove circulating autoantibodies in patients, would be beneficial. Interestingly, PLEX proved to be a highly successful treatment for NMO arguing in favor of an immune-mediated pathogenesis of this disease [81]. In line with the autoimmune-mediated hypothesis, humoral immunity-suppressing drugs such as mitoxantrone hydrochloride [82] (a synthetic anthracenedione that was approved for the treatment of worsening relapsing-remitting and secondary progressive MS), mycophenolate mofetil [83] (an immunosuppressive therapy), and rituximab [84] (a B-cell depleting therapy) were demonstrated to be beneficial for treatment of NMO. De Parratt and Prineas recently described an abrupt destruction of perivascular astrocytes in patients with NMO that preceded oligodendrocyte apoptosis in early lesions. Their findings add to the experimental evidence that serum antibody directed against astrocytes present in a high proportion of patients with NMO is pathogenic. In addition, their data supports a new definition of the disease based on pathology: NMO is a demyelinating disease characterized pathologically by multifocal lesions disseminated in time and space and in which demyelination is secondary to acute destruction of perivascular astrocytes [85]. However, Takano et al. reported that astrocytic damage is far more severe than demyelination in NMO [86]. It is now considered that NMO is an inflammatory autoimmune disorder of the CNS.
7. An Alternate Hypothesis for MS Pathogenesis
An attractive hypothesis to explain the immune-mediated pathogenesis of MS is that it is induced by an infectious agent. Even though no infectious agent has convincingly been demonstrated in MS, there is experimental evidence to support the hypothesis.
8. Experimental Evidence for Virus-Induced Demyelination
Experimental infection of laboratory animals with various viruses induces demyelination in the CNS. The most studied viral animal model of MS is the disease induced by Theiler's murine encephalomyelitis virus (TMEV), a mouse enteric pathogen that belongs to the single-stranded RNA picornaviruses [87]. The disease model is chronic-progressive in susceptible mice, a striking contrast to the much-used autoimmune EAE model. Two salient features make it the best-suited model for studying MS. There is evidence of an immune response to virally infected cells [88, 89] as well as autoimmune response triggered by viral infection in the CNS [90], both of which are potentially similar to MS. Miller et al. reported that TMEV infection leads to CNS autoimmunity via epitope spreading [91]. TMEV infection of oligodendrocytes results in cell lysis and liberation of more virions [92]. On the contrary, infection of TMEV in macrophages is restricted and results in their apoptosis. Virus spreads from macrophages to other macrophages and oligodendrocytes, adding to the immunopathological destruction of myelin. Demyelination is in part the result of direct virus destruction of oligodendrocytes but also the consequence of immune and inflammatory responses. Other viral models of demyelination include mice with JHM and MHV-4 virus (coranoviruses) infection, dogs with canine distemper virus, and sheep and goats with Visna virus and caprine arthritis-encephalitis virus. An animal model of virus-induced demyelination with no relapses is the Semliki Forest virus (SFV) infection of mice [93]. All viruses are capable of establishing persistent viral infection over a long period without inducing mortality of the host. All of these examples make a case for the viral hypothesis of CNS demyelination.
9. Evidence for a Virus-Induced Etiology of MS
Epstein-Barr virus (EBV), human herpes virus 6 (HHV-6), varicella zoster virus (VZV), and Chlamydia pneumonia are some of the proposed infectious agents in humans implicated in MS. Many studies have demonstrated antibody titers to a broad range of pathogens in MS patients; however, many of these findings remain solitary and unconfirmed. EBV is a B-lymphotropic human DNA herpes virus that infects most individuals asymptomatically but causes infectious mononucleosis (IM) in some [95, 96]. Cepok et al. identified EBV proteins as putative targets of the immune response in MS [97]. Another study demonstrated the increased risk of MS in individuals with a clinical history of IM [98, 99]. Recently, researchers from the United Kingdom studied the prevalence of MS and infectious mononucleosis (IM) and how they relate to ultraviolet B (UVB) exposure [100]. As previously shown in other studies, MS highly correlated with IM [101, 102]. As a control, the authors also examined correlations of MS with cytomegalovirus prevalence and varicella prevalence, respectively, both of which were not correlated with MS. Of note, the authors found that UVB in any season correlated closely with MS and EBV infection. These results fit well with the EBV hypothesis because there may be a mechanism through which UVB radiation mediates MS risk.
It has been suggested that low vitamin D levels result in immunosuppression that lead to an increase in EBV infection. It is also known that a low amount of UVB decreases vitamin D levels. The geographical variation in the MS prevalence, with a higher prevalence of the disease in northern latitudes and a lower prevalence at the equator, is well established [103–105]. This variation in MS prevalence correlates positively with changes in the serum concentrations of 25-hydroxyvitamin D [106–108]. Several, but not all, studies show an inverse correlation between serum 25-hydroxyvitamin D concentrations and the incidence of MS, the severity, and progression of disease [109–120]. Vitamin D, and its biologically active metabolite 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), not only plays an important role in the regulation of calcium and phosphorus homeostasis but also is an important modulator of immune function. 1,25(OH)2D3 functions by associating with the vitamin D receptor that is widely distributed in a number of calcium-transporting tissues, neural tissues, and immune cells (dendritic cells, T-lymphocytes, B lymphocytes and macrophages) [121–128]. 1,25(OH)2D3 increases macrophage activity, inhibits dendritic cell maturation, inhibits B-cell functions, and favors the production of T-helper 2 cells, thereby shifting the ratio of Th1/Th2 cells in favor of Th2 helper cells (Figure 3) [94, 129–134]. The polarization of activated CD4+ T cells to a Th-1 phenotype (IL-2, IFNγ, TNFα secretion) or to a Th-2 phenotype (IL-4, 5, 13, 10 secretion) represents a major determinant of the nature of subsequent cellular and humoral immune responses. It is a self-perpetuating process in that one subtype inhibits the generation of the other [131, 132]. The primary generation of Th-1-type T-cell responses is potently inhibited by 1,25(OH)2D3 both in vitro and in vivo. 1,25(OH)2D3 also induces production of human cathelicidin, LL-37, which is particularly effective against respiratory viruses such as influenza [135]. The lack of vitamin D may result in persistent infection, for example, EBV infection causing IM, thereby leading to a higher risk for MS [136].
