

Abstract
Recent evidence showed the role of peroxisome proliferator-activated receptors (PPARs) in cardiac function. Cardiac contraction induced by various agents is critical in restoring the activity of peroxisome proliferator-activated receptors δ (PPARδ) in cardiac myopathy. Because dobutamine is an agent widely used to treat heart failure in emergency setting, this study is aimed to investigate the change of PPARδ in response to dobutamine. Neonatal rat cardiomyocytes were used to examine the effects of dobutamine on PPARδ expression levels and cardiac troponin I (cTnI) phosphorylation via Western blotting analysis. We show that treatment with dobutamine increased PPARδ expression and cTnI phosphorylation in a time- and dose-dependent manner in neonatal rat cardiomyocytes. These increases were blocked by the antagonist of β1-adrenoceptors. Also, the action of dobutamine was related to the increase of calcium ions and diminished by chelating intracellular calcium. Additionally, dobutamine-induced action was reduced by the inhibition of downstream messengers involved in this calcium-related pathway. Moreover, deletion of PPARδ using siRNA generated the reduction of cTnI phosphorylation in cardiomyocytes treated with dobutamine. Thus, we concluded that PPARδ is increased by dobutamine in cardiac cells.
1. Introduction
Dobutamine is one of the most widely used agents for heart failure in clinic settings [1]. Dobutamine acts through the activation of β1-adrenoceptor which is linked to a guanine nucleotide regulatory cascade via heterotrimeric G proteins. This activation results in an increase of adenylyl cyclase activity for the conversion of adenosine triphosphate (ATP) to cyclic AMP (cAMP). An increase in intracellular cAMP concentration causes the release of calcium from sarcoplasmic reticulum. This increased calcium is used by contractile proteins to increase stroke volume [2–6].
Troponin I (TnI) is an inhibitory unit of the troponin complex associated with thin filaments and acts by inhibiting actomyosin interactions in the presence of low levels of intracellular calcium ions (Ca2+) during diastole [7, 8]. Modulation of myofilament properties via changes in TnI phosphorylation has profound effects on cardiac contractility and pumping [9]. Phosphorylation of TnI by protein kinase A results in a reduction in myofilament sensitivity to Ca2+ and an increase in the cross-bridge cycling rate, resulting in the acceleration of relaxation and an increase in power output, but a reduced economy of contraction [8, 9]. Ca2+ is involved in muscle contraction and is an intracellular messenger that activates a wide variety of cellular responses including gene transcription [7–10].
Various studies have shown that the activation of calcineurin (Cn) and calcium/calmodulin-dependent protein kinase (CaMK) signaling pathways serves a major role in the regulation of gene expression in cardiac muscles [11, 12]. These same genes have been shown to also be regulated by peroxisome proliferator-activated receptors (PPARs) [13].
PPARs are ligand-activated transcription factors that regulate expression of genes involved in lipid metabolism and inflammation [13]. The three subtypes of PPARs (PPARα, PPARγ, and PPARδ) modulate expression of different genes and exert various bioactivities [13]. Previous studies also showed that metabolic modulators can have beneficial effects in both experimental and clinical heart failure settings [14]. PPARδ-dependent maintenance of cardiac function is crucial for cardiomyocytes [15–17]. The deletion of cardiac PPARδ results in decreased contraction, increased left ventricular end-diastolic pressure, lowered cardiac output, and increased incidences of cardiac failure [15]. Our previous study demonstrated that digoxin enhanced cardiac output by increasing PPARδ expression [18].
Dobutamine is the widely used cardiac agent for patients with heart failure. However, the theory is that the action of dobutamine occurs via the activation of PPARδ remained obscure. In this study, we used the neonatal rat cardiomyocytes to investigate the role of PPARδ in dobutamine-induced action. Moreover, we determined the possible signaling pathways for increase of PPARδ induced by dobutamine.
2. Methods
2.1. Materials
Dobutamine, atenolol, butoxamine, and cyclosporine A were purchased from Sigma-Aldrich (St Louis, MO, USA). BAPTA-AM and KN93 were purchased from Calbiochem-Novabiochem Corp (La Jolla, CA, USA). The fluorescent probe, Fura2-AM, was obtained from Molecular Probes (Eugene, OR, USA). The Opti-MEM I Reduced Serum Medium, Stealth Select RNAi (siRNA-PPARδ), scramble siRNA (siRNA-control), and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies to PPARδ and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to cardiac TnI and phospho-TnI (Ser 23/24) were purchased from Cell Signaling Technology (Beverly, MA, USA).
