

Summary
The endoplasmic reticulum chaperone GRP94 is essential for early embryonic development and in particular affects differentiation of muscle lineages. To determine why a ubiquitously expressed protein has such a specific effect, we investigated the function of GRP94 in the differentiation of established myogenic cell lines in culture. Using both genetic suppression of expression, via RNA interference, and inhibition of function, via specific chemical inhibitors, we show that GRP94 expression and activity are needed for the in vitro fusion of myoblasts precursors into myotubes and the expression of contractile proteins that mark terminal differentiation. The inhibition can be complemented by addition of insulin-like growth factors to the cultures. GRP94 is not needed for the initial steps of myogenesis, only for the steps downstream of MyoD up-regulation, coinciding with the known need for synergistic input from growth factor signaling. Indeed, GRP94 is needed for the production of insulin-like growth factors I and II (IGF-I and IGF-II) by the differentiating cells. Moreover, the depletion of the chaperone does not increase the rate of apoptosis that always accompanies myogenic differentiation. Thus, the major effect of GRP94 on muscle differentiation is mediated by its regulation of IGF production.
Keywords: GRP94, chaperone, muscle differentiation, IGF, endoplasmic reticulum, secretion
1. Introduction
A number of lines of evidence point to an important role for the ER chaperone GRP94 in muscle physiology. First, grp94 −/− ES cells cannot differentiate into any of the muscle sub-lineages [1] and reduction of GRP94 levels in skeletal myoblasts leads to loss of myocyte fusion competence [2]. Second, GRP94 expression is up-regulated during prenatal development of rabbit skeletal myocytes and displays different expression patterns in skeletal and cardiac muscle [3]. Third, GRP94 functions as a stress protein in muscle: its expression was observed to increase transiently in fibrillating atrial myocytes [4] and over-expression of GRP94 in stressed cardiomyocytes protected them from cell death [5]. The stress-protecting role is specific to GRP94 and is not shared by other endoplasmic reticulum chaperones like BiP or calreticulin [5].
GRP94 may exert its effects on muscle cells because of two mutually non-exclusive activities: due to its essential role in the production of IGF [1, 6], on whose activity muscle fibers are dependent [7–10], or because of its importance for calcium homeostasis [11] in this excitatory cell type. We were particularly intrigued by the possibility that GRP94 is important for muscle growth through its chaperone activity towards IGF. Over-expression of insulin-like growth factor-II in mouse embryonic stem cells promotes their myogenic differentiation [12]. Localized IGF-I transgene expression enhances muscle hypertrophy [13, 14], sustains hypertrophy and regeneration in senescent skeletal muscle [7], accelerates muscle regeneration [15] and counters muscle decline in mdx mice [14]. Finally, muscle produces specific isoforms of IGF-I whose function is yet unclear [16].
This work shows that GRP94 activity is indeed crucial for myogenic cell differentiation and that GRP94 is important at an intermediate phase of myogenesis, because it promotes the production of locally acting IGF.
2. Materials and Methods
2.1. Materials
β estradiol, tunicamycin and thapsigargin were purchased from Sigma Chemicals (St. Louis, MO). Lipofectamine 2000 transfection reagent was from Invitrogen (Carlsbad, CA). Recombinant IGF-II was purchased from GroPep (Adelaide, Australia), recombinant IGF-I was from R&D systems (Minneapolis, MN). 17-allylamino-17-demethoxygeldanamycin (17AAG) was from InvivoGen (San Diego, CA) and the XTT viability kit from Roche Applied Science (Indianapolis, IN). DMEM was from Mediatech, Inc. (Manassess, VA), fetal bovine serum was from Gemini (West Sacramento, CA) and horse serum and media supplements were from Gibco-Invitrogen (Grand Island, NY).
2.2. Antibodies
Mouse anti-myosin heavy chain (MHC) mAb MF-20 and anti-troponin T mAb CT-3 were obtained from the Developmental Studies Hybridoma Bank (at the Univ. of Iowa, Iowa City, IA); mouse anti-sarcomeric actin from Sigma; mouse mAb anti-p21 from BD (Pharmingen (CT); rabbit anti-MyoD (C-20), rabbit anti-14-3-3 (C16) and mAb anti-myogenin (F5D) were purchased from Santa Cruz, Biotechnology, Santa Cruz, CA. mAb anti-Desmin (D33) was from Imgenex (San Diego CA); mAb anti-KDEL was from StressGen (Vancouver, BC anti-HSP90 was from BD Transduction Laboratories (San Jose, CA) and anti-caspase 3 and anti-cleaved caspase 3 (Asp175) were from Cell Signaling; secondary antibodies conjugated to HRP, rhodamine or Cy3 were from Jackson ImmunoResearch Laboratories (West Grove, PA). Biotinylated anti-mouse IGF-II (#BAF792), monoclonal anti-IGF-II (MAB792) and polyclonal anti-mouse IGF-II (AF792) were from R&D Systems (Minneapolis, MN). The anti-GRP94 monoclonal antibody 9G10 (SPA-850) from Stressgen Biotechnologies (Victoria, BC), recognizes an epitope located in the charged linker of GRP94 and is a conformation-sensitive antibody [17].
2.3. Cells and differentiation conditions
C2C12 and 10T1/2 were from the ATCC, and 10T1/2 MD:ER cells were a generous gift from Dr. S. Tapscott (Univ. of Washington, WA). These cell lines were grown in DMEM in the presence of 10%FBS and induced to differentiation either by complete withdrawal of serum or by shifting to medium supplemented with 2% house serum. 10T1/2MD:ER were induced to differentiate as previously reported [18]. 17AAG at concentrations of 10–15μM was used to inhibit GRP94 activity. Recombinant IGF-II (R&D Systems) at 100–1000 ng/ml, or conditioned media of differentiated C2C12 cells, were used to complement differentiation. For proliferation assays using the XTT formazan colorimetric assay (Roche), cells were grown in 3% serum, to limit the background of the assay and to enable minimal growth of the shRNA25-containing cells, which otherwise do not grow.