Figure 3.
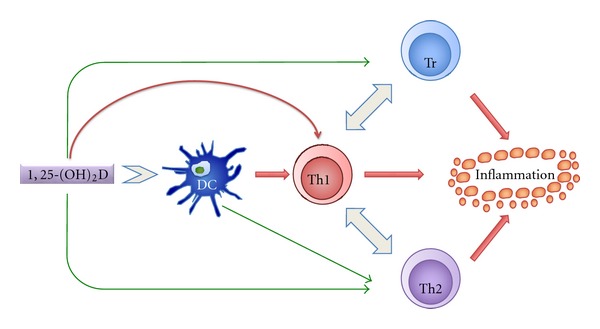
The in vitro effects of 1,25(OH)2D on the immune system. The effects of 1,25(OH)2D either directly or indirectly are depicted by arrows. While a green arrow represents positive influence, a red arrow represents the negative influence. The negative influence on inflammation indicates dampening of the inflammatory response. DC: dendritic cell; Th1: T helper type 1 lymphocyte; Th2: T helper type 2 lymphocyte; Tr: regulatory T lymphocyte [94].
10. Conclusions
In MS, for an oligodendrocyte to be injured by inflammatory cells, it must express MHC class I or class II genes. CD8+ T cells can then engage a novel protein that is expressed in the context of class I MHC. CD8+ T cells would then secrete perforin, granzyme, or other factors that may directly injure or kill the oligodendrocyte resulting in demyelination. Antibodies, through molecular mimicry, may recognize autoantigens of the CNS and can also injure the oligodendrocyte by binding to the surface of the cell and, in association with complement, may induce direct injury to myelin or the oligodendrocytes. This partially leads us to the autoimmune hypothesis. In addition, the B cells may also present virus antigens in the context of class II MHC molecules. The oligodendrocyte or microglia itself may also express class II MHC. This presentation of the viral antigen must be processed, which allows the CD4+ T cells to be engaged with class II MHC to induce injury, the common mechanism of injury presumed to be present in experimental autoimmune encephalomyelitis. The oligodendrocyte may die as a consequence of direct and persistent virus infection. These mechanisms of injury may be independent or occur concurrently in each brain. All of these mechanisms lead to demyelination that the host may correct by transient remyelination. Ultimately, the demyelination process overtakes remyelination resulting in axonal damage, thus leading to permanent neurologic deficits. At the present time, there is no clear evidence that these patterns of injury relate to various stages of the disease course and do not correlate with the clinical subtypes of relapsing-remitting multiple sclerosis, secondary progressive multiple sclerosis, or primary progressive multiple sclerosis, although this is yet to be determined.
Many of the observed findings have subsequently led investigators to false conclusions regarding MS pathogenesis. Immune cells are present in the MS plaque, and the immune system is important in the pathogenesis of the disease because a number of immunomodulatory and immunosuppressive therapies do decrease relapses and the number of gadolinium-enhancing lesions in MS brain. However, the long-term consequences of immunosuppression on disease course are unknown because most published clinical trials end after two years of observation, an insufficient period of time to address the long-term consequences of these treatments. It is increasingly evident that CD8+ T cells and their effector molecules may directly affect the disease process. Unfortunately, despite years of documentation of involvement of CD8+ T cells in MS lesions, scant experimentation has been performed on this aspect of the immune response. This is probably due to the bias of the experimental model, EAE, in which CD8+ T cells play only a regulatory role whereas CD4+ T cells play a major effector role in disease pathogenesis. Once we move away from the experimental model and begin to investigate MS in humans, it becomes apparent that the MHC class II CD4+ T-cell immune response yields less important critical data of the MHC class I CD8+ T-cell immune response. The most important diagnostic test for MS continues to be the presence of increased CSF IgG and the presence of specific oligoclonal bands in the CSF but not in the serum. Therefore, it is critical to identify the specificity of these bands. Ultimately, it may be proven that CSF oligoclonal IgG bands play a neuroprotective rather than a pathologic role [137–140].
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01s-GM092993, NS024180, NS032129, NS048357, R21-NS073684), the National Multiple Sclerosis Society (CA1060A11), the Applebaum Foundation, the Hilton Foundation, the Peterson Foundation, Minnesota Partnership for Biotechnology and Medical Genomics, and the European Regional Development Fund—Project FNUSA-ICRC (no. CZ.1.05/1.1.00/02.0123). The authors also thank the McNeilus Family and a High-Impact Pilot and Feasibility Award (HIPFA) from the Mayo Clinic Center for Translational Science Activities (CTSA) for financial support in this project. B. Wootla is supported through a Collaborative Multiple Sclerosis Research Award from the National Multiple Sclerosis Society. M. Eriguchi is the recipient of a fellowship from Japan Brain foundation. The technology for remyelination-promoting antibody rHIgM22 has been licensed to Acorda Therapeutics, Inc. No royalties have accrued to Dr. M. Rodriguez or Mayo Clinic to date, but both have rights to receive future royalties.