2.2. Cell Culture
Primary cultures of neonatal rat cardiomyocytes were prepared by the modification of a previously described method [14]. Briefly, under anesthesia with 2% isoflurane, hearts of 1-to-2-day-old Wistar rats were excised, cut into 1-2 mm pieces, and predigested with trypsin to remove red blood cells. The heart tissue was then digested with 0.25% trypsin and 0.05% collagenase. The dissociated cells were placed in uncoated 100 mm dishes and incubated at 37°C in a 5% CO2 incubator for at least 1 hour to remove the nonmyocytic cells. This procedure caused fibroblasts to predominantly attach to the dishes while most of the cardiomyocytes remained in suspension. The cardiomyocyte-enriched population was collected and counted. The cells were cultured in Dulbecco/Vogt modified Eagle's minimal essential medium (DMEM) with 1 mmol/L pyruvate, 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 units/mL streptomycin. Over 95% of the collected cells were characterized cardiomyocytes on the basis of the sarcomeric myosin content. On the second day, the medium was replaced. After 3 to 4 days in culture, the cells were exposed to hyperglycemic conditions. The high glucose-treated cardiomyocytes were generated by applying 30 mmol/L glucose to the cells for 24 hours [14]. This animal experiment was approved and conducted in accordance with local institutional guidelines for the care and use of laboratory animals in the Chi-Mei Medical Center (number 100052307) and followed the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication number 85-23, revised 1996), as well as the guidelines of the Animal Welfare Act.
2.3. Drug Treatment of Cardiomyocytes
Stock solutions of dobutamine were prepared with normal media. Cells were treated with varying concentrations of dobutamine (0.01–10 μmol/L) for 4 hours, washed twice with PBS, and removed by trypsinization. The treated cells were then collected and subjected to a gene expression assay. In addition, pretreatment with various inhibitory agents (β1-adenocepotor antagonist (10 μmol/L atenolol) [19, 20], β2-adenocepotor antagonist (10 μmol/L butoxamine) [21], calcium chelator (25 mmol/L BAPTA-AM), calcineurin inhibitor (1 μmol/L cyclosporine A) [22], or CaMK inhibitor (1 μmol/L KN-93) [22]) was applied for 30 minutes before the addition of dobutamine.
2.4. Western Blotting Analysis
Protein was extracted from tissue homogenates and cell lysates using ice-cold radio-immunoprecipitation assay (RIPA) buffer supplemented with phosphatase and protease inhibitors (50 mmol/L sodium vanadate, 0.5 mmol/L phenylmethylsulphonyl fluoride, 2 mg/mL aprotinin, and 0.5 mg/mL leupeptin). Protein concentrations were determined with the Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total protein (30 μg) was separated by SDS/polyacrylamide gel electrophoresis (10% acrylamide gel) using the Bio-Rad Mini-Protein II system. Protein was transferred to expanded polyvinylidene difluoride membranes (Pierce, Rockford, IL, USA) with a Bio-Rad Trans-Blot system. After the transfer, the membranes were washed with PBS and blocked for 1 hour at room temperature with 5% (w/v) nonfat dry milk (NFDM) in PBS. Blots were incubated overnight at 4°C with an immunoglobulin-G polyclonal rabbit anti-mouse antibody (Affinity BioReagents, Inc., Golden, CO, USA) diluted 1 : 500 in 5% (w/v) NFDM dissolved in PBS/Tween 20 (0.5% by volume). The blots were also incubated with goat polyclonal antibody (1 : 1000) targeted to actin, which served as an internal control. After the removal of the primary antibody, the blots were extensively washed with PBS/Tween 20. The blots were then incubated for 2 hours at room temperature with the appropriate peroxidase-conjugated secondary antibody diluted in 5% (w/v) NFDM dissolved in PBS/Tween 20. The blots were developed by autoradiography using the ECL-Western blotting system (Amersham International, Buckinghamshire, UK). The immunoblots were quantified with a laser densitometer.
2.5. Measurement of Intracellular Calcium Concentration
The changes in intracellular calcium were detected using the fluorescent probe Fura2-AM [23]. Primary cultured cardiomyocytes were placed in a buffered physiological saline solution containing 140 mmol/L NaCl, 5.9 mmol/L KCl, 1.2 mmol/L CaCl2, 1.4 mmol/L MgCl2, 11.5 mmol/L glucose, 1.8 mmol/L Na2HPO4, and 10 mmol/L HEPES-Tris. A final concentration of 5 μmol/L Fura-2AM was added to the cells which were incubated for 1 hour in humidified 5% CO2 and 95% air at 37°C. The cells were washed and incubated for an additional 30 minutes in PSS. The neonatal rat cardiomyocytes were inserted into a thermostatic (37°C) cuvette containing 2 mL of calcium-free PSS and various doses of dobutamine or inhibitor as previously indicated. The fluorescence was continuously recorded using a fluorescence spectrofluorometer (Hitachi F-2000, Tokyo, Japan). The values of [Ca2+]i were calculated from the ratio R = F340/F380 by the formula: [Ca2+]i = KdB (R − R min)/(R max − R), where Kd is 225 nM, F is the fluorescence, and B is the ratio of the fluorescence of the free dye to that of the Ca2+-bound dye measured at 380 nm. R max and R min were determined in separate experiments by using Dobutamine to equilibrate [Ca2+]i with ambient [Ca2+] (R max), and the addition of 0.1 mmol/L MnCl2 and 1 mmol/L EGTA (R min). Background autofluorescence was measured in unloaded cells and subtracted from all experimental measurements.
