

Abstract
Eukaryotic genomes exist in the cell nucleus as an elaborate three-dimensional structure which reflects various nuclear processes such as transcription, DNA replication and repair. Next-generation sequencing (NGS) combined with chromosome conformation capture (3C), referred to as 3C-seq in this article, has recently been applied to the yeast and human genomes, revealing genome-wide views of functional associations among genes and their regulatory elements. Here, we compare the latest genomic approaches such as 3C-seq and ChIA-PET, and provide a condensed overview of how eukaryotic genomes are functionally organized in the nucleus.
Keywords: Next-generation sequencing, 3C-seq, Genome organization, Genome structure
1. Introduction
Next-generation sequencing (NGS) has been utilized for re-sequencing of various genomes from bacteria to mammals [1,2]. Sequencing of the human genome has already identified millions of single-nucleotide variations [3]. We anticipate that in the near future personal genome projects will yield sequences for a large number of human individuals, ultimately allowing us to statistically assess predispositions toward respective diseases. In addition, the application of NGS offers alternatives for microarray-based technologies. For instance, RNA-seq provides a gene expression profile of the entire transcriptome, while ChIP-seq can profile the genome-wide distributions of histone modifications and chromatin-associated proteins [4–7].
Chromatin fibers are non-randomly organized in the nucleus and three-dimensional (3D) organization of the genome is involved in the various nuclear processes such as transcription, DNA replication, and repair [8]. NGS has been recently applied to understanding global genome organization in yeast and human nuclei. Four groups have independently reported methods to analyze in vivo genome organization by combining NGS and the molecular biology procedure called chromosome conformation capture (3C), here referred to as 3C-seq [9–12]. The first report from the O’Sullivan laboratory showed that their method, referred to as genome conformation capture (GCC), successfully combined NGS and 3C, and identified genomic associations throughout the budding yeast genome [9]. Several months later, the Lander and Dekker laboratory reported their modified version of GCC, referred to as Hi-C, and mapped long-range genomic associations throughout the human genome [10]. Subsequently, the Noble laboratory at the University of Washington applied their modified method to the budding yeast genome and modeled the three-dimensional genome structure [11]. Finally, our group has developed a relatively simple approach, named enrichment of ligation products (ELP), and applied it to the fission yeast genome [12]. Our study suggests that chromosomal territories and transcription factories, proposed to exist in mammalian cells, are also present in this model organism.
2. 3C-seq methods to capture global genome organizations: a comparison of the treatments
Four different 3C-seq methods were applied to budding yeast, fission yeast or human cells (Fig. 1). The GCC and biotin-based methods were applied to budding yeast cells [9,11]. The ELP method was applied to fission yeast cells [12]. In each of the studies the yeast cells can be considered as wild type, because mutations are only present at the marker genes and the mating-type loci. The Hi-C approach was applied to human cell lines, lymphoblastoid cells (GM06990) and erythroleukemia cells (K562) [10]. In both the yeast and human experiments, the cells are cultured in nutrient-rich liquid media. For fixation of the freshly growing cells, 1% of formaldehyde is added to the cultures, and cells are cross-linked at room temperature for 10 min. Our ELP method instead employs a Zymolyase treatment, which digests yeast cell walls, before the fixation with 4% paraformaldehyde (pFA) at 18 °C for 30 min. We carefully investigated the different fixation conditions ranging from 1 to 4% of pFA concentration, and it is clear that 4% pFA most efficiently captures genomic associations in the fission yeast cells. In the biotin-based budding yeast method, cells are digested by Zymolyase after the fixation. In the Hi-C method, the human cells are lysed by the Dounce homogenizer. At this point, all the samples contain fixed nuclei and cellular debris.
Fig. 1.

Application of NGS for capturing genome-wide associations. The experimental procedures for GCC, Hi-C, biotin-based yeast method, ELP and ChIA-PET are schematically illustrated. All the methodologies start with fixation of in vivo chromatin structures. The 3C-seq method employs 3C, followed by the procedures designed for concentration of small hybrid DNA molecules reflecting genomic associations. In the ChIP-PET method, the fixed samples are immunoprecipitated (ChIP) and the hybrid DNA molecules are enriched by biotin–streptavidin bead purification. The resultant samples are processed by paired-end sequencing.
After removing the uncross-linked proteins from the nuclei by treating the samples with SDS solution, permeable nuclei are subjected to restriction enzyme digestion. The GCC uses 4 bp cutter MspI, and DNA is digested for 2 h. The biotin-based budding yeast method uses HindIII or EcoRI for overnight digestion. The ELP employs HindIII for 2 h digestion. The Hi-C uses HindIII or NcoI for overnight digestion. The shorter digestion time is likely better if the restriction enzymes efficiently cut a major population of genomic DNA in the fixed samples, because endogenous nucleases derived from the samples and exonucleases co-purified with restriction enzymes can potentially cause deleterious DNA digestion. In the Hi-C method, the overhangs derived from the restriction enzyme digestion are filled with biotin-labeled nucleotides. Samples are diluted 16–20 times with T4 DNA ligase buffer, followed by DNA ligation at 16 °C for 1–4 h. More dilution is theoretically better, because it prevents inter-molecular random ligation between non-associating DNA fragments. The cross-links in the ligation products are then reversed by incubating the samples containing proteinase K at 65 beads. We note here that the biotin C overnight, followed by phenol extraction and ethanol precipitation.