References
- 1.Kurtzke JF, Page WF, Murphy FM, Norman JE., Jr. Epidemiology of multiple sclerosis in US veterans. 4. Age at onset. Neuroepidemiology. 1992;11(4–6):226–235. doi: 10.1159/000110935. [DOI] [PubMed] [Google Scholar]
- 2.Sadovnick AD, Baird PA. Sex ratio in offspring of patients with multiple sclerosis. The New England Journal of Medicine. 1982;306(18):1114–1115. doi: 10.1056/NEJM198205063061821. [DOI] [PubMed] [Google Scholar]
- 3.Wallin MT, Page WF, Kurtzke JF. Multiple sclerosis in US veterans of the vietnam era and later military service: race, sex, and geography. Annals of Neurology. 2004;55(1):65–71. doi: 10.1002/ana.10788. [DOI] [PubMed] [Google Scholar]
- 4.Orton SM, Herrera BM, Yee IM, et al. Sex ratio of multiple sclerosis in Canada: a longitudinal study. Lancet Neurology. 2006;5(11):932–936. doi: 10.1016/S1474-4422(06)70581-6. [DOI] [PubMed] [Google Scholar]
- 5.Ramagopalan SV, Yee IM, Dyment DA, et al. Parent-of-origin effect in multiple sclerosis: observations from interracial matings. Neurology. 2009;73(8):602–605. doi: 10.1212/WNL.0b013e3181af33cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. The New England Journal of Medicine. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 7.Lassmann H. Acute disseminated encephalomyelitis and multiple sclerosis. Brain. 2010;133(2):317–319. doi: 10.1093/brain/awp342. [DOI] [PubMed] [Google Scholar]
- 8.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Annals of Neurology. 2001;50(1):121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 9.Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Annals of Neurology. 2000;47(6):707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 10.Frohman EM, Racke MK, Raine CS. Medical progress: multiple sclerosis—the plaque and its pathogenesis. The New England Journal of Medicine. 2006;354(9):942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 11.Lassmann H, Brück W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends in Molecular Medicine. 2001;7(3):115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- 12.Wekerle H. Immune pathogenesis of multiple sclerosis. Neurological Sciences. 2005;26(supplement 1):S1–S2. doi: 10.1007/s10072-005-0386-9. [DOI] [PubMed] [Google Scholar]
- 13.Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. The Journal of Clinical Investigation. 1942;21(5):571–577. doi: 10.1172/JCI101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez M. Have we finally identified an autoimmune demyelinating disease? Annals of Neurology. 2009;66(5):572–573. doi: 10.1002/ana.21838. [DOI] [PubMed] [Google Scholar]
- 15.Saeki Y, Mima T, Sakoda S, et al. Transfer of multiple sclerosis into severe combined immunodeficiency mice by mononuclear cells from cerebrospinal fluid of the patients. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(13):6157–6161. doi: 10.1073/pnas.89.13.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Q, Saida T, Nishimura M, Ozawa K, Saida K. Failure to transfer multiple sclerosis into severe combined immunodeficiency mice by mononuclear cells from CSF of patients. Neurology. 1994;44(1):163–165. doi: 10.1212/wnl.44.1.163. [DOI] [PubMed] [Google Scholar]
- 17.Lo R, Feasby TE. Multiple sclerosis and autoimmune diseases. Neurology. 1983;33(1):97–98. doi: 10.1212/wnl.33.1.97. [DOI] [PubMed] [Google Scholar]
- 18.Warren S, Warren KG. Multiple sclerosis and associated diseases: a relationship to diabetes mellitus. Canadian Journal of Neurological Sciences. 1981;8(1):35–39. doi: 10.1017/s0317167100042815. [DOI] [PubMed] [Google Scholar]
- 19.Wynn DR, Rodriguez M, O’Fallon WM, Kurland LT. A reappraisal of the epidemiology of multiple sclerosis in Olmsted County, Minnesota. Neurology. 1990;40(5):780–786. doi: 10.1212/wnl.40.5.780. [DOI] [PubMed] [Google Scholar]
- 20.Pittock SJ, Lennon VA, de Seze J, et al. Neuromyelitis optica and non-organ-specific autoimmunity. Archives of Neurology. 2008;65(1):78–83. doi: 10.1001/archneurol.2007.17. [DOI] [PubMed] [Google Scholar]
- 21.Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurological Research. 2006;28(3):348–353. doi: 10.1179/016164106X98260. [DOI] [PubMed] [Google Scholar]
- 22.Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. Journal of Clinical Investigation. 1998;102(5):1045–1050. doi: 10.1172/JCI3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Annals of Neurology. 2004;55(4):458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- 24.Barnett MH, Parratt JDE, Cho ES, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Annals of Neurology. 2009;65(1):32–46. doi: 10.1002/ana.21524. [DOI] [PubMed] [Google Scholar]
- 25.Breij ECW, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Annals of Neurology. 2008;63(1):16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- 26.Barnett MH, Sutton I. The pathology of multiple sclerosis: a paradigm shift. Current Opinion in Neurology. 2006;19(3):242–247. doi: 10.1097/01.wco.0000227032.47458.cb. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez M, Karnes WE, Bartleson JD, Pineda AA. Plasmapheresis in acute episodes of fulminant CNS inflammatory demyelination. Neurology. 1993;43(6):1100–1104. doi: 10.1212/wnl.43.6.1100. [DOI] [PubMed] [Google Scholar]