2.6. Small Interfering RNA (siRNA)
Duplexed RNA oligonucleotides for rat PPARδ (Stealth RNAi) were synthesized by Invitrogen using our previous method [24]. The neonatal rat cardiomyocytes were transfected with 40 pmol of PPARδ-specific siRNA (siRNA-PPARδ) or scramble siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. These cardiomyocytes were subjected to experimental conditions as described above for 48 hours posttransfection. The sequences of the siRNA-PPARδ are UUGCAGAUCCGAUCGCACUUCUCGU (sense strand) and ACGAGAAGUGCGAUCGGAUCUGCAA (antisense strand) as described previously [24].
2.7. Statistical Analysis
Statistical analysis was carried out using an ANOVA and the Newman-Keuls post-hoc analysis. Statistical significance was set as P < 0.05. The results were expressed as mean ± SEM.
3. Results
3.1. Increase of PPARδ Expression by Dobutamine in Neonatal Rat Cardiomyocytes
The neonatal rat cardiomyocytes were treated with dobutamine to identify the changes in PPARδ expression. Treatment with dobutamine at 0.1 μmol/L increased PPARδ protein expression level in a time-dependent manner (Figure 1(b)) and the levels in these cells were increased to maximum at 4 hours later of drug treatment. Dobutamine was then incubated for 4 h at various concentrations ranging from 0.01 to 10 μmol/L. The PPARδ protein expression levels in neonatal rat cardiomyocytes were increased by dobutamine in a concentration-dependent manner (Figure 1(a)).
Figure 1.
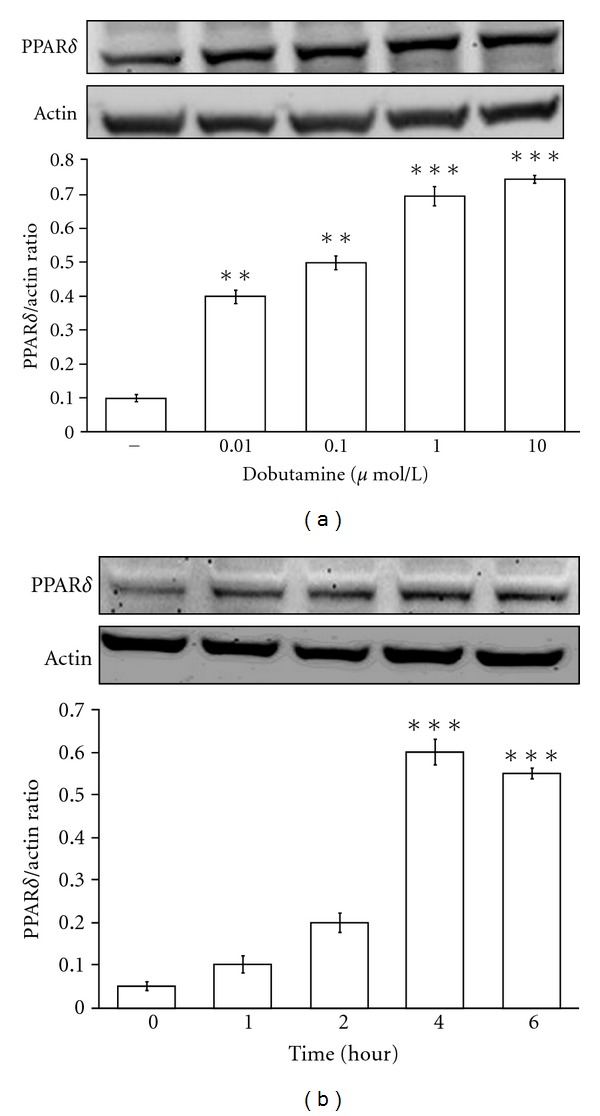
Effects of dobutamine on PPARδ expression in neonatal rat cardiomyocytes. The neonatal rat cardiomyocytes were treated with dobutamine at various concentrations for 4 hours (a) or at 1 μmol/L during various time points (b). The treated cells were harvested to determine the protein levels by Western blotting analysis. All values are presented as mean ± SEM (n = 6 per group). *P < 0.05 and **P < 0.01 as compared with the vehicle-treated control group.
