

Abstract
Neutrophil gelatinase associated lipocalin (NGAL), also known as oncogene 24p3, uterocalin, siderocalin or lipocalin 2, is a 24 kDa secreted glycoprotein originally purified from a culture of mouse kidney cells infected with simian virus 40 (SV-40). Subsequent investigations have revealed that it is a member of the lipocalin family of proteins that transport small, hydrophobic ligands. Since then, NGAL expression has been reported in several normal tissues where it serves to provide protection against bacterial infection and modulate oxidative stress. Its expression is also dysregulated in several benign and malignant diseases. Its small size, secreted nature and relative stability have led to it being investigated as a diagnostic and prognostic biomarker in numerous diseases including inflammation and cancer. Functional studies, conducted primarily on lipocalin 2 (Lcn2), the mouse homologue of human NGAL have revealed that Lcn2 has a strong affinity for iron complexed to both bacterial siderophores (iron binding proteins) and certain human proteins like norepinephrine. By sequestering iron-laden siderophores, Lcn2 deprives bacteria of a vital nutrient and thus inhibits their growth (bacteriostatic effect). In malignant cells, its proposed functions range from inhibiting apoptosis (in thyroid cancer cells), invasion and angiogenesis (in pancreatic cancer) to increasing proliferation and metastasis (in breast and colon cancer). Ectopic expression of Lcn2 also promotes BCR-ABL induced chronic myelogenous leukemia in murine models. By transporting iron into and out of the cell, NGAL also regulates iron responsive genes. Further, it stabilizes the proteolytic enzyme matrix metalloprotease-9 (MMP-9) by forming a complex with it, and thereby prevents its autodegradation. The factors regulating NGAL expression are numerous and range from pro-inflammatory cytokines like interleukins, tumor necrosis factor-α and interferons to vitamins like retinoic acid. The purpose of this review article is to examine the expression, structure, regulation and biological role of NGAL and critically assess its potential as a novel diagnostic and prognostic marker in both benign and malignant human diseases.
Keywords: NGAL, lipocalin 2, 24p3, uterocalin, glycoprotein, secretion, neutrophil, bacteriostatic, siderophore, diagnosis, prognosis
1. INTRODUCTION
Glycoproteins play a key role in the body’s defense against multiple diseases. From being structural components of the cell membrane to antigenic determinants on immune cells, glycoproteins serve an important functional role in the body. Circulating glycoproteins are also commonly used as blood-based biomarkers to detect and follow the progression of both benign and malignant diseases. Examples include CA19-9 (carbohydrate antigen 19-9) in pancreatic cancer, CEA (carcinoembryonic antigen) in multiple solid tumors and CA125 (carbohydrate antigen 125) in ovarian cancer. Most of these glycoproteins are large molecules. However, there is a family of small, secreted glycoproteins that are important in the maintenance of health and in combating diseases effectively. This family of proteins is called “lipocalins”. A prototype of this family called Neutrophil gelatinase associated lipocalin or NGAL (also called lipocalin 2 or 24p3) has emerged in recent years as a biomarker in several benign and malignant diseases. Further, studies in cultured cells and in murine models have revealed a pivotal role for this molecule both in health and disease. A search of the PubMed database with the terms “NGAL”, “Lipocalin 2” and “24p3” identified a total of 2,177 articles from 1961 till date, suggesting considerable interest in this molecule. Work from our laboratory and others have shown that NGAL is not just an important molecule from the functional point of view, but also a very promising biomarker to diagnose, follow-up and predict outcome in both benign and malignant diseases. Previous reviews on this molecule have focused on its role as a biomarker alone, specifically, in renal injury [1–10], anemia [11] and cancer [12]. However, an in depth assessment of its biology, role in cell signaling and its role as a biomarker in other benign and malignant diseases though needed, is lacking. In this review, we have sought to address the biology of NGAL, its functional role in physiological conditions and in pathologic states, and explored its potential as a novel biomarker in inflammation and cancer. The article reveals that though small in size, NGAL mediates, through elegant pathways, processes that are crucial for our survival. Further, its small size makes it an attractive target as a molecular imaging tool and for clinical application as a diagnostic and follow-up marker in several diseases.
1.1 The Lipocalin family
Lipocalins are a diverse family of small secreted proteins that act as carriers, transporting predominantly small lipophilic molecules. In recent years, several additional functions have been discovered for these proteins, including regulation of cell division (e.g. α1-microglobulin), differentiation, cell to cell adhesion and survival (e.g. Purpurin). Unlike most other protein families, whose members are identified on the basis of similarities in their amino acid sequence, the members of the lipocalin family share much less sequence identity, in some cases as low as 20%. However, they all share a common secondary and tertiary structural feature- called as the “lipocalin fold”. The lipocalin fold, depicted schematically in Figure 1 comprises of an antiparallel beta barrel structure comprising eight beta sheets that are extensively hydrogen bonded to one another, resulting in a cup-shaped cavity that can bind to specific ligands. The beta sheets are connected to one another by seven short loops (L1-L7), of which the loop L1 forms a lid-like structure to close the ligand binding cavity. The difference in specific amino acids within the lipocalin fold gives rise to the wide diversity in ligands that can be bound by lipocalins. While the overall sequence identity between different lipocalin proteins is low, they share three regions of significant sequence and structural conservation. These regions, termed as structurally conserved regions or SCRs are useful to classify all lipocalins into two broad categories-the kernel and the outlier lipocalins. While the former possess all three SCRs, the latter have only one or two, but never all three SCRs. Examples of the kernel and outlier lipocalins are summarized in Table 1. Thus, the lipocalin family is characterized by structural similarity in the absence of significant sequence identity.
Figure 1. Schematic representation of the lipocalin fold.

The characteristic feature of lipocalins is the “lipocalin fold” which comprises of an N-terminal 3–10 helix followed by eight beta sheets (A–I) arranged in an anti-parallel orientation. The eighth beta sheet is connected to an alpha helix (denoted as α1), which is in turn connected to a C-terminal beta sheet. The beta sheets are connected by loops (L1-L7). Loops L1, L3, L5 and L7 form the open end of the molecule (i.e. the opening to the ligand binding site of NGAL). The portion of the lipocalin fold that are structurally conserved between different lipocalins is indicated by the blue boxed regions while the region that shows significant conservation in amino acid sequence is indicated by the black boxed region.
Table 1.
List of known Kernel and Outlier Lipocalin proteins
| Lipocalin (Abbreviation) | Molecular Weight (kDa) | Properties and Function(s) |
|---|---|---|
| Kernel Lipocalins | ||
| α1-microglobulin (A1M) | 33 | Heme Scavenger, an anti-oxidant and immunoregulator. |
| Apolipoprotein D (ApoD) | 29–32 | Member of the HDL, Apo D has binding affinity for cholesterol, progesterone, pregnenolone, bilirubin and arachidonic acid. It is proposed to be involved in maintenance and repair of central and peripheral nervous systems. |
| α2-microglobulin (A2U) | 18.7 | Major urinary protein of the male rat with extensive sequence homology to many lipid binding proteins. |
| Bilin binding protein (BBP) | 19.6 | A blue pigment protein abundant in the butterfly Pieris brassicae. |
| β1-Lactoglobulin (Blg) | 18 | Homolog of serum retinol-binding protein, Blg is thought to facilitate the absorption of Vitamin A from milk. |
| C8γ (subunit of human C8 complement) | 22 | As a component of the C8 complement, functions in the formation of membrane attack complex. |
| Choroid plexus protein (CPP) | 20 | Proposed to transport substances across the blood brain barrier. |
| Cellular Retinoic acid binding protein (CRABP2) | 18.5 | A retinoic acid (RA) binding protein, induced by RA in RA- responsive cells and enhances transcriptional activity of RA. |
| α-Crustacyanin (ACC) | 350 | Responsible for the blue–black coloration of lobster carapace by causing bathochromic shifts. |
| Major Urinary Protein (MUP) | 17.8 | Abundant protein of urine and other secretions of animal, functions as pheromones, transporters of organic ions, regulators of metabolism and as potent human allergens. |
| Neutrophil gelatinase associated Lipocalin (NGAL) | 24 | Discussed in text. |
| Prostaglandin D synthase (PGDS) | 27 | Involved in isomerization of PGH2 to PGD2. |
| Pregnancy protein 14 (PP14) | 56 | Involved in implantation of the embryo and immunosuppression. |
| Purpurin (PURP) | 20 | Abundant protein of the neural retina, proposed to play prominent role in retinol transport across the inter-photoreceptor cell matrix. |
| Lazarillo | 45 | GPI anchored surface glycoprotein, involved in the axonal growth, the regulation of lifespan, stress resistance and neuro-degeneration. |
| Outlier Lipocalins | ||
| α1-Acid glycoprotein (AAAG) | 40 | Acute phase serum protein secreted by the liver in response to inflammation, stress, and various malignancies and affects pharmacokinetics of drugs. |
| Aphrodisin | 17 | A component of hamster vaginal secretions, triggers mating behavior of naive males. |
| Odorant binding protein (OBP) | 37–40 | Bind to specific odorants including pheromones. |
| Probasin | 20 | Androgen regulated prostate specific protein, functions as pheromone carrier. |
| von Ebner’s-gland protein (VEGP) | 18 | Salivary protein secreted by the Von Ebner’s glands located around the circumvallate and foliate papilla of the human tongue, it has a large variety of ligands and secreted in response to stress, infection, and inflammation. It is suggested to inhibit cysteine proteases that are important for embryo hatching and implantation. |
HDL, high density lipoproteins; PGH2, Prostaglandin H2; GPI, glycosylphosphatidyl inositol.
Several elegant reviews have described the structure and function of the lipocalin family and of specific lipocalins [13,14]. However, in this review, we will focus on a member of the kernel lipocalins, called neutrophil gelatinase associated lipocalin, which has emerged as a significant mediator of several physiological processes and pathological states including benign and malignant conditions. We will review its structure, biology regulation and clinical significance in depth and discuss its role as a modulator of both health and disease.
1.2 NGAL-Isolation and genomic organization
Human neutrophil gelatinase associated lipocalin (NGAL), also known as neutrophil glucosaminidase-associated lipocalin, 24p3, oncogene 24p3, p25, migration stimulating factor inhibitor (MSFI), human neutrophil lipocalin (HNL), α1-microglobulin related protein, siderocalin, or uterocalin, is a 198 amino acid long secreted glycoprotein encoded by a gene located at the chromosome locus 9q34.11. The NGAL gene has seven exons that produce at least five functional transcripts (i.e. mRNAs that are translated into protein), the most common of which encodes for a 198 amino acid secreted protein (Figure 2). The mouse homologue of NGAL is called lipocalin 2 (Lcn2). It is denoted by lower case (Lcn2 or Ngal) to distinguish it from its human counterpart (LCN2 or NGAL). Lcn2 is also called SV-40 induced 24P3 protein, secreted inducible protein 24, superinducible protein 24 (SIP24) and is encoded by a gene on chromosome locus 2 27.0 cM. [15–17]. The Lcn2 gene has six exons and codes for two functional transcripts (Figure 2).
Figure 2. Transcripts encoded by the human and mouse NGAL genes.

The boxes represent exons while the connecting lines represent introns. Filled in boxes represent conding sequences, while empty (unfilled) boxes represent the untranslated region (UTR). The number above the transcript is the length of the mature transcript (indicated as number of base pairs). The number of amino acids corresponds to the number of residues that are translated. The length of each transcript is proportional to the length of the genomic DNA (Source: http://www.ebi.ac.uk)
Lcn2 was first purified from a culture of murine kidney cells infected with the simian virus (SV-40) or the polyoma virus [18]. Triebel and colleagues isolated it as a 25 kDa protein that was associated with the monomeric form of matrix metalloproteinase-9 (MMP-9), a gelatinase secreted by neutrophils that degrades several basement membrane and extracellular matrix components (including cartilage proteoglycan, type I gelatin and collagens type I, IV, V and XI) [19]. They called it α2-microglobulin related protein (α2-MRP) as the protein had a sequence homology to the rat α2-microglobulin protein. The association between α2-MRP and MMP-9 appeared to occur through a disulfide bond that could be broken under reducing conditions. Further, this association did not appear to have a significant effect on the enzymatic activity (of MMP-9) against a synthetic substrate, thus suggesting that α2-MRP (or Lcn2) had a role in modulating the stability rather than the enzymatic activity of MMP-9. The molecular weight of the novel protein was observed to decrease from 22 to 25 kDa after treatment with endoglycosidase F, an enzyme that removes N-liked oligosaccharide side chains, suggesting that α2-MRP was a heavily N-glycosylated protein.
Axelsson and colleagues in 1995 subsequently identified NGAL as a protein present in preparations of another neutrophil protein, NP-4 (neutrophil proteinase 4) [20]. They noted that some NP-4 preparations when used to immunize rabbits produced antibodies that recognized two proteins- NP4 and a second unknown protein. After eliminating NP-4 using a specific monoclonal antibody, they identified the second protein as NGAL using antiserum raised a couple of years earlier against NGAL by Borregaard and co-workers [21]. They also developed the earliest enzyme linked immunosorbent assay (ELISA) for detection of NGAL, using a rabbit polyclonal antibody raised against the partially purified protein. Using this assay, they demonstrated that NGAL was present, albeit at a low level, in the plasma of healthy humans, the mean level being 72 ng/ml (range 40–109 ng/ml). They also observed two forms of NGAL on immunoblotting- a 25 kDa monomer and a 50 kDa dimer. Upon intravenous injection of radioactively labeled (I131) NGAL into rats, they observed two distinct phases of its clearance from the body- an initial phase (within 1 hour post-injection) where the monomeric form was more rapidly cleared than the dimeric form (half-life: 10 and 20 minutes respectively), and a second phase where the two forms were cleared off at a similar rate. Further, the labeled monomeric and dimeric NGAL accumulated mostly in the kidney suggesting that renal clearance is by far the predominant mechanism for excretion of NGAL from the body [20].
1.3 Domain structure of NGAL
A comparison of the amino acid sequence of NGAL homologues expressed in different species reveals that the human and chimpanzee proteins share the greatest similarity, being nearly 98% identical at the sequence level (Table 2). Human NGAL however has little similarity to either the mouse (62%) or the rat Lcn2 proteins (63%). This fact is important as most of the studies (discussed later) into the functions of NGAL in vivo have been carried out in mouse models. Despite limited sequence identity, there is significant conservation of short stretches of amino acid residues between the different NGAL homologues. These conserved regions, mostly comprising of short stretches of hydrophobic amino acids have been suggested to be responsible for the conservation of ligands (e.g. bacterial siderophores) among lipocalins.
Table 2.
Comparison of NGAL homologues found in different species
| Species | Gene Symbol | Other aliases | Accession number of Protein | Chromosomal location | Length of protein (amino acids) | Percent similarity to human LCN2 |
|---|---|---|---|---|---|---|
| Human | LCN2 | 24p3, MSF1, NGAL | NP_005555.2 | 9q34 | 198 a.a. | - |
| Mouse | Lcn2 | RP23-161B9, 11- 003, 24p3, AW212229, Sip24, OTTMUSP00000013 951, Ngal, SV40- I24p3P | NP_032517.1 | 2A3, 2 27.0 cM (Chr. 2 - NC_000068.6) | 200 a.a | 62% |
| Rat | Lcn2 | Ngal, p25, lipocalin- 2, oncogene 24p3, α-2U-GRP, α-2-MRP | NP_570097.1 | 3p11 | 198 a.a | 80% |
| Chimpanzee | LCN2 | NGAL, Oncogene 24p3 |
XP_001153985.1 (isoform 1) XP_001154043.1 (isoform 2) XP_520287.1 (isoform 3) |
Chr. 9 - NC_006476.2 | 198 a.a. (isoforms 1-3) | 98% |
| Dog | LCN2 | Predicted similar to NGAL, p25, Oncogene 24p3, lipocalin 2 |
XP_548441.1 (isoform 1) XP_862322.1 (isoform 2) |
Chr. 9 - NC_006591.2 | 198 a.a. (isoform 1) 207 a.a. (isoform 2) |
66% |
| Cow | LCN2 | NGAL | XP_605012.3 | Chr.11 - NC_007309.4 | 200 a.a | 68% |
| Horse | LOC1000 70310 | similar to lipocalin 2 (oncogene 24p3) | XP_001501198.2 | Chr. 25 - NC_009168.2 | 296 a.a | 66% |
| Rabbit | LOC1003 52980 | lipocalin 2-like | XP_002723019.1 | NW_00315947 3.1 | 198 a.a | 67% |
| Wild hog | LCN2 | none | XP_001927681.2 | Chr. 1 - NC_010443.2 | 243 a.a | 70% |
A bioinformatics analysis of the protein sequence of human NGAL reveals two main features- a 20 amino acid N-terminal signal peptide and a lipocalin domain (amino acids 48–193) which makes up most of the length of the molecule. The lipocalin domain (also called the lipocalin fold) is the characteristic feature of the lipocalin family and contains the ligand binding region that binds to and transports small lipophilic ligands (including retinoids, steroids and iron). The equine and porcine homologues differ from human NGAL in not possessing a signal peptide. Additionally, they possess a second lipocalin domain (termed lipocalin-2) and in case of the porcine homologue, an N-terminal transmembrane domain. Whether these additional domains translate into differences in function of these homologues is still unclear.
1.4 Three dimensional structure of NGAL
The earliest studies aimed at elucidating the three-dimensional structure of NGAL were carried out by Chu and co-workers (1998) who used circular dichroism (CD) to investigate the structure of the mouse Lcn2 protein [22]. They observed that the two hydrophobic tryptophan residues in Lcn2 (at positions 31 and 81) are in a restricted conformation. Binding assays using triturated (H3) retinol revealed that Lcn2 binds to the hydrophobic form of vitamin A (retinol). Scatchard plot analysis subsequently revealed that retinol, a hydrophobic compound, bound more strongly with Lcn2 than its hydrophilic counterpart retinoic acid (association constants for retinol and retinoic acid being 4.9 X105 M−1 and 1.17X105 M−1 respectively). This finding suggested that Lcn2 may transport hydrophobic ligands like retinol. Further, the maximum binding capacity of Lcn2 for retinol was nearly 3-fold higher than that for retinoic acid (5.87 nmole for retinol vs. 1.91 nmole for retinoic acid per mg of Lcn2 respectively), suggesting that the binding pocket of Lcn2 has a much stronger affinity for hydrophobic than for hydrophilic ligands. Analysis of the binding affinity of Lcn2 for other ligands revealed that while it did not bind significantly to cholesterol, it had a strong affinity for cholesterol oleate, an intensely hydrophobic cholesteryl ester. The suggested mechanism for NGAL binding as a strong hydrophobic interactions between the aliphatic side chains of the cholesteryl ester with the hydrophobic residues in the binding pocket of Lcn2. Significantly, oleic acid, a molecule with both hydrophobic and hydrophilic ends, was as effective in binding to Lcn2 as cholesterol oleate, while other hydrophobic small molecules like α-aminoacaproic acid and undecanoic acid did not [22]. These observations suggest that the binding pocket of Lcn2 has a preference for small hydrophobic ligands. Further, it is not just the functional groups attached to the ligand but also their three dimensional conformations that influence its affinity for Lcn2.