2.4. Knockdown of GRP94
Seven predicted 21-mer sequences were tested, and three were effective in knocking down GRP94 expression to at least 50% of wild type levels. The shRNA clones were from the Mission shRNA library (Sigma), numbers TRCN 0000071923-27. For plasmid-based RNAi, the cells were transfected using Lipfectamine 2000 according to the manufacturer’s guidelines. After 48 hrs they were selected with 2 μg/ml puromycin and maintained under selection. For viral-based RNAi, the cells were cultured in 6-well plates and transduced with lentiviral particles encoding the same shRNA sequences, packaged using the VeraPower kit (Invitrogen). The MOIs were in the range of 107–108. After 24 hrs the cells were selected with puromycin as above.
2.5. Immunostaining
Cells were fixed in 4% paraformaldehyde and permeabilized in 0.25% Triton X-100 for 15 min. Subsequently, the cells were stained with either anti-myosin heavy chain (MHC) mAb MF-20, or any-MyoD, or anti-Desmin, followed by a secondary antibodies conjugated to rhodamine or Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA). The cells were counterstained to visualize nuclei with 1 ng/ml 4,6-diamidino-2-phenylindole (DAPI) in phosphate-buffered saline, and then mounted. Cells were imaged on a Zeiss Axiovert 200M microscope equipped with a Roper Coolsnap FX-HQ camera. Image analysis was performed with the Slidebook v4.0 software (Intelligent Imaging Innovations, Denver, CO).
2.6. Western blot analysis
Cells were lysed with a 0.5% Nonidet P-40 detergent solution as described in [6]. Equal amounts of extracted proteins (15–100 μg) were loaded per lane, separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated in blocking buffer (20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% Tween 20, and 3% (w/v) nonfat dry milk) and then in 20 mM Tris-HCl (pH 7.4), 150 mM NaCl and 2% (w/v) nonfat dry milk, with the primary and secondary antibodies. Antibodies were diluted as follows: anti-GRP94 (1:1000), anti-desmin (1:500), anti-MyoD (1:500), anti-myogenin (1:1000), anti-p21 (1:500), anti-myosin heavy chain (1:1000), anti-troponin T (1:1000), anti-sarcomeric actin (1:2000), anti-KDEL (1:1000), anti-HSP90 (1:1000), anti-cleaved caspase 3 (1:500), anti-caspase 3 (1:2000) and anti-14-3-3 (1:2000). Proteins were visualized using the enhanced chemiluminescence kit (Pierce, Rockford, IL). Images were recorded using an Alpha Inotech imager.
2.7. Quantitation of IGF-I and -II
Both growth factors were detected by ELISA. For IGF-II, plates were coated with anti-IGF-II (MAb 792, R&D Systems) and incubated with conditioned media of the indicated cells. The bound IGF-II was detected with a biotinylated anti-IGF-II antibody (BAF792, R&D Systems) and developed with streptavidin-HRP (R&D Systems) according to the manufacturer’s recommended procedure. Optical density units were converted to concentrations of the hormone with a standard curve generated with recombinant IGF-II (792-MG)(R&D Systems). Total IGF-I in the media was measured by a commercially available ELISA kit (MG100, R&D Systems, Minneapolis, MN) as previously described (Pfeffer et al, 2009). This kit detects total rodent IGF-I and is not affected by IGF-I binding proteins or IGF-II. It does not detect equine IGF-I from the horse serum, but can detect endogenous IGF-I production by murine cells. The assay can detect IGF-I at 30 – 2000 pg/mL with an intra-assay precision of 4.3%, and an inter-assay precision of 5.9%. Data was acquired in duplicate on a microtiter-plate reader (Dynatech Laboratories, Chantilly, VA) at 450 nm.
3. Results
3.1. Specific silencing of GRP94
To test the mechanism of GRP94 action during muscle development, we used two complementary tools: down-regulation of GRP94 expression with RNAi and pharmacological inhibition of its activity with 17AAG. Out of seven potential sequences predicted by RNAi algorithms, three inhibited GRP94 expression (shRNA24, 25 and 27). As shown in Fig. 1A, the three shRNA vectors provided a graded series of knockdown of GRP94 expression in C2C12 cells, with sh25 being the most effective (usually >90% knockdown), sh24 partially effective (around 60% knockdown), and sh27 intermediate between them. This rank order of activity held true in a number of cells tested, both human and murine, muscle and non-muscle cells. Efficient knockdown via transfection required more than 60 hrs after transfection, consistent with the long half-life of GRP94 (>48 hrs). Because of the poor transfectability of C2C12, we subsequently delivered the shRNAs by lentiviral infection, which proved to be a highly efficient method for knockdown of GRP94 (Fig. 1B). The specificity of the shRNA reagents towards GRP94 is demonstrated by their failure to silence BiP (78kD), another ER luminal chaperone. Both GRP94 and BiP are co-detected in the same blots when using the anti-KDEL monoclonal antibody (Fig. 1A). Specificity of knockdown is also demonstrated by the failure of the anti-GRP94 RNAi to suppress expression of tubulin, 14.3.3 protein or even the closely related, cytoplasmic homologue of GRP94, HSP90 (Fig. 1C). ShRNA depletion of GRP94 in C2C12 or 10T1/2 cells lasted for weeks, enabling us to follow muscle differentiation with or without GRP94. ShRNA depletion of GRP94 caused some BiP induction (Fig. 1B), but follow-up experiments show that this induction does not reflect an unfolded protein response (Eletto et al, paper in preparation, and see also Fig. 6 below).
Figure 1. RNAi silencing of GRP94 expression.

A. C2C12 cells were stably transfected with plasmids encoding shRNA 24, 25 or 27, directed against the GRP94 sequence. Detergent lysates of pools of transfected cells (15 or 50 μg total cell protein for each treatment) were resolved by SDS PAGE and immunoblotted with anti-KDEL monoclonal antibody. A representative experiment is shown. Untreated, non-transfected C2C12 cells. DC, 50 μg of murine dendritic cells proteins, shown for comparison. The positions of GRP94 and BiP are indicated. Quantitation of this and other blots by densitometry is indicated by the numbers below the blot, which enumerate the immuno-reactivity remaining relative to untreated cells.