3.2. Effects of Atenolol and Butoxamine on Dobutamine-Induced Actions in Neonatal Rat Cardiomyocytes
To determine the receptor involved in dobutamine-induced the expressions of PPARδ and the phosphorylation of cTnI, we treated the cells with atenolol at a concentration sufficient to block the β1-adrenoceptor [19, 20] and butoxamine to block the β2-adrenoceptor [21]. The increases in PPARδ expression and cTnI phosphorylation due to dobutamine were inhibited by 10 μmol/L of atenolol (Figures 2(a) and 2(b)); however, butoxamine failed to modify the actions of dobutamine (Figures 2(a) and 2(b)).
Figure 2.
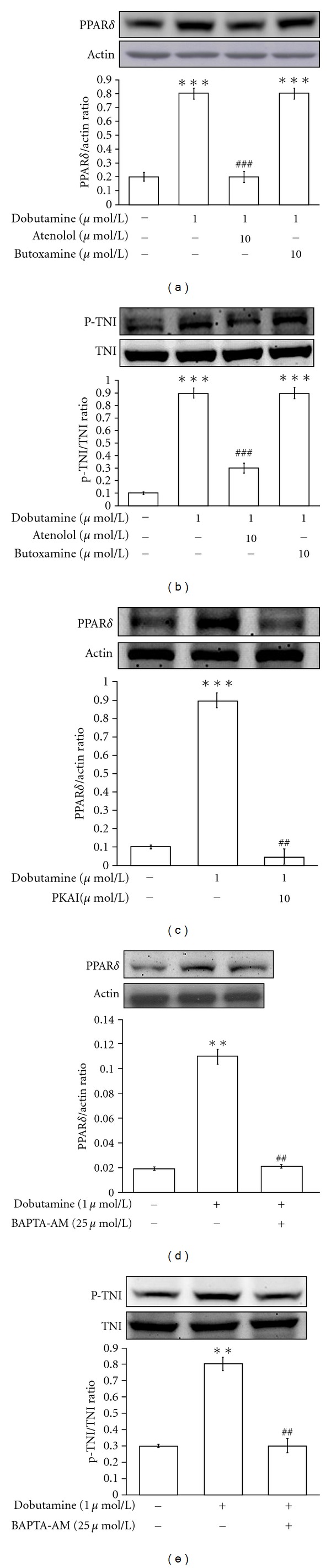
Effects of receptor antagonists, protein kinase A inhibitor, and calcium chelater on dobutamine-induced actions in neonatal rat cardiomyocytes. The neonatal rat cardiomyocytes were treated with atenolol, butoxamine, PKAI or BAPTA-AM at indicated concentration for 30 minutes prior to incubation with 1 μmol/L dobutamine. All drugs were dissolved in normal medium. Cells treated with the same volume of normal medium only are indicated as the vehicle-treated control. The treated cells were then harvested to measure PPARδ protein expression (a, c, d) and cTnI phosphorylation (b, e) using Western blotting analysis. All values are expressed as mean ± SEM (n = 6 per group). *P < 0.05 as compared with the vehicle-treated control. # P < 0.05 as compared to cells treated with dobutamine only.
3.3. Effect of Protein Kinase A Inhibitor on Dobutamine-Induced Actions in Neonatal Rat Cardiomyocytes
The β1-adrenoceptor is known to couple to adenylyl cyclase [2–6]. An increase of cyclic AMP causes protein kinase A (PKA) activation which results in higher levels of PPARδ protein expression [25, 26]. Thus, we used the specific inhibitor of protein kinase A (PKAI) to verify the roles of PKA in changes of PPARδ expressions. The dobutamine-induced increases of PPARδ expression were reduced by treatment with 10 μmol/L of PKAI (Figure 2(c)).
3.4. Effects of Intracellular Calcium Levels on Dobutamine-Induced Actions in Neonatal Rat Cardiomyocytes
The fluorescent probe, Fura2-AM is used to detect the intracellular calcium concentration in neonatal rat cardiomyocytes. Dobutamine increased the intracellular calcium levels from 155.4 + 11.4 nmol/L to 484.7 + 22.4 nmol/L (n = 8) at 0.1 μmol/L and increased in a concentration-dependent manner. Pretreatment of the cells with the calcium chelator BAPTA-AM (BAPTA) at an effective concentration (25 μmol/L) [27] reduced the actions of dobutamine on the increases of PPARδ expression and cTnI phosphorylation (Figures 2(d) and 2(e)).
3.5. Calcium-Related Pathway as the Possible Signal for Dobutamine-Induced Actions in Neonatal Rat Cardiomyocytes
Cyclosporine A (CsA, a calcineurin inhibitor) and KN93 (a calcium/calmodulin kinase inhibitor) were used to investigate whether dobutamine-induced PPARδ expression is mediated through the activation of calcium-related pathway [22]. The dobutamine-induced increases in PPARδ expression and cTnI phosphorylation were markedly inhibited by either 1 μmol/L of CsA or 1 μmol/L of KN93 (Figures 3(a) and 3(b)).