- 28.Weinshenker BG, O'Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Journal of Clinical Apheresis. 1999;46(6):878–886. doi: 10.1002/1531-8249(199912)46:6<878::aid-ana10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 29.Keegan M, König F, McClelland R, et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. The Lancet. 2005;366(9485):579–582. doi: 10.1016/S0140-6736(05)67102-4. [DOI] [PubMed] [Google Scholar]
- 30.Cross AH, Wu GF. Multiple sclerosis: oligoclonal bands still yield clues about multiple sclerosis. Nature Reviews Neurology. 2010;6(11):588–589. doi: 10.1038/nrneurol.2010.142. [DOI] [PubMed] [Google Scholar]
- 31.Paolino E, Fainardi E, Ruppi P, et al. A prospective study on the predictive value of CSF oligoclonal bands and MRI in acute isolated neurological syndromes for subsequent progression to multiple sclerosis. Journal of Neurology Neurosurgery and Psychiatry. 1996;60(5):572–575. doi: 10.1136/jnnp.60.5.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amato MP, Ponziani G. A prospective study on the prognosis of multiple sclerosis. Neurological Sciences. 2000;21(4, supplement 2):S831–S838. doi: 10.1007/s100720070021. [DOI] [PubMed] [Google Scholar]
- 33.Joseph FG, Hirst CL, Pickersgill TP, Ben-Shlomo Y, Robertson NP, Scolding NJ. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. Journal of Neurology, Neurosurgery and Psychiatry. 2009;80(3):292–296. doi: 10.1136/jnnp.2008.150896. [DOI] [PubMed] [Google Scholar]
- 34.Mattson DH, Roos RP, Arnason BG. Isoelectric focusing of IgG eluted from multiple sclerosis and subacute sclerosing panencephalitis brains. Nature. 1980;287(5780):335–337. doi: 10.1038/287335a0. [DOI] [PubMed] [Google Scholar]
- 35.Mattson DH, Roos RP, Arnason BGW. Oligoclonal IgG in multiple sclerosis and subacute sclerosing panencephalitis brains. Journal of Neuroimmunology. 1982;2(3-4):261–276. doi: 10.1016/0165-5728(82)90059-5. [DOI] [PubMed] [Google Scholar]
- 36.Whitacre CC, Mattson DH, Paterson PY, et al. Cerebrospinal fluid and serum oligoclonal IgG bands in rabbits with experiment allergic encephalomyelitis. Neurochemical Research. 1981;6(1):87–96. doi: 10.1007/BF00963909. [DOI] [PubMed] [Google Scholar]
- 37.Walsh MJ, Tourtellotte WW. Temporal invariance and clonal uniformity of brain and cerebrospinal IgG, IgA, and IgM in multiple sclerosis. Journal of Experimental Medicine. 1986;163(1):41–53. doi: 10.1084/jem.163.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt S, Haase CG, Bezman L, et al. Serum autoantibody responses to myelin oligodendrocyte glycoprotein and myelin basic protein in X-linked adrenoleukodystrophy and multiple sclerosis. Journal of Neuroimmunology. 2001;119(1):88–94. doi: 10.1016/s0165-5728(01)00345-9. [DOI] [PubMed] [Google Scholar]
- 39.Karni A, Bakimer-Kleiner R, Abramsky O, Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Archives of Neurology. 1999;56(3):311–315. doi: 10.1001/archneur.56.3.311. [DOI] [PubMed] [Google Scholar]
- 40.Arnon R, Crisp E, Kelley R, et al. Anti-ganglioside antibodies in multiple sclerosis. Journal of the Neurological Sciences. 1980;46(2):179–186. doi: 10.1016/0022-510x(80)90076-3. [DOI] [PubMed] [Google Scholar]
- 41.Endo T, Scott DD, Stewart SS, Kundu SK, Marcus DM. Antibodies to glycosphingolipids in patients with multiple sclerosis and SLE. The Journal of Immunology. 1984;132:1793–1797. [PubMed] [Google Scholar]
- 42.Freedman MS, Laks J, Dotan N, Altstock RT, Dukler A, Sindic CJM. Anti-α-glucose-based glycan IgM antibodies predict relapse activity in multiple sclerosis after the first neurological event. Multiple Sclerosis. 2009;15(4):422–430. doi: 10.1177/1352458508101944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz M, Spector L, Gortler M, et al. Serum anti-Glc(α1,4)Glc(α) antibodies as a biomarker for relapsing-remitting multiple sclerosis. Journal of the Neurological Sciences. 2006;244(1-2):59–68. doi: 10.1016/j.jns.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Sriram S, Steiner I. Experimental allergic encephalomyelitis: a misleading model of multiple sclerosis. Annals of Neurology. 2005;58(6):939–945. doi: 10.1002/ana.20743. [DOI] [PubMed] [Google Scholar]
- 45.Costantino CM, Baecher-Allan C, Hafler DA. Multiple sclerosis and regulatory T cells. Journal of Clinical Immunology. 2008;28(6):697–706. doi: 10.1007/s10875-008-9236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + regulatory T cells in the human immune system. Nature Reviews Immunology. 2010;10(7):490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 47.Huan J, Culbertson N, Spencer L, et al. Decreased FOXP3 levels in multiple sclerosis patients. Journal of Neuroscience Research. 2005;81(1):45–52. doi: 10.1002/jnr.20522. [DOI] [PubMed] [Google Scholar]