B. 10T1/2 cells infected with lentiviruses encoding either irrelevant RNAi sequence (shCTRL), or anti-GRP94 shRNAs 23–27. Cell lysates were prepared 7 days after the initial infection. 40 μg total protein were loaded for each lysate. GRP94 and BiP were detected with the same anti-KDEL monoclonal antibody. Numbers below the top panel indicate quantitation of the remaining immuno-reactivity, relative to cells expressing shRNA-CTRL.α-tubulin and 14-3-3 were detected with their respective antibodies, and served as loading controls.
C. C2C12 and 10T1/2MD:ER cells stably expressing either shRNA25, an irrelevant shRNA as control (shCTRL), or untransduced (U) were lysed, and 40 μg of total protein each were resolved by SDS PAGE and immunoblotted. The blots were probed with anti-HSP90 antibody, anti-KDEL or anti-14-3-3 antibodies.
Figure 6. GRP94 depletion affects differentiation but does not increase apoptosis.

A. 10T1/2 MD:ER cells expressing either sh25 or shCTRL were grown in the presence of 15% FCS and allowed to differentiate by provision of β estradiol. The cells were harvested at the indicated times and 40 μg of total protein each were resolved by SDS-PAGE and analysed by western blotting with the indicated antibodies. To control for detection of cleaved caspase 3 the cells were treated with 1μM thapsigargin for 24 hrs, a dose known to induce ER stress and cause apoptosis. Dying cells that detached from the plates and expressed active caspase 3 were easily detected in the thapsigargin-treated cultures, but were not detectable in the other conditions, including the sh25-expressing cells. Note that the induction of BiP in GRP94-depleted cells is sub-maximal in comparison to the induction following thapsigargin treatment.
B. C2C12 cells expressing either shRNA25 or shRNACTRL were allowed to differentiate as described in Fig. 2 and stained with DAPI. Nuclear size was determined by quantifying their circumference using the analysis module of SlideBook, and the distribution of xxx nuclei in each sample was plotted. Bar, the mean nuclear
3.2. Inhibition of GRP94 in myoblasts reduces muscle differentiation
When GRP94 levels were depleted, the ability of C2C12 cells to differentiate was inhibited. Fewer cells fused into myotube-like syncytia (Fig. 2A), fewer cells expressed myosin heavy chain (Fig. 2A) or desmin (Fig. 2C), and the total amount of contractile proteins produced by the cells was dramatically reduced: induction of myosin heavy chain, sarcomeric actin, troponin T and desmin expression was inhibited (Figs. 2B, D). The severity of the differentiation defect correlated with the extent of knockdown in multiple experiments, providing good evidence that C2C12 myogenic differentiation is proportional to the activity of GRP94.
Figure 2. GRP94 silencing results in lack of myogenic differentiation.

A. C2C12 cells transfected either with empty vector (control), or with shRNA25 (sh25) or shRNA27 (sh27) constructs were grown in the presence of serum and induced to differentiate by serum depletion. 48 hrs later, the cells were fixed and stained with the anti-myosin heavy chain antibody MF-20 followed by rhodamine-anti-mouse Ig and also counterstained with DAPI (blue) to visualize nuclei. All cells shown were cultured at the same density. Note the syncitium of nuclei within a fused myotube in the control cells. Magnification bar, 20μm. N=4.
B. C2C12 cells infected with lentiviruses encoding shRNA25 (sh25) or control shRNA (shCTRL) were grown in the presence of serum for 5 days after viral infection. They were then placed in differentiation medium (2% horse serum) (day 0) for the indicated times and equal aliquots of cells were harvested for immunoblots on each of days 0-4. Fifty μg of total protein was loaded on each gel lane. Blots were probed for expression of GRP94. Differentiation was assessed by the induced expression of myosin heavy chain (MHC), sarcomeric actin or troponin T. A representative experiment out of three is shown.
C. C2C12 cells transfected either with empty vector or with shRNA25 were induced to differentiate and 48hs later stained with anti-desmin antibody and rhodamine-anti-rabbit secondary antibody (red, top panels). DAPI staining was used (blue, bottom panels) to highlight nuclei. Magnification bar represents 20μm. N=2.
D. Equal amounts of detergent lysates of the same cultures as in C (15 and 50 μg each sample) were resolved by SDS-PAGE and probed with anti-desmin antibody, to show a second muscle differentiation marker. The membrane stained with ponceau S is shown in the bottom panel, not only to serve as a loading control, but also to show that the patterns of total cell proteins were not affected by the RNAi.
When GRP94 expression was silenced using shRNA24 or shRNA25, not only expression of muscle proteins, but also production of IGF-II by differentiating C2C12 cells was strongly inhibited (Fig. 3A). Similar inhibition of IGF-II secretion was observed in 10T1/2 MD:ER, another cell line capable of muscle differentiation in culture, because it expresses estradiol-activatable MyoD [18] (Fig. 3B). The requirement for GRP94 is not limited to IGF-II, but is shared by the homologous growth factor IGF-I. When GRP94 expression is silenced, IGF-I secretion by 10T1/2 MD:ER cells is also inhibited (Fig. 3C).
Figure 3. GRP94 silencing leads to inhibition of IGF-II and IGF-I secretion.
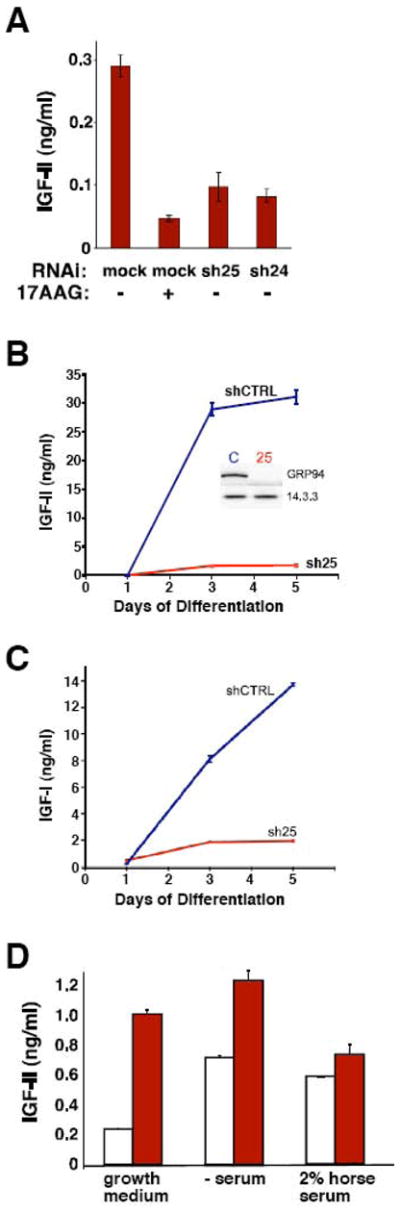
A. C2C12 cells expressing shRNA25 (sh25), shRNA24 (sh24) or empty vector (mock) were switched to differentiation medium for 36 hrs, in the presence or absence of 10μM of 17AAG. The culture media were collected and the concentrations of IGF-II in the media were determined by ELISA, as described in Materials and Methods. All cells were cultured at the same density. The results represent the mean ± S.D. of three independent experiments.