In 1999, the three-dimensional structure of human NGAL in solution was elucidated by Coles and co-workers [23]. NMR (nuclear magnetic resonance) analysis revealed that NGAL contains an N-terminal 310-helix, followed by eight antiparallel beta strands, an alpha helix and a C-terminal beta strand (depicted schematically in Figure 1). The beta strands form a barrel like structure whose walls are formed by two beta sheets- the first by the strands β2-β4 and the second by the strands β6-β8. Three beta bulges are also observed- one in the 1st, and two in the 6th β strands. These bulges have been suggested to contribute to the ligand binding site of NGAL, which itself is located at the base of the barrel and comprised predominantly of hydrophobic residues (Trp 31, Trp 33, Val 66, Phe 83, Phe 92, Phe 94, Val 108, Val 110, Val 121 and Phe 123). On the other hand, the region closer to the opening of the barrel is comprised of polar residues (Tyr 52, Thr 54, Tyr56, Tyr 106, Thr 136, Tyr138). Near the mouth of the barrel, side chains of three highly polar residues (Lys 125, Lys134 and Arg81) project into the cup-like ligand binding cavity of NGAL. A negatively charged patch (formed by three amino acids Asp 34, Glu 60 and Asp 61) is present in a “pit” like region at the floor of the barrel close to an unpaired cysteine residue (Cys 87). This cysteine residue forms an intermolecular disulfide bond with the gelatinase MMP-9. While the negatively charged patch at the floor of the cup has been suggested to be the actual site of interaction between NGAL and MMP-9, it has also been suggested that the open end of the molecule, with its greater conformational flexibility is likely to bind to a cell surface receptor that shuttles the protein (either free or bound to its ligand) in and out of cells (receptors for NGAL have been discussed in Section 3.2.4).
More recently, the NMR structure of the ligand binding cavity of NGAL was elucidated. It emerged that the cavity in NGAL is distinct from that in other lipocalins in being significantly polar [24]. Further, it is large enough to accommodate macromolecular ligands like proteins. This suggests a possible mechanism to explain how NGAL interacts with bacterial (and possibly mammalian) proteins which have a significant number of polar residues. NGAL specifically interacts with bacterial proteins termed siderophores (the term “siderophore” is a Greek word meaning an “iron carrier protein”) that bind to circulating and intracellular free iron. These are relatively low molecular weight proteins produced by microorganisms (including bacteria and fungi) that bind specifically to the ferric (Fe3+) form of iron. Siderophores are essential for the survival of many microorganisms in the human body as they are exposed to conditions of severe iron deficiency in vivo, primarily due to the extremely low circulating levels of free iron [25]. Owing to their very high affinity for iron, siderophores can abstract free iron from the surrounding milieu and make it available to the microorganism [26]. There are chiefly two classes of siderophores- the phenolate/catecholate type (produced by gram negative Enterobacteria), which are significantly polar, and carboxymycobactin (CMB) type (produced by mycobacteria like Mycobacterium tuberculosis), which are more hydrophobic. It is interesting to note that NGAL only binds to iron complexed with siderophores but not to free iron [27]. Co-crystallization of NGAL with enterochelin (a phenolate type siderophore) has revealed that despite tight binding, the siderophore fits poorly into the ligand binding cavity of NGAL [16]. On the other hand, the complex of NGAL with iron bound CMBs filled the cavity (of NGAL) more completely. The difference between the occupancy of the ligand binding cavity by the two siderophores is attributed to the formation of a larger number of van der Waal interactions and more extensive hydrogen bonding with the residues lining the ligand binding pocket of NGAL by the Fe-CMB complex than by the Fe-enterochelin complex. The importance of hydrophobic interactions in the association of NGAL with CMB was further strengthened by the observation that deletion of even one methylene group (from an eight methylene group-long aliphatic linker that helps CMB bind to the binding pocket of NGAL) significantly decreased the binding between the two proteins [16]. Based on these results, it has been suggested that polar residues that make up the cup-like ligand binding pocket (of NGAL) are responsible for its interaction with the phenolate/catecholate type siderophores while a different set of residues mediate its binding to the more hydrophobic CMBs [16]. It is believed that through this dual mechanism, NGAL is able to bind to a wide variety of siderophores (the property is termed as the “ligand plasticity” of NGAL), and thus mediate its physiologic role as a broad specificity siderophore binding protein of the innate immune system.
2. EXPRESSION PROFILE OF NGAL
2.1 EXPRESSION OF NGAL IN NORMAL TISSUES
2.1.1 Adult Human Tissues
NGAL is normally synthesized as a component of the late granules of neutrophils [17]. Cabec and coworkers first demonstrated that NGAL was located in the azurophilic [or myloperoxidase peroxidase (MPO) positive] neutrophil granules where it co-localized with MPO [28]. After this, various groups analyzed the expression of NGAL by in situ hybridization, northern blot analyses as well as immunohistochemistry as detailed Table 3. Apart from tissue expression, NGAL is also been detected in supernatants from cultured neutrophils and in culture media from human oral and gingival keratinocytes but not in supernatants from healthy gingival [29]. A significant observation was that the amount of NGAL secreted into the culture medium (by unstimulated A549, NHBE and NHEK cells) was more than 200-fold higher than that present within the cells [30]. This suggested that a mechanism might exist in these cells wherein the NGAL synthesized is constitutively secreted out. Cabec and co-workers sought to solve this puzzle by investigating the fate of exogenously transfected and constitutively transcribed human NGAL in HL-60 promyelocyte cells [28]. HL-60 cells are arrested at the promyelocyte stage of neutrophil maturation. At this stage of maturation, only azurophilic granules (containing MPO) but not specific granules (containing gelatinases) have been synthesized. Following transfection of HL-60 cells with full length NGAL cDNA under the control of a cytomegalovirus promoter (CMV), it was observed that NGAL co-localized with MPO [31]. When granulocytic differentiation was induced in these cells [by treatment with DMSO (dimethyl sulfoxide) and retinoic acid] there was a significant and progressive time-dependent downregulation of NGAL protein in the transfected cells, until it eventually disappeared completely. This suggests that NGAL is synthesized during the early stage of neutrophil maturation but its synthesis stops with induction of neutrophil maturation. It was further uncovered that this disappearance of NGAL was not due to the breakdown of the protein or exocytosis of azurophilic granules (containing NGAL) but rather due to secretion of the ectopically expressed NGAL from transport vesicles into the culture medium. This was supported by observations that while NGAL levels decreased in the transfected HL-60 cells (upon induction of maturation), there was no change in the expression of MPO, a companion of NGAL in the azurophilic granules. As differentiated granulocytes do not possess the ability to synthesize specific granule proteins (like NGAL) de novo [32], the results of this study suggested that the ectopically expressed and constitutively transcribed NGAL protein fails to get retained in the granules and is thus secreted. This suggests that differentiated neutrophils have a defect both in the synthesis and storage of NGAL. From the standpoint of diagnosis (and prognosis), it would be of great interest to investigate whether a similar defect exists in other cell types and tissues and the proportion of NGAL synthesized in different cell types that is secreted into the bloodstream.
Table 3.
Expression of NGAL in Adult, Fetal Tissues and in Stem cells
| ADULT HUMAN TISSUE | ||
|---|---|---|
| Strategy | NGAL expression status | Reference |
| In situ hybridization: | Ductal epithelium of the breast, bone marrow, circulating macrophages, kidney, liver, trachea, lungs (both bronchial goblet cells and alveolar type-II pneumocytes), small intestine, salivary glands, thymus, prostate and adipocytes expresses NGAL. | [30,267–269] |
| Northern blot analysis: |
|
[79,88,268, 269] |
| Immunohistochemistry: |
|
[31,34,170] |
| FETAL HUMAN TISSUE | ||
|
[34] | |
| EXPRESSION OF NGAL IN STEM CELLS | ||
| Normal Stem Cells | Stimulation of rat bone marrow stem cells (BMSCs) under in vitro conditions for osteogenic differentiation resulted in >2.5 fold upregulation in differentiated BMSCs (vs. the undifferentiated). | [111] |
| Cancer Stem Cells | Side population (SP) cells isolated from the human squamous cell carcinoma cell line A431 revealed a significant downregulation (2- fold) of NGAL compared to the non-side population (NSP) cells by microarray analysis. | [113] |
Compared to adults, much less is known about expression of NGAL in children, particularly infants. Urine NGAL levels were observed to decrease with increasing gestational age in premature infants (nearly 4-fold decrease from ≤26 to 36 weeks gestational age). When corrected for urine creatinine excretion, urine NGAL showed a nearly 6-fold decrease with increasing gestation age. Further, urine NGAL levels in newborns showed a significant positive correlation with female gender but not with race [33].
2.1.2. Fetal Human Tissues
An analysis of NGAL expression in various human fetal tissues revealed that different tissues express NGAL at different weeks of gestation (Table 3). A focal staining appeared in the epidermis in the 20th week of gestation and this spread to the stratum granulosum and stratum corneum around 24 weeks. With further advancement of gestation however, immunoreactivity for NGAL in the fetal skin became progressively more concentrated towards the hair follicles [34]. Barring these few studies, not much is known about the time course and pattern of NGAL expression during in utero development in humans.
2.1.3. Mouse Tissues
The tissue expression of mouse Lcn2 has also been well studied. In the fetus, Lcn2 is expressed in the hypertrophic and perihypertrophic zones of the developing cartilage, with the expression shifting to the proliferating zone chondrocytes 10-days after parturition. With advancing age, Lcn2 expression becomes more intense in the proliferating and hypertrophic zones and in the articular chondrocytes of the articular cartilage [35]. Lcn2 is also expressed by the luminal epithelium and glands of the mouse uterus during the estrous and proesterous phases of the estrous cycle [36] in the uterine luminal fluid and by the uterine surface epithelium immediately following fertilization (days 1 and 2). However, it is not detectable in the stroma or the uterine smooth muscle [36]. Lcn2 is also strongly expressed in the bone marrow, with much weaker expression in the spleen, lung and granulocytes and no expression in the liver, heart, kidney, small bowel or thymus [37]. Rojas and co-workers, in one of the earliest studies on Lcn2, reported that the mRNA was expressed in several adult (3 weeks old) mouse tissues including the liver, spleen, testis and lungs and in the kidney of young (10-days old) mice, while no expression was detectable in the adult murine kidney, brain, thymus or muscle, or in the embryonic liver. Further analysis revealed that Lcn2 mRNA expression in adult mice progressively declines with advancing age, particularly in the liver, kidney and the spleen with complete disappearance by the time the mice are about 75-days (i.e. 2.5 months) old [38].
These studies taken together suggest that NGAL is expressed in adult healthy tissues derived from all the three germ layers- ectoderm (e.g. hair follicles of adult skin), mesoderm (kidney, blood cells) and endoderm (e.g. epithelial lining of the bronchi, lungs, gut and the thymus). While limited, available data also suggests that NGAL expression begins in utero, and is either maintained or lost with development. What triggers the induction of NGAL expression and what factor(s) modulate its appearance and disappearance in various tissues however, is still an unsolved mystery.
2.2 EXPRESSION OF NGAL IN BENIGN DISEASES
NGAL expression is significantly upregulated both in the tissues and in the body fluids in several benign conditions including inflammatory, ischemic, and metabolic disorders.
2.2.1 Inflammatory diseases
NGAL expression is upregulated in several acute and chronic inflammatory diseases (Table 4). NGAL was expressed at a higher level in the skin of patients with psoriasis compared to patients with atopic dermatitis or eczema. A significant negative co-relation was observed between the expressions of NGAL and the degree of differentiation of keratinocytes [34]. Staining of skin tissues underneath areas of parakeratosis (i.e abnormal differentiation) revealed a strong positivity for NGAL while that for filaggrin, a marker of terminal epidermal differentiation was absent, suggesting that NGAL is expressed at a higher level by undifferentiated epidermal cells [40]. Notably, topical treatment of psoriatic patients with calcipotriol (a derivative of vitamin D) for upto 14 days produced no significant change in NGAL expression in these lesions. However, once the lesions healed, NGAL expression disappeared on its own, suggesting that its expression is closely related to (and regulated by) the disease process. An interesting observation was that (skin) lesions with a positive staining for MMP9 were negative for NGAL [34].
Table 4.
Expression of NGAL in benign diseases
| Disease | Change in NGAL expression |
|---|---|
| Inflammatory diseases | |
| Psoriasis | 10-fold upregulation in the skin of psoriasis patients in comparison to atopic dermatitis cases [270]. |
| Parakeratosis | Strong expression of NGAL in the epidermis of affected skin tissue with the strongest staining noted underneath areas of parakeratosis [34]. |
| Eczema | No expression in the epidermal cells in acute eczema while superficial keratinocytes (specifically in the areas of parakeratosis) shows strong expression of NGAL in biopsies from patients with chronic eczema. |
| Periodontitis | Strong upregulation of NGAL in the neutrophils from alveolar tissue specimens in patients of AdP and LJP. Staining extended to tissue in the area of neutrophil extravasation [29]. |
| Myocarditis | Significant upregulation of NGAL, NGAL receptor as well as IL-2 (the regulator of NGAL) expression was observed in the cardiomyocytes, endothelial cells, pericytes, smooth muscle cells, fibroblasts, leucocytes in both animals and patients of myocarditis in comparison to control tissues [39,40]. |
| HIV | Higher in HIV negative healthy individual (n=21) in comparison to HIV-positive patients (n=37). Treatment of HIV patients with highly active anti-retroviral therapy (HAART) led to a progressive increase in NGAL levels reaching to near normal levels after 12 months of therapy [41]. |
| Ulcerative colitis |
|
| Ischemic diseases | |
| Cerebrovascular accident/Strokes |
|
| Myocardial infarction (MI) | Significant elevation (p<0.0001) in NGAL expression in patients with acute MI (146±23 ng/ml) as compared to those with stable CAD (101±53ng/ml) [49]. |
| Chronic venous wounds (CVW) | NGAL levels were significantly lower in the wound exudates from healing chronic venous wound (CVW) patients compared to the Non-healing-CVWs. Further, in the healing-CVW patients, the levels of NGAL in the exudates showed a decline with time, which correlated with progressive healing of the wound [50]. |
| Ischemia Reperfusion Injury (IRI) | Leads to increase in urine NGAL levels. Sphingosine-1-phosphate (SIP), a lipid which reduces the severity of IRI counteracts upregulation of urine NGAL [51]. |
| Metabolic diseases |
|
| Renal diseases |
|
| Drugs and intoxicants | |
| Organ transplants |
|
AdP, adult periodontitis; LJP, localized juvenile periodontitis; ADPKD, adult polycystic kidney disease.
Alveolar tissue specimens from patients with both adult (AdP) and localized juveline periodontitis (LJP) revealed a strong upregulation of NGAL (and MMP-9) in the neutrophils [29]. Immunohistochemical staining of clinically healthy alveolar mucosa revealed that NGAL (and MMP-9) is expressed by resident neutrophils, but not by the healthy alveolar epithelium. Further, staining for NGAL was localized to the cytoplasm of the neutrophils when they were located within the blood vessels. However, when they extravasated into the surrounding tissue, NGAL staining could be seen both inside the cells and in the adjacent connective tissue [29]. A significant increase in serum Lcn2 levels was also seen in a rat model of autoimmune myocarditis following immunization with porcine myosin suggesting that Lcn2 is involved in a variety of inflammatory processes [39,40].
Serum NGAL levels are lower in treatment naive HIV positive patients and increase with initiation of highly active anti-retroviral therapy (HAART) [41]. When mononuclear cells (MNCs) from the bone marrow of the treatment naïve HIV positive patients or controls were treated with phytohemagglutinin (PHA) in vitro, MNCs from the latter (but not the HIV infected patients) showed a significant increase in NGAL release into the medium. Following HAART therapy for 26 weeks (but not at 4 weeks) however, MNCs from HIV positive patients also demonstrated a significant induction of NGAL following treatment with PHA [41]. This suggests that NGAL may be a surrogate marker of immune competence in HIV positive patients and also useful to monitor response to and adherence to HAART therapy.
2.2.2 Ischemic diseases
A second group of diseases associated with significant elevation in NGAL levels are ischemic disorders, i.e. diseases characterized by a decrease in blood supply to a particular organ with resultant hypoxia and either temporary (e.g. fatty change) or permanent (e.g. apoptosis and necrosis) tissue damage. The major ischemic diseases associated with an elevation in NGAL include cerebrovascular accidents and myocardial infarction.
Cerebrovascular accident (or stroke) is the third leading cause of death in the United States with an estimated 143,000 patients dying each year from this condition [42]. A major cause of stroke is atherosclerosis affecting the carotid arteries [43]. Anwaar and colleagues reported that the median plasma NGAL levels in subjects with asymptomatic carotid atherosclerosis was 97.5 ng/ml (range: 42ng/ml-291 ng/ml) with no significant difference between males and females [44]. However, a weak positive correlation was observed between plasma NGAL and diastolic pressure and age (Table 4). While NGAL levels were not significantly different between smokers and non-smokers, the levels were significantly higher in hypertensive compared to normotensive women [45]. NGAL levels were also elevated in the atherosclerotic plaques themselves, particularly unstable plaques [46]. NGAL was strongly expressed in the plaque associated macrophages, endothelial cells and smooth muscle cells. NGAL expression and NGAL/MMP9 gelatinolytic activity were both higher in fibrous plaques and in plaques with higher levels of the pro-inflammatory cytokines IL-6 and IL-8. NGAL released from the plaque lesions also produced a local increase in blood NGAL levels suggesting that the NGAL-MMP9 association was involved in the disruption of the fibrous plaque [46]. Investigations in a rat model of carotid atherosclerosis revealed that while NGAL mRNA is not expressed by the uninjured arterial tissue, its expression is significantly upregulated 2 weeks after balloon-induced endothelial injury [47].
Acute myocardial infarction (AMI) is a leading cause of mortality worldwide and an estimated 17 million deaths every year are attributable to coronary artery disease [48]. An analysis of plasma NGAL levels in patients with coronary artery disease (CAD) revealed that NGAL levels were significantly elevated (p<0.0001) in patients with AMI (146±23 ng/ml) compared to those with stable CAD (101±53ng/ml). There was no significant difference in NGAL levels between patients with ST elevation vs. non-ST elevation MI. There was also no correlation with age, serum creatinine or number of coronary arteries with a >50% luminal obstruction. Patients with AMI had significantly higher neutrophil count than the stable CAD group. In multivariate analysis, plasma NGAL above a cut-off >127ng/ml was an independent predictor of the risk of AMI (odds ratio 12.2, p=0.003) [49].
NGAL levels in wound exudate correlated inversely with healing of chronic venous wounds (CVWs) suggesting its potential as a marker of healing in such skin lesions [50]. Urine NGAL levels were also increased in patients with ischemia reperfusion injury (IRI) to the liver and kidney [51]. Taken together, these observations suggest that ischemia is a potent stimulus that increases levels of tissue and circulating NGAL. Apart from potential in diagnosis of ischemic diseases, it also reveals a much broader function for NGAL beyond inflammation and iron transport.
2.2.3 Metabolic diseases
An important group of disorders associated with significant morbidity (rather than mortality) are metabolic disorders. In recent years, there has been an exponential increase in the prevalence of these disorders, particularly obesity and its most widespread associated chronic disease, type-II diabetes mellitus. NGAL expression is also differentially altered in diabetes. For instance, Lcn2 levels were significantly higher in adipose tissues of obese mice in a mouse model of human obesity [52]. Further, NGAL levels were increased in human subjects upon a 26 hour continuous infusion of insulin suggesting that it is upregulated in response to insulin [53]. Serum NGAL levels were also higher in women with gestational diabetes [54] and pre-eclampsia [54,55] (Table 4). Diabetes is associated with chronic inflammation and microvasculopathy. However, whether the increase in NGAL levels reflects a response or contributes to the pathogenesis of this disease is still being explored.