At this stage, the samples contain purified hybrid DNA molecules reflecting genomic associations. To obtain short DNA molecules suitable for NGS, DNA samples must be digested into smaller sizes ranging from approximately 100–700 bp. To accomplish this the GCC and Hi-C uses sonication and the ELP and biotin-based budding yeast method employs a restriction enzyme with a 4 bp sequence specificity. After shearing the hybrid DNA molecules into small fragments, a majority of DNA fragments do not correspond to hybrid DNA fragments including the restriction enzyme site. For example, if sample DNA is sheared into around 200 bp in length and HindIII sites appear, on average, every 4 kb, then only 1 of 20 DNA molecules (~5%) contains HindIII sites, indicating genomic associations. Therefore, small hybrid DNA molecules containing restriction enzyme sites must be enriched before being applied to NGS. This enrichment can be achieved by three methods (Fig. 1). In brief, the Hi-C method uses streptavidin beads to purify the biotin-labeled fragments. The ELP method employs the simple trick using self-ligation and further treatment with another restriction enzyme used for 3C (HindIII in our experiment). The biotin-based budding yeast method takes additional steps compared to the ELP. It involves ligation of biotinylated adaptor containing the EcoP15I restriction enzyme site and EcoP15I digestion, followed by purification of biotin-labeled DNA fragments using streptavidin beads. We note here that the biotin–streptavidin purification and the ELP method appear to be comparable in terms of their efficiency in recovering hybrid DNA molecules.
After concentrating the small hybrid DNA molecules, DNA fragments are fused to adaptors and sequenced by NGS with the Paired End (PE) module. The NGS with PE module can determine the sequences present at both ends of the single DNA molecule, and can also determine whether or not they are derived from the same contiguous DNA fragment. Therefore, the PE sequencing allows us to detect hybrid DNA molecules reflecting genomic associations.
3. Processing of sequencing data to detect genomic associations
A single sequencing run of NGS can provide more than 10 million paired reads for hybrid DNA molecules generated by the processes described above. Those paired reads are first mapped to genomic positions, followed by several filtering processes (Fig. 2). To obtain a sufficient number of paired reads to cover the entire genomes, the samples were sequenced 3–10 times. The number of paired reads recovered determines the resolution of the genomic data. It is clear that more sequencing data increases the mapping resolution. For example, the ELP sample was sequenced three times and its derived genomic data achieved a 20 kb resolution for the fission yeast genome. Two independent experiments showed a clear correlation at a 20 kb resolution. The biotin-based budding yeast sample was sequenced 4–10 times for each library, which was sufficient for a 5 kb resolution. The Hi-C sample was sequenced twice, which offered a 1 Mb resolution for the human genome. The resolution is dependent upon the amount of PE sequencing data and the size of the target genome (Fig. 2). Every combination of genomic sections, whose sizes are dependent upon the level of resolution, is scored according to the number of assigned paired reads. The number of paired reads assigned to the combination of genomic sections correlates with the average association frequency in the cell population. Therefore, the numbers of the paired reads assigned to the genomic combinations can be used to estimate genomic associations (Fig. 2).
Fig. 2.

Processing of 3C-seq raw data to detect genome-wide associations. NGS with PE module provides more than 10 million paired sequence information for hybrid DNA molecules, which reflect genomic associations. Those paired-end reads are mapped to genomic positions, and reads are eliminated based on four different filtering processes (labeled a–d). The paired-end reads reflect short-range associations (a), and associations involving repetitive DNA sequences (b) are eliminated. The paired reads assigned to regions where restriction enzyme sites are not present nearby are also eliminated, because it is not clear how these hybrid DNA molecules are produced during the 3C-seq procedures (c). The redundant paired-end reads with the exact same sequences are counted as a single hybrid molecule to avoid PCR bias (d). The remaining paired-end reads are then examined to identify long-range genomic associations. Numbers of paired-end reads assigned to genomic combinations are used for score calculation.
We performed a control experiment, in which purified genomic DNA that was not cross-linked was digested by a restriction enzyme and ligated, allowing ligation to occur between any DNA fragments. This randomly ligated (RL) control sample, which does not reflect in vivo associations between genomic loci, was also processed with the ELP method. We noticed that paired reads from the RL control are not evenly distributed throughout the genome, indicating that there are obvious sequencing biases which also appear to affect the distribution of paired reads from the 3C samples. Therefore, numbers of paired reads from the 3C samples need to be normalized based on the RL data.