B. 10T1/2 MD:ER expressing sh25 or control shRNA were grown in differentiation medium (2% horse serum in the presence of 100 nM of β-estradiol) and at the indicated time points the levels of IGF-II in the culture media were determined by ELISA as in A. The results represent the mean ± S.D. of four independent experiments. The inset shows the blots of shCTRL (C) and sh25 (25) cells, to illustrate the efficiency of silencing.
C. IGF-I secretion is also inhibited by GRP94 knockdown. Cumulative secretion of IGF-I from 10T 1/2 MD:ER cells into the media was measured by ELISA at 1, 3 and 5 days after differentiation. Data is presented as mean ± SD for 3 measurements. *, P<0.05 for comparisons to Day 1 in the same condition. †, P<0.05 for comparisons between conditions on the same day. 1-way ANOVA followed by Tukey’s multiple comparision test.
D. IGF-II secretion is stimulated by tunicamycin treatment. IGF-II secretion was measured in the medium collected from C2C12 cells after 48hrs treatment with 0.25μg/ml tunicamycin, in either growth medium, serum-free differentiation medium or 2% horse serum differentiation medium. Note that the low dose of tunicamycin was chosen to minimize toxicity and enable the measurement of IGF-II secretion over 48hrs. Blank bars, mock treatment. Red bars, tunicamycin treatment.
The inhibition of both myogenic differentiation and IGF secretion were confirmed by pharmacological, rather than genetic inhibition of GRP94 activity. When treated with 17AAG, C2C12 cells did not fuse, did not express myosin heavy chain (Fig. 4D), and failed to produce significant levels of IGF-II (Fig. 3A). While 17AAG affords the advantage of a rapid assay, it is a less specific agent, because it inhibits cytosolic HSP90 just as efficiently as GRP94. The combination of the drug effects and the RNAi phenotype, however, confirms that GRP94 is essential for myogenesis because of its chaperone activity towards IGF.
Figure 4. Complementation of the GRP94 defect by exogenous IGF.

A. C2C12 cells that were transfected with shRNA25 (sh25) or with vector alone (control) were induced to differentiate for 72 hr in low serum, either in the absence or presence of 1μg/ml of IGF-II (right panel). The cells were stained with anti-myosin heavy chain antibody MF20 (red) and counterstained with DAPI (blue). All cells were cultured at the same density. Bar, 20μm.
B. Frequency of myosin heavy chain (MHC)-positive cells. The number of MHC-positive cells per field in images such as shown in A is plotted for each condition. Data represent a total of 41 fields from three independent experiments, two of which involved complementation with IGF-II and one with IGF-I. Horizontal lines are the mean values.
C. The extent of cell fusion was quantified by counting the number of nuclei in each MHC-positive cell in the samples described in A. Only four MHC-positive cells were found in shRNA25-treated cells, and all of them were mononuclear.
D. C2C12 cells were induced to differentiate for 24 hr. Then, control cultures received DMSO (0.1% final concentration) and experimental cultures received either 15μM 17AAG, or 15μM 17AAG plus 100nM of IGF-II (right panel), and all cultures were allowed to differentiate for additional 48 hrs. The cells were stained as in A. The images represent data from three independent experiments. Bar, 20μm.
E. The number of myosin heavy chain-positive cells per field is plotted for the samples illustrated in panel D. Data represent a total of 40 fields from two independent experiments.
F. Quantitation of cell fusion by counting the number of nuclei in each MHC-positive cell in the samples described in D.
3.3. Complementation with exogenous IGF
Importantly, addition of exogenous IGF to myoblasts with knocked-down or inhibited GRP94 partially rescued differentiation. Both the expression of contractile proteins and cell fusion were restored in the presence of IGF (Fig. 4A–C for RNAi silencing and Fig. 4D–F for 17AAG inhibition of activity). The rescue shows that inhibition of GRP94 activity does not cause the cells to be unresponsive to IGF (and thus receptor expression is likely intact). Rather, the rescue with IGF is consistent with it being the main effector through which GRP94 influences muscle differentiation. Either recombinant IGF-I or recombinant IGF-II was effective in restoring myogenesis, consistent with the common receptor, IGF-R1, and signaling pathway that the two hormones share [19]. C2C12 and 10T1/2MD:ER produce both IGFs, but much more IGF-II than IGF-I (Fig. 3). It should be noted that the rescue of myogenesis by exogenous IGFs is incomplete: the median number of MHC-positive cells observed per field increases from near zero to approximately 4 in the presence of IGF-II, compared to approximately 7 in untreated C2C12 cells (Fig. 4B). Similarly, when another parameter is quantified, the number of cells fused to yield a myotubes – the median number is 4 nuclei per myotube in the presence of IGF, as compared to 8 in control myotubes (Fig. 4C). The rescue maybe incomplete because we did not achieve the optimal concentration of active IGF, or more likely, because other GRP94 clients that are not complemented are involved in myogenesis. Nonetheless, the improvement in both parameters when exogenous IGFs is added to GRP94-depleted cells is impressive. In addition, a quantitative correlation between the level of IGF-II in the medium and the quality of muscle differentiation was consistently observed in multiple silencing experiments (e.g. Figs. 3 and 4). Thus, we conclude that the major reason for inhibition of muscle differentiation by silencing GRP94 is the inability of myoblasts to produce IGF. As we showed previously, GRP94 is an obligatory chaperone for production of signaling-competent IGF-I and -II and that it binds to ER forms of the pro-hormones, before they are processed proteolytically into the mature growth factors [6].