Figure 3.
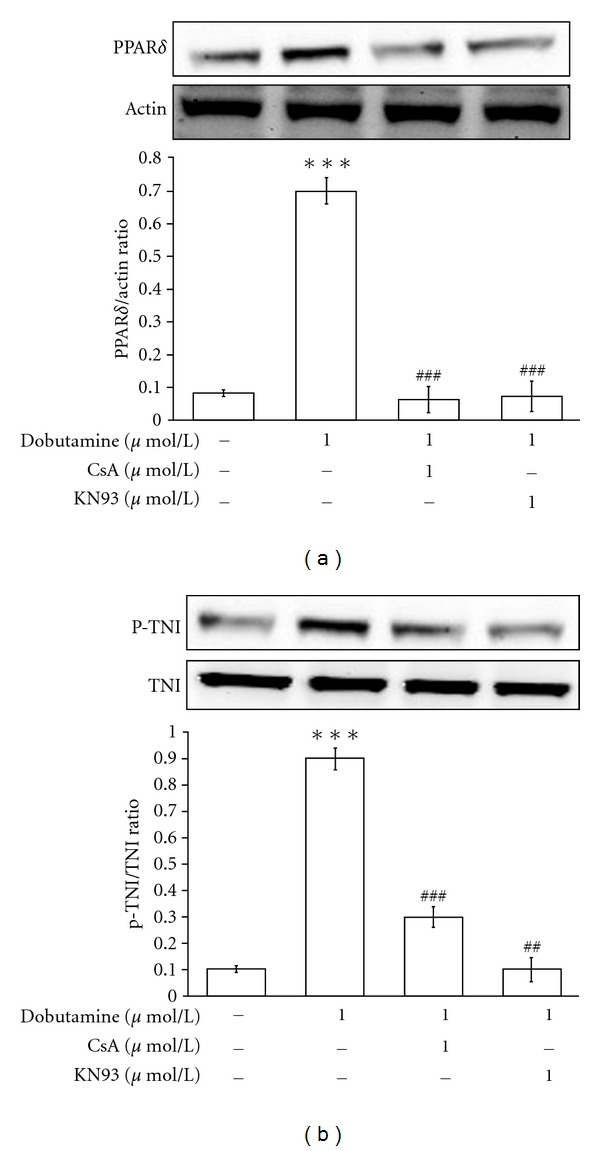
Effects of calcium-mediated signaling inhibitors on dobutamine-induced actions in neonatal rat cardiomyocytes. The neonatal rat cardiomyocytes were treated with 1 μmol/L cyclosporine A (CsA, calcineurin inhibitor) or 1 μmol/L KN93 (CaMK inhibitor) at 30 minutes prior to incubation with 1 μmol/L dobutamine for 4 hours. The cells were then harvested to measure PPARδ protein expression (a) and cTnI phosphorylation (b) using Western blotting analysis. All values are expressed as mean ± SEM (n = 6 per group). **P < 0.01 as compared with control cells. ## P < 0.01 as compared to the control cells incubated with dobutamine only.
3.6. Effects of siRNA-PPARδ on Dobutamine-Induced Actions in Neonatal Rat Cardiomyocytes
The neonatal rat cardiomyocytes were transfected with either siRNA targeted to PPARδ or a scramble control for 48 hours, as described previously [24]. After treatment with dobutamine, PPARδ expression was markedly reduced in the cardiomyocytes that were transfected with the siRNA targeted to PPARδ (Figure 4(a)). The cells transfected the siRNA-scramble did not affect dobutamine-induced increase of PPARδ expression (Figure 4(a)). Moreover, dobutamine-induced TnI phosphorylation was also reduced in cells treated with siRNA-PPARδ but remained unchanged in the cells transfected with siRNA-scramble (Figure 4(b)).
Figure 4.