4. ChIA-PET
The ChIA-PET method was designed to capture associations among genomic regions bound by a specific protein such as the estrogen receptor α (ER-α) in the published study (Fig. 1; [13]). Human breast adenocarcinoma cells (MCF-7) are treated with estrogen E2 and fixed with 1% formaldehyde for 10 min. The sonicated sample is immunoprecipitated with the antibody recognizing ER-α. The chromatin immunoprecipitation (ChIP) DNA fragments are ligated to the biotinylated adaptor containing the MmeI restriction enzyme site, followed by a second ligation reaction mediating adaptor–adaptor ligation. The ligation product is then digested by MmeI, which cuts 20 bp downstream of the enzyme site, producing two adaptor sequences flanked by two different DNA fragments reflecting genomic associations. The DNA fragments are recovered by using streptavidin beads and are fused to the sequencing adaptors.
Since this procedure employs ChIP, which selectively enriches specific genomic DNA fragments, even random ligation events occasionally occur between highly enriched DNA fragments, although those ligation events do not reflect true genomic associations. Therefore, a random control experiment is critical for the ChIA-PET method. In brief, the second ligation producing adaptor–adaptor DNA molecules is carried out after reverse cross-linking in SDS and proteinase K solution. This step is essential for constructing the random ligation control. Otherwise, the whole procedure is the same, as described above. The significance of genomic associations is validated by comparing the numbers of paired reads from the sample data and the random control.
The ChIA-PET study demonstrated that ER-α-mediated chromatin associations are essentially local, because less than 1% of the data correspond to the association among genomic regions located more than 1 Mb apart. The ChIA-PET study detected gene-gene associations and local chromatin loops probably including enhancer–promoter interactions. The ChIA-PET method can be used to comprehensively map genomic associations mediated by specific proteins of interest in any organisms. Indeed, ChIA-PET successfully disclosed a comprehensive view of CTCF-mediated genomic associations in mouse embryonic stem (ES) cells [14].
5. Genome organizations from three points of view
Large-scale DNA sequencing of a variety of organisms has led to a detailed annotation of genes and regulatory elements dispersed throughout their genomes. These genetic elements are embedded in the DNA fibers packaged in the nucleus, and distant genomic loci are also non-randomly localized in the nucleus. The orchestration of multiple genome organizations contributes to the assembly of functional genome architecture in the cell nucleus. Here, we discuss the in vivo genome organizations in terms of three different aspects: chromosome territories, nuclear domains, and genomic associations, taking into consideration genomics data from 3C-seq and a wealth of microscopic observations.
5.1. Chromosome territories
The largest organizing unit of DNA fibers present within the nucleus is the chromosome, which occupies a specific subnuclear domain, referred to as a chromosome territory (Fig. 3A; [15–17]). For instance, the gene-rich human chromosome 19 tends to be located toward the interior of the nucleus, but the relatively gene-poor chromosome 18 preferentially associates with the nuclear periphery [18]. The correlation between the gene densities and the radial arrangements of chromosomes is observed for all human chromosomes and evolutionarily conserved among higher-eukaryotes [17,19]. While the 3C-seq studies have indicated the existence of chromosome territories in yeast and human nuclei, significant inter-chromosomal associations are also detected [10–12]. The comprehensive mapping of these inter-chromosomal contacts allows us to speculate on the relative positioning of chromosomes within the nucleus. Interestingly, the 3C-seq data have revealed that small chromosomes tend to associate with one another in yeast and human [10,11]. It has been consistently shown by a multicolor 3D FISH approach that small chromosomes are distributed toward the center of the nucleus [20]. The observations from 3C-seq and microscopic studies agree that chromatin fibers from different chromosomes are principally separated, but intermingled at the junctions of chromosome territories (Fig. 3A). What is the biological significance of the inter-chromosomal contacts? It has been shown that transcriptionally active genes are looped out from chromosome territories, which can contribute to inter-chromosomal associations [21]. Moreover, accumulating evidence indicates that transcriptional regulatory elements such as enhancers, locus control regions (LCRs), and the imprinting control region (ICR) regulate genes on different chromosomes through inter-chromosomal associations [22–26]. Therefore, an uncharacterized mechanism arranging adjacent positioning of specific chromosomes is probably required for functional inter-chromosomal associations between genes and their regulatory elements. Alternatively, those functional genomic associations might contribute to the relative positioning of the respective chromosomes.
Fig. 3.

Genome organization from three points of view. (A) Chromosome territories are depicted in different colors. An enlarged view indicates intermingling chromatin fibers derived from two chromosome territories. (B) Nuclear domains: Multiple architectural components and various nuclear bodies are involved in global genome organization. (C) Cohesin mediates enhancer–promoter interaction through its interaction with mediator (left), and also associations among genes and their regulatory elements via CTCF (right).
5.2. Nuclear domains
Chromatin fibers exist in the nucleus, which consists of multiple architectural components and various nuclear bodies. These nuclear domains are known to be involved in genome organization (Fig. 3B; [27]).
5.2.1. Nucleolus
The tandemly repeated ribosomal RNA genes are transcribed by RNA polymerase I (RNA Pol I) in a subnuclear compartment, the nucleolus, where RNA Pol I and rRNA genes are concentrated [28,29]. The 3C-seq method revealed that chromosome XII of budding yeast is divided into two large domains at the position of rDNA repeats, and intra-chromosomal associations can occur within the respective domains [11]. This study indicates that nucleolar localization of rDNA repeats creates an absolute physical barrier, which allows two domains from the same chromosome to act as if those domains are present on different chromosomes.