2.2.4 Renal diseases
While NGAL expression is altered in several of the aforementioned conditions, its elevation in response to kidney damage, both acute and chronic, has gained considerable prominence in recent years.
Acute kidney injury (AKI) due to a variety of insults ranging from radiologic contrast to post-surgical stress can cause a significant increase in the expression of NGAL in the kidneys [56–59]. Nearly 6% of all critically ill patients with AKI require renal replacement therapy (RRT) in the form of dialysis or a kidney transplant. The mortality in these patients can reach as high as 60%. The early diagnosis of AKI is currently limited by the poor sensitivity of creatinine as a marker of renal injury [60]. In a single center prospective study involving 109 patients, serum NGAL levels were found to be significantly elevated in AKI patients receiving RRT who died vs. those who survived during the hospital stay. Further, NGAL levels correlated positively with the severity of AKI and an elevated NGAL was an independent predictor of increased 28-day mortality (hazards ratio (H.R. 1.6, 95% C.I. 1.15–2.23) [60]. NGAL levels however did not show any variation during the process of continuous renal replacement therapy (CRRT) in another study [61]. The results of these studies, supported by several others (discussed later) suggest that NGAL is a novel early diagnostic and prognostic marker in patients with renal injury. The diagnostic and prognostic potential of NGAL in renal diseases is discussed in detail in Section 6.1.5.
Studies conducted in animal models suggest that at least in AKI, the source of urinary NGAL is the ischemic renal tubules themselves (Table 4) [62]. The main source of NGAL in the ischemic kidney appears to be cells lining the thick ascending limb of the loop of Henle and collecting ducts. A second suggested source for urinary NGAL is protein that is filtered through the glomeruli but fails to get reabsorbed into the proximal tubules. In extrarenal diseases however, the source of NGAL, particularly in the urine is unclear. One proposed mechanism is that the NGAL is released into systemic circulation (from the sites of inflammation and/or malignancy) and is filtered by the glomeruli. Most of the NGAL (in the glomerular filtrate) is then reabsorbed by the proximal tubules expressing the NGAL receptor (megalin) and thus rises in the bloodstream. This hypothesis is supported by studies in megalin deficient mice who demonstrate significantly higher levels of Lcn2 in their urine [63]. A third potential source of systemic NGAL is neutrophils and macrophages. In support of the last hypothesis, a large single center study observed a significant positive correlation (r=0.9, p<0.001) between urine NGAL levels in patients with AKI and serum neutrophil myeloperoxidase levels [64].
Chronic kidney disease (CKD), defined as albuminuria with/without a decrease in the glomerular filtration rate (GFR) affects between 10%–13% of the population worldwide. CKD is also associated with significant elevation in tissue, blood and urine NGAL levels (Table 4) [65,66]. Lcn2 was strongly expressed in the proximal tubules and to a lesser extent in the ascending limb of the loop of Henle and collecting ducts following loss of functioning kidney mass. Further, the expression of Lcn2 mRNA and protein in the kidney tissues correlated with the extent of renal tubular damage, and urine Lcn2 levels correlated with tissue Lcn2 expression, suggesting that the damaged kidneys secrete Lcn2 into the bloodstream, which is then excreted in the urine. Lcn2 levels were also significantly elevated in the jck2 (juvenile cystic kidney) mouse model of human adult polycystic kidney disease (APKD). APKD is an autosomal dominant inherited disorder characterized primarily by development of cysts bilaterally in the kidneys, liver, pancreas, seminal vesicles and the arachnoid membrane in the nervous system [67]. The expression of NGAL was significantly increased in dilated cysts among APKD patients. Further, urine NGAL levels correlated positively with the rate of disease progression and inversely with the residual GFR [66]. Both the incidence and severity of renal lesions and their functional effects (elevation of serum creatinine and hypertension) were significantly reduced in Lcn2−/− mice (compared to their wild type littermates) 2 months after 75% nephron reduction, suggesting that Lcn2 is a specific promoter of pathological proliferation of renal tubular and interstitial cells following glomerular loss.
2.2.5 Drugs and intoxicants
NGAL expression is also affected by the intake of drugs and intoxicants like alcohol [68], methamphetamine and phencyclidine [69] (Table 4) Significantly, the expression of Lcn2 was nearly 3-fold higher in ethanol fed mice in which both copies of the gene for C3 complement were silenced, suggesting that C3 may be a novel negative regulator the of Lcn2 expression [68].
NGAL expression has also been shown to be elevated following administration of hepatotoxic agents. A study in Winstar rats revealed that serum NGAL levels rose within 24 hours after administration of the hepatotoxic drug BAY16, and increased progressively (~16-fold and 37-fold upregulation after 3 days and 12 days following repeated administrations of BAY16). Serum and liver NGAL protein levels correlated with the severity of liver injury [70]. Specifically, the increased expression of NGAL was noted in the hepatocytes, biliary epithelial cells and proximal tubular epithelial cells of the kidney. NGAL protein was seen expressed on the apical side of the proximal tubular epithelium suggesting that the kidneys reabsorbed NGAL from the glomerular filtrate. Since these animals also had elevated serum NGAL, it is possible that the urinary NGAL is derived from extra-renal organs (e.g. liver) in response to tissue damage.
2.2.6 Organ transplants
NGAL levels are also significantly upregulated in transplanted organs following reperfusion of the graft. Two organs where this has been well studied are the heart and the kidney (Table 4) [37,71,72]. Studies using a murine model of cardiac transplantation have revealed that the upregulation of Lcn2 is in fact a reaction of the recipient’s immune system to the allograft. This is suggested by the observation that when heart from an Lcn2+/+ mouse is transplanted into an Lcn2−/− recipient, the number of granulocytes infiltrating the recipient heart is decreased by nearly 54% (compared to that when an Lcn2+/+ recipient that receives a heart from an Lcn2+/+ donor). However, there was no difference in the percentage of apoptotic cells (in the transplanted heart) between the Lcn2 +/+ and the Lcn2−/− recipient mice, suggesting that Lcn2 does not modulate apoptosis in the transplanted heart tissues. In vitro results also showed no correlation between Lcn2 expression and the percentage of apoptotic cells in the cardiac myocytes. However, there was a systemic elevation of Lcn2, both in the serum and in the proximal renal tubules, in recipient mice following reperfusion of the graft. It is suggested that Lcn2 released from granulocytes (infiltrating the transplanted heart) is filtered into the proximal renal tubules and contributes to the rise in serum Lcn2 levels in the recipient mice [37]. However, whether Lcn2 knockout affects the ability of granulocytes to mount an inflammatory response or prevents the grafted heart (from an Lcn2−/− donor to an Lcn2+/+ recipient) from providing the appropriate microenvironment for establishment of granulocytes is still an open question.
Microarray analysis of kidneys from brain dead donors harvested either prior to (1 hour) or immediately after transplantation (1 hour-5 days) revealed a significant upregulation of pro-inflammatory genes including NGAL. Pathway analysis revealed that the p53 and NFκB signaling pathways were the most prominently altered in both brain dead and ischemia-reperfusion affected donor kidneys [72]. It is possible that ischemia resulting from brain death upregulates NGAL through the NF-kB (and/or the p53 pathway). NGAL in turn serves as a signal to recruit inflammatory cells to the kidney. The resulting inflammation may subsequently damage the grafted organ in the post-transplant period. Further studies particularly employing mice deficient in NF-κB and p53 are needed to elucidate the functional relevance of these pathways in regulating NGAL expression in transplanted organs.
2.3 EXPRESSION OF NGAL IN MALIGNANT DISEASES
2.3.1. Expression of NGAL in solid tumor malignancies
NGAL has been reported to be expressed in malignant tumors arising from several organs including the skin [34], thyroid, breast [73,74], ovary [75,76], endometrium [77], colon [78–80], lung [81], liver, bile ducts, esophagus [79], stomach [82,83] and pancreas [79,84–86] as summarized in Table 5.
Table 5.
Expression of NGAL in malignant conditions
| Cancer | Expression | Correlation between NGAL expression and clinicopathological characteristics [reference] |
|---|---|---|
| Thyroid | Elevated expression in papillary, follicular and anaplastic thyroid carcinoma. | Negative correlation: Degree of differentiation [142]. |
| Ovarian cancer | Significantly elevated expression in borderline and grade 1 tumors (in both tissues and serum) in comparison to high grade tumors. Elevated expression in OSPC cell lines in comparison to normal HOSE cells. |
Positive correlation: Degree of differentiation [74–76,89]. |
| Breast | Elevated expression in ductal carcinoma of the breast while absent in lobular and tubular breast cancer. Elevated in primary (2-fold) and metastatic breast cancer tissues 3-fold compared to the non-neoplastic breast tissue. |
Positive correlation: Grade of differentiation, lymph node metastasis, cellular proliferation and HER-2 expression status Negative correlation: Estrogen and progesterone receptor expression. [73,74,88]. |
| Endometrium | Significant elevation in endometrial hyperplasia and endometrial carcinoma (in tissue by IHC and microarray analysis of microdissected cells) in comparison to the normal endometrial glands. Elevated expression in HHUA and low level expression in HEC1A, HEC1B and KLE endometrial carcinoma cell lines. |
Negative correlation: Nuclear staining of NGAL with degree of differentiation and stage [77,92]. |
| Colorectal Cancer (CRC) | Significant elevation of NGAL expression in visceral adipose tissues of CRC patients (at mRNA level) in comparison to HC. Aberrant expression in low grade dysplasia and expression increases progressively through high grade adenoma to carcinoma and metastatic CRC. |
Positive correlation: TNM staging [94] and expression of inflammatory molecules, TNF-α, MCP-1, YKL40, osteopontin, HIF-1α and MMP-2 [93]. Negative correlation: Expression of adiponectin [93]. |
| Pancreatic cancer (PC) | Weak to no-expression in non-neoplastic tissues, moderate expression in chronic pancreatitis cases and strong expression in pancreatic adenocarcinoma cases [79,84–86,96,155]. High expression in well-differentiated PC cell lines while absent in poorly differentiated or transformed normal pancreatic epithelial cells. |
Positive correlation Increasing degree of dysplasia (from PanIN-1 to PanIN-3). Negative correlation: Progressive decrease in expression in established PC with loss of differentiation [79,84–86,96]. |
| Gastric cancer | Expressed by 67% of non-neoplastic gastric mucosa (epithelial cells and neutrophils), 9% of dysplasia and 11% of gastric cancer and 92% of H.pylori positive gastritis cases and 50% of H.pylori negative gastritis cases [83, 101]. | Positive correlation: Expression of NGAL/MMP-9 complex correlated with severity and worse survival [271]. Significantly high in females, larger tumors, diffuse type cancer, moderate and poorly differentiated tumors, advanced stage tumors and tumors with either lymph node metastases, concomitant vascular invasion, or distant metastases [82]. |
| Hepatocellular cancer (HCC) | Elevated expression in HCC cases in comparison to normal liver tissue. | [154]. |
| Rectal cancer (RC) | Elevated expression in RC cases in comparison to adjacent normal tissue. | Positive correlation: Invasion, lymph node metastasis and advanced pTNM stage [272]. |
| Oesophageal squamous cell carcinoma | Heterogeneous expression varying from focal to strong staining in malignant cells. | Positive correlation: Cell differentiation and invasiveness [273]. |
| Lung | Heterogeneous expression with strong staining in tumor cells. | [271]. |
| Chronic myeloid leukemia (CML) | Significantly upregulated in CML patients compared to HC or patients with non- malignant leukocytosis. Strongly downregulated in cytogenetically confirmed remission (CCR) patients. | Absolute level of NGAL mRNA was significantly higher in patients with higher disease activity-indicated by the ratio of BCR-ABL mRNA to total ABL mRNA. |
OSPC, ovarian serous papillary carcinoma; HOSE, human ovarian surface epithelial cells; MCP-1, macrophage chemoattractant protein-1, MMP-2, Matrix metalloprotease-2.
It is pertinent to mention here that Stoesz and Gould identified NGAL as a gene that was specifically and significantly overexpressed in breast cancers overexpressing the receptor tyrosine kinase HER-2 (or neu), hence the name Neu Related Lipocalin for NGAL [87]. The specificity of NGAL for HER-2 driven breast cancer was further suggested by the observation that NGAL expression is not upregulated in breast cancers induced by other carcinogens (including chemical carcinogens N-nitroso-N-methyl urea and dimethylbenz(a)anthracene) or by the oncogene v-Ha-Ras.
2.3.1.A Endocrine gland tumors
NGAL expression is differentially altered in tumors arising from several endocrine glands including the thyroid, ovaries, breast and uterine endometrium (Table 5). In one study, NGAL was differentially expressed in 94% of ductal carcinomas tissues of the breast [73,74]. The aberrant expression of NGAL in breast cancer tissues was also evident on western blotting where 44% of the 250 breast cancer tissues expressed NGAL [88].
NGAL expression was also significantly upregulated in borderline and grade 1 malignant ovarian tumors [89] and in cell lines derived from ovarian serous papillary carcinoma (OSPC) cells, serous (YDOV-157) and mucinous ovarian carcinoma [75,76,90]. Immunostaining of tissue sections confirmed the findings in cell lines demonstrating no expression in the non-neoplastic ovarian surface epithelium (and stroma) while 73% of benign ovarian tumors, 100% of borderline ovarian tumors and 98% of ovarian cancers were positive for NGAL. NGAL expression in ovarian cancer was positively correlated with differentiation grade but not with tumor stage or histology [76]. In a recent study however, Emmanuel and co-workers reported that NGAL was neither expressed in the normal ovarian epithelium nor in the ovarian cancer tissues [91]. Positive staining (for NGAL) was however seen in a few inclusion cysts and in some intracytoplasmic vacuoles. A possible reason for the difference could be the difference in antibodies used by the two studies. The role of NGAL in ovarian cancer remains to be better elucidated.
NGAL was one of the most highly upregulated genes in both endometrial hyperplasia and endometrial carcinoma compared to the non-neoplastic endometrium [77,92]. At the subcellular level, NGAL was also expressed in all endometrial carcinoma cell lines tested. Subcellular fractionation confirmed the dual cytoplasmic and nuclear localization of NGAL in the endometrial carcinoma cells. Interestingly, all the endometrial cancer cell lines expressed two isoforms of NGAL, the well known 25 kDa isoform, and a second 30 kDa isoform which has been suggested to result from differential glycosylation of the protein [77].
2.3.1.B Gastrointestinal tumors
NGAL expression is significantly upregulated in patients with several GI malignancies including colorectal, pancreatic, hepatocellular and gastric cancer (Table 5)
Studies in human colon tissues suggest that the normal colon does not express NGAL [93,94]. However, its expression appears during low grade dysplasia and increases progressively through high grade adenoma to cancer [94]. Both tissue and plasma NGAL expression correlated positively with advanced disease stage suggesting that NGAL may play a role in the progression of colorectal cancer.
Pancreatic cancer is one of the most lethal malignancies with a 5-year survival rate of less than 3.5% [95]. Furutami and co-workers using the signal sequence trap method (SST) identified NGAL as one of the secreted proteins significantly upregulated in pancreatic cancer cells. NGAL was also expressed in 8/8 PC cell lines while a weak expression of NGAL was detected in some of the non-neoplastic pancreas [79]. We observed that NGAL expression in the normal pancreatic ducts (by IHC) ranged from weak to complete absence, while a moderate degree of expression in the tissues from patients with chronic pancreatitis [86,96]. In contrast, all of the pancreatic adenocarcinoma tissues examined expressed NGAL [86]. NGAL mRNA was detected in 75% of chronic pancreatitis patients, 100% of pancreatic cancer patients but in none of the normal pancreatic tissues. NGAL expression was also noted to progressively increase with increasing degree of pancreatic ductal dysplasia pointing to a differential induction of NGAL during the transformation of normal ductal epithelium to adenocarcinoma [86].
Primary cancer of the liver is among the most common malignancies worldwide, ranking fifth in terms of incidence and third in terms of mortality globally. Hepatocellular carcinoma (HCC) is the most common type of liver cancer accounting for upto 90% of all primary liver cancers [97]. Studies in animal models of HCC suggest that NGAL may be upregulated early on during the process of hepatic carcinogenesis and might be a target of several hepatic carcinogens. For instance, Lcn2 was the strongly upregulated (≈11-fold) in spontaneous liver tumors arising in mice deficient in the enzyme peroxisomal fatty acyl CoA oxidase (AOX). The deficiency of AOX, a H2O2 generating enzyme, leads to accumulation of its substrates that in turn act as a natural ligand for peroxisome proliferator-activated receptor α (PPARα), a nuclear receptor that upon activation dimerizes with the retinoid X receptor (RXR), and drives the transcription of target genes. Prolonged activation of PPARα in the liver has been shown to promote development of HCCs in rodents [98,99]. Lcn2 expression was also strongly upregulated in tumors developing in AOX expressing mice fed either a genotoxic carcinogen diethylnitrosamine (DENA) or a non-genotoxic agonist of PPARα, ciprofibrate (64-fold and 22-fold upregulation respectively) [100]. The strong upregulation of Lcn2 in HCCs due to multiple agents suggests that Lcn2 may be involved at a point of convergence downstream of multiple carcinogenic stimuli. This in turn raises the possibility that Lcn2 could be a novel diagnostic marker for HCC in high risk patients (e.g. chronic alcoholics and those with Hepatitis B or C infection).
The issue of NGAL’s expression in gastric cancer is somewhat controversial, with two groups reporting nearly opposite findings. Alpizar and co-workers reported that NGAL is strongly expressed both in the non-neoplastic gastric tissues (67% cases positive) and in gastritis (100% positive with 83% showing intense NGAL expression) [83] but significantly downregulated in dysplasia and invasive gastric carcinoma (9% and 11% strongly positive respectively). A study by Wang and colleagues however reported that while NGAL was expressed by neutrophils invading the lamina propria, no expression was detectable in the non-neoplastic gastric epithelium [82]. In chronic gastritis also, NGAL expression was restricted to neutrophils invading the inflamed mucosa. NGAL immunopositivity was also noted in neutrophils invading the necrotic tissue in areas of gastric ulceration. In gastric dysplasia however, a weak expression of NGAL was observed in the epithelial cells while in gastric cancer NGAL expression was strongly upregulated. NGAL expressions was higher in females, tumors larger than 4cm, diffuse type cancer, moderate and poorly differentiated cancers and tumors with either lymph node, vascular or distant metastasis [82]. Gastritis, particularly that associated with Helicobacter pylori infection has been strongly associated with an increased risk of gastric cancer [101]. A strong expression of NGAL was observed in 92% cases of H.pylori positive gastritis in comparison to 50% in H.pylori negative gastritis [83].
Taken together, these studies in a variety of solid organ malignancies suggests that NGAL is generally expressed by epithelial malignancies and correlates with clinicopathologic characteristics including disease stage and degree of tumor differentiation. These features together with its small size, secreted nature and availability of robust quantitative assays has made it an extremely attractive target both as a diagnostic and prognostic biomarker in solid organ malignancies. The functional significance of NGAL expression in malignant (vs. benign or inflammatory conditions) is discussed in depth in Section 3.