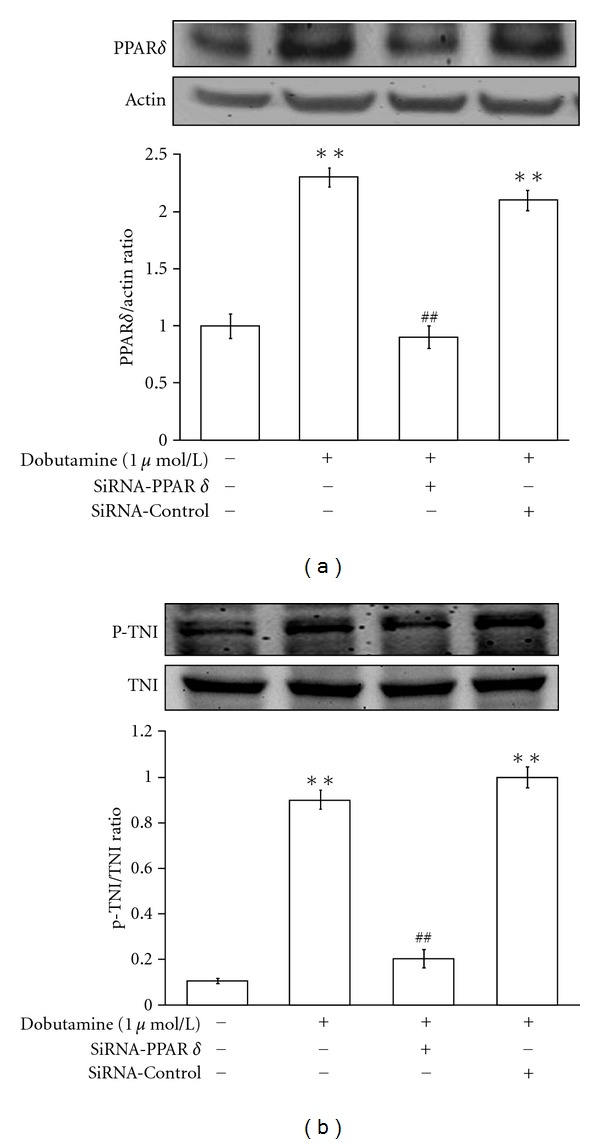
Effects of PPARδ-targeted siRNA on dobutamine-induced actions in neonatal rat cardiomyocytes. The neonatal rat cardiomyocytes transfected with siRNA-targeted to PPARδ or siRNA-scramble (siRNA-control) were incubated with 1 μmol/L dobutamine. All cells were then harvested to measure PPARδ expression (a) or cTnI phosphorylation (b) using Western blotting analysis. All values are expressed as mean ± SEM (n = 6 per group). *P < 0.05 as compared with the vehicle-treated control and # P < 0.05 as compared with the dobutamine-treated cells transfected with the scramble siRNA (siRNA-control).
4. Discussion
In the present study, treatment with dobutamine in neonatal rat cardiomyocytes caused a concentration-dependent increase in both PPARδ protein expression and cTnI phosphorylation. These increases induced by dobutamine were blocked by pretreatment with atenolol, PKAI, KN93, CsA, or BAPTA. Moreover, the dobutamine-induced increases in PPARδ protein expression and cTnI phosphorylation were markedly reduced in neonatal rat cardiomyocytes that were transfected with siRNA targeted to PPARδ. Thus, the mediation of PPARδ in dobutamine-induced cardiac action can be considered. It has been reported that PPARδ is involved in excitation-transcription coupling [28] and that calcineurin-mediated skeletal muscle reprogramming induces the expression of several transcriptional regulators, including PPARδ [29]. Taken together, we suggest that the increase in PPARδ expression by dobutamine is mainly induced by an activation of the β1-adrenoceptor, which results in an increase of intracellular cAMP and calcium. This leads to an increase in heart contractility.
Regulation of PPARδ expression in cardiac muscles through the intracellular Ca2+ signaling pathway has been established [22, 30, 31]. We show that treatment with BAPTA suppressed dobutamine-induced PPARδ protein expression. We also show that the inhibition of PKA reduced dobutamine-induced expression of PPARδ. This result is consistent with the finding that the activation of PKA induces intracellular calcium release [32]. Thus, dobutamine exerts its effects on PPARδ expression in a calcium-dependent manner via the activation of PKA in cardiac cells.
It has been demonstrated that cTnI phosphorylation most likely occurs due to an enhanced off rate during Ca2+ exchange with the cardiac troponin calcium binding site, leading to an acceleration of relaxation and an increase in cardiac output [31, 33–36]. Similarly, we found that cTnI phosphorylation is elevated in neonatal rat cardiomyocytes after treatment with dobutamine. We also observed that pretreatment with calcium chelater (BAPTA) decreased the levels of cTnI phosphorylation in dobutamine-treated cardiomyocytes. Therefore, we suggest that the increase in intracellular calcium is responsible for the increase of cTnI phosphorylation by dobutamine. This explanation is consistent with previously published reports [31, 33–36].
The role of PPARδ in the phosphorylation of cTnI in cardiomyocytes remains unclear. Thus, we applied PPARδ-targeted siRNA to better characterize this possible relationship. In this study, a 48 hours transfection of siRNA-PPARδ in cardiomyocytes suppressed dobutamine-induced cTnI phosphorylation. Thus, it is suggested that PPARδ is involved in dobutamine-induced cTnI phosphorylation in cardiomyocytes.