- 48.Cvetanovich GL, Hafler DA. Human regulatory T cells in autoimmune diseases. Current Opinion in Immunology. 2010;22(6):753–760. doi: 10.1016/j.coi.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dejaco C, Duftner C, Grubeck-Loebenstein B, Schirmer M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology. 2006;117(3):289–300. doi: 10.1111/j.1365-2567.2005.02317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torgerson TR. Regulatory T cells in human autoimmune diseases. Springer Seminars in Immunopathology. 2006;28(1):63–76. doi: 10.1007/s00281-006-0041-4. [DOI] [PubMed] [Google Scholar]
- 51.Lovett-Racke AE, Yang Y, Racke MK. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochimica et Biophysica Acta. 2011;1812(2):246–251. doi: 10.1016/j.bbadis.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wucherpfennig KW, Ota K, Endo N, et al. Shared human T cell receptor V(β) usage to immunodominant regions of myelin basic protein. Science. 1990;248(4958):1016–1019. doi: 10.1126/science.1693015. [DOI] [PubMed] [Google Scholar]
- 53.Hafler DA, Saadeh MG, Kuchroo VK, Milford E, Steinman L. TCR usage in human and experimental demyelinating disease. Immunology Today. 1996;17(4):152–159. doi: 10.1016/0167-5699(96)80611-6. [DOI] [PubMed] [Google Scholar]
- 54.Burns J, Rosenzweig A, Zweiman B, Lisak RP. Isolation of myelin basic protein-reactive T-cell lines from normal human blood. Cellular Immunology. 1983;81(2):435–440. doi: 10.1016/0008-8749(83)90250-2. [DOI] [PubMed] [Google Scholar]
- 55.McLaurin J, Hafler DA, Antel JP. Reactivity of normal T-cell lines to MBP isolated from normal and multiple sclerosis white matter. Journal of the Neurological Sciences. 1995;128(2):205–211. doi: 10.1016/0022-510x(94)00224-c. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. The EMBO Journal. 2005;24(17):2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brinkman CJ, Nillesen WM, Hommes OR, Lamers KJ, de Pauw BE, Delmotte P. Cell-mediated immunity in multiple sclerosis as determined by sensitivity of different lymphocyte populations to various brain tissue antigens. Annals of Neurology. 1982;11(5):450–455. doi: 10.1002/ana.410110503. [DOI] [PubMed] [Google Scholar]
- 58.Wucherpfennig KW, Hafler DA, Strominger JL. Structure of human T-cell receptors specific for an immunodominant myelin basic protein peptide: positioning of T-cell receptors on HLA-DR2/peptide complexes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(19):8896–8900. doi: 10.1073/pnas.92.19.8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Severson C, Hafler DA. T-cells in multiple sclerosis. Results and Problems in Cell Differentiation. 2010;51:75–98. doi: 10.1007/400_2009_12. [DOI] [PubMed] [Google Scholar]
- 60.Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-γ-expressing TH17 cells in multiple sclerosis. Annals of Neurology. 2009;66(3):390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 61.Sinha S, Subramanian S, Proctor TM, et al. A promising therapeutic approach for multiple sclerosis: recombinant T-cell receptor ligands modulate experimental autoimmune encephalomyelitis by reducing interleukin-17 production and inhibiting migration of encephalitogenic cells into the CNS. Journal of Neuroscience. 2007;27(46):12531–12539. doi: 10.1523/JNEUROSCI.3599-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montes M, Zhang X, Berthelot L, et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clinical Immunology. 2009;130(2):133–144. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friese MA, Fugger L. Pathogenic CD8+ T cells in multiple sclerosis. Annals of Neurology. 2009;66(2):132–141. doi: 10.1002/ana.21744. [DOI] [PubMed] [Google Scholar]
- 64.McDole J, Johnson AJ, Pirko I. The role of CD8+ T-cells in lesion formation and axonal dysfunction in multiple sclerosis. Neurological Research. 2006;28(3):256–261. doi: 10.1179/016164106X98125. [DOI] [PubMed] [Google Scholar]
- 65.Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. The FASEB Journal. 2009;23(11):3659–3673. doi: 10.1096/fj.09-136200. [DOI] [PubMed] [Google Scholar]
- 66.Malmeström C, Lycke J, Haghighi S, et al. Relapses in multiple sclerosis are associated with increased CD8+ T-cell mediated cytotoxicity in CSF. Journal of Neuroimmunology. 2008;196(1-2):159–165. doi: 10.1016/j.jneuroim.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 67.van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR- monitored phase II trial. Neurology. 1997;49(2):351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- 68.Moreau T, Coles A, Wing M, et al. CAMPATH-IH in multiple sclerosis. Multiple Sclerosis. 1996;1(6):357–365. doi: 10.1177/135245859600100616. [DOI] [PubMed] [Google Scholar]
- 69.Jacobsen M, Cepok S, Quak E, et al. Oligoclonal expansion of memory CD8+ T cells in cerebrospinal fluid from multiple sclerosis patients. Brain. 2002;125(3):538–550. doi: 10.1093/brain/awf059. [DOI] [PubMed] [Google Scholar]
- 70.Junker A, Ivanidze J, Malotka J, et al. Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain. 2007;130(11):2789–2799. doi: 10.1093/brain/awm214. [DOI] [PubMed] [Google Scholar]
- 71.Skulina C, Schmidt S, Dornmair K, et al. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(8):2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richert JR, Robinson ED, Deibler GE, Martenson RE, Dragovic LJ, Kies MW. Human cytotoxic T-cell recognition of a synthetic peptide of myelin basic protein. Annals of Neurology. 1989;26(3):342–346. doi: 10.1002/ana.410260306. [DOI] [PubMed] [Google Scholar]