It has been shown that RNA Pol III-transcribed genes such as tRNA and 5S rRNA genes tend to cluster in budding yeast and fission yeast [30–32]. For instance, tRNA genes, dispersed throughout chromosomal arm regions, cluster in the nucleolus of budding yeast [30]. 5S rRNA genes arranged as contiguous repeats in many eukaryotes often localize to the nucleolar periphery [33,34]. While 5S rRNA genes are dispersed in fission yeast and a few other organisms, they still co-localize at the nucleolar periphery [31]. Likewise, 3C-seq also supports that tRNA genes associate with one another [11]. Interestingly, tRNA genes tend to associate with either centromeres or rDNA repeats present in the nucleolus [11], consistent with previous findings that tRNA genes co-localize with centromeres in fission yeast [31,32].
5.2.2. Transcription factories
Classes of Pol II-transcribed genes are clustered at transcription factories or nuclear speckles [35–38]. The 3C-seq approach revealed that highly expressed genes, but not poorly expressed genes, tend to cluster in the fission yeast nucleus [12]. How transcription factories function in terms of transcriptional regulation remains unclear, but the concentration of genes, transcription machinery and RNA-processing enzymes within the subnuclear domains presumably contribute to efficient transcription and processing of transcripts [38]. We further discuss the role of transcription factories in the following section of transcription factor-mediated gene clustering (Section 6).
5.2.3. Replication factories
DNA replication occurs at nuclear domains referred to as replication factories, which were initially observed in rat fibroblast during S phase more than 20 years ago [8,35,39]. The replication factories consist of replication origins and the entire replication machinery. A 3C-seq study points out that those replication origins tend to cluster in budding yeast [11]. Moreover, Hi-C reveals that early and late-replicating genomic domains associate with the same replication timing domains, respectively, implying that subnuclear positioning of genomic loci predisposes the respective loci to replicate at the same timing [40]. It has been suggested that replication factories are self-organizing structures, based on observations that formation of replication factories is solely dependent upon the replication process [8,41].
5.2.4. Repressive nuclear domains
The nuclear lamina consisting of Lamin A/C and B exhibits a mesh-like structure beneath the inner nuclear membrane [42,43]. It has been shown that transcriptionally repressive chromatin domains possessing features of heterochromatin associate with the nuclear lamina in the fly and human nuclei [44,45]. The Polycomb group (PcG) proteins preferentially localize at discrete subnuclear domains termed PcG bodies and mediate gene silencing from fly to mammals [46–48]. The PcG bodies associate with pericentromeric heterochromatin in the human nucleus [46]. The polycomb response elements (PREs) and PcG target genes associate at the PcG bodies, and those associations function in gene silencing [49,50]. It is also shown that heterochromatin domains, including satellite repeats, often localize at the nucleolar surface of human cells [51,52]. Tethering and clustering of repressive chromatin to these nuclear domains are advantageous for insulation of repressed genes from being exposed to proteins involved in gene activation. To this end, the Hi-C study revealed that active and inactive chromatin domains tend to associate throughout the human genome [10].
5.2.5. Other nuclear domains
There are other nuclear domains including nuclear pore complexes (NPCs), Cajal bodies, promyelocytic leukemia (PML) bodies, nuclear stress bodies, insulator bodies, nuclear matrix (SATB1), DNA repair centers, and so on [27,42,53,54]. Several nuclear domains are linked to genome organization and gene regulation. For instance, genomic regions are known to be tethered to NPCs from yeast to human, and those regions are generally implicated in transcriptional activation [55–58]. SATB1 was originally identified as a protein binding to nuclear matrix attachment regions (MARs) and, interestingly, shows cage-like nuclear distribution in thymocytes [59,60]. The SATB1 network serves as chromatin-looping bases for a large number of genes and is involved in transcriptional regulation [60,61]. The PML bodies co-localize with SATB1 and are also involved in chromatin-looping [62].
Matching analyses of two different genomic datasets, which indicate either comprehensive maps of long-range genomic associations (3C-seq) or distributions of genomic regions associating with distinct nuclear domains (ChIP-seq), will lead to further understanding of global genome organizations mediated by the nuclear domains.
5.3. Genomic associations
Genomic associations can be categorized into various subtypes by considering functions of distinct genomic regions. For example, microscopic and 3C-seq studies reveal that centromeres and telomeres associate at the nuclear periphery [11,12,63–65]. Enhancer and other transcriptional regulatory elements associate with their target genes at the β-globin locus [66,67], at the T-helper type 2 cytokine locus [68], at the immunoglobulin κ locus [69], and at the Igf2/H19 locus [70] (for reviews, see [26,71]). It has recently been shown by the Young laboratory that cohesin, which is known to mediate sister chromatid cohesion, interacts with mediator and physically connects enhancers and their target gene promoters in mouse (Fig. 3C; [72–74]). Cohesin is also recruited to CTCF binding sites through its interaction with CTCF, and mediates associations among genes and their regulatory elements (Fig. 3C; [75–79]). Moreover, cohesin functions in transcriptional insulation through its interaction with CTCF [80]. Therefore, cohesin is independently loaded onto genomic regions by either mediator or CTCF, but once recruited it mediates chromatin looping in both cases, suggesting that cohesin acts as one of the key genome-organizers. To this end, analyses of the Hi-C and ChIA-PET data revealed that CTCF is a major genome organizer for intra- and inter-chromosomal associations [14,81]. Cohesin should have a greater impact on genome organization than CTCF because of its additional binding to genomic regions through mediator.