2.3.1.C Nervous system tumors
NGAL/MMP-9 complex was detected in the urine and cerebrospinal fluid (CSF) of patients with ependymomas, primitive neuroendocrine tumors (PNETs) and glioblastomas (GBMs) but not in healthy control subjects. The levels of NGAL/MMP-9 complex (measured by ELISA) were also significantly higher in the urine of patients with brain tumors than in controls. Immunohistochemical analysis revealed that while NGAL was not expressed in the normal brain or in benign brain tumors (ependymomas), its expression was significantly elevated in PNET and GBMs [102]. A follow-up study noted that NGAL was expressed in 100%glioblastomas, but in only 14% of anaplastic oligoastrocytomas. A comparison of NGAL immunostaining in primary vs. metastatic brain tumors revealed that none of the metastatic tumors expressed the protein. NGAL expression showed a significant positive association with proliferation of brain tumor cells, being more frequently positive in tumors with higher Ki-67 staining (a marker of proliferation) [103]. Both these studies, based on small number of tissue samples nonetheless suggest that NGAL may play a role in the pathogenesis of specific, highly malignant subtypes of brain tumors. Further studies are needed to validate these results and uncover NGAL’s functional role in brain tumor development and progression.
2.3.1.D Genitourinary system tumors
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) is an inherited syndrome characterized by an increased tendency to develop cutaneous and uterine tumors (leiomyomas) and aggressive papillary and collecting duct renal cell carcinomas. Mutations in the Kreb’s cycle associated enzyme Fumarate hydratase (FH) are associated with a strong risk of developing HLRCC [104]. Analysis of global gene expression profile of renal epithelial cysts from FH deficient mice revealed that Lcn2 was the most differentially expressed gene (nearly 63-fold upregulated) in FH null renal epithelial cysts. NGAL was also nearly 19-fold upregulated in kidney tissue from a patient with HLRCC [105]. This observation suggests that NGAL may play a role in the development of renal cancers in individuals with heterozygous germline mutations in the FH gene.
2.3.2 Expression of NGAL in hematologic malignancies
NGAL expression is elevated in a number of hematologic malignancies including chronic myeloid leukemia (CML), polycythemia vera and essential thrombocythemia.
NGAL is significantly upregulated in CML patients compared to healthy controls and patients with non-malignant leukocytosis [106]. Significantly, NGAL levels were strongly downregulated in patients who were in cytogenetically confirmed remission (CCR), while no significant elevation (compared to controls) was observed in patients who were either in blast crisis or resistant to Imatinib (an inhibitor of the BCR-ABL tyrosine kinase specific to CML cells). Serum NGAL levels were also significantly elevated in patients with CML, although no change was evident in patients who were in CCR. Further, no correlation was noted between NGAL expression in the blood and any other hematologic parameters [106]. The absolute level of NGAL mRNA was however found to be significantly higher in patients with higher disease activity- indicated by the ratio of BCR-ABL mRNA to total ABL mRNA with a ratio of >1 indicating active disease. These studies suggested an association between NGAL expression and disease activity in CML [107].
The mechanistic role of NGAL in leukemia was subsequently elucidated in studies employing the mouse homologue (Lcn2) in work done chiefly by Arlinghaus and co-workers [107–109]. Their studies revealed that Lcn2 is important both in the induction and in determining the severity of leukemia produced by BCR-ABL+ leukemia cells. The mechanism involves secretion of large quantities of Lcn2 by leukemia cells which then induces apoptosis in normal hematopoietic cells but not in the BCR-ABL+ leukemia cells. The differential effect of Lcn2 on normal vs. leukemic cells was traced to differential expression of the Lcn2 (or 24p3) receptor (called 24p3R) by normal hematopoietic cells, but not by the BCR-ABL+ cells [110]. The pro-apoptotic effect of Lcn2 on normal hematopoietic cells could be blocked by a monoclonal antibody against Lcn2, suggesting that the effect was specific to Lcn2. Inhibition of the BCR-ABL tyrosine kinase activity by Imatinib mesylate significantly decreased the production of Lcn2 by the blast cells suggesting that BCR-ABL directly regulates the expression of this glycoprotein [108]. When Lcn2 expression was stably downregulated in BCR-ABL+ cells, the resultant cells produced a significantly attenuated form of leukemia characterized by a normal sized spleen, normal platelet count, lack of bone marrow (BM) infiltration and absence of ascites upon injection into non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice. In comparison, cells expressing the non-specific short hairpin RNA when injected into mice produced features of severe leukemia including a massively enlarged spleen, thrombocytopenia, marked bone marrow infiltration by leukemic cells and marked ascites rich in leukemic cells [109].
When leukemic cells from BCR-ABL+ and either Lcn2 null (Lcn2−/−) or Lcn2 expressing (wild-type or Lcn2+) mice were injected into immunocompetent mice, 8/8 mice in the Lcn2+ group developed severe leukemic features while only 1/8 in the Lcn2−/− group showed severe symptoms [108]. Interestingly, when the recipient mice were irradiated prior to injection of the leukemic cells (to destroy most of their normal marrow), even the Lcn2−/− derived leukemic cells produced features of severe disease [108]. However, features of severe leukemia (massive splenic enlargement and infiltration of leukemic cells into the bone marrow, spleen and liver) were observed only in those mice injected with a high (1 X 106 cells) but not a low dose (0.3 X 106 cells) dose of the Lcn2−/− donor cells, suggesting that Lcn2 is a key requirement for the establishment and development of leukemia particularly when the number of leukemic cells is low. Further, a second, Lcn2 independent but BCR-ABL dependent mechanism of CML progression appears to exist at higher doses [107].
Lcn2 appears to be important not just important for the establishment of leukemia cells in the bone marrow, but also appears to affect their tumorigenic potential elsewhere. For instance, Lcn2 expressing (but not the Lcn2 null) transformed marrow cells (transformed with BCR-ABL) produced tumors following subcutaneous injection in immunocompetent mice. Further, following intravenous injection, the Lcn2+ (but not the Lcn2 null) cells produced signs of cachexia (significant weight loss) in the recipient mice and in one case, anemia together with tumors in the kidney, small intestine and bone marrow [107]. The myelosuppressive role of NGAL was further confirmed when human chronic myeloid leukemia (CML) cells expressing high levels of NGAL induced severe myelosuppression (measured by splenic and bone marrow atrophy, disappearance of myeloid progenitor cells and significant apoptosis in the spleen) upon injection into irradiated NOD/SCID mice. In comparison, CML cells expressing 5-times lower level of NGAL induced only mild disease with significantly longer survival [107].
These studies suggest that while BCR-ABL is the driving force behind leukemia, the severity of the disease is determined by Lcn2 and modulated by the presence or absence of a functioning healthy marrow. In this process, Lcn2 complements the oncogenic activity of BCR-ABL. Lcn2 secreted by leukemic cells functions to induce apoptosis in pre-existing normal hematopoietic cells (expressing the Lcn2R) and thus creates space for colonization of the bone marrow by the leukemic cells which themselves do not express the Lcn2R and hence are resistant to induction of apoptosis (by Lcn2) [108]. Non-radiated recipient mice inoculated with BCR-ABL+ Lcn2+ donor marrow cells demonstrated significantly elevated level of circulating Lcn2 (in addition to other features of CML) compared to undetectable levels in the untreated animals, suggesting the possible utility of Lcn2 as a diagnostic biomarker in patients suspected of CML [107]. Further, the ability to block the pro-apoptotic effect of Lcn2 on normal hematopoietic cells with an anti-Lcn2 antibody raises the possibility of using Lcn2 targeting as a novel approach to treat CML in the future.
2.3.3 Expression of NGAL in Normal and Cancer stem cells
Bone marrow derived mesenchymal stem cells (BMSCs), which can differentiate into bone cells (osteoblasts), cartilage (chondrocytes), fat (adipocytes) and muscle cells (myoblasts) have been the most attractive sources for synthesizing these tissues. When rat BMSCs were stimulated in vitro to undergo osteogenic differentiation, Lcn2 was one of the 12 genes that showed at least a 2.5 fold or higher upregulation in the differentiating (vs. the undifferentiated) BMSCs [111].
Cancer stem cells (CSCs) or tumor initiating cells (TICs) comprise an extremely small sub-population of cells within the tumor mass of predominantly poorly differentiated solid tumors that are characterized by the properties of long-term self-renewal and high degree of chemoresistance. CSCs are divided into two sub-populations- side population (SP) and the non-side population (NSP). SP cells have been shown to possess the ability to give rise to both SP and non-SP cells, thus suggesting that these are in fact “multipotent cancer stem cells” [112]. SP cells isolated from the human squamous cell carcinoma cell line A431 revealed a significant downregulation (2-fold) of LCN2/NGAL compared to the NSP cells by microarray analysis. In vitro, the SP cells grew significantly faster while in vivo they formed significantly larger tumors than the NSP cells [113]. The functional role of NGAL in CSCs and the mechanics underlying the regulation of its expression (in them) however remains a mystery.
3. FUNCTIONS OF NGAL IN HEALTH AND DISEASE
3.1 HEALTHY TISSUES
One of the earliest clues to the function of NGAL came from observations that incubation of heparinized human blood with opsonized yeast leads to a significant increase in its synthesis and release. Interestingly, the monomeric form of NGAL is released first followed by a combination of the monomer and dimer [20]. The experiment suggested that NGAL might be important in the body’s immune response. Subsequently, it was discovered that under non-reducing conditions, NGAL exists in three forms: as a monomer of 25 kDa, a dimer of 46 kDa and a homotrimer of 70 kDa. Immunoprecipitation assays revealed that NGAL interacts with the gelatinase MMP-9. However, this interaction only occurs in the extracellular space following secretion of both proteins [47]. NGAL and MMP-9 have been shown to interact via a disulfide linkage, however such a linkage is not observed in the murine Lcn2 which lacks the corresponding cysteine residues [16]. It can however form a 135 kDA heterodimer with neutrophil gelatinase via a disulfide linkage [114]. The interaction of NGAL with MMP-9 sequesters and thus inhibits the proteolytic activity of MMP-9 [47]. Nitrogen cavitation followed by fractionation of neutrophil extracts on a Percoll gradient, identified NGAL as a receptor for the potent neutrophil chemoattractant N-formylmethionyl-leucyl-phenylalanine (fMLP). NGAL was detected in the fraction that contains the specific granules (termed the β band) as a complex with gelatinase, and in that containing the cell membrane and secretory vesicles (termed the γ band) [115]. When Lcn2 was overexpressed in murine chondrocytes, their proliferation in vitro was significantly inhibited suggesting that NGAL may be a negative regulator of cartilage formation [35]. The specific functions of NGAL in healthy tissues are discussed below.
3.1.1 Modulation of intracellular iron stores and bacteriostatic function
Microbes, chiefly bacteria that require iron have evolved to survive within the severely iron-poor environment of the human body (estimated concentration of free iron in the body is estimated to be as low as 10−24 M) [116]. The exceedingly low availability of free iron is attributable to iron binding proteins like transferrin, ferritin and lactoferrin which form complexes with any available free iron molecules. Bacteria have developed special proteins called siderophores that have an affinity for iron (particularly ferric iron or Fe3+) several times higher than that of the endogenous iron chelators. This enables siderophores to not only bind available free iron, but also extract iron from iron-binding proteins of the host [117]. For instance, the logarithmic association constant of transferrin (Kf) is about 22, while that of catecholate type siderophores is about 45 suggesting a stronger affinity of these siderophores for iron than the host iron binding proteins [117,118]. Of the various siderophores, the catecholate type siderophores (e.g. enterobactin from E.coli and bacillibactin from Bacillus species) are particularly well known for their strong affinity for metallic iron (Fe3+) [117,119].
Work done on Lcn-2, the murine homologue has revealed that NGAL can modulate iron stores within mammalian cells. Iron has both protective and harmful effects on the immune system. On one hand, it is required for the generation of reactive oxygen species that mediates killing of bacteria inside lysosomes, while on the other hand it inhibits the expression of the major histocompatibility protein MHC-II, and thus negatively regulates immune response to bacterial lipopolysaccharides (LPS). The mechanism underlying Lcn2 mediated scavenging of iron involves binding of Lcn2 to bacterial iron-binding proteins (or siderophores) like enterochelin (or enterobactin) and carboxymycobactin. These proteins produced by intracellular gram negative bacteria (mostly belonging to the genus Enterobactericiae) or mycobacteria bind to free iron (specifically ferric iron) both in the intestinal fluid and inside macrophages and dendritic cells. The iron-siderophore complex is then transported into the bacteria where the iron is released and drives bacterial proliferation [16]. Lcn2 binds to these siderophores both in their iron-laden and iron-free state, and transports them through the Lcn2 receptor (LcnR or 24p3R) into mammalian cells where the iron is stored. Bacteria require iron for their growth; hence, by depleting iron stores, Lcn2 inhibits bacterial growth (i.e. has a bacteriostatic effect). When Lcn-2 binds to iron-empty siderophores and transports them into mammalian cells however, the siderophore binds to intracellular iron (within the mammalian cell) and transports it out of the cell. This in turn induces apoptosis in these cells as iron is required for several key cellular metabolic processes [120]. In macrophages, Lcn2 expression is upregulated in response to infection with Salmonella and serves both to sequester iron (from the bacteria) and also to transport free iron out of the cell (Figure 3) [120].
Figure 3. Regulation of Lcn2 expression in macrophages.
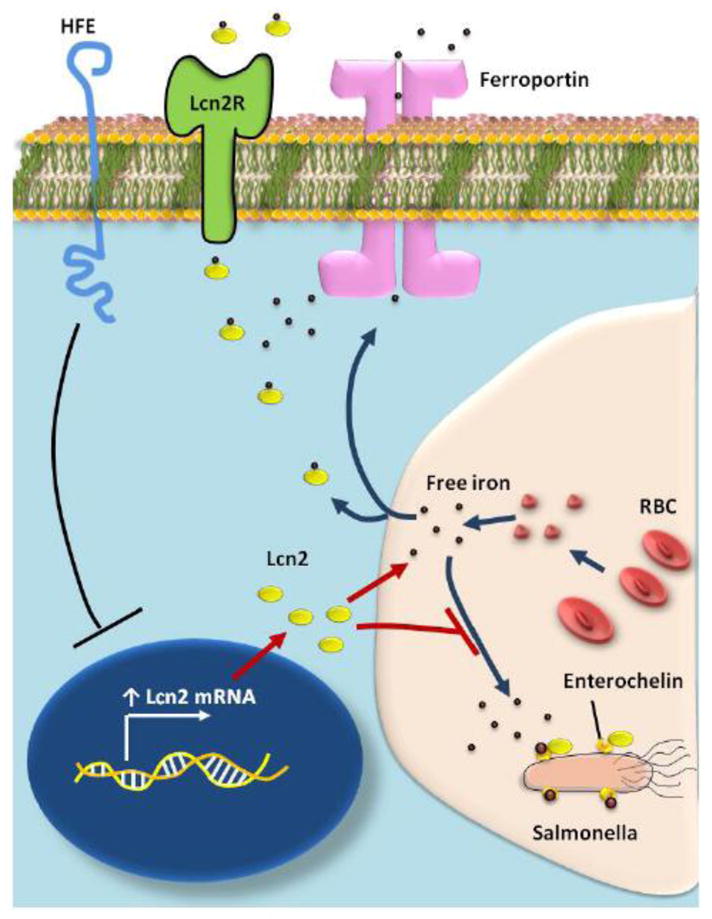
Macrophages break down senescent red blood cells (RBCs) releasing free iron from the hemoglobin contained within them. This free iron is then exported out of the macrophages by two pathways: a) by a transmembrane iron exporting protein ferroportin and b) as a complex with lipocalin 2 (Lcn2). Macrophages are also the site of infection by Salmonella, a gram negative intracellular bacteria. The bacteria require iron for their growth and survival. They express enterochelin, a siderophore on their outer membrane which binds to the free iron (released from the RBCs) and transports it into the bacterial cell. Infection of macrophages by Salmonella upregulates the expression of the anti-microbial protein lipocalin 2 (Lcn2). This binds to the iron-laden siderophores, thereby preventing the bacteria from using the free iron for their proliferation. This, together with the increased export of iron from the macrophages deprives bacteria of iron and induces growth arrest. The gene product of HFE (HFE) is a transmembrane protein similar to major histocompatibility antigen-1. It is expressed on the macrophage cell membrane and regulates both the uptake and efflux of iron from these cells. It is also a negative regulator of Lcn2 expression. Loss of functional HFE1 (most commonly by mis-sense mutations in patients with hereditary hemochromatosis) leads to an upregulation of Lcn2 in the macrophages thus conferring resistance to the host against infection by Salmonella.
Several pathogenic and even commensal bacteria including Escherichia coli, Salmonella enterica, Shigella sonnei, Klebsiella pneumoniae, Pseudomonas aeruginosa, Enterobacter species, Staphylococcus auerus, Vibrio parahemolyticus, Listeria species and Bordetella bronchiseptica have evolved a mechanism to use host catecholamines (L-norepinephrine (NE), L-epinephrine (EPI) and their progenitors, L-3,4- dihydroxyphenylalanine (L-DOPA) and dopamine) to abstract iron bound to host iron proteins (like transferrin and lactoferrin). The mechanism involves forming of a Fe3+-catecholamine complex, which is then scavenged by an extracellular bacterial lipoprotein (e.g. FeuA in Bacillus subtilis) followed by its active uptake by a membrane transporter (FeuBC/YusV permease/ATP-binding cassette complex) [121,122]. In vitro studies reveal that NGAL can bind to the complex of Fe3+ with NE (but not that of Fe3+ with EPI or L-DOPA). This interaction is quite strong (dissociation constant (KD) value nearly 51 nM), but still weaker than the affinity of NGAL for its known ligands-Fe3+ complexed enterobactin and bacillobactin (KD of 0.4 and 0.5 nM respectively). E.coli and B.subtilis (representative of Gram negative and Gram positive bacteria) that express siderophores (wild-type or WT strain) show increased proliferation in presence of holo-transferrin (holo-Tf, i.e. transferrin loaded with iron), a process that can be inhibited by adding NGAL to the cultures. Strains that are deficient in isochorismate synthase, the enzyme needed to synthesize siderophores respond neither to holo-Tf nor NGAL, but show a significant increase in proliferation when incubated with a combination of holo-Tf and NE. This suggests that the mutant strains utilize NE to abstract iron from holo-Tf for their growth. When NGAL is added to this combination (holo-Tf + NE), the growth of the mutant strains is significantly decreased (albeit at a higher concentration of NGAL than was required to inhibit the WT strain), suggesting that NGAL interferes with the uptake of NE-Fe3+ complex by the mutant strain of bacteria. When the iron transporter (FeuABC system in B. subtilis) was deleted in the siderophore deficient mutants, no growth stimulation was observed with either the EPI-Fe3+ or NE-Fe3+ complexes. This suggests that this iron scavenging transport system is responsible for the uptake of catecholamine- iron complexes into the bacterial cells and thus helps the bacteria in acquiring this essential growth nutrient even in the absence of siderophores. NGAL directly competes with this scavenger (by binding to the NE-Fe3+ complex) and thereby deprives the microbes of iron, thus blocking their proliferation. Interestingly, this mechanism appears to operate only under conditions where iron is available in limited quantities. In the presence of an abundant iron source (e.g. ferric chloride), bacterial growth increased independent of exogenous NGAL [117].