- 73.Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. American Journal of Pathology. 2008;172(1):146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Buckle GJ, Höllsberg P, Hafler DA. Activated CD8+ T cells in secondary progressive MS secrete lymphotoxin. Neurology. 2003;60(4):702–705. doi: 10.1212/01.wnl.0000048204.89346.30. [DOI] [PubMed] [Google Scholar]
- 75.Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Annals of Neurology. 2010;67(5):625–638. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 76.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69(24):2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 77.Hinson SR, Roemer SF, Lucchinetti CF, et al. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down- regulating EAAT2. Journal of Experimental Medicine. 2008;205(11):2473–2481. doi: 10.1084/jem.20081241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. Journal of Experimental Medicine. 2005;202(4):473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain. 2002;125(7):1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130(5):1194–1205. doi: 10.1093/brain/awl371. [DOI] [PubMed] [Google Scholar]
- 81.Bonnan M, Valentino R, Bonnan M, Mehdaoui H, Smadja D, Cabre P. Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Multiple Sclerosis. 2009;15(4):487–492. doi: 10.1177/1352458508100837. [DOI] [PubMed] [Google Scholar]
- 82.Weinstock-Guttman B, Ramanathan M, Lincoff N, et al. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease) Archives of Neurology. 2006;63(7):957–963. doi: 10.1001/archneur.63.7.957. [DOI] [PubMed] [Google Scholar]
- 83.Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Archives of Neurology. 2009;66(9):1128–1133. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- 84.Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64(7):1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 85.De Parratt J, Prineas JW. Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Multiple Sclerosis. 2010;16(10):1156–1172. doi: 10.1177/1352458510382324. [DOI] [PubMed] [Google Scholar]
- 86.Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology. 2010;75(3):208–216. doi: 10.1212/WNL.0b013e3181e2414b. [DOI] [PubMed] [Google Scholar]
- 87.Lipton HL, Dal Canto MC. Theiler’s virus induced demyelination: prevention by immunosuppression. Science. 1976;192(4234):62–64. doi: 10.1126/science.176726. [DOI] [PubMed] [Google Scholar]
- 88.Dal Canto MC, Lipton HL. Multiple sclerosis. Animal model: Theiler’s virus infection in mice. American Journal of Pathology. 1977;88(2):497–500. [PMC free article] [PubMed] [Google Scholar]
- 89.Lipton HL. Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infection and Immunity. 1975;11(5):1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rodriguez M, Oleszak E, Leibowitz J. Theiler’s murine encephalomyelitis: a model of demyelination and persistence of virus. Critical Reviews in Immunology. 1987;7(4):325–365. [PubMed] [Google Scholar]
- 91.Miller SD, Vanderlugt CL, Begolka WS, et al. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nature Medicine. 1997;3(10):1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 92.Rodriguez M, Leibowitz JL, Lampert PW. Persistent infection of oligodendrocytes in Theiler’s virus-induced encephalomyelitis. Annals of Neurology. 1983;13(4):426–433. doi: 10.1002/ana.410130409. [DOI] [PubMed] [Google Scholar]
- 93.Atkins GJ, Sheahan BJ, Dimmock NJ. Semliki Forest virus infection of mice: a model for genetic and molecular analysis of viral pathogenicity. Journal of General Virology. 1985;66, part 3:395–408. doi: 10.1099/0022-1317-66-3-395. [DOI] [PubMed] [Google Scholar]
- 94.Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R. Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochemical and Biophysical Research Communications. 2000;270(3):701–708. doi: 10.1006/bbrc.2000.2490. [DOI] [PubMed] [Google Scholar]
- 95.Bailey RE. Diagnosis and treatment of infectious mononucleosis. American Family Physician. 1994;49(4):879–885. [PubMed] [Google Scholar]
- 96.Jenson HB. Virologic diagnosis, viral monitoring, and treatment of Epstein-Barr virus infectious mononucleosis. Current Infectious Disease Reports. 2004;6(3):200–207. doi: 10.1007/s11908-004-0009-2. [DOI] [PubMed] [Google Scholar]
- 97.Cepok S, Zhou D, Srivastava R, et al. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. Journal of Clinical Investigation. 2005;115(5):1352–1360. doi: 10.1172/JCI23661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. Plos ONE. 2010;5(9):1–5. doi: 10.1371/journal.pone.0012496. Article ID e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thacker EL, Mirzaei F, Ascherio A. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Annals of Neurology. 2006;59(3):499–503. doi: 10.1002/ana.20820. [DOI] [PubMed] [Google Scholar]