6. Transcription factor-mediated gene clustering
The 3C-seq approach revealed that co-regulated genes during cell-cycle progression and functionally related genes from particular gene ontology groups tend to co-localize in the in vivo structure of the fission yeast genome [12]. Remarkably, those associating genes contain the same DNA motifs at promoter regions, suggesting that transcription factors likely play a role in gene associations. ChIA-PET also revealed that the estrogen receptor α (ER-α), which functions mainly as a transcription factor, mediates chromatin looping to bring genes together [13]. Moreover, several transcription factors are implicated in chromatin looping [68,69,82–85]. It has been recently suggested that the transcription factor Klf1 is involved in the association of genes with transcription factories in the mouse [86]. Similar to the observations in fission yeast, Hi-C indicates that co-expressed genes and functionally related genes tend to associate in the human nucleus [87]. Therefore, it is likely that binding of transcription factors to DNA motifs at gene promoter regions may trigger gene associations within the RNA Pol II-enriched subnuclear domain allocated to the specific transcription factor, which is suitable for coordinated expression of a group of genes dispersed throughout the genome (Fig. 4). RNA Pol II, transcription factors, and their genomic binding sites could become enriched at subnuclear domains by a self-organization process [88,89]. Notably, these recent studies in fission yeast and mammals suggest evolutionary conservation of functional genome organization through transcription factors.
Fig. 4.
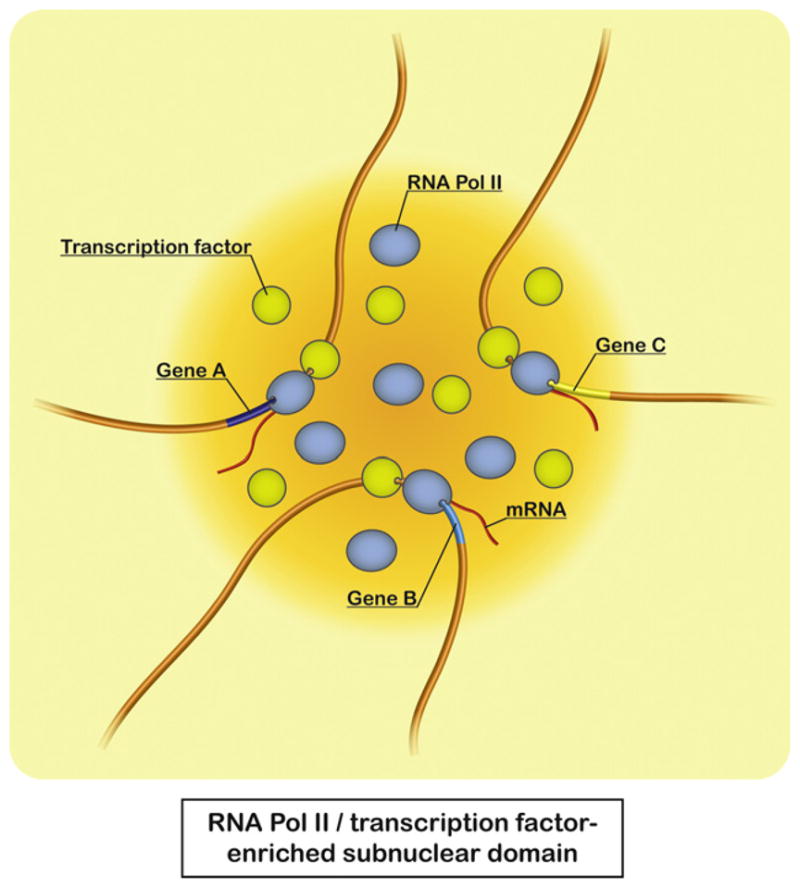
Gene associations mediated by transcription factors. Recent studies from fission yeast and mammals imply that transcription factors mediate colocalization of their target genes at RNA Pol II-enriched subnuclear domains. This gene arrangement potentially facilitates coordinated regulation and efficient expression of a group of genes dispersed throughout the genome.
7. Concluding remarks
The 3C-seq approaches have provided comprehensive maps of genomic associations in yeast and in human cells. Similar methodologies can be applied to any organisms, from bacteria to mammals, and even viruses containing dsDNA. The above-described methods have recaptured the previously identified genome organizations ranging from functional gene associations to chromosome territories. Since the large genomic data offer a comprehensive view of genomic associations, we expect to find a series of novel genome organizations as the data are more intensely studied and explored. Indeed, we found that specific gene arrangements, for example the tandem array of convergent genes, are favorable for genomic association [12]. Once all the genomic associations are defined, it will be critical to characterize the dynamic nature of those associations. Our results in fission yeast suggest that centromeric and telomeric associations are more stable than other associations including gene associations. It is important to study by FISH analysis and preferably by live-cell imaging the dynamic nature of the functionally different genomic associations. This is the essential step for understanding “real” genome organization in the nucleus. The next important challenge after the characterization of all the genomic associations is obviously to decipher the functional significance of various genome organizations for different nuclear processes such as transcription. So far, enhancer–promoter association has clearly been demonstrated to be essential for gene activation by utilizing in vitro and in vivo assays [90,91]. Similar efforts should be undertaken to prove the functional significance of other aspects of genome organization.