In conclusion, we demonstrated that treatment with dobutamine in neonatal rat cardiomyocytes can increase PPARδ expression by an activation of the β1-adrenoceptor through cAMP to activate PKA and increase intracellular calcium levels. This increase may induce calmodulin and calcineurin activation which could result in higher PPARδ protein expression. Taken together, increases in PPARδ protein expression and cTnI phosphorylation are responsible for dobutamine-induced cardiac action, suggesting a new mechanism for dobutamine-induced cardiac contraction.
Author's Contribution
The first two authors contributed equally to this work (M. T. Chou & S. H. Lo).
Abbreviations
- PPARδ:
Peroxisome proliferator-activated receptors δ
- cTnI:
Cardiac troponin I
- ATP:
Adenosine triphosphate
- cAMP:
Cyclic adenosine monophosphate
- TnI:
Troponin I
- Cn:
Calcineurin
- CaMK:
Calcium/calmodulin-dependent protein kinase
- PPARs:
Peroxisome proliferator-activated receptors
- DMEM:
Dulbecco/Vogt modified Eagle's minimal essential medium
- FBS:
Foetal bovine serum
- RIPA:
Radio-immuno-precipitation assay
- BAPTA:
BAPTA-AM
- IBMX:
3-isobutyl-1-methylxanthine
- PKA:
Protein kinase A
- PKAI:
Inhibitor of protein kinase A
- CsA:
Cyclosporine A.
References
- 1.Opasich C, Russo A, Mingrone R, Zambelli M, Tavazzi L. Intravenous inotropic agents in the intensive therapy unit: do they really make a difference? European Journal of Heart Failure. 2000;2(1):7–11. doi: 10.1016/s1388-9842(99)00061-6. [DOI] [PubMed] [Google Scholar]
- 2.Schulz R, Guth BD, Pieper K, Martin C, Heusch G. Recruitment of an inotropic reserve in moderately ischemic myocardium at the expense of metabolic recovery: a model of short-term hibernation. Circulation Research. 1992;70(6):1282–1295. doi: 10.1161/01.res.70.6.1282. [DOI] [PubMed] [Google Scholar]
- 3.Fu ZL, Feng YB, Xu HX, Zhang XP, Shi CZ, Gu X. Role of norepinephrine in development of short-term myocardial hibernation. Acta Pharmacologica Sinica. 2006;27(2):158–164. doi: 10.1111/j.1745-7254.2006.00245.x. [DOI] [PubMed] [Google Scholar]
- 4.Heusch G, Rose J, Skyschally A, Post H, Schulz R. Calcium responsiveness in regional myocardial short-term hibernation and stunning in the in situ porcine heart: inotropic responses to postextrasystolic potentiation and intracoronary calcium. Circulation. 1996;93(8):1556–1566. doi: 10.1161/01.cir.93.8.1556. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhry FA, Tauke JT, Alessandrini RS, Vardi G, Parker MA, Bonow RO. Prognostic implications of myocardial contractile reserve in patients with coronary artery disease and left ventricular dysfunction. Journal of the American College of Cardiology. 1999;34(3):730–738. doi: 10.1016/s0735-1097(99)00252-1. [DOI] [PubMed] [Google Scholar]
- 6.The treatment of heart failure. Task Force of the Working Group on Heart Failure of the European Society of Cardiology. European Heart Journal. 1997;18(5):736–753. doi: 10.1093/oxfordjournals.eurheartj.a015339. [DOI] [PubMed] [Google Scholar]
- 7.Ohtsuki I, Morimoto S. Troponin: regulatory function and disorders. Biochemical and Biophysical Research Communications. 2008;369(1):62–73. doi: 10.1016/j.bbrc.2007.11.187. [DOI] [PubMed] [Google Scholar]
- 8.Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circulation Research. 2004;94(2):146–158. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- 9.Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovascular Research. 2005;66(1):12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83(5):675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 11.Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. Journal of Biological Chemistry. 2000;275(7):4545–4548. doi: 10.1074/jbc.275.7.4545. [DOI] [PubMed] [Google Scholar]
- 12.Passier R, Zeng H, Frey N, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. Journal of Clinical Investigation. 2000;105(10):1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Q, Li Y. Roles of PPARs on regulating myocardial energy and lipid homeostasis. Journal of Molecular Medicine. 2007;85(7):697–706. doi: 10.1007/s00109-007-0170-9. [DOI] [PubMed] [Google Scholar]
- 14.deGoma EM, Vagelos RH, Fowler MB, Ashley EA. Emerging therapies for the management of decompensated heart failure. From bench to bedside. Journal of the American College of Cardiology. 2006;48(12):2397–2409. doi: 10.1016/j.jacc.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 15.Cheng L, Ding G, Qin Q, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nature Medicine. 2004;10(11):1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 16.Cheng L, Ding G, Qin Q, et al. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochemical and Biophysical Research Communications. 2004;313:277–286. doi: 10.1016/j.bbrc.2003.11.127. [DOI] [PubMed] [Google Scholar]