- 100.Ramagopalan SV, Handel AE, Giovannoni G, Siegel SR, Ebers GC, Chaplin G. Relationship of UV exposure to prevalence of multiple sclerosis in England. Neurology. 2011;76(16):1410–1414. doi: 10.1212/WNL.0b013e318216715e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goldacre MJ, Wotton CJ, Seagroatt V, Yeates D. Multiple sclerosis after infectious mononucleosis: record linkage study. Journal of Epidemiology and Community Health. 2004;58(12):1032–1035. doi: 10.1136/jech.2003.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nielsen TR, Rostgaard K, Nielsen NM, et al. Multiple sclerosis after infectious mononucleosis. Archives of Neurology. 2007;64(1):72–75. doi: 10.1001/archneur.64.1.72. [DOI] [PubMed] [Google Scholar]
- 103.Ebers GC, Sadovnick AD. The geographic distribution of multiple sclerosis: a review. Neuroepidemiology. 1993;12(1):1–5. doi: 10.1159/000110293. [DOI] [PubMed] [Google Scholar]
- 104.Poser CM, Benedikz J, Hibberd PL. The epidemiology of multiple sclerosis: the Iceland model. Onset-adjusted prevalence rate and other methodological considerations. Journal of the Neurological Sciences. 1992;111(2):143–152. doi: 10.1016/0022-510x(92)90062-p. [DOI] [PubMed] [Google Scholar]
- 105.Rosen LN, Livingstone IR, Rosenthal NE. Multiple sclerosis and latitude: a new perspective on an old association. Medical Hypotheses. 1991;36(4):376–378. doi: 10.1016/0306-9877(91)90014-p. [DOI] [PubMed] [Google Scholar]
- 106.Cantorna MT. Vitamin D and autoimmunity: is vitamin D status an environmental factor affecting autoimmune disease prevalence? Proceedings of the Society for Experimental Biology and Medicine. 2000;223(3):230–233. doi: 10.1046/j.1525-1373.2000.22333.x. [DOI] [PubMed] [Google Scholar]
- 107.Cantorna MT. Vitamin D and multiple sclerosis: an update. Nutrition Reviews. 2008;66(supplement 2):S135–S138. doi: 10.1111/j.1753-4887.2008.00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.VanAmerongen BM, Dijkstra CD, Lips P, Polman CH. Multiple sclerosis and vitamin D: an update. European Journal of Clinical Nutrition. 2004;58(8):1095–1109. doi: 10.1038/sj.ejcn.1601952. [DOI] [PubMed] [Google Scholar]
- 109.Goswami R, Marwaha RK, Gupta N, et al. Prevalence of vitamin D deficiency and its relationship with thyroid autoimmunity in Asian Indians: a community-based survey. British Journal of Nutrition. 2009;102(3):382–386. doi: 10.1017/S0007114509220824. [DOI] [PubMed] [Google Scholar]
- 110.Grant WB, Cross HS, Garland CF, et al. Estimated benefit of increased vitamin D status in reducing the economic burden of disease in western Europe. Progress in Biophysics and Molecular Biology. 2009;99(2-3):104–113. doi: 10.1016/j.pbiomolbio.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 111.Kragt JJ, van Amerongen BM, Killestein J, et al. Higher levels of 25-hydroxyvitamin D are associated with a lower incidence of multiple sclerosis only in women. Multiple Sclerosis. 2009;15(1):9–15. doi: 10.1177/1352458508095920. [DOI] [PubMed] [Google Scholar]
- 112.Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. Journal of the American Medical Association. 2006;296(23):2832–2838. doi: 10.1001/jama.296.23.2832. [DOI] [PubMed] [Google Scholar]
- 113.Orton SM, Morris AP, Herrera BM, et al. Evidence for genetic regulation of vitamin D status in twins with multiple sclerosis. American Journal of Clinical Nutrition. 2008;88(2):441–447. doi: 10.1093/ajcn/88.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raghuwanshi A, Joshi SS, Christakos S. Vitamin D and multiple sclerosis. Journal of Cellular Biochemistry. 2008;105(2):338–343. doi: 10.1002/jcb.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smolders J, Damoiseaux J, Menheere P, Tervaert JWC, Hupperts R. Association study on two vitamin D receptor gene polymorphisms and vitamin D metabolites in multiple sclerosis. Annals of the New York Academy of Sciences. 2009;1173:515–520. doi: 10.1111/j.1749-6632.2009.04656.x. [DOI] [PubMed] [Google Scholar]
- 116.Smolders J, Damoiseaux J, Menheere P, Tervaert JWC, Hupperts R. Fok-I vitamin D receptor gene polymorphism (rs10735810) and vitamin D metabolism in multiple sclerosis. Journal of Neuroimmunology. 2009;207(1-2):117–121. doi: 10.1016/j.jneuroim.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 117.Smolders J, Peelen E, Thewissen M, et al. The relevance of vitamin D receptor gene polymorphisms for vitamin D research in multiple sclerosis. Autoimmunity Reviews. 2009;8(7):621–626. doi: 10.1016/j.autrev.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 118.Smolders J, Thewissen M, Peelen E, et al. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. Plos ONE. 2009;4(8) doi: 10.1371/journal.pone.0006635. Article ID e6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Soilu-Hanninen M, Laaksonen M, Laitinen I, Eralinna JP, Lilius EM, Mononen I. A longitudinal study of serum 25-hydroxyvitamin D and intact parathyroid hormone levels indicate the importance of vitamin D and calcium homeostasis regulation in multiple sclerosis. Journal of Neurol Neurosurg Psychiatry. 2008;79(2):152–157. doi: 10.1136/jnnp.2006.105320. [DOI] [PubMed] [Google Scholar]
- 120.van der Mei IAF, Ponsonby AL, Dwyer T, et al. Vitamin D levels in people with multiple sclerosis and community controls in Tasmania, Australia. Journal of Neurology. 2007;254(5):581–590. doi: 10.1007/s00415-006-0315-8. [DOI] [PubMed] [Google Scholar]