3.4. GRP94 activity is needed for intermediate steps during myogenic differentiation
We next asked whether the requirement for GRP94 extends throughout the myogenic program, or whether it is needed at a particular stage of differentiation. Importantly, silencing GRP94 expression did not prevent the induction of expression of MyoD, the master transcription factor that initiates the myogenic program; this was shown by both immunoblots (Fig. 5A) and immunofluorescence (Fig. 5B). In contrast to MyoD, the induction of expression of another muscle-specific transcription factor, myogenin, was inhibited in GRP94-deficient C2C12 cells (Fig. 5A). The failure to induce myogenin is not an exclusive property of C2C12, because myogenin is also undetectable in muscle differentiation cultures derived from grp94−/− ES cells [1]. Even in 10T1/2MD:ER cells that over-express MyoD, the induction of myogenin expression is attenuated by silencing GRP94 (data not shown). This is consistent with the known synergism that leads to myogenin promoter activation, which reflects the integrated action of inputs from MyoD and from other growth factors [20–22]. In fact, the parental 10T1/2 cell line, which does not express MyoD, cannot differentiate [18] and does secretes only negligible amount of IGF-II (0.94+−0.013 ng/ml after 48hrs, compared to the 30 ng/ml level secreted by 10T1/2MD:ER; and see Fig. 3B). Since myogenin acts downstream of MyoD, its dependence on GRP94 activity suggests that the chaperone is not required for the commitment to myogenesis nor for its initiation, but rather at an intermediate step in the differentiation program.
Figure 5. GRP94 depletion does not affect MyoD, but inhibits myogenin induction.
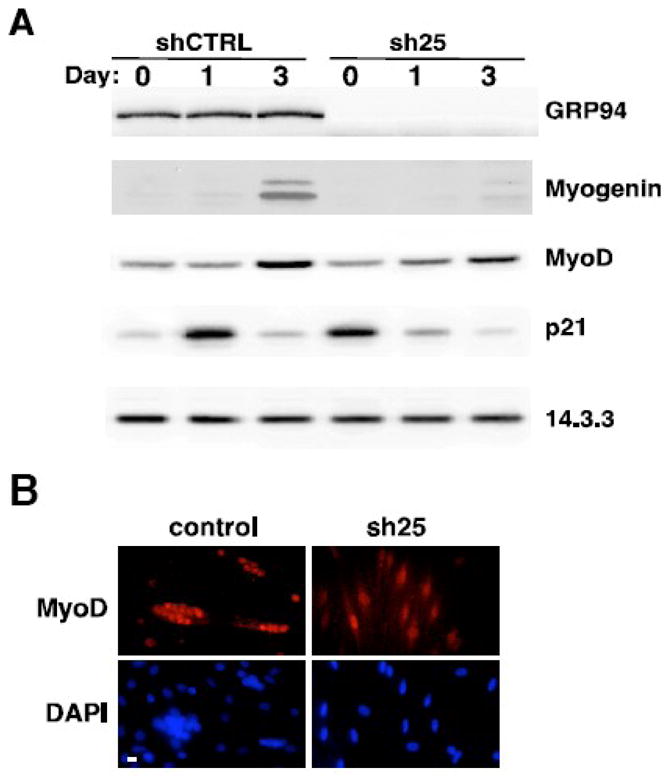
A. Western blot analysis showing a time course of differentiation of C2C12 cells expressing either shRNA25 or control shRNA. Blots were probed for expression of GRP94 and for expression of myogenic transcription factors and cell cycle regulators. 14.3.3 levels were used as a loading control. A representative experiment out of more than four is shown.
B. C2C12 cells stably expressing sh25 or control shRNA plasmids were placed in differentiation medium for 24hs then stained with anti-MyoD (red, top panels) and with DAPI (blue, bottom panels) to highlight nuclei. Note the concentration of MyoD in nuclei under both conditions. Shown are images representative of three independent experiments. Bar, 20μm.
3.5. Depletion of GRP94 does not cause ER stress and does not change apoptosis
Silencing of GRP94 in either C2C12 or 10T1/2MD:ER cells leads to up-regulation of BiP expression (Figs. 1 and 6). This phenomenon is not the same as typical ER stress response. First, the magnitude of BiP induction is sub-maximal, as can be seen when compared with BiP induction using the ER stress inducer thapsigargin (Fig. 6). Second, many of the targets of ER stress, such as calreticulin and protein disulfide isomerase, are not induced when GRP94 is lost (Eletto et al., manuscript in preparation). Furthermore, the effect of ER stress on IGF-II secretion is opposite from the effect of silencing GRP94. As shown in Fig. 3D, IGF-II secretion is enhanced, not inhibited, by treatment with low concentrations of tunicamycin, either in growth or differentiation conditions.
It is known that during myogenic differentiation in culture a fraction of cells dies by apoptotis induced by ER stress [23, 24]. We uncoupled cell differentiation from apoptosis by using the 10T1/2MD:ER cells, which can be induced to differentiate with estradiol even in the presence of serum. When induced with estradiol, these cells differentiate as shown by up-regulation of MHC expression (Fig. 6A). Without GRP94, the differentiation is largely inhibited. Under these conditions, there is no detectable cleavage of caspase 3, the hallmark of apoptosis (Fig. 6A). If there was significant apoptosis, it would have been easily detected, as shown by activation of caspase 3 when the same cells are treated with the ER stress inducer thapsigargin (Fig. 6A) [24]. Furthermore, we quantified nuclear size from DAPI-stained images such as those shown in Fig. 2, where C2C12 cells differentiate after the stress of serum withdrawal. There was no increased fraction of cells with condensed nuclei when sh25-expressing cells were compared to control cells (Fig. 6B). This is consistent with the conclusion that depletion of GRP94 does not direct the cells to the apoptotic pathway.