The aforementioned studies suggest that Lcn2 exerts its bacteriostatic effect by binding to bacterial siderophores. Further, Lcn2 is released from the liver and spleen in response to an acute bacterial infection. However, an interesting question still persists- do siderophore-like proteins exist in mammals that can interact with and modulate the functions of NGAL? The answer to this question came in 2010, when Bao and his colleagues discovered that a family of commonly occurring metabolites called “catechols” can bind to NGAL inside the human body. The study also showed that while catechol alone bound poorly to NGAL (association constant in the micromolar range) the addition of ferric iron to it significantly improved its affinity for NGAL (decreased the association constant to the nanomolar range) [123]. Catechols are metabolites derived from plant sources like quinic, shikimic and 34-dihydroxybenzoic acid and plant hydroxybenzenes like caffeic acid, chlorgenic acid, catechin and epicatechin. They are also synthesized de novo in the body from dietary proteins through a phenol intermediate. This process is affected through the NADPH- dependent liver microsomal cytochrome P450 enzyme. The resident microflora of the gut appears to be crucial for synthesis of endogenous catechol as sterilization of the gastrointestinal tract by neomycin (in mice) is observed to significantly decrease the excretion of catechol in the urine [124]. Catechols (catechol, 3-methyl catechol, 4-methylcatechol and pyrogallol) are normally abundantly excreted in the urine (concentrations ranging from 30 μM for 4-methylcatechol to 500 μM for pyrogallol). The interaction of catechols with iron could be abrogated by O-methylation or O-sulfonation of their hydroxyl groups suggesting a specific mechanism of interaction. Interestingly, enterobactin, the gram negative siderophore was shown to inhibit the binding of catechol to Lcn2, supporting the hypothesis that bacterial siderophores compete with endogenous mammalian siderophores (like catechols) to bind free iron. The interaction of Lcn2 with catechol requires the presence of the cationic amino acids Lys 125 and Lys 134. Mutation of both these residues to alanine led to a loss of Lcn2’s ability to bind to both the enterobactin-iron(III) or the catechol-iron(III) complexes. Further analysis revealed that different catechols exhibited varying affinities for binding to Lcn2 with catechol binding with the highest, pyrogallol the lowest and 3-methyl catechol an intermediate affinity.
It has been proposed that the recruitment of the iron(III)-catechol complex (i.e Fe3+-catechol) to the Lcn2 ligand binding pocket occurs in a two-step process- in the first step which occurs at physiological pH (7.4), a bicatecholate-iron complex (2 catechols complexed to one iron molecule) is recruited to the calyx. This is followed by the addition of a third catechol moiety to the complex in a pH independent manner to create a tris-catecholate complex. This configuration leads to the iron molecule being stabilized in a hexadentate co-ordination complex owing to the formation of stable cationic-π interactions and coulombic (electrostatic) interactions between the ferric iron (Fe3+) and the catechols. In the absence of these stabilizing interactions, iron remains insoluble and little if any (iron) binds to the Lcn2 calyx in solution. Binding of the Fe3+-catechol complex to Lcn2 does not significantly alter its structure (as assessed by X-ray diffraction).
When Fe3+-catechol complex and Lcn2 were administered separately to immunocompetent mice, a Lcn2- Fe3+-catechol complex was detectable in the serum after only 5 minutes post-injection. Over time, the complex accumulated in various organs, most abundantly in the liver, kidneys (maximum) and lungs. This suggests that catechol is a mammalian siderophore that binds free iron. Once bound to iron, the Fe3+-catechol complex has a high affinity for binding to circulating Lcn2. This complex is then trapped in various organs of the body, particularly in the kidneys where it is filtered through the proximal convoluted tubules. The filtered complex (Lcn2-Fe3+-catechol complex) is subsequently reabsorbed in the proximal tubules through the Lcn2 receptor megalin (or Lcn2R) [123]. The importance of megalin (Lcn2R) in regulating iron levels is evidenced by the observation that megalin knockout mice have a significantly higher urine excretion of Lcn2 [125,126].
When the Lcn2-Fe3+-catechol complex was added to polarized proximal tubule epithelial cells or to kidney stromal cells in vitro, the complex was demonstrated to enter the cells and donate iron intracellularly. This was detectable as a fluorescent signal following activation of an iron responsive fluorescent probe. The uptake of the Fe3+-catechol complex is believed to occur via endocytosis. This is suggested from the observation the Lcn2-Fe3+-catechol complex is stable at neutral pH (7.0) but dissociates to release iron at an acidic pH (<6.0). The mechanism involves the protonation of either the hydroxyl groups of catechol or the amino acids within the calyx of Lcn2. Different catechols dissociate at different pH ranges- catechol and 3-methylcatechol release iron when the pH falls below 6.0 while pyrogallol releases iron at a pH below 4.0. The release of Lcn2 from the catechol-Fe3+-catechol complex was significantly more sensitive to pH than that from the enterobactin, occurring at a pH under 6.0 and 5.5 respectively [123]. Among the cellular organelle, the late endosome and lysosome have a pH of ≤6.0 and ≤5.5 [127]. We can thus hypothesize that the Lcn2-Fe3+-catechol complex following endocytosis is transported to and releases Iron in the late endosome from where Lcn2 (±catechol) is either translocated to the trans Golgi network (and secreted out) or degraded in the lysosomes [128]. Further, the higher sensitivity of catechol to low pH suggests that catechol releases iron into the cells much more readily than enterobactin, and thus supports its role as an endogenous ligand for Lcn2.
Studies conducted in mice and rhesus macaques have revealed that Lcn2 expression is induced de novo during cecal infection with Salmonella typhimurium (a common cause of human gastroenteritis) [129]. This induction is mediated by the inflammatory cytokines IL-17 and IL-22. The mechanism (summarized in Figure 4) involves the activation of antigen presenting cells following phagocytosis of the bacteria. The activated cells release T-lymphocyte activating cytokines such as IL-18 and 23 which in turn stimulate the T-cells to release IL-17 and 22. Interestingly, IL-17, but not IL-22 is absolutely required to induce synthesis of Lcn2 by the inflamed intestinal epithelium. However, IL-22 synergizes with IL-17 to cause further increase (>2-fold) in Lcn2 levels compared to that by IL-17 alone. Interestingly, neither incubation with the purified bacterial flagellar protein nor treatment with the bacteria had any effect on NGAL production by human colon cancer cells. However, these cells demonstrated a strong upregulation of NGAL following treatment with IL-22 [129]. IL-22 was also shown to upregulate Lcn2 expression in polarized primary mouse tracheal epithelial cells (MTECs). Treatment of MTECs with either 20 ng/ml of IL-22 or 10ng/ml of IL-17 led to a significant upregulation of Lcn2 levels. Pre-treatment of MTECs with either IL-17 or IL-22 also significantly decreased proliferation of Klebsiella pneumoniae on the apical epithelium in vitro with the combination being more effective than either cytokine alone. Significantly, this effect was dependent on Lcn2 as MTECs from Lcn2−/− mice failed to inhibit K.pneumoniae growth despite pre-treatment with either or both cytokines [130]. These studies reveal a complex network of signaling events involving antigen presenting cells, T-cells and cytokines that upregulates NGAL expression in intestinal epithelial cells upon infection by an enteropathogen like Salmonella. This information in turn is useful to understand the physiologic function of NGAL as an antibacterial agent on multiple epithelial surfaces.
Figure 4. Mechanism of bacteriostatic action of NGAL.

Gram negative bacteria (like Salmonella typhimurium) trigger an immune response characterized by the activation of antigen presenting cells (macrophages and dendritic cells) upon engulfing the bacteria. These activated cells then release cytokines like IL-18 and IL-23 that in turn activate T-lymphocytes. These activated T-cell in turn release IL-17 and IL-22. These two cytokines act on the intestinal cells and stimulate the de novo synthesis of lipocalin 2 (Lcn2). The Lcn2 is secreted into the intestinal lumen and binds to bacterial iron binding proteins (siderophores) like enterochalin. Since iron is essential for the growth of bacteria, the sequesteration of bacterial siderophores by Lcn2 has a bacteriostatic effect. In some gram negative bacteria (like Salmonella typhimurium), a cluster termed as the iroBCDEiroN cluster is present which encodes for salmochelin, a gycosylated derivative of enterochelin which does not bind Lcn2. Hence, gram negative bacteria containing this cluster are resistant to the bacteriostatic effects of Lcn2. Commensals like certain species of E.coli lack the iroBCDEiroN cluster and hence are targeted by Lcn2 secreted during intestinal inflammation.
The human hemochromatosis protein (HFE), a transmembrane protein similar to major histocompatibility antigen 1 (MHC1) is a potent negative regulator of Lcn2 in macrophages. Studies in Hfe (mouse homologue of human HFE) and Lcn2 single and double knockout mice have revealed that loss of even one allele of Hfe is enough to increase Lcn2 levels in peritoneal macrophages and protect mice from enteric infection by Salmonella [120] This work has led to a suggestion that patients with a non-functioning HFE (i.e. those with hemochromatosis) have a natural resistance to enterochelin expressing Salmonella typhimurium infection, providing a natural evolutionary advantage and possibly accounting for the high prevalence of hereditary hemochromatosis.
While bacteria produce more than 500 different siderophores (reviewed in [131]), Lcn2 appears to have an affinity to bind only specific types of these iron chelators. Elegant experiments by Flo, Aderem and colleagues demonstrated that Lcn2 specifically sequesters only catecholate type bacterial siderophores like enterochelin, but neither hydroxamate type (e.g. aerobactin and ferrichrome) nor polycarboxylate (e.g. rhizoferrin) type siderophores [132]. They observed that when mice were injected intraperitoneally with Staphylococcus aureus, a gram positive bacterium whose uptake of iron is not circumvented by Lcn2, all the mice injected with the bacterium died irrespective of whether they expressed Lcn2 (wild type) or not (Lcn2−/− mice). On the other hand, E.coli synthesizes and uses the siderophore enterochelin, which is a target of Lcn2. Although the strain of E.coli used in their experiments (H9049) does not produce hydroxamate type siderophores like ferrichrome, it has receptors for ferrichrome (but not aerobactin). This receptor serves as an alternate (enterochelin independent) pathway of iron uptake in these bacteria. When these authors injected wild type mice intraperitoneally with the H90949 strain of E.coli either in the presence or absence of ferrichrome, they observed that animals who received both E.coli and ferrichrome had a significantly greater mortality than those who received E.coli alone. Ferrichrome itself had no significant effect on the survival of these mice. These observations suggested that in presence of ferrichrome, the E.coli were able to take up iron through an Lcn2 independent pathway and subsequently proliferate to cause lethal sepsis. The importance of Lcn2 as a pivotal component of the body’s antibacterial defense mechanism was further strengthened by their observation that injection of E.coli into Lcn2 null mice resulted in nearly 80% mortality 42 hours post-injection at a dose that caused no mortality in the Lcn2 wild type mice. The specificity of Lcn2 for enterochelin type siderophores is highly significant as several bacterial including Salmonella, Brucella, Bacillus, Burkholderia, Corynebacterium and Paracoccus which cause disease in humans use enterochelin to take up iron from the host tissues [132].
3.1.2 Role in inflammation and neutrophil chemotaxis
The inflammatory response is a natural defense mechanisms used by the body to clear irritants and pathogens. NGAL has been shown to be a pro-inflammatory molecule causing some to call it a cytokine. Studies conducted in mouse models of pulmonary inflammation for instance have revealed that Lcn2 mRNA and protein are strongly upregulated following exposure to pro-inflammatory stimuli [133]. A similar upregulation was also seen in the lungs of patients with bronchial inflammation both in the epithelial cells and in the type-II alveolar pneumocytes [30]. The pro-inflammatory cytokine IL-1β but neither TNF-α, IL-6 nor bacterial LPS induced significant upregulation of NGAL in normal human lung epithelial cells [30]. While this suggests a mechanism for upregulation of NGAL in pulmonary inflammation, its functional role in this process is still unclear. Several hypotheses have however been suggested to explain the functional role of Lcn2 in inflammation. One such hypothesis is that acute (predominantly) or chronic inflammation (e.g. bronchial asthma) leads to the accumulation of granulocytes at the sites of inflammation. These granulocytes undergo apoptosis, release their granules (containing NGAL) and thereby mediate local tissue injury. In support of its pro-inflammatory function, Lcn2 appears to be a chemoattractant for neutrophils. Studies in a murine model of allogenic cardiac transplant have shown that there was a significant reduction in the number of neutrophils infiltrating the transplanted heart among Lcn2−/− mice (compared to the Lcn2+/+ mice) [37].
The importance of Lcn2 as an acute phase protein was directly demonstrated by the observation that following an intraperitoneal injection of E.coli, Lcn2 levels are elevated in the serum and in liver tissue within 4 hours, and in the spleen (primarily in the red pulp, macrophages, endothelial cells and fibroblasts) within 6 hours. The increase in serum levels of Lcn2 was preceded by an increase in mRNA synthesis in the peripheral blood cells [132]. Serum Lcn2 levels rose by nearly 22-fold (from 100 ng/ml to 2200 ng/ml) within 8 hours following injection (of E.coli), reached a peak by 24 hours and then gradually returned to baseline levels.
The complement system provides a link between the innate and adaptive immune systems. The complement component C3 is cleaved into C3a and C3b by C3 convertase. C3b can in turn promote further cleavage of C3 and C5 generating C3b and C5b as well as other inflammatory mediators C3a and C5a. The latter two are important for recruiting inflammatory cells under physiological conditions and have also been implicated in several acute and chronic inflammatory diseases [134,135]. CD21 and CD35 are membrane bound proteins that act as co-receptors to ensure optimum B-cell function. In mice, the two proteins are encoded by the same locus (Cr2) and are co-expressed in both B-lymphocytes and follicular dendritic cells. Animals lacking the CD21/CD35 co-receptors exhibit a specific defect in the synthesis of IgG3 compared to their wild type counterparts. Subsequently, these mice are highly susceptible to lethal infections by the gram positive bacterium Streptococcus pneumoniae [136]. When mice deficient in CD21/CD35 are vaccinated with an immunogen, they show a significant impairment in the ability to produce antigen-specific IgG3 antibodies. The production of other antibody isotypes (IgG1, IgG2a and IgG2b) is also reduced when vaccinated with low doses of the immunogen, but unlike IgG3 returns to near normal levels upon immunization with a higher dose of the immunogen. When the global gene expression in the spleen from CD21/CD35−/− mice was compared with that in the spleen from wild type mice, a significant upregulation of pro-inflammatory genes was observed in the former. The expression of Lcn2 specifically was upregulated nearly 3 fold. Analysis of the different leucocyte fractions revealed that the upregulation in Lcn2 occurred in all fractions including B-cells (B220+), monocytes (CD11B+) and neutrophils (CD11b+GR1high). The magnitude of upregulation (fold change measured as expression in CD21/35−/− expression in wild type mice) varied for different subsets and ranged from 3.2 fold (B220−) vs. 22 fold (B220+ cells), to 5.6-fold (CD11B−) vs.3-fold (CD11B+). The results suggested that there was in general an increased accumulation of inflammatory cells and mediators (e.g. Lcn2) in the spleen of the CD21/CD35 deficient mice. This upregulation could be reversed by depleting the mice of either neutrophils (using an anti-GR1 antibody) or C3 (using cobra venom factor). The upregulation of Lcn2 (in the CD21/CD35−/− mice) could also be decreased to levels close to that in the wild type (CD21/CD35+/+) mice upon pre-treatment with a pharmacological inhibitor of C3aR (receptor for C3a). This suggests a mechanism of CD21/CD35 regulated and C3 mediated regulation of Lcn2 expression in the spleen resident immune cells of mice. CD35 serves as a co-factor for factor I that in turn degrades C3b. Deficiency of CD35 leads to increased half-life of C3b (C5 convertase) in the circulation, in turn leading to increased generation of C3a and C5a and in turn enhanced recruitment of inflammatory cells to the spleen. The answer to the functional significance of the elevated Lcn2 expression in splenic immune cells could be the key to unraveling the role of Lcn2 in the body’s immune response.
3.1.3 Role in Oxidative stress response
Oxidative stress is defined as the imbalance between the pro-oxidant and anti-oxidant machinery in the cell [137]. It is responsible for a wide array of diseases ranging from coronary heart disease to cancer. Aberrant expression of NGAL in Chinese hamster ovary (CHO) and human embryonic kidney (HEK293T0 cells resulted in an upregulation of the mRNA and protein levels of the antioxidant enzymes superoxide dismutase (SOD1 and SOD2) and heme oxygenase (HO1 and HO2) together with decrease in expression of NF-κB. Silencing of NGAL in A549 lung cancer cells had the opposite effect on HO1 and NFκB expression but increased the expression of SOD1 and SOD2. Further, the cells ectopically expressing NGAL were more resistant to the cytotoxic effects of hydrogen peroxide in vitro [138]. These results suggest that NGAL by inducing the expression of anti-oxidant enzymes helps cells counteract oxidative damage. Other studies have also suggested that NGAL may have a protective effect against cellular injury mediated by reactive oxygen species (ROS) [139–141].
A clue to the mechanism underlying Lcn2’s role as an antioxidant comes from the observation that catechol, the only Lcn2 binding mammalian siderophore identified so far binds to Iron (III) (in absence of Lcn2) and reduces Iron(III) to Iron(II) (Fenton’s reaction). This accelerates the generation of hydroxyl free radicals which are extremely active and can activate ROS mediated signaling cascades. However, when Lcn2 is added (in a stoichiometric ratio of 1:3 of Lcn2: catechol), this reaction is inhibited. The hydroxyl groups of catechol are indispensible for its reducing activity as their O-hydroxylation or O-sulfonation prevents the reduction of iron and generation of hydroxyl free radicals [123].
3.2 BENIGN DISEASES
NGAL has been shown to modulate the pathogenesis of several benign conditions particularly vascular diseases (e.g. atherosclerosis) and renal diseases. In atherosclerotic plaques, NGAL expression correlated with presence of inflammatory changes, and signs of plaque instability like intra-plaque hemorrhage and presence of a luminal thrombus. NGAL has been shown to associate with gelatinases, particularly matrix metalloproteinase-8 (MMP8) and 9 (MMP9) [46]. Through this association, it prolong the proteolytic activity of the MMPs.
Lcn2 also mediates proliferation in renal tubular cells but not glomeruli. Downregulation of Lcn2 in mouse renal tubular cells led to significant decrease in EGF-induced cell proliferation in vitro while Lcn2 silencing in mice significantly inhibited renal tubular (but not glomerular) proliferation as assessed by positive staining with Proliferating cell nuclear antigen (PCNA) [66]. Further, Lcn2 silencing significantly inhibited apoptosis of both glomeruli and renal tubules in partially nephrectomized mice (compared to Lcn2+/+ nephrectomized mice). The differential effects of NGAL on the renal tubules vs. glomeruli may be important in the pathogenesis of chronic renal diseases, particularly those associated with disturbed tubular proliferation (discussed in Section 6.1.5).
3.3 MALIGNANT DISEASES
A clue to the role of NGAL in solid tumors came from the observation in thyroid cancer. Stable silencing of NGAL in thyroid cancer cells leads to a decrease in anchorage independent clonogenic growth in vitro and decreased tumorigenecity and tumor size upon subcutaneous (s.c.) injection into nude mice. NGAL appears to be required for survival of thyroid cancer cells in serum deprived conditions in vitro, as evident from a nearly 5-fold increase in apoptosis in the NGAL knockdown cells (compared to control cells) upon serum withdrawal [142]. A similar pro-proliferative effect was also noted when NGAL was ectopically expressed in endometrial cancer cells (Ishikawa and HEC1B). In vitro rate of proliferation was significantly increased (range: 27%–37% over control) in the NGAL expressing cells. Further, these cells exhibited enhanced invasive abilities (between 90%–645% higher than control cells) suggesting that NGAL may be involved in the growth and metastasis of endometrial cancer in humans [77].