- 121.Craig TA, Sommer S, Sussman CR, Grande JP, Kumar R. Expression and regulation of the vitamin D receptor in the zebrafish, Danio rerio. Journal of Bone and Mineral Research. 2008;23(9):1486–1496. doi: 10.1359/JBMR.080403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gross M, Kumar R. Physiology and biochemistry of vitamin D-dependent calcium binding proteins. American Journal of Physiology. 1990;259(2):F195–F209. doi: 10.1152/ajprenal.1990.259.2.F195. [DOI] [PubMed] [Google Scholar]
- 123.Haussler MR, Haussler CA, Bartik L, et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutrition Reviews. 2008;66(2):S98–S112. doi: 10.1111/j.1753-4887.2008.00093.x. [DOI] [PubMed] [Google Scholar]
- 124.Johnson JA, Kumar R. Renal and intestinal calcium transport: roles of vitamin D and vitamin D-dependent calcium binding proteins. Seminars in Nephrology. 1994;14(2):119–128. [PubMed] [Google Scholar]
- 125.Jurutka PW, Bartik L, Whitfield GK, et al. Vitamin D receptor: key roles in bone mineral pathophysiology, molecular mechanism of action, and novel nutritional ligands. Journal of Bone and Mineral Research. 2007;22(supplement 2):V2–V10. doi: 10.1359/jbmr.07s216. [DOI] [PubMed] [Google Scholar]
- 126.Jurutka PW, Whitfield GK, Hsieh JC, Thompson PD, Haussler CA, Haussler MR. Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Reviews in Endocrine & Metabolic Disorders. 2001;2(supplement 2):203–216. doi: 10.1023/a:1010062929140. [DOI] [PubMed] [Google Scholar]
- 127.Kumar A. Metabolism of 1,25-dihydroxyvitamin D3. Physiological Reviews. 1984;64(2):478–504. doi: 10.1152/physrev.1984.64.2.478. [DOI] [PubMed] [Google Scholar]
- 128.Veenstra TD, Prüfer K, Koenigsberger C, Brimijoin SW, Grande JP, Kumar R. 1,25-dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Research. 1998;804(2):193–205. doi: 10.1016/s0006-8993(98)00565-4. [DOI] [PubMed] [Google Scholar]
- 129.Dong X, Lutz W, Schroeder TM, et al. Regulation of relB in dendritic cells by means of modulated association of vitamin D receptor and histone deacetylase 3 with the promoter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16007–16012. doi: 10.1073/pnas.0506516102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Griffin MD, Dong X, Kumar R. Vitamin D receptor-mediated suppression of RelB in antigen presenting cells: a paradigm for ligand-augmented negative transcriptional regulation. Archives of Biochemistry and Biophysics. 2007;460(2):218–226. doi: 10.1016/j.abb.2007.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Griffin MD, Kumar R. Effects of 1α,25(OH)2D3 and its analogs on dendritic cell function. Journal of Cellular Biochemistry. 2003;88(2):323–326. doi: 10.1002/jcb.10335. [DOI] [PubMed] [Google Scholar]
- 132.Griffin MD, Kumar R. Multiple potential clinical benefits for 1alpha,25-dihydroxyvitamin D3 analogs in kidney transplant recipients. The Journal of Steroid Biochemistry and Molecular Biology. 2005;97(1-2):213–218. doi: 10.1016/j.jsbmb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 133.Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1α,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo . Proceedings of the National Academy of Sciences of the United States of America. 2001;98(12):6800–6805. doi: 10.1073/pnas.121172198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xing. N, Maldonadoa ML, Bachmana LA, McKeanb DJ, Kumara R, Griffin MD. Distinctive dendritic cell modulation by vitamin D(3) and glucocorticoid pathways. Biochemical and Biophysical Research Communications. 2002;297(3):645–652. doi: 10.1016/s0006-291x(02)02262-3. [DOI] [PubMed] [Google Scholar]
- 135.Cannell JJ, Zasloff M, Garland CF, Scragg R, Giovannucci E. On the epidemiology of influenza. Virology Journal. 2008;5, article 29 doi: 10.1186/1743-422X-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grant WB. Latitude and multiple sclerosis prevalence: vitamin D reduces risk of Epstein-Barr virus infection. Multiple Sclerosis Journal. 2010;16(3):p. 373. doi: 10.1177/1352458509358093. [DOI] [PubMed] [Google Scholar]
- 137.Warrington AE, Asakura K, Bieber AJ, et al. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Warrington AE, Bieber AJ, Ciric B, Pease LR, Van Keulen V, Rodriguez M. A recombinant human IgM promotes myelin repair after a single, very low dose. Journal of Neuroscience Research. 2007;85(5):967–976. doi: 10.1002/jnr.21217. [DOI] [PubMed] [Google Scholar]
- 139.Warrington AE, Bieber AJ, Van Keulen V, Ciric B, Pease LR, Rodriguez M. Neuron-binding human monoclonal antibodies support central nervous system neurite extension. Journal of Neuropathology and Experimental Neurology. 2004;63(5):461–473. doi: 10.1093/jnen/63.5.461. [DOI] [PubMed] [Google Scholar]
- 140.Watzlawik J, Holicky E, Edberg DD, et al. Human remyelination promoting antibody inhibits apoptotic signaling and differentiation through Lyn kinase in primary rat oligodendrocytes. GLIA. 2010;58(15):1782–1793. doi: 10.1002/glia.21048. [DOI] [PMC free article] [PubMed] [Google Scholar]