3.6. GRP94 is important for cell cycle control
Myogenesis involves not only commitment to lineage differentiation and expression of muscle-specific genes, but also programmed withdrawal from the cell cycle, via down-regulation of cell cycle activators and up-regulation of cell cycle inhibitors [25]. Indeed, in addition to inhibiting muscle-specific gene expression, silencing of GRP94 slows down the cell cycle of myogenic cells. C2C12 with graded reduced expression exhibit progressively slower proliferation (Fig. 7A). This is not a property unique to one cell line, because we observed the reduced proliferation with MCF-7 RD, 293T and CHO cells, as well as primary chondrocytes (data with MCF-7 shown in Fig. 7B, representative of the other cells). Silencing of GRP94 reproducibly led to higher expression of the cell cycle inhibitor p21 even in growth medium, before the induction of differentiation (Fig. 5A). Consistent with the levels of p21, cyclin D1 expression was lower in C2C12 whose GRP94 expression was silenced (data not shown). These data correlate well with the observed reduced proliferation rate.
Figure 7. GRP94 depletion inhibits cell proliferation.
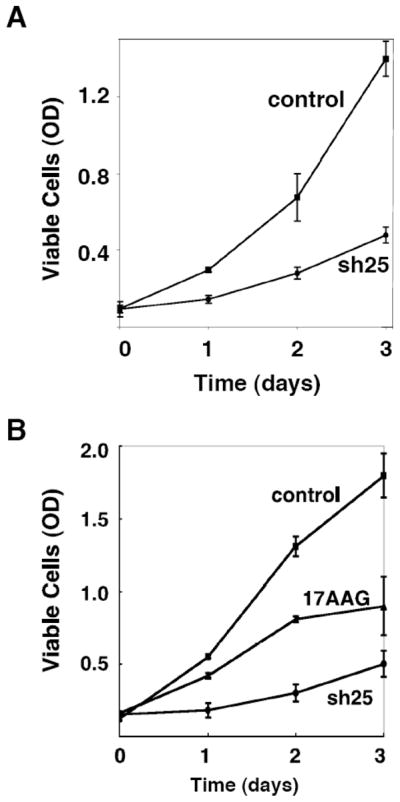
A. C2C12 cells expressing either sh25 or control RNAi were grown in the presence of 3% FCS (see Materials and Methods) for different time points. Proliferation rate of the each cell line were measured by XTT assays according to the manufacturer’s instructions. A representative experiment is shown.
B. MCF-7 cells stably expressing sh25 or treated with 10 μM 17AAG were grown for the indicated time points and their proliferation was measured by XTT assays as above. DMSO treatment (the solvent control for 17AAG) did not differ from the vector control shown. N=3.
4. Discussion
This work demonstrates that myogenic differentiation in culture requires the activity of the ER stress protein GRP94 and provides a molecular explanation for this requirement. We show that GRP94 is needed primarily through its effect on the production of IGFs, growth factors that are very important for muscle development as well as for maintenance of muscle mass [26, 27]. As we recently showed using another experimental system, GRP94 acts as a chaperone towards IGF-II, and without its activity pro-IGF-II is not processed to the mature, active hormone, and is consequently not secreted [6]. It has long been known that differentiation of myoblasts into contractile myotubes involves autocrine secretion of IGF, both IGF-I and IGF-II, and we show here that GRP94 is needed for this essential step in myogenesis. We further show that the requirement for GRP94 is not for initiation of myogenesis, but rather during a downstream step in the differentiation program, after the induction of MyoD and before the increase of myogenin.
The stage(s) of myogenesis that require GRP94 coincides with the timing of the requirement for IGF, as shown before [28, 29]. IGF signaling integrates environmental cues with the intrinsic genetic program controlled by MyoD- and MEF2-family transcription factors, creating an autocrine network where MyoD enhances later differentiation steps by induction of local action of a growth factor [21]. On the other hand – growth factor signaling enhances MyoD expression by affecting transcriptional co-regulators that are essential co-factors for MyoD, thus establishing an amplification cascade for sustaining early events in muscle differentiation [29].
Although a priori the failure of myogenesis in the absence of GRP94 can be attributed to multiple client proteins whose production maybe needed for terminal differentiation, the activity of GRP94 towards IGF plays a dominant role in this process, because supplying the hormone exogenously complements myoblast fusion and terminal differentiation even in cells lacking GRP94. Therefore, even if other proteins also rely on GRP94 activity, their role in differentiation is not essential. For example, GRP94 was shown to be required for proper surface expression of some, but not all integrins [30] and integrins are known to be required for fusion-competence of myoblasts [31]. Nonetheless, GRP94-depleted cells can still fuse when provided only with IGF. Therefore, there is apparently sufficient compensatory activity provided by GRP94-independent integrins. This conclusion is in line with the data on integrin-deficient myoblasts reviewed in [31].
Another line of evidence pointing to the major role of the GRP94-IGF axis is the stage of myogenesis that requires GRP94 activity. In GRP94-depleted myogenic cells there was a normal level of MyoD, but expression of myogenin, desmin, myosin heavy chain, troponin T, and sarcomeric actin was undetected. The timing of the requirement for IGF is also after MyoD induction and before myogenin expression [28, 29]. IGF signaling integrates environmental cues with the intrinsic genetic program controlled by the MyoD- and MEF2-family transcription factors, creating an autocrine network where MyoD enhances later differentiation steps by induction of local action of IGF [21]. On the other hand, IGF signaling enhances MyoD expression by affecting transcriptional co-regulators that are essential cofactors for MyoD, thus establishing an amplification cascade for sustaining early events in muscle differentiation [29]. Readouts of IGF actions are shown to be critical for activation of myogenic specific promoters, protein synthesis, apoptosis, regeneration and hypertrophy in muscle [8, 29, 32, 33]. Since GRP94-depleted muscle cells are not able to produce IGF, MyoD-initiated differentiation in these cells fails to proceed to the terminal stages.