It has been shown that epigenetic chromatin regulation through histone modification and DNA methylation is tightly linked to global genome organization. For example, active and repressive chromatin domains tend to associate, respectively [10]. Moreover, histone H4-K16 acetylation is known to disrupt the assembly of condensed chromatin structure [92,93]. Collating genomic association maps to genome-wide views of epigenetic marks in different organisms can further help to characterize the epigenetic regulation of functional genome organization. To this end, several groups have mapped the histone modifications in the human genome, and pointed out that monomethylation of histone H3-K4 is preferentially associated with enhancer elements [94,95]. As described above, enhancer elements are involved in functional genome organization, thus providing an additional link between epigenetic regulation and genome organization. Importantly, disease-related single nucleotide polymorphisms are often positioned within enhancer elements, suggesting that genome organization is severely perturbed in certain human diseases including cancer [96].
Global genome disorganization is often linked to various human diseases [97]. For instance, SATB1, which forms a cage-like matrix associating with a large number of genes, promotes breast tumor and metastasis through alteration of gene expression profiling [60,98]. CTCF, which is known as an insulator protein and believed to be a major contributor of global genome organization, has also been implicated in various human diseases including trinucleotide repeat expansion diseases and cancer [81,97,99]. Moreover, the Misteli laboratory demonstrated that genes can become re-positioned in breast cancer [100]. Arrangements of chromosome territories are different between normal and tumor cell nuclei [101]. Considering that many cancers are diagnosed by morphological changes in nuclear architecture, defective organization of higher-order genome structure is likely to be involved in oncogenesis [102]. How genome disorganization contributes to pathological processes of respective human diseases remains to be characterized. In order to solve this problem, further elucidation of the molecular mechanisms that direct functional genome organization is essential.
Acknowledgments
We apologize to all the colleagues whose work could not be cited due to space constraints. We thank Louise Showe for critically reading the manuscript and Marion Sacks for editorial assistance. Our work was supported by the National Institutes of Health grant CA010815 and funded by the NIH Director’s New Innovator Award Program DP2-OD004348, the G. Harold & Leila Y. Mathers Foundation, the V foundation, the Edward Mallinckrodt Jr. foundation, and the Wistar Pilot Project Funds. The project used the Wistar Genomics and Bioinformatics facilities.
References
- 1.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 2.Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–6. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 4.Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–64. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilhelm BT, Landry JR. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009;48:249–57. doi: 10.1016/j.ymeth.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 7.Marguerat S, Bahler J. RNA-seq: from technology to biology. Cell Mol Life Sci. 2010;67:569–79. doi: 10.1007/s00018-009-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 9.Rodley CD, Bertels F, Jones B, O’Sullivan JM. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genet Biol. 2009;46:879–86. doi: 10.1016/j.fgb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 10.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, et al. A three-dimensional model of the yeast genome. Nature. 2010;465:363–7. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanizawa H, Iwasaki O, Tanaka A, Capizzi JR, Wickramasinghe P, Lee M, et al. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010;38:8164–77. doi: 10.1093/nar/gkq955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–8. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cremer T, Cremer M, Dietzel S, Muller S, Solovei I, Fakan S. Chromosome territories—a functional nuclear landscape. Curr Opin Cell Biol. 2006;18:307–16. doi: 10.1016/j.ceb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet. 2007;8:104–15. doi: 10.1038/nrg2041. [DOI] [PubMed] [Google Scholar]
- 17.Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2:a003889. doi: 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol. 1999;145:1119–31. doi: 10.1083/jcb.145.6.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum Mol Genet. 2001;10:211–9. doi: 10.1093/hmg/10.3.211. [DOI] [PubMed] [Google Scholar]
- 20.Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chambeyron S, Bickmore WA. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004;18:1119–30. doi: 10.1101/gad.292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–45. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 23.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–13. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–7. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 25.Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, et al. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–72. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- 26.Williams A, Spilianakis CG, Flavell RA. Interchromosomal association and gene regulation in trans. Trends Genet. 2010;26:188–97. doi: 10.1016/j.tig.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spector DL. Nuclear domains. J Cell Sci. 2001;114:2891–3. doi: 10.1242/jcs.114.16.2891. [DOI] [PubMed] [Google Scholar]
- 28.Long EO, Dawid IB. Repeated genes in eukaryotes. Annu Rev Biochem. 1980;49:727–64. doi: 10.1146/annurev.bi.49.070180.003455. [DOI] [PubMed] [Google Scholar]
- 29.Shaw PJ, Jordan EG. The nucleolus. Annu Rev Cell Dev Biol. 1995;11:93–121. doi: 10.1146/annurev.cb.11.110195.000521. [DOI] [PubMed] [Google Scholar]