NGAL also appears to be a marker of cellular stress. For instance, in vitro studies in human breast (MCF-7) and lung cancer (A549) cells have revealed that NGAL expression is significantly upregulated in response to multiple apoptosis inducing agents [114]. Notably, NGAL upregulation in these cells appears to be an attempt to survive the apoptotic stimulus rather than a pro-apoptotic response. This is based on the observation that ectopic expression of NGAL significantly reduces sensitivity of A549 cells to the apoptosis inducing agent OSU03012 (celecoxib-derived inhibitors of the phosphoinositide-dependent kinase-1 enzyme devoid of a COX-2 inhibitory effect), while an opposite effect is observed upon stable silencing of its mRNA [114].
While change in endogenous NGAL levels appears to modulate cellular response to stress, the addition of exogenous recombinant NGAL to NGAL non-expressing breast and lung cancer cells had no effect. Furthermore, neither its ectopic expression nor its stable silencing had any effect on proliferation of breast cancer cells, suggesting that NGAL does not affect cell proliferation or survival in these cells. Rather, it is upregulated as a response to an extrinsic apoptotic stimulus [114]. The effects of NGAL on untransformed but immortalized breast epithelial cells however appears to be different from those on breast cancer cells. Addition of recombinant NGAL to the immortalized normal breast epithelial cell line MCF10A (NGAL+) for instance led to a significant increase in the number of colonies, suggesting that it may stimulate proliferation in normal breast epithelial cells [143].
Aside from exogenous administration, ectopic overexpression of Lcn2 (mouse lipocalin 2) in 4T1 murine breast cancer cells significantly inhibited their ability to migrate and invade in vitro but had no effect on their rate of proliferation or clonogenic potential in soft agar. Upon implantation into mice, the Lcn2 expressing tumors did not differ significantly from the mock transfected cell induced tumors in mean weight, but showed a significant reduction in the number of visible liver metastasis [144].
In colon cancer too, NGAL does not appear to affect tumor cell proliferation (either in vitro or in vivo). However, when ectopically expressed, NGAL significantly suppressed the invasiveness of colon cancer cell lines in vitro. Further, addition of recombinant human NGAL too significantly decreased invasiveness of the highly metastatic KM12SM colon cancer cell line in vitro. NGAL expressing cells also exhibited a significant decrease in the incidence of liver metastases (compared to NGAL silenced cells) upon splenic injection into nude mice, an effect that could also be replicated by either intraperitoneal injection of recombinant human NGAL or ectopic expression of NGAL in a cell line that normally expresses low endogenous levels of the protein [145]. Ectopic expression of NGAL led to increased growth of KM12C colon cancer cells upon injection into the cecum of athymic mice. However, there was no metastasis in any mouse in this experiment. When the NGAL expressing cells were injected into the spleen, the NGAL expressing cells caused significant increase in splenic size compared to control cells [94].
An ectopic overexpression of NGAL in poorly differentiated pancreatic cancer cells significantly reduced the ability of these cells to adhere to fibronectin coated plates and to invade through an extracellular matrix-like material [96]. An opposite effect was observed when NGAL expression was stably silenced in the well and moderately differentiated pancreatic cancer cells. A clue to the underlying mechanism came from the observation that the ectopic expression of NGAL significantly downregulated the level of expression of both the active (phosphorylated on tyrosine 397) and total focal adhesion kinase (FAK). FAK, a 125-kDa molecule has been shown to be a potent suppressor of invasion and metastasis in pancreatic cancer cells [146,147]. One mechanism by which it may affect this function is through its interaction with cell-surface integrins and integrin-liked kinases (reviewed in [148]). Whether NGAL modulates the expression of FAK or its interactions and thus regulates the migratory behavior of pancreatic cancer cells is an outstanding question. Interestingly, neither the overexpression of NGAL nor its silencing had any effect on the proliferation of pancreatic cancer cells or their sensitivity to chemotherapeutic drugs (Gemcitabine and 5-fluorouracil) in vitro. However, when implanted orthotopically into the pancreas of athymic mice, pancreatic cancer cells ectopically expressing NGAL produced significantly smaller tumors, and had a significantly smaller incidence of metastasis to the liver, spleen and omentum/mesentery. Similar to the observation in vitro, no significant difference was noted in the proliferation index (measured by Ki-67 positivity) of the tumor cells obtained from the two groups of animals (vector control vs. NGAL overexpressing) [96].
In addition to inhibiting invasion and metastasis, NGAL also appears to be a negative regulator of angiogenesis in pancreatic cancer cells. In vitro, addition of conditioned media from poorly differentiated pancreatic cancer cells ectopically expressing NGAL to human vascular endothelial (HUVEC) cells reduced tubule formation by nearly 70% with an opposite (i.e. pro-angiogenic) effect observed when conditioned medium from NGAL-knockdown pancreatic cancer cells was applied [96]. In vivo, tumors from NGAL overexpressing MiaPaca cells showed significantly reduced microvessel density (measured by positive staining for the vascular marker CD31) compared to tumors from the empty vector transfected cells. Vascular endothelial growth factor (VEGF) is a key positive modulator of angiogenesis. We have reported in a recent study that NGAL overexpressing pancreatic cancer cells secrete significantly lower amounts of VEGF compared to the vector transfected control cells with an opposite effect seen in the NGAL knockdown cells (i.e. NGAL knockdown increases VEGF production). Interestingly, addition of recombinant VEGF to HUVEC cells could not rescue the ant-angiogenic effect of conditioned medium from NGAL overexpressing cells. Previous studies in Ras transformed 4T1 cells have shown that NGAL can inhibit Ras induced synthesis of VEGF [149]. In addition, NGAL could interfere with the interaction between VEGF and its receptor. Further, through inhibition of FAK activation, NGAL could inhibit angiogenesis as an interaction between FAK and integrins has been shown to be important for endothelial tube formation by HUVEC cells [150]. Angiogenesis is of particular importance in pancreatic cancer since the tumor becomes progressively hypovascular making it an extremely difficult tumor to treat medically [151]. The mechanism by which NGAL blocks signaling by exogenous VEGF will be important to discern in future studies.
Melatonin is a hormone produced by the pineal gland and functions to regulate hormone secretion by other endocrine glands according to the time of the day (termed as the circadian rhythm). Low levels of melatonin has been linked to several types of cancer including breast and prostate cancer [152]. Downregulation of NGAL particularly in the brain and liver has been suggested to be one of the mechanisms by which melatonin supplementation decreased the incidence of spontaneous tumors in older (≥26 months) mice [153].
A stable ectopic expression of NGAL in Chang liver and SK-Hep1 hepatocellular carcinoma (HCC) cells resulted in a significant inhibition of their proliferation (by 37% and 12% respectively), anchorage independent clonogenecity and their migratory and invasive potential (using both serum and in presence of a combination of EGF and TGFβ1) in vitro. The NGAL expressing cells showed a significant reduction in tumor growth upon subcutaneous implantation in athymic mice. The NGAL expressing HCC cells showed a significant reduction in both the expression and gelatinolytic activity of MMP-2 (compared to the vector control cells) [154]. However, unlike the upregulation of the epithelial marker E-cadherin induced by NGAL in Ras transformed 4T mammary cells and pancreatic cancer cells [144,155], the ectopic expression of NGAL had no effect on the expression of E-cadherin in HCC cells.
The aforementioned studies suggest that the functional role of NGAL varies from one malignancy to the other. In some cancer types (e.g. thyroid cancer), it is a potential target for downregulation to arrest tumor growth, while in others (e.g. breast and lung cancer) its downregulation could be a potentially useful mechanism to increase sensitivity to chemotherapeutic agents.
4. REGULATION OF NGAL EXPRESSION
Axelsson and co-workers were among the first to investigate the regulation of NGAL. In 1995, after identifying the protein from neutrophil extracts, they demonstrated that the hematopoietic cytokine granulocyte monocyte colony stimulating factor (GMCSF) when added to neutrophils (dose 50 pM) in vitro induced a strong upregulation of NGAL protein and increased its secretion into the medium [20]. An analysis of the gene structure of 24p3 (mouse Ngal) has revealed a remarkable conservation in the size of exons as well as the position of exon-intron junctions among various lipocalin-encoding genes [38]. This has strengthened the theory that all lipocalins arose from a duplication of one common ancestor. Further, the mouse 24p3 has several cis regulatory elements in its 5′-upstream region including a TATA box-like element (between nucleotides −28 to −23 relative to the transcription start site) and cis-binding elements for several transcription factors including SP1, PEA3, LFA-A1 and glucocorticoid receptors (GRs). In vitro treatment of mouse L cells transfected with an 845 bp fragment comprising the whole 5′-flanking region (nucleotide −793 to +50) with dexamethasone led to a significant upregulation in 24p3 mRNA levels. Interestingly, a strong time dependent upregulation of 24p3 was observed when cells were treated once (with the drug) followed by collection of mRNA over time but not when they were treated daily with the drug. This suggested that a protein, possibly 24p3 itself is released from the cells into the culture medium, accumulates therein and promotes its own synthesis. Analysis of the 24p3 upstream region through progressive deletion mutagenesis revealed that not only were the two GREs predicted in the 5′-flanking region of the gene important, but additional regions of the promoter, possibly important for binding of other proteins or a protein-complex were crucial for responsiveness of these cells to dexamethasone. 24p3 was also upregulated by retinoic acid (Vitamin A), a vitamin with known therapeutic applications in several diseases like acne and even as cancer chemopreventive and therapeutic agents [38]. What the alteration of 24p3 (and its human homologue) by various chemotherapeutic agents means in terms of the efficacy of or response to the chemotherapeutic is a question that still begs an answer.
In vitro studies on freshly isolated adipose tissue explants cultured in presence of insulin (10 pM– 100 nM) revealed that NGAL expression (both in tissues and in the conditioned medium) was increased significantly in a dose dependent manner by insulin. This upregulation was blocked by adding either a MEK or a PI-3K inhibitor suggesting that both these pathways are involved in insulin mediated regulation of NGAL expression [53]. Proteomic analysis of conditioned medium from FRO thyroid cancer cells expressing IκBαM, a super-repressor of NFκB revealed that NGAL was the most downregulated secretory protein in these cells (compared to parental FRO cells). This suggests that NFκB is a positive regulator of NGAL expression [142]. Chromatin immunoprecipitation analysis confirmed that the NFκB inducible factor IκBζ binds to the NGAL promoter together with the p50 and p52 subunits of NFκB and thus drives its transcription [142]. Recently, it has been shown that NF-κB is a positive regulator of the microRNA miR-146a in FRO cells [156]. Silencing of miR-146a in the FRO cells led to singnificant decrease in their tumorigenecity. It is possible that miR146a and other microRNAs may be involved in NFκB mediated regulation of NGAL. NF-κB also regulates the expression of NGAL by the injured vascular endothelium. In a rat model of carotid arterial injury, it was observed that the expression of NGAL by smooth muscle cells in an injured blood vessel (intimal NGAL > medial NGAL expression) could be blocked by the expression of a dominant negative form of IKKβ [47].
NGAL expression can be induced by several cytokines and growth factors including interleukin-1 (IL-1α and β) [30,47,157], IL-17, IL-22 [129,130], insulin like growth factor-1 (IGF-1), transforming growth factor alpha (TGF-α) [158] and tumor necrosis factor alpha (TNF-α) [41]. IL-1β induced expression of NGAL and its interacting partner MMP-9 is dependent on activation of NF-κB transcriptional activity [47]. It appears that in response to several kinds of stimuli all of which can activate NF-κB, both MMP-9 and NGAL expression is modulated similarly, resulting in a change in proteolytic activity of MMP-9. The transcription factor NF-κB is actually a dimer composed of two proteins of the NF-κB family- p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), RelB and c-Rel. In absence of stimulation, this dimer exists in the cytoplasm in an inactive state, sequestered by one of the two IκB proteins (IκBα or IκBβ). In presence of the appropriate stimulus, IκB is phosphorylated and then degraded (in the proteosome), This frees the NFκB complex which then translocates into the nucleus where it binds to and induces the transcription of its target genes [159]. Interestingly, while both the inflammatory cytokines IL-1β and TNF-α induce NF-κB mediated transcription, only the former induces NGAL in lung cancer cells (Figure 5). The specificity of IL-1β for NGAL involves the upregulation of IκB-ζ by IL-1β (but not TNF-α), a co-factor that binds to the NFκB dimer and induces transcription of NGAL mRNA [47]. In vitro treatment of immortalized normal epithelial cells (A549, NHBE and NHEK) with different cytokines (IL-1β, TNF-α, IL-6 and LPS) led to significant increase in both intracellular and secreted NGAL (10–12 fold, 3-fold and 5–6 fold compared to un-stimulated A549, NHBE and NHEK cells respectively) with IL-1β (but not the other cytokines). The cell lines differed in the rapidity of response (A549 > NHEK > NHBE), probably owing to differential expression of the IL-1 receptor between them. The IL-1β responsive element was localized to the region −183 to −153 5′-upstream of the transcription start site. Bioinformatics analysis revealed the presence of an NF-κB response element (5′-GGGAATGTCC-3′) with high degree of similarity to the consensus sequence (5′-GGG(A/G)NNT(C/T)CC-3′). A mutation of the GGG triplet in the consensus sequence to AAA completely abolished the induction of the reporter gene following treatment with IL-1β. Similarly, co-transfection with IκBα and IκBM abolished the IL-1β induced upregulation of the reporter gene induced by upregulation of IκBα alone. Further deletion mutagenesis narrowed the IL-1β responsive region to between −155 and −195 upstream of the transcription start site. Toll-like receptor 4 (TLR4), which is induced by LPS treatment and activates a similar downstream signaling cascade as IL-1β was shown to be important for mediating LPS induced expression of NGAL. Toll like receptors (TLRs) comprise a family of ten microbe-recognizing receptors that form a part of the innate immune system (reviewed in [160]). Most of the ligands for TLRs are bacterial in origin- for instance lipoproteins constitute the ligand for TLR2, LPS for TLR4 and flagellin for TLR5. TLR2 and TLR4 have previously been shown to share the similar signaling as IL-1 [161,162]. Transfection of TLR4 and its co-factor MD2 (required for recognition of LPS by TLR4 and for its proper distribution to the cell surface [163,164]) led to significant upregulation of NGAL expression (compared to empty vector transfected controls) in A549 cells following treatment with LPS. This upregulation was abolished by co-transfection with a dominant negative form of MyD88, an adaptor protein that is required for activation of NF-κB mediated transcription. NF-κB was also important for LPS induced NGAL expression as evidenced by the abolition of NGAL synthesis in presence of a reporter plasmid with a mutated NF-kB site [30]. These results shed new light on the regulation of NGAL expression by inflammatory cytokines and suggest that the regulation of NGAL expression in response to various cytokines is dependent on the expression of specific receptors and co-factor proteins, which are tissue specific. In vitro treatment of whole blood cultures (but not PBMCs) with TNFα, LPS or recombinant HIV-tat was also demonstrated to significant increase the release of NGAL into the culture medium [41].
Figure 5. Regulation of NGAL expression by NF-κB.

NF-κB is a dimer composed of one or more of five members of the NF-κB family (p50, p52, p65, Rel-A, Rel-B or c-Rel). Under resting state, the complex remains inactive in the cytoplasm bound to the inhibitor of IκB (IκB α or β). (A) NGAL inducers like IL-1β activate the I-kappa kinase (IKK) which phosphorylates IκB. This promotes ubiquitylation and subsequent degradation of IκB, thus freeing the NF-κB complex. (B) This complex now translocates to the nucleus where it upregulates the expression of the co-activator IκB-ζ (through a κB response element on its promoter) (Broken yellow arrows). This cofactor then binds to the NF-κB dimer (in the nucleus) and the complex in turn binds to the κB response element (κB RE) located at position −181/−171 on the NGAL promoter. This leads to activation of transcription of NGAL. IκB-ζ itself however, does not bind to the κB-RE on the NGAL promoter. (C) Incidentally, the NF-κB inhibitor IκB also has a κB RE on its promoter. Thus, its expression is also paradoxically upregulated by NF-κB. IκB in turn binds to the NF-κB transcription complex and transports it out of the nucleus (D), thus terminating its transcriptional activity. (E)TNF-α on the other hand binds to TNF-receptor (type 1 present in most tissues or type 2 present only on immune cells), which activates the TNF receptor type 1 associated DEATH domain protein (TRADD in case of TNFR-1). TRADD then recruits TRAF2 (TNF receptor associated factor) and the serine threonine kinase RIP (receptor interacting protein). TRAF2 also recruits IKK (existing as a complex of IKKα,β and γ) which is phosphorylated (and thus activated) by RIP. IKK then phosphorylates and thus targets IκBα for degradation. However, unlike IL-1β, the activated NF-κB induced by TNF-α does not induce transcription of the co-factor IκB-ζ and thus does not induce NGAL expression upon its transport into the nucleus. The differential effect of TNF-α and IL-1β on IκB-ζ is suggested to occur by increased stabilization (and therefore translation) of the IκB-ζ mRNA by IL-1β (but not TNFα). (F) TLR signaling and NGAL regulation. Toll-like receptors (TLRs) are a family of evolutionary conserved proteins that function as part of the body’s innate immune system defense mechanism to recognize pathogen associated molecular signatures. Ten TLR proteins have been identified so far in humans, each of which has a specific ligand. Activation of TLR signaling occurs in response to the binding of specific ligands (microbial lipopeptides for TLR2 and lipopolysaccharide of gram negative bacteria for TLR4). Myeloid differentiation primary response gene (MyD88), an adaptor protein used universally by all TLRs for signaling, possesses a C-terminal Toll/IL-1 receptor (TIR) domain that binds to the cytoplasmic portion of the TLRs. Following this, the IL-1 receptor-associated protein kinase (IRAK) proteins (IRAK-1 and 4) are recruited to the TLRs by interaction between the death domains of MyD88 and IRAK. IRAK-1 is then phosphorylated (activated) and in turn associates with the TNF receptor associated factor 6 (TRAF6). The IRAK-1/TRAF6 complex then dissociates from the TLR receptor and associates with TGF-beta activated kinase 1 (TAK1) and the TAK1 binding proteins (TAB1 and TAB2). Till this step the complex is attached to the cell membrane. The IRAK-1 protein is then degraded allowing the TRAF6/TAK1/TAB1/TAB2 complex to move from the cell membrane into the cytoplasm where it associates with, among other proteins with the E3 ubiquitin ligases (Ubc13 and Uec1A). The ubiquitin ligases catalyze the synthesis of a Lys-63 linked polyubiquitin chain on TRAF6. This induces TRAF6 to activate TAK1 which in turn phosphorylates the IKK complex (IKK a,b,g) or the MAP kinases (e.g. JNK) thus leading to transcription of NF-kB and AP-1 regulated genes respectively (including NGAL).
The critical role of TLRs in regulating Lcn2 expression was demonstrated when intraperitoneal injection of LPS into wither wild type or Tlr4−/− mice resulted in a significant (>300 fold upregulation in Lcn2 expression both in the peripheral blood cells and in the peritoneal cells. Lcn2 expression is also significantly upregulated in macrophages upon treatment with other TLR ligands including Pam3CSK4 (ligand for TLR1 and TLR2) and flagellin (ligand for TLR5) [132].