Our data are complementary to the previous observations by Gorza et al. that increased expression of GRP94 serves as a protective mechanism against muscle stress, such as calcium overload in cardiomyocytes [4, 5, 34]. We suggest that increased GRP94 expression is needed to promote the production of local IGF by the stressed myocytes, enabling them to better cope with the stress. Interestingly, Frasson et al. recently reported that early in myocyte differentiation, a fraction of GRP94 is phosphorylated and then exported from the endoplasmic reticulum to the Golgi complex [35]. Phosphorylation of GRP94 had also been reported in cardiac myocytes [36]. The functional relation of this phosphorylated pool of GRP94 molecules to the IGF-dependent promotion of muscle differentiation is currently unknown and experiments are now in progress to pursue this intriguing relationship. Our data also add to the published work that argues that the unfolded protein response has a positive influence on myogenesis. For example, Nakanishi et al. showed that pre-treatment of C2C12 cells with tunicamycin or thapsigargin actually promotes differentiation of the cells that survived the apoptotic fate [37]. We find that low-dose tunicamycin treatment not only induces some unfolded protein response, but also promotes IGF-II secretion, even in growth conditions without induction of the complete differentiation program. Nonetheless, the effects of the unfolded protein response are distinct from the effects of depletion of GRP94, even if the two processes share the induction of BiP expression.
Although we focus on IGF in this work, our data do not exclude other activities or targets of GRP94 that may influence muscle differentiation. For example, GRP94 is known to be a high capacity calcium-binding protein [38] and this activity is necessary for calcium homeostasis [11]. It is quite likely that without the calcium-binding capacity of GRP94, sarcoplasmic reticulum differentiation is compromised. Non-IGF mediated effects are also suggested by the incomplete complementation of the phenotype with IGF. Furthermore, our data show that GRP94 activity is important also for cell cycle control even before differentiation commences, and withdrawal from the cell cycle is known to be required for myogenic differentiation [39, 40]. Even though GRP94-deficient cells have higher level of p21 under growth conditions, they fail to execute the differentiation program. At present we do not have a molecular explanation for the intersection of GRP94 with cell cycle regulators. Additional mechanisms are also worth exploring; GRP94 is known to be important for proper expression of some integrins [30] and this may be important in the fusion-competence of myoblasts.
The evidence reported in this paper indicates that GRP94 is essential for muscle formation primarily because its chaperone activity is required for the production of IGF. We propose that not only IGF-II, but also IGF-IA and other related isoforms are dependent on GRP94. Therefore, a method to tune the activity of GRP94 would affect a number of target proteins simultaneously and would be advantageous over modulation of each client protein separately in order to improve muscle physiology.
Acknowledgments
We thank Amy Ziober for excellent help throughout this work, Dr. Shara Kabak (the University of Pennsylvania) for help with the C2C12 line, Ms. Rennell Dupree for help with microscopy, Julian Ostrovsky for tissue culture help. Dr. S. Tapscott (Fred Hutchinson Cancer Research Center, Seattle) generously provided the MD:ER cell line and Yagun Zou (Children’s Hospital of Philadelphia) – the C2C12 cell line. Supported by grants from the NIH (AI-30178, AG-18001 and NS-059367 to Y.A. and AR056480 to E.R.B) and the Commonwealth of Pennsylvania (to Y.A.). O. O. was supported by postdoctoral fellowships from the Juvenile Diabetes Research Foundation and the Arthritis Foundation. The authors have no conflicting interests.
Abbreviations used
- ES cell
embryonic stem cell
- GRP
glucose regulated protein
- IGF
Insulin-like growth factor
- 17-AAG
17-allylamino-17-demethoxygeldanamycin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wanderling S, Simen BB, Ostrovsky O, Ahmed NT, Vogen S, Gidalevitz T, Argon Y. GRP94 is essential for mesoderm induction and muscle development because it regulates IGF secretion. Mol Biol Cell. 2007;18:3764–3775. doi: 10.1091/mbc.E07-03-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorza L, Vitadello M. Reduced amount of the glucose-regulated protein GRP94 in skeletal myoblasts results in loss of fusion competence. FASEB J. 2000;14:461–475. doi: 10.1096/fasebj.14.3.461. [DOI] [PubMed] [Google Scholar]
- 3.Vitadello M, Colpo P, Gorza L. Rabbit cardiac and skeletal myocytes differ in constitutive and inducible expression of the glucose-regulated protein GRP94. Biochem J. 1998;332:351–359. doi: 10.1042/bj3320351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vitadello M, Ausma J, Borgers M, Gambino A, Casarotto DC, Gorza L. Increased myocardial GRP94 amounts during sustained atrial fibrillation: a protective response? Circulation. 2001;103:2201–2206. doi: 10.1161/01.cir.103.17.2201. [DOI] [PubMed] [Google Scholar]
- 5.Vitadello M, Penzo D, Petronilli V, Michieli G, Gomirato S, Menabo R, Di Lisa F, Gorza L. Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. Faseb J. 2003;17:923–925. doi: 10.1096/fj.02-0644fje. [DOI] [PubMed] [Google Scholar]
- 6.Ostrovsky O, Ahmed NT, Argon Y. GRP94 protects cells grown without serum from apoptotis by regulating the secretion of insulin-like growth factor II. Mol Biol Cell. 2009;20:1855–1864. doi: 10.1091/mbc.E08-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 8.Philippou A, Maridaki M, Halapas A, Koutsilieris M. The role of the insulin-like growth factor 1 (IGF-1) in skeletal muscle physiology. In Vivo. 2007;21:45–54. [PubMed] [Google Scholar]
- 9.Goldspink G. Mechanical signals, IGF-I gene splicing, and muscle adaptation. Physiology (Bethesda) 2005;20:232–238. doi: 10.1152/physiol.00004.2005. [DOI] [PubMed] [Google Scholar]
- 10.Musaro A. Growth factor enhancement of muscle regeneration: a central role of IGF-1. Arch Ital Biol. 2005;143:243–248. [PubMed] [Google Scholar]