- 17.Barish GD, Narkar VA, Evans RM. PPARδ: a dagger in the heart of the metabolic syndrome. Journal of Clinical Investigation. 2006;116(3):590–597. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su JC, Li ZD, Yu BQ, Cao LH, Zhang CC. Diagnosis and treatment of peripheral nerve injury in Wenchuan earthquake: a report of 14 cases. Zhongguo Gu Shang. 2008;21(10):739–740. [PubMed] [Google Scholar]
- 19.Spallarossa P, Fabbi P, Manca V, et al. Doxorubicin-induced expression of LOX-1 in H9c2 cardiac muscle cells and its role in apoptosis. Biochemical and Biophysical Research Communications. 2005;335(1):188–196. doi: 10.1016/j.bbrc.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 20.Spallarossa P, Garibaldi S, Altieri P, et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. Journal of Molecular and Cellular Cardiology. 2004;37(4):837–846. doi: 10.1016/j.yjmcc.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 21.Costa SKP, Hyslop S, Nathan LP, et al. Activation by Phoneutria nigriventer spider venom of autonomic nerve fibers in the isolated rat heart. European Journal of Pharmacology. 1998;363(2-3):139–146. doi: 10.1016/s0014-2999(98)00767-5. [DOI] [PubMed] [Google Scholar]
- 22.Schaeffer PJ, Wende AR, Magee CJ, et al. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. Journal of Biological Chemistry. 2004;279(38):39593–39603. doi: 10.1074/jbc.M403649200. [DOI] [PubMed] [Google Scholar]
- 23.Halaq HA, Haupert GT., Jr. Positive inotropic effects of the endogenous Na+/K+-transporting ATPase inhibitor from the hypothalamus. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(24):10080–10084. doi: 10.1073/pnas.86.24.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu BC, Chang CK, Ou HY, Cheng KC, Cheng JT. Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovascular Research. 2008;80(1):78–87. doi: 10.1093/cvr/cvn172. [DOI] [PubMed] [Google Scholar]
- 25.Ji Z, Mei FC, Cheng X. Epac, not PKA catalytic subunit, is required for 3T3-L1 preadipocyte differentiation. Frontiers in Bioscience. 2010;2:392–398. doi: 10.2741/e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Ju D, Zhang M, Yang G. Interleukin-6 stimulates lipolysis in porcine adipocytes. Endocrine. 2008;33(3):261–269. doi: 10.1007/s12020-008-9085-7. [DOI] [PubMed] [Google Scholar]
- 27.Vlasblom R, Muller A, Musters RJP, et al. Contractile arrest reveals calcium-dependent stimulation of SERCA2a mRNA expression in cultured ventricular cardiomyocytes. Cardiovascular Research. 2004;63(3):537–544. doi: 10.1016/j.cardiores.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Lunde IG, Ekmark M, Rana ZA, Buonanno A, Gundersen K. PPARδ expression is influenced by muscle activity and induces slow muscle properties in adult rat muscles after somatic gene transfer. Journal of Physiology. 2007;582(3):1277–1287. doi: 10.1113/jphysiol.2007.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long YC, Glund S, Garcia-Roves PM, Zierath JR. Calcineurin regulates skeletal muscle metabolism via coordinated changes in gene expression. Journal of Biological Chemistry. 2007;282(3):1607–1614. doi: 10.1074/jbc.M609208200. [DOI] [PubMed] [Google Scholar]
- 30.Shkryl VM, Shirokova N. Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. Journal of Biological Chemistry. 2006;281(3):1547–1554. doi: 10.1074/jbc.M505024200. [DOI] [PubMed] [Google Scholar]
- 31.Tate CA, Hyek MF, Taffet GE. The role of calcium in the energetics of contracting skeletal muscle. Sports Medicine. 1991;12(3):208–217. doi: 10.2165/00007256-199112030-00005. [DOI] [PubMed] [Google Scholar]
- 32.Reiken S, Lacampagne A, Zhou H, et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor)-in skeletal muscle: defective regulation in heart failure. Journal of Cell Biology. 2003;160(6):919–928. doi: 10.1083/jcb.200211012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Takeda N, Dhalla NS. Troponin I phosphorylation in heart homogenate from diabetic rat. Biochimica et Biophysica Acta. 1996;1316(2):78–84. doi: 10.1016/0925-4439(96)00007-5. [DOI] [PubMed] [Google Scholar]
- 34.Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. Journal of Molecular and Cellular Cardiology. 2007;42(1):247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in β-adrenergic-induced acceleration of cardiac relaxation. American Journal of Physiology. 2000;278(3):H769–H779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- 36.Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circulation Research. 2002;90(6):649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]