Polyinosinic-polycytidylic acid (poly-I:C or pIC) a synthetic analog of double stranded RNA that mimics the molecular pattern associated with viral infection, and unmethylated CpG oligonucleotide sequences (CpGs) are potent ligands for the TLR3 and TLR9 respectively [165]. When macrophages are stimulated with these ligands in vitro, they release pro-inflammatory cytokines including TNF-α, IL-12 and IL-6. Whitmore and colleagues observed that treatment of bone marrow derived macrophages with pIC and CpG also stimulated the synthesis of Lcn2 (>2 fold compared to basal levels) [166]. Further, pIC was more potent at stimulating Lcn2 transcription than CpG and a combination of the two more potent than either ligand alone. The upregulation of Lcn2 appeared to be independent of the interferon (IFN) mediated pathway as macrophages derived from IFN-α/IFN-β receptor knockout mice retained the strong induction of Lcn2 following treatment with a combination of pIC and CpG ligands.
IL-1α is produced by keratinocytes and regulates their differentiation [167]. Treatment of human keratinocyte (HaCaT) cells with 10 ng/ml IL-1α for 24 hours led to a significant upregulation of NGAL mRNA (6.5 fold). This upregulation was both dose and time dependent and associated with an upregulation of other antimicrobial proteins (S100A7, S100A8, S100A9 and secretory leukocyte protease inhibitor (SLIP)) [157]. In another study, while primary human keratinocytes had a low basal level of NGAL, its expression was upregulated by treatment with IGF-1, IL-1β and TGF-α. Maximum induction was observed at 48 hours and a combination of IGF-1 and TGF-α had a synergistic effect compared to either growth factor alone in inducing NGAL expression. Further, treatment with one or both of these cytokines also resulted in a significant increase in the amount of NGAL secreted into the medium. However, treatment with TGF-β, basic fibroblast growth factor (bFGF), IL-6 and TNF-α had no significant effect on the expression of NGAL in these cells [158]. Treatment with EGF led to a slight increase in NGAL expression. These results are intriguing as TNF-α has been shown to induce NGAL expression while EGF has been demonstrated to inhibit NGAL expression in other studies [41,50,155]. The mechanisms for these differential effects to the same stimulus is unknown, but could be related to the differential expression of the cognate receptors or other downstream effectors. Calcium is a known factor that influences differentiation of primary human keratinocytes (PHKs). When PHKs are maintained in low calcium (30 nM) containing media, they do not differentiate. However, when the same is increased above 100 μM, they begin to differentiate and express markers of terminal differentiation (like involucrin, loricrin and keratins 1 and 10) [168]. Treatment of PHKs with 1.2 mM calcium induced their differentiation together with a strong induction of NGAL beginning on day 3 [169]. Differentiated keratinocytes form the most superficial layer of the human skin and constitute the body’s first line of defense against microbial invasion. It is possible that the induction of NGAL represents a step in the process of preparing the cells to counter microbial pathogens. Treatment of cultured NHEKs with both gram positive (lipotechoic acid and peptidoglycan) and gram negative (lipopolysaccharide) cell wall components led to a significant upregulation of NGAL mRNA and protein expression (while unstimulated cells showed no NGAL mRNA or protein) [170]. To investigate the pathways regulating LCN2 expression in HCC cells, Huh7 cells (express LCN2 under basal conditions) cells were treated with identical concentrations of several inflammatory cytokines including IL-1β, IL-2, IL-4 and TGFβ1 for 24 hours. Of these, only IL-1β elicited a strong induction of LCN2 mRNA and protein [154].
Activator protein 1 (AP-1) is a transcription factor complex (both homo and heterodimers) comprised of members of four transcription factor families-JUN, FOS (FBJ murine osteosarcoma viral oncogene homolog), ATF (activating transcription factor) and MAF (musculoaponeurotic fibrosarcoma) (reviewed in [171]). Upon occurrence of a wound on the skin, the keratinocytes (the predominant cell type found in the epidermis or outer layer of the skin) release the cytokine IL-1. This in turn induces the formation of a dimer between the AP-1 subunits c-Jun and JunB in skin fibroblasts and AP-1 mediated transcriptional activation or repression. The co-ordinate action of keratinocytes, fibroblasts and immune cells helps to close the wound. Analysis of Lcn2 mRNA expression in JunB and c-Jun deficient MEFs revealed an elevated basal Lcn2 expression in the JunB−/− MEFs suggesting that under basal conditions JunB is a negative regulator of Lcn2 expression. However, silencing of c-Jun (c-Jun−/− MEFs) had no effect on the basal expression of Lcn2. Treatment of the MEFs with IL-1 however significantly induced the expression of Lcn2 in both wild-type and in JunB and c-Jun deficient cells. Given that AP-1 is a target of IL-1 signaling pathway through JNK activation, a possible mechanism that has been suggested is that IL-1 co-activates both the JNK MAPK and the NF-κB pathways, and that a crosstalk occurs between the two pathways at the level of the target gene promoters (in response to IL-1 stimulation), thus permitting an upregulation of Lcn2 (via NF-κB) [172].
TH17 cells are a subtype of CD4+ helper T-cells that have been found to play an important role in the pathogenesis of several autoimmune diseases (e.g. collagen induced arthritis) and inflammatory conditions (e.g. experimental autoimmune encephalitis) [173,174]. These cells also produce two cytokines IL-17A and IL-17F, both of which positively regulate NGAL expression. While IL-17A binds to its cognate receptor (IL-17R) and activates many of the classical receptor tyrosine kinase pathways (MEK-ERK, JNK and p38), JAK/STAT and protein kinase C, IL-17F does not bind to the receptor. However, the IL-17R has been shown to be required for the functioning of both cytokines. Both IL’s stimulate the release of pro-inflammatory cytokines IL-6, IL-8 and G-CSF. TRAF6, an E3 ubiquitin ligase has been demonstrated to bind IL-17R and ubiquitinate it in response to IL-17F (but not IL-17A). Further, a dominant negative form of TRAF6 significantly decreased IL-17F (but not IL-17A) induced Lcn2 transcription. These results suggest that, in response to IL-17F, TRAF2 is recruited to and ubiquitilates the IL-17R, followed by increased transcription of target genes like Lcn2 [175]
Lcn2 expression has been demonstrated to be induced by the anti-inflammatory glucocorticoid dexamethasone [35]. A nearly 12-fold upregulation of NGAL mRNA was observed when murine chondrocytes were treated in vitro with 1μM dexamethasone for 24 hours. Another steroid hormone demonstrated to regulate NGAL expression is estrogen. SAGE analysis revealed the presence of NGAL tags in non-neoplastic luminal human mammary epithelial cells expressing the estrogen receptor (ER), while these tags were not detected in SAGE libraries generated from ductal carcinoma in situ and invasive breast cancer tissues [143]. Further investigation revealed that all of the ER positive breast cancer cells (BT20, T47D, BT474, ZR75-1 and MCF-7) expressed low to undetectable levels of NGAL, while it was strongly expressed in 5/12 ER negative cell lines. Lcn2 levels were slightly upregulated following estrogen administration to virgin ovariectomized mice, while a marked upregulation was noted in lactating mice. The Lcn2 levels were also observed to increase progressively in the mammary gland of pregnant mice during the progression of pregnancy, suggesting a strong link to hormone mediated regulation of Lcn2 expression in vivo. An estrogen response element (ERE) was identified in the NGAL promoter of both Lcn2 (−2503 to −2491) and NGAL (−823 to −811). Interestingly, this promoter showed considerable activity even in the absence of exogenous estrogen (in presence of ER) and this activity could be abrogated by estrogen receptor antagonists [143].
The role of iron in mediating the beneficial (and deleterious) effects of NGAL has been controversial. Deferoxamine, an iron chelating agent was observed to significantly increase Lcn2 mRNA and protein levels in the kidneys of a mouse model of CKD (discussed earlier [176]). While this correlated with worsening of the renal disease in the FVB/N mice, there was no difference in iron staining in the kidneys of the Lcn−/− and wild type mouse suggesting that modulation of iron levels might not be a mechanism underlying alteration in Lcn2 levels in mice (and patients) with CKD [66] .
The epidermal growth factor receptor-1 (EGFR-1 or EGFR) has been demonstrated to be involved in the development and progression of several benign and malignant diseases (The biology of EGF receptors and the significance of EGFR mediated signaling pathways in cancer has been well reviewed in several articles [177,178]). We noted that in pancreatic cancer cells, treatment with epidermal growth factor (EGF), the ligand for EGFR significantly downregulated NGAL mRNA and protein expression (Figure 6). This was associated with a downregulation of the epithelial marker E-cadherin. EGF-mediated downregulation of NGAL appeared to involve an upregulation of the transcription factor Zeb-1 (a negative regulator of E-cadherin) and a repression of NF-κB mediated transcription from the NGAL promoter. In contrast, treatment of mouse renal tubular cells with EGF in vitro led to a significant upregulation of Lcn2 expression. Further, inhibition of EGFR prevented the anticipated rise in Lcn2 levels following nephron reduction in a mouse model of human CKD, while mice expressing a dominant negative form of EGFR had significantly reduced severity of kidney lesions together with a decrease in the levels of Lcn2 mRNA and protein (compared to mice expressing the wild type EGFR) [66]. EGF induced Lcn2 expression was found to be mediated through stabilization of the transcription factor hypoxia inducible factor-1 alpha (HIF-1α). These studies suggest that the mechanisms regulating NGAL are quite complex. The effect of a particular regulatory factor appears to involve tissue and disease specific mechanisms which may reflect the physiologic or pathobiologic role of altered NGAL expression in that tissue.
Figure 6. Dual regulation of NGAL expression by EGFR.

(A) In pancreatic cancer cells, treatment with recombinant EGF results in a downregulation of both NGAL mRNA and protein. The mechanism involves upregulation of Zeb1, a transcription factor that inhibits E-cadherin synthesis. E-cadherin helps maintain NGAL expression in pancreatic cancer cells by activating NF-kappaB mediated transcription of NGAL. (B) In renal tubular epithelial cells, EGF treatment leads to a significant upregulation of NGAL mRNA and protein. This occurs through an upregulation of the transcription factor HIF-1 alpha, which in turn promotes transcription of NGAL. This induction of HIF-1 alpha is specific to EGF and independent of hypoxia.
NGAL expression is also regulated by hormones, chiefly estrogen. In vitro treatment of aortic segments from ovariectomized mice with 17-β estradiol (E2) led to a significant (~4-fold) downregulation of Lcn2 mRNA levels [179]. However, in vitro treatment of breast cancer cell lines selected for positive estrogen receptor (ER) expression with 10 nM estradiol led to a significant upregulation of Lcn2 mRNA after 24 hours [143]. SAGE analysis revealed that normal breast epithelial cells expressing the estrogen receptor had significant levels of Lcn2 expression compared to the estrogen receptor negative breast cancer cells. NGAL mRNA was also highly expressed in organoids (breast ducts composed of luminal and myoepithelial cells). While Lcn2 was expressed in 5/2 ER negative breast cancer cells, its level was significantly lower than that in normal epithelial cells and organoids. Estrogen also appears to regulate the expression of Lcn2 in vivo. Lcn2 levels were significantly decreased in the mammary glands of ovariectomized virgin mice compared to animals with intact ovaries. Further, supplementation with estrogen led to a restoration of Lcn2 protein levels to nearly that of the non-ovariectomized controls suggesting that ovarian steroids regulate Lcn2 expression in vivo. An analysis of Lcn2 levels in the mammary glands during gestation and lactation revealed that the strongest expression was observed in the lactating mammary gland. Lcn2 levels also increased progressively during pregnancy, however the exact role of the complex hormonal and physiological changes in regulating Lcn2 expression, the sites of Lcn2 upregulation in the mammary gland and the significance of its upregulation remain unanswered [143]. Further, the expression of Lcn2 was completely lost from the uteri of ovariectomized mice (compared to non-ovariectomized controls) suggesting the dependence of Lcn2 expression on ovarian steroid hormones [143]. The role of hormones in regulating Lcn2 expression is further evidenced by the observation that while Lcn2 was not expressed in murine uterine tissue in the diestrous and metestrous phases, it was strongly expressed in the estrous and proestrous phases of the estrous cycle. The diestrous and metestrous phases are characterized by the formation of the corpus luteum while the proestrous and estrous phases occur under the influence of ovarian estrogens. The uterine luminal fluid was also found to express high levels of Lcn2 possible through secretion of the protein into the lumen. In situ hybridization and immunohistochemistry both revealed that Lcn2 was expressed by the luminal epithelium and glandular epithelial cells of the proestrous endometrium (but not that in diestrous and metestrous). Further, Lcn2 expression was restricted to the epithelium, being absent from the stroma and myometrium [36]. A strong expression of Lcn2 was also observed in the luminal epithelium and uterine glands on day 1 of pregnancy. By day 2, the strong Lcn2 staining was limited to the luminal epithelium, and disappeared completely from day 3 onwards. These results suggest that Lcn2 expression is induced in the mouse uteri in a brief window period from just before fertilization to immediately after fertilization. A direct evidence for the regulation of Lcn2 by ovarian estrogens comes from the observation that exogenous administration of either 17-β estradiol (E2) or diethylstilbestrol to sexually immature mice (30 μg/g body weight) leads to a strong upregulation of Lcn2 expression in the luminal epithelium of the treated mice. Further, administration of E2 (but not Progesterone) for 3 consecutive days led to significant restoration of Lcn2 expression in the uterine epithelium of ovariectomized mice. Significantly, when E2 and Progesterone were given together, it led to a disappearance of Lcn2 mRNA expression (compared to the strong upregulation with E2 treatment alone) [36]. These studies suggest that estrogen promotes while progesterone is a negative regulator of Lcn2 expression in the uterus.
Bioinformatics analysis of the NGAL/Lcn2 promoters revealed the presence of an estrogen response element (ERE- from position −823 to −811 (GGTCtCAGTGACC) for NGAL and from position −2503 to −2491 (GGTCACTCTGcCC) for mouse Lcn2) with nearly complete identity to the consensus ERE sequence. Treatment of HepG2 hepatocellular carcinoma cells transfected with a construct bearing the proximal Lcn2 promoter (contains the ERE) led to a significant increase in activity of the downstream luciferase gene. This effect was significantly abrogated upon transfection with the promoter bearing a deletion of the ERE, providing evidence for direct regulation of Lcn2 by estrogen [143]. A clue to the functional relevance of Lcn2 expression in breast epithelial cells came from the observation that incubation of ER- immortalized MCF10A cells with conditioned media from Lcn2 expressing cells led to significant increase in cell growth [143]. This suggests a possible paracrine mechanism underlying Lcn2 mediated cell proliferation.
When endometrial carcinoma cells were treated with 5-aza-cytidine, a demethylating agent, there was a significant induction of NGAL mRNA in those cells that normally express a low basal level of NGAL (HEC1A, HEC1B and KLE). However, NGAL expression was not significantly altered in cells (HHUA, Ishikawa, and RL-95-2) that had a high basal expression of the gene [77].
Tissue engineering, a technique to replace diseased and damaged tissues with new tissue has witnessed significant advances in recent years. Bone marrow derived mesenchymal stem cells (BMSCs), which can differentiate into bone cells (osteoblasts), cartilage (chondrocytes), fat (adipocytes) and muscle cells (myoblasts) have been the most attractive sources for synthesizing these tissues. When rat BMSCs were stimulated in vitro to undergo osteogenic differentiation, Lcn2 was one of the 12 genes that showed at least a 2.5 fold or higher upregulation in the differentiating (vs. the undifferentiated) BMSCs [111]. Several secreted proteins including Insulin like growth factor (IGF), fibroblast growth factor (FGF), Wnt, Hedgehog, TGF-β, Runx2 and osterix have been demonstrated to be involved in the differentiation of BMSCs into osteoblasts [180]. While specific role of Lcn2 in BMSC differentiation are yet to be elucidated, it raises exciting possibilities including modulation of Lcn2 expression to achieve a specific line of stem cell differentiation.
Cancer stem cells (CSCs) or tumor initiating cells (TICs) comprise an extremely small sub-population of cells within the tumor mass of predominantly poorly differentiated solid tumors that are characterized by the properties of long-term self-renewal and high degree of chemoresistance. Identification, characterization and elucidation of CSCs and the molecular pathways in them is crucial to understand the process underlying both tumor formation and maintenance. CSCs are divided into two sub-populations- side population (SP) and the non-side population (NSP). The former have a characteristic ability to actively pump out the Hoechst 33342 dye (a vital DNA dye) while the latter lack this property. Further, SP cells have been shown to possess the ability to give rise to both SP and non-SP cells, thus suggesting that these are in fact “multipotent cancer stem cells” [112]. SP cells isolated from the human squamous cell carcinoma cell line A431 revealed a significant downregulation (2-fold) of LCN2/NGAL compared to the NSP cells by microarray analysis. In vitro, the SP cells grew significantly faster while in vivo they formed significantly larger tumors than the NSP cells [113]. The functional role of NGAL in CSCs and the mechanics underlying the regulation of its expression (in them) however remains a mystery.
Several single nucleotide polymorphisms are also known to occur in the human NGAL mRNA that can alter the amino acid sequence of the protein and in the NGAL promoter that can alter the transcription factor binding sites. The significance of these SNPs has not been explored so far. Whether they contribute to an altered function of the protein or are responsible for the differential expression of the protein in health and disease will be an important question to answer in future studies.
MicroRNAs, small, 11–25 nucleotide RNAs have emerged as novel, negative regulators of gene expression. While it is not yet known if one or more specific microRNAs target NGAL, a few candidates exist. Search of the available microRNA target prediction tools reveals that NGAL can be targeted by hsa-miR-491-5p (human), mmu-miR-761 (mouse), rno-miR-125a-3p (rat) (www.microrna.org), hsa-miR-138 (www.targetscan.org), hsa-miR-17, hsa-miR-92a (human) (miRWalk: http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/index.html), hsa-miR-5589-3p, hsa-miR-374b-3p, hsa-miR-3166 (human) and rno-miR-92a-2 (rat) (MiRtarget2: http://mirdb.org/cgi-bin/search.cgi). Experimental evidence to link one or more of these candidate microRNAs to regulation of NGAL expression however is pending.
5. ROLE OF NGAL IN CELLULAR SIGNALING
While NGAL expression is modulated by a wide array of exogenous factors as detailed in the previous section, some insight has been gained in recent years into the role of NGAL itself in modulating intracellular signaling events. A literature search for interactors of NGAL has revealed that several proteins have been suggested to interact with NGAL (summarized in Table 6).
Table 6.