- 30.Thompson M, Haeusler RA, Good PD, Engelke DR. Nucleolar clustering of dispersed tRNA genes. Science. 2003;302:1399–401. doi: 10.1126/science.1089814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki O, Tanaka A, Tanizawa H, Grewal SI, Noma K. Centromeric localization of dispersed Pol III genes in fission yeast. Mol Biol Cell. 2010;21:254–65. doi: 10.1091/mbc.E09-09-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwasaki O, Noma KI. Global genome organization mediated by RNA polymerase III-transcribed genes in fission yeast. Gene. 2011 doi: 10.1016/j.gene.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matera AG, Frey MR, Margelot K, Wolin SL. A perinucleolar compartment contains several RNA polymerase III transcripts as well as the polypyrimidine tract-binding protein, hnRNP I. J Cell Biol. 1995;129:1181–93. doi: 10.1083/jcb.129.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haeusler RA, Engelke DR. Spatial organization of transcription by RNA polymerase III. Nucleic Acids Res. 2006;34:4826–36. doi: 10.1093/nar/gkl656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook PR. The organization of replication and transcription. Science. 1999;284:1790–5. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- 36.Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–12. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 37.Chakalova L, Debrand E, Mitchell JA, Osborne CS, Fraser P. Replication and transcription: shaping the landscape of the genome. Nat Rev Genet. 2005;6:669–77. doi: 10.1038/nrg1673. [DOI] [PubMed] [Google Scholar]
- 38.Sutherland H, Bickmore WA. Transcription factories: gene expression in unions? Nat Rev Genet. 2009;10:457–66. doi: 10.1038/nrg2592. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura H, Morita T, Sato C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–7. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- 40.Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, et al. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–70. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitamura E, Blow JJ, Tanaka TU. Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell. 2006;125:1297–308. doi: 10.1016/j.cell.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao R, Bodnar MS, Spector DL. Nuclear neighborhoods and gene expression. Curr Opin Genet Dev. 2009;19:172–9. doi: 10.1016/j.gde.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–53. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B. Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. 2006;38:1005–14. doi: 10.1038/ng1852. [DOI] [PubMed] [Google Scholar]
- 45.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–51. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 46.Saurin AJ, Shiels C, Williamson J, Satijn DP, Otte AP, Sheer D, et al. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J Cell Biol. 1998;142:887–98. doi: 10.1083/jcb.142.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet. 2011;12:123–35. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 49.Lanzuolo C, Roure V, Dekker J, Bantignies F, Orlando V. Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat Cell Biol. 2007;9:1167–74. doi: 10.1038/ncb1637. [DOI] [PubMed] [Google Scholar]
- 50.Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, et al. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 2011;144:214–26. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 51.Sullivan GJ, Bridger JM, Cuthbert AP, Newbold RF, Bickmore WA, McStay B. Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli. EMBO J. 2001;20:2867–74. doi: 10.1093/emboj/20.11.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, et al. Initial genomics of the human nucleolus. PLoS Genet. 2010;6:e1000889. doi: 10.1371/journal.pgen.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gurudatta BV, Corces VG. Chromatin insulators: lessons from the fly. Brief Funct Genomic Proteomic. 2009;8:276–82. doi: 10.1093/bfgp/elp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dundr M, Misteli T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000711. doi: 10.1101/cshperspect.a000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell. 2004;117:427–39. doi: 10.1016/s0092-8674(04)00448-9. [DOI] [PubMed] [Google Scholar]
- 56.Schmid M, Arib G, Laemmli C, Nishikawa J, Durussel T, Laemmli UK. Nup-PI: the nucleopore–promoter interaction of genes in yeast. Mol Cell. 2006;21:379–91. doi: 10.1016/j.molcel.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol Cell. 2006;21:811–23. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 58.Brown CR, Kennedy CJ, Delmar VA, Forbes DJ, Silver PA. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 2008;22:627–39. doi: 10.1101/gad.1632708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dickinson LA, Joh T, Kohwi Y, Kohwi-Shigematsu T. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell. 1992;70:631–45. doi: 10.1016/0092-8674(92)90432-c. [DOI] [PubMed] [Google Scholar]
- 60.Cai S, Han HJ, Kohwi-Shigematsu T. Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat Genet. 2003;34:42–51. doi: 10.1038/ng1146. [DOI] [PubMed] [Google Scholar]
- 61.Cai S, Lee CC, Kohwi-Shigematsu T. SATB1 packages densely looped, transcriptionally active chromatin for coordinated expression of cytokine genes. Nat Genet. 2006;38:1278–88. doi: 10.1038/ng1913. [DOI] [PubMed] [Google Scholar]
- 62.Kumar PP, Bischof O, Purbey PK, Notani D, Urlaub H, Dejean A, et al. Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol. 2007;9:45–56. doi: 10.1038/ncb1516. [DOI] [PubMed] [Google Scholar]