- 11.Biswas C, Ostrovsky O, Makarewich CA, Wanderling S, Gidalevitz T, Argon Y. The peptide binding activity of GRP94 is regulated by Calcium. Biochem J. 2007;405:233–241. doi: 10.1042/BJ20061867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prelle K, Wobus AM, Krebs O, Blum WF, Wolf E. Overexpression of insulin-like growth factor-II in mouse embryonic stem cells promotes myogenic differentiation. Biochem Biophys Res Commun. 2000;277:631–638. doi: 10.1006/bbrc.2000.3737. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Barton ER, Sweeney HL, Farrar RP. Viral expression of insulin-like growth factor-I enhances muscle hypertrophy in resistance-trained rats. J Appl Physiol. 2004;96:1097–1104. doi: 10.1152/japplphysiol.00479.2003. [DOI] [PubMed] [Google Scholar]
- 14.Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL. Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol. 2002;157:137–148. doi: 10.1083/jcb.200108071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelosi L, Giacinti C, Nardis C, Borsellino G, Rizzuto E, Nicoletti C, Wannenes F, Battistini L, Rosenthal N, Molinaro M, Musaro A. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. Faseb J. 2007;21:1393–1402. doi: 10.1096/fj.06-7690com. [DOI] [PubMed] [Google Scholar]
- 16.Barton ER. The ABCs of IGF-I isoforms: impact on muscle hypertrophy and implications for repair. Appl Physiol Nutr Metab. 2006;31:791–797. doi: 10.1139/h06-054. [DOI] [PubMed] [Google Scholar]
- 17.Vogen SM, Gidalevitz T, Biswas C, Simen BS, Stein E, Gulmen F, Argon Y. Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J Biol Chem. 2002;277:40742–40750. doi: 10.1074/jbc.M205323200. [DOI] [PubMed] [Google Scholar]
- 18.Hollenberg SM, Cheng PF, Weintraub H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc Natl Acad Sci U S A. 1993;90:8028–8032. doi: 10.1073/pnas.90.17.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 20.Tureckova J, Wilson EM, Cappalonga JL, Rotwein P. Insulin-like growth factor-mediated muscle differentiation: collaboration between phosphatidylinositol 3-kinase-Akt-signaling pathways and myogenin. J Biol Chem. 2001;276:39264–39270. doi: 10.1074/jbc.M104991200. [DOI] [PubMed] [Google Scholar]
- 21.Wilson EM, Hsieh MM, Rotwein P. Autocrine growth factor signaling by insulin-like growth factor-II mediates MyoD-stimulated myocyte maturation. J Biol Chem. 2003;278:41109–41113. doi: 10.1074/jbc.C300299200. [DOI] [PubMed] [Google Scholar]
- 22.Klover P, Hennighausen L. Postnatal body growth is dependent on the transcription factors signal transducers and activators of transcription 5a/b in muscle: a role for autocrine/paracrine insulin-like growth factor I. Endocrinology. 2007;148:1489–1497. doi: 10.1210/en.2006-1431. [DOI] [PubMed] [Google Scholar]
- 23.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 24.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitzmann M, Fernandez A. Crosstalk between cell cycle regulators and the myogenic factor MyoD in skeletal myoblasts. Cell Mol Life Sci. 2001;58:571–579. doi: 10.1007/PL00000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul AC, Rosenthal N. Different modes of hypertrophy in skeletal muscle fibers. J Cell Biol. 2002;156:751–760. doi: 10.1083/jcb.200105147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Musaro A, McCullagh KJ, Naya FJ, Olson EN, Rosenthal N. IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature. 1999;400:581–585. doi: 10.1038/23060. [DOI] [PubMed] [Google Scholar]
- 28.Sarbassov DD, Stefanova R, Grigoriev VG, Peterson CA. Role of insulin-like growth factors and myogenin in the altered program of proliferation and differentiation in the NFB4 mutant muscle cell line. Proc Natl Acad Sci U S A. 1995;92:10874–10878. doi: 10.1073/pnas.92.24.10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson EM, Rotwein P. Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by insulin-like growth factor-II. J Biol Chem. 2006;281:29962–29971. doi: 10.1074/jbc.M605445200. [DOI] [PubMed] [Google Scholar]
- 30.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 31.Mayer U. Integrins: redundant or important players in skeletal muscle? J Biol Chem. 2003;278:14587–14590. doi: 10.1074/jbc.R200022200. [DOI] [PubMed] [Google Scholar]
- 32.Palsgaard J, Brown AE, Jensen M, Borup R, Walker M, De Meyts P. Insulin-like growth factor I (IGF-I) is a more potent regulator of gene expression than insulin in primary human myoblasts and myotubes. Growth Horm IGF Res. 2009;19:168–178. doi: 10.1016/j.ghir.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Noguchi S. The biological function of insulin-like growth factor-I in myogenesis and its therapeutic effect on muscular dystrophy. Acta Myol. 2005;24:115–118. [PubMed] [Google Scholar]
- 34.Tarricone E, Ghirardello A, Zampieri S, Elisa RM, Doria A, Gorza L. Cell stress response in skeletal muscle myofibers. Ann N Y Acad Sci. 2006;1069:472–476. doi: 10.1196/annals.1351.046. [DOI] [PubMed] [Google Scholar]
- 35.Frasson M, Vitadello M, Brunati AM, La Rocca N, Tibaldi E, Pinna LA, Gorza L, Donella-Deana A. Grp94 is Tyr-phosphorylated by Fyn in the lumen of the endoplasmic reticulum and translocates to Golgi in differentiating myoblasts. Biochim Biophys Acta. 2008;1793:239–252. doi: 10.1016/j.bbamcr.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Cala SE, Jones LR. GRP94 resides within cardiac sarcoplasmic reticulum vesicles and is phosphorylated by casein kinase II. J Biol Chem. 1994;269:5926–5931. [PubMed] [Google Scholar]
- 37.Nakanishi K, Dohmae N, Morishima N. Endoplasmic reticulum stress increases myofiber formation in vitro. Faseb J. 2007;21:2994–3003. doi: 10.1096/fj.06-6408com. [DOI] [PubMed] [Google Scholar]
- 38.Macer DPJ, Koch GLE. Identification of a set of calcium-binding protein in reticuloplasm, the luminal content of the endoplasmic reticulum. J Cell Sci. 1988;91:61–70. doi: 10.1242/jcs.91.1.61. [DOI] [PubMed] [Google Scholar]
- 39.Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol Cell Biol. 1999;19:5073–5082. doi: 10.1128/mcb.19.7.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Q, Paterson BM. Regulation of MyoD function in the dividing myoblast. FEBS Lett. 2001;490:171–178. doi: 10.1016/s0014-5793(01)02120-2. [DOI] [PubMed] [Google Scholar]