- 63.Funabiki H, Hagan I, Uzawa S, Yanagida M. Cell cycle-dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J Cell Biol. 1993;121:961–76. doi: 10.1083/jcb.121.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gotta M, Laroche T, Formenton A, Maillet L, Scherthan H, Gasser SM. The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae. J Cell Biol. 1996;134:1349–63. doi: 10.1083/jcb.134.6.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jin Q, Trelles-Sticken E, Scherthan H, Loidl J. Yeast nuclei display prominent centromere clustering that is reduced in nondividing cells and in meiotic prophase. J Cell Biol. 1998;141:21–9. doi: 10.1083/jcb.141.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W. Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell. 2002;10:1453–65. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 67.Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;35:190–4. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 68.Spilianakis CG, Flavell RA. Long-range intrachromosomal interactions in the T helper type 2 cytokine locus. Nat Immunol. 2004;5:1017–27. doi: 10.1038/ni1115. [DOI] [PubMed] [Google Scholar]
- 69.Liu Z, Garrard WT. Long-range interactions between three transcriptional enhancers, active Vkappa gene promoters, and a 3′ boundary sequence spanning 46 kilobases. Mol Cell Biol. 2005;25:3220–31. doi: 10.1128/MCB.25.8.3220-3231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–93. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 71.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–39. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–5. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nasmyth K, Haering CH. The structure and function of SMC and kleisin complexes. Annu Rev Biochem. 2005;74:595–648. doi: 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 74.Losada A, Hirano T. Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev. 2005;19:1269–87. doi: 10.1101/gad.1320505. [DOI] [PubMed] [Google Scholar]
- 75.Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, et al. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–3. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nativio R, Wendt KS, Ito Y, Huddleston JE, Uribe-Lewis S, Woodfine K, et al. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5:e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mishiro T, Ishihara K, Hino S, Tsutsumi S, Aburatani H, Shirahige K, et al. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 2009;28:1234–45. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hou C, Dale R, Dean A. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc Natl Acad Sci U S A. 2010;107:3651–6. doi: 10.1073/pnas.0912087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin-dependent insulation activity. Mol Cell Biol. 2011;31:2174–83. doi: 10.1128/MCB.05093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 81.Botta M, Haider S, Leung IX, Lio P, Mozziconacci J. Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol Syst Biol. 2010;6:426. doi: 10.1038/msb.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Drissen R, Palstra RJ, Gillemans N, Splinter E, Grosveld F, Philipsen S, et al. The active spatial organization of the beta-globin locus requires the transcription factor EKLF. Genes Dev. 2004;18:2485–90. doi: 10.1101/gad.317004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vakoc CR, Letting DL, Gheldof N, Sawado T, Bender MA, Groudine M, et al. Proximity among distant regulatory elements at the beta-globin locus requires GATA-1 and FOG-1. Mol Cell. 2005;17:453–62. doi: 10.1016/j.molcel.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 84.Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, et al. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol Cell. 2008;29:232–42. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sexton T, Bantignies F, Cavalli G. Genomic interactions: chromatin loops and gene meeting points in transcriptional regulation. Semin Cell Dev Biol. 2009;20:849–55. doi: 10.1016/j.semcdb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 86.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dong X, Li C, Chen Y, Ding G, Li Y. Human transcriptional interactome of chromatin contribute to gene co-expression. BMC Genomics. 2010;11:704. doi: 10.1186/1471-2164-11-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Misteli T. The concept of self-organization in cellular architecture. J Cell Biol. 2001;155:181–5. doi: 10.1083/jcb.200108110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cook PR. Predicting three-dimensional genome structure from transcriptional activity. Nat Genet. 2002;32:347–52. doi: 10.1038/ng1102-347. [DOI] [PubMed] [Google Scholar]
- 90.Mueller-Storm HP, Sogo JM, Schaffner W. An enhancer stimulates transcription in trans when attached to the promoter via a protein bridge. Cell. 1989;58:767–77. doi: 10.1016/0092-8674(89)90110-4. [DOI] [PubMed] [Google Scholar]
- 91.Mahmoudi T, Katsani KR, Verrijzer CP. GAGA can mediate enhancer function in trans by linking two separate DNA molecules. EMBO J. 2002;21:1775–81. doi: 10.1093/emboj/21.7.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL, et al. H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 93.Shogren-Knaak M, Peterson CL. Switching on chromatin: mechanistic role of histone H4-K16 acetylation. Cell Cycle. 2006;5:1361–5. doi: 10.4161/cc.5.13.2891. [DOI] [PubMed] [Google Scholar]
- 94.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 96.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Misteli T. Higher-order genome organization in human disease. Cold Spring Harb Perspect Biol. 2010;2:a000794. doi: 10.1101/cshperspect.a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008;452:187–93. doi: 10.1038/nature06781. [DOI] [PubMed] [Google Scholar]
- 99.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Meaburn KJ, Gudla PR, Khan S, Lockett SJ, Misteli T. Disease-specific gene repositioning in breast cancer. J Cell Biol. 2009;187:801–12. doi: 10.1083/jcb.200909127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cremer M, Kupper K, Wagler B, Wizelman L, von Hase J, Weiland Y, et al. Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J Cell Biol. 2003;162:809–20. doi: 10.1083/jcb.200304096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4:677–87. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]