Summary of proteins interacting with NGAL
| Interacting Protein | Function(s) |
|---|---|
| Matrix Metalloprotease 9 (MMP9) | Degrades Collagen IV and V. Involved in IL-8 induced mobilization of hematopoietic cells from bone marrow (rhesus monkeys). |
| Cystatin C (CST3) | Belongs to the cystatin family of cysteine protease inhibitors, gene mutation is associated with amyloid angiopathy, found in nearly all body fluids. Decreased expression observed in the vascular wall in atherosclerosis and aortic aneurysms, possibly contributing to formation of plaques. |
| Interleukin 18 (IL18) | IL1B is a pro-inflammatory cytokine that induces IFN- mediated T-cell production. Combination of IL1B and IL12 inhibits IL4 dependent IgE and IgG1 production and enhances IgG2a production by B-lymphocytes. IL1B binding protein interacts with IL1B and negatively regulates its activity. |
| Hepatitis A Virus Cellular Receptor 1 (HAVCR1) | Acts as a cell membrane associated receptor for human hepatitis A virus (HHAV). Suggested to reduce severity of the allergic reaction in asthama and atopy. The protein contains an MTTVP sequence that has been shown to confer protection against atopy in HHAV seropositive individuals. |
| Tumor Necrosis Factor (TNF) | TNF is a pro-inflammatory cytokine secreted predominantly by macrophages. Acts by binding to its receptors TNFR1 and 2 and modulates cell proliferation, differentiation, apoptosis, lipid metabolism and coagulation. Suggested to be involved in pathogenesis of multiple diseases including autoimmune diseases, insulin resistance and cancer. Knockout studies in mice suggest a neuro-protective role. |
| Interleukin 1β (IL-1β) | IL-1B is a pro-inflammtory cytokine and regulates cell proliferation, differentiation and apoptosis. Induces expression of cycloxygenase-2 (PTGS2/COX2) in the central nervous system that contributes to hyper-senstivity to pain in acute inflammatory conditions. |
| Nuclear Factor of Kappa Light Polypeptide Gene Enhancer In B-Cells (NFKB1) | Synthesized as a 105 kDa protein that undergoes cotranslational processing in the 26S proteasome to produce a 50 kDa protein. The 105 kD protein is a Rel protein-specific transcription inhibitor while the 50 kD protein is the DNA binding subunit of NF-kB protein complex. Possesses transcription regulatory function that is activated by several intra and extra cellular stimuli including cytokines, oxygen, free radicals, UV radiation, bacterial and viral products. Inappropriate activation is associated with inflammatory diseases while persistent inhibition leads to inappropriate immune cell development or delayed cell growth. |
| NFKBIZ | Induced by Lipopolysaccharide produced by Gram negative bacteria. Activates transcription of the pro-inflammatory cytokine IL-6 in response to LPS. |
| Interleukin 6 (IL-6) | IL6 is a pro-inflammatory cytokine that also regulates B-cell maturation. Produced in both acute and chronically inflamed tissues and secreted into the serum. Induces transcription of inflammatory response genes via IL6 receptor alpha (IL6Rα) binding. Involved in several inflammatory diseases and suggested to modulate susceptibility to diabetes mellitus. |
| Retinol Binding Protein 4 (RBP4) | Lipocalin family protein that transports retinol from the liver to the peripheral tissues. RBP4 interacts with transthyretin in plasma, thus preventing its own excretion through the kidneys. Secretion is blocked in case of retinol deficiency. |
| C-Abl Oncogene 1 (ABL1) | Encodes a tyrosine kinase that has been implicated in regulating cell division, differentiation, adhesion and response to stress. Possesses an SH3 domain that negatively regulates Abl1 protein function. Deletion of this domain turns this protein into an oncogene. The T(9;22) translocation results in the head-to-tail fusion of the BCR (MIM:151410) and ABL1 genes present in many cases of chronic myelogeneous leukemia. |
| Matrix Metallopeptidase 2 (MMP2) | 72 KDa gelatinase that degrades collagen IV present in the basement membrane. Plays an important role in role in endometrial menstrual breakdown, regulation of vascularization and the inflammatory response. Mutations in this gene have been associated with Winchester syndrome and Nodulosis-Arthropathy-Osteolysis (NAO) syndrome. |
| Pentraxin-Related C- Reactive Protein (CRP) | Member of pentaxin family of proteins and is involved in host defense mechanisms. Recognizes foreign pathogens and damaged host cells and initiates their elimination by interaction with host immune effector cells in the blood. |
| S100 Calcium Binding Protein A9 (S100A9) | Regulation of cell cycle progression and differentiation. Inhibits function of the enzyme casein kinase and its deficiency is associated with cystic fibrosis. |
| Interleukin 3 (IL-3) | A potent growth promoting cytokine that supports proliferation of a broad range of hematopoeitic cell types. Regulates cell growth, differentiation, apoptosis and possesses neurotrophic activity. |
| Interleukin 8 (IL-8) | Member of CXC family of chemokines, major mediator of the inflammatory response Secreted by several cell types, it functions as a potent chemoattractant and pro- angiogenic factor and believed to be a key player in the pathogenesis of viral bronchiolitis. |
| Insulin (INS) | Potent activator of glucose uptake. |
| Interleukin 17A (IL17A) | Pro-inflammatory cytokine produced by activated T-lymphocytes. Regulates the activities of NF-kappa B and Mitogen activated protein kinase. Stimulates transcription of other cytokines (IL6), cyclooxygenase-2 (COX-2) and nitric oxide (NO). Elevated expression of IL17A is observed in several inflammatory diseases including rheumatoid arthritis, psoriasis and multiple sclerosis. |
| Myeloperoxidase (MPO) | A heme protein and major component of neutrophil azurophilic granules. Produced as a single chain precursor that is cleaved into a light and a heavy chain. Mature MPO is a tetramer of two light and two heavy chains. Produces hypohalous acid that is essential for the microbicidal activity of neutrophils. |
| Liver Fatty Acid Binding Protein 1 (FABP1) | Belongs to the family of conserved cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands (FABP1 and FABP6 also bind bile acids). Regulates fatty acid uptake, transport and metabolism. |
| Hepcidin Antimicrobial Peptide (HAMP) | Maintains iron homeostasis by regulating iron storage in macrophages and intestinal absorption of iron. Synthesized as a pre-pro-protein that is post-translationally cleaved into mature peptides which in turn form intramolecular bonds that stabilize their beta sheet structures. The peptides exhibit anti-microbial activity. Mutations in this gene causes hemochromatosis type 2B (juvenile hemochromatosis) characterized by severe iron overload that results in cardiomyopathy, cirrhosis, and endocrine failure. |
| Proteinase 3 (PRTN3) | No summary found. |
| Secreted Phospho-protein 1 (SPP1) | No summary found. |
| Secretory Leukocyte Peptidase Inhibitor (SLPI) | Protects epithelial tissues from endogenous serine proteases and present in seminal plasma, cervical mucus and bronchial secretions. Has affinity for trypsin, leukocyte elastase and cathepsin G and suggested to have broad-spectrum antibiotic activity |
| Chemokine (C-C Motif) Ligand 2 (CCL2) | A secretory protein related structurally to the CXC family of chemokines. Acts as a potent chemoattractant for monocytes and basophils. Involved in the pathogenesis of diseases characterized by monocytic infiltration including psoriasis, rheumatoid arthritis and atherosclerosis. Binds to the chemokine receptors CCR2 and CCR4 |
5.1 Effects of NGAL silencing on cell signaling
Considerable insight into the effect of NGAL on intracellular pathways has been gained through studying the effects of altered NGAL expression- either ectopic expression or silencing of the gene. Stable silencing of NGAL expression in FRO thyroid cancer cells for instance led to activation of both initiating (2 and 9) and execution caspases (3 and 7) associated with an increase in apoptosis under serum deprived conditions. A similar pattern i.e., activation of pro-apoptotic proteins together with a decrease in mitochondrial membrane potential and enhanced release of cytochrome c was also observed when parental FRO cells were cultured in the presence of the iron-chelator deferoxamine (DFO). The importance of iron in modulating NGAL regulated apoptosis was further proven as antagonism of DFO induced apoptosis by the exogenous addition of ferric chloride (an iron donor). Further, colorimetric analysis revealed a decrease in intracellular iron concentration in NGAL silenced cells suggesting that NGAL may bind to extracellular iron and transport it into the cell, where it might help maintains mitochondrial membrane potential and thereby ensure cell survival [142].
Autophagy, a process by which a cell consumes its own cellular material is used by all cells as a housekeeping process to clear away defective cellular components and aggregated proteins. In cancer cells, autophagy appears to be a double edged sword. On one hand, it may allow cancer cells to survive conditions of nutrient deprivation, hypoxia and oxidative stress created by chemotherapy and radiation treatment. On the other hand, it has pro-apoptotic functions as evidenced by a strong suppression of tumor formation upon deletion of autophagy specific genes like beclin1 [181]. Autophagy also appears to regulate the function of mitochondria, the energy powerhouses of a cell. Knockout of both alleles of Lcn2 in mice was associated with a decrease in the oxidative activity in the mitochondria, suggesting a possible role of Lcn2 in regulating mitochondrial function. Mouse embryonic fibroblasts (MEFs) in which the endogenous Lcn2 gene was stably silenced, exhibited a significant decrease in the rate of cell proliferation in vitro. Further analysis revealed that the Lcn2 deficient MEFs had a significantly lower mitochondrial DNA content than the wild type cells. A possible mechanism for the decreased mitochondrial biogenesis was uncovered when it was discovered that there was a significant decrease in the expression of PGC-1α (Peroxisome proliferator-activated receptor gamma coactivator-1 alpha), a co-activator that regulates mitochondrial biogenesis. Consistent with the decrease in mitochondrial DNA, there was also a significant reduction in the amount of mitochondrial COX-2 (cytochrome c oxidase-2) mRNA, reflecting a decrease in mitochondrial function. Additionally, there was a significant decrease in the expression of LC3 (microtubule associated protein 1 light chain 3), a marker of autophagy, in the Lcn2 deficient MEFs suggesting a defect in the autophagic process. Activation of NF-κB is an important pathway that inhibits autophagy in cancer cells, particularly that induced by cytokines like TNF-α [182]. The Lcn2 deficient cells exhibited a significant increase in the rate of degradation of IκBα (the inhibitor whose degradation is an important pre-requisite to the activation of NF-κB), suggesting that the NF-κB pathway is activated in Lcn2 silenced cells and may be an important mediator of Lcn2 regulated autophagy. Insulin-receptor substrate-1 (IRS-1), whose degradation correlates positively with tumor cell proliferation (in breast and lung cancer [183,184]) was also observed to accumulate in the Lcn-2 null MEFs suggesting another mechanism for Lcn-2 mediated inhibition of cell proliferation. Interestingly, re-introduction of Lcn2 into the Lcn2 deficient MEFs failed to restore either the expression of LC3, or block TNF-α induced degradation of IκBα, suggesting that while Lcn2 does mediate proliferation, mitochondrial biogenesis and autophagy in MEFs, this might actually be an indirect effect.
Secreted NGAL is known to form a complex with the enzyme matrix metalloproteinase-9 (MMP-9), a gelatinase first identified in neutrophils as an enzyme that extensively degrades gelatin and collagen V (but not collagen I) [185]. Analysis of broncholalveolar lavage (BAL) fluid from patients with emphysema (n=10) and control subjects (n=11) revealed that a complex of NGAL and MMP-9 was expressed at a significantly higher level in emphysematous patients but not in the control subjects [186]. Neutrophils and macrophages have been suggested as the potential sources of the NGAL-MMP9 complex in the BAL of emphysematous patients. NGAL forms a covalently linked complex with the inactive (proenzyme) form of the metalloproteinase MMP-9. While this does not affect the activity of the enzyme, it stabilizes it by protecting it against inactivation by its natural inhibitor TIMP-1 (Tissue inhibitor of metalloproteinase-1). The mechanism of NGAL mediated stabilization of MMP-9 involves the binding of NGAL with pro-MMP9 followed by the formation of a ternary complex between the C-terminal hemopexin domain of MMP-9 and the non-inhibitory C-terminal domain of TIMP-1. In contrast, the binding of the N-terminal domain of TIMP-1 to the active site of MMP-9 inhibits its gelatinolytic activity [187]. This property of NGAL to stabilize MMP-9 has been suggested to be responsible for its function as a modulator of the invasiveness of malignant cells through the basement membrane [145]. The functional effect of NGAL-MMP-9 interaction on the pathogenesis and progression of emphysema and other chronic inflammatory diseases however remains to be elucidated.
5.2 Effect of ectopic expression of NGAL on intracellular signaling
Ectopic expression of Lcn2 in murine mammary carcinoma cells led to a decrease in phosphorylation of Akt (at both Serine-473 and Threonine-308 residues) and a decrease in phosphorylation of PTEN while the level of phosphorylated ERK remained unchanged [144]. When expressed in Chinese hamster ovary (CHO) and human embryonic kidney (HEK293T) cells, NGAL upregulated the expression of the antioxidant enzymes HO-1, SOD-1 and SOD-2 together with an upregulation of NF-κB [138]. This is interesting as it suggests that NGAL and NF-κB may regulate each other. Whether a feedback loop exists to mutually regulate their expression would certainly be interesting to explore. An insight into the pathways regulated by NGAL in HCC cells came from the observation that ectopic expression of the gene significantly inhibited the activation of c-jun N-terminal kinase (JNK) and PI3K/Akt pathways in the HCC cells. Studies using pharmacological inhibitors further revealed that blocking the JNK and PI3K pathways, but not the ERK or p38 MAPK pathways significantly inhibited proliferation and invasion in the HCC cells and decreased their expression of MMP2 [154].
5.3 NGAL receptors
While it is known that exogenous NGAL (or Lcn2 in mouse) can produce effects similar to that produced by overexpression of the protein, it is not yet clear if there is a definite receptor for NGAL. Two candidate proteins have been however come to the forefront. The first, termed as solute carrier family 22 member 17 (SLC22A17 or Lcn2R) has been demonstrated to bind to mouse Lcn2 while the second termed as low density lipoprotein receptor related protein (LRP2 or megalin) has been shown to bind human NGAL protein.
Slc22a17, like its ligand Lcn2 has homologues in other species. Unlike LCN2 however, human SLC22A17 is nearly 97% identical in amino acid sequence to the mouse and rat proteins. Two isoforms of human SLC22A17 are known, namely isoform a and b. The two are nearly 99% identical in their sequence except for a short stretch of 18 amino acids (EHPIFPTVWAQQGNPNRD) present in the longer isoform. SLC22A17 shares relatively poor sequence identity (38%) to homologous proteins from invertebrate species like the zebrafish. Alignment of SLC22A17 homologues by degree of conservation of their amino acid residues reveals conservation of certain hydrophobic amino acids (chiefly Valine, Leucine, Isoleucine, Phenylalanine, Tryptophan and Cysteine). The significance of these hydrophobic residues in the interaction of SLC22A17 with the hydrophobic calyx of NGAL remains to be investigated.
LRP2 (or megalin) also has homologues in other species including rodents, chimpanzees, zebrafish and Drosophila. Significantly, its amino acid sequence appears to be highly conserved among higher mammals, being 99% identical to the homologue in chimpanzees. In other mammals however, sequence identity is much lower – about 76% identities to its mouse and rat homologues. Similar to the SLC22A17 however, LRP2 too shares little similarity in sequence with the homologues present in invertebrates. Multiple sequence alignment of the amino acid sequence of various LRP2 homologues reveals short stretches of amino acids, particularly several cysteine residues, that are conserved across all species. Given that NGAL utilizes an unpaired cysteine residue (Cys87) to form a disulfide bond with MMP-9, the significance of these conserved residues in binding of megalin (LRP2) to NGAL will be an important question to answer in future studies.
Based on the degree of conservation of the NGAL receptors across species, we can hypothesize that SLC22A17 which is highly conserved in all mammals may mediate critical functions of NGAL (e.g. immune response, neutrophil maturation) LRP2 on the other hand shows a stronger conservation between related species (e.g. between human and chimpanzee, rat and mouse) suggesting that it may mediate different effects in lower (e.g. mouse and rat) vs. higher mammals (humans and chimpanzees).
6. NGAL AS A DIAGNOSTIC AND PROGNOSTIC MARKER
Recent years have seen a tremendous increase in the number of studies that have investigated NGAL as a biomarker for both diagnosis and prognosis. Its secreted nature and the availability of commercially available robust immunoassays have contributed to NGAL emerging as a potential biomarker in a wide array of benign and malignant human diseases.
6.1 Benign diseases
6.1.1 Inflammatory diseases
In one of the earliest studies on the diagnostic potential of NGAL, a strong (nearly 10-fold) upregulation of NGAL was observed in the plasma of patients suffering from acute generalized peritonitis (n=37, mean peritoneal fluid NGAL being 37.2±6.2 μg/ml) [20]. There was a good linear correlation between the levels of NGAL and that of leucocyte elastase (r2=0.7, p=0.00011) and with NP4 (r2=0.62, p=0.0001). Plasma NGAL levels were also nearly 3-fold higher during acute peritonitis (791ng/ml±130 ng/ml) compared to that during convalescence (276ng/ml± 42ng/ml) and nearly 10 fold higher than the mean levels in healthy individuals. Significantly, by the end of the first post-operative week, the plasma levels of all three markers (NGAL, NP4 and leucocyte elastase) showed a downward trend towards normal levels.
Sepsis, particularly when associated with multi-organ dysfunction (combination termed as “severe sepsis) is associated with significantly higher mortality (12% vs. 0.9% in those without severe sepsis in one study [188]). A large multicenter prospective study (total 971 patients meeting inclusion criteria) investigating a multi-biomarker panel (nine potential markers selected from an initial set of about 150 potential markers) to diagnose “severe sepsis” at an early stage in adult patients found that a combination of plasma NGAL, interleukin-1 receptor antagonist (IL-1ra) and protein C was the best predictor of severe sepsis [188]. Patients were included in this study if they were 18 years or older and presented to the emergency department (ED) with a presumptive source of infection or had an elevated serum lactic acid (>2.5 mM) and met at least two criteria for “severe inflammatory response syndrome” (SIRS) (temperature >38°C or <36°C; respiratory rate >20/minute; partial pressure of carbon dioxide <32 mm Hg; heart rate >90/min; white blood cell count >12,000/cc or <4,000/cc and >10% immature neutrophils). Severe sepsis was defined as the presence of a suspected infection together with evidence of organ dysfunction including either renal (increase in serum creatinine of >0.5 mg/dl between two measurements, requirement of acute dialysis or an initial creatinine >2mg/dl with a decrease of >50%), cardiac (systolic pressure<90 mm Hg, mean arterial pressure <70 mm Hg or need for vasopressors). pulmonary (oxygen saturation <90%, a PaO2/FiO2 ratio <300 or the need for mechanical ventilation) hepatic (total bilirubin >4mg/dl), metabolic (serum lactic acid >1.5 times the upper limit of normal) or hematologic (platelet count <100,000/cc on a single measurement, prothrombin time >15 seconds, active partial thromboplastin time >60 seconds or an international normalized ratio (INR) >1.5) dysfunction. Employing this definition, 506 (52.1%) patients had severe sepsis, 359 of whom met the criteria at the time of presentation and 140 who developed severe sepsis within 72 hours after presentation to the ED. Both mean and median plasma NGAL levels were significantly higher in patients who developed severe sepsis at some point during the study (381 ng/ml and 249 ng/ml respectively) compared to those who did not meet criteria for severe sepsis (144 ng/ml and 71 ng/ml respectively). Plasma NGAL alone was fairly accurate in discriminating between these two groups of patients (AUC 0.75, 95% C.I. 0.72–0.78). Using multivariate regression, a model was constructed using the quartile values each of the three markers. This model when applied to critically ill patients was a good predictor of the risk of developing septic shock (within the next 24 hours) or death (AUC being 0.77 and 0.79 respectively). The mean “septic score” created using the tri-marker panel (NGAL, IL-1ra and Protein C) was observed to progressively increase with increasing severity of illness- from no organ dysfunction (37.7), through severe sepsis (59.0) to septic shock (64.9) and finally death (75.7). Interestingly, among patients who did not initially present with sepsis, the “septic score” was significantly higher in those who subsequently progressed to severe sepsis (56.3) compared to those that did not (38.0). In a multiple regression analysis, the septic score remained the most significant independent predictor of severe sepsis among patients without organ dysfunction at the time of presentation to the ED (odds ratio 1.51, 95% C.I. 1.25–1.51).
Crohn’s disease is a chronic inflammatory disease of the gastrointestinal tract characterized by periodic remissions and relapses with a tendency to progress leading to several complications including luminal strictures, abscesses and fistulas. Infliximab, an IgG1 monoclonal antibody against TNF-α has been shown to be highly effective in the management of CD patients, particularly in combination with azathioprine [189]. A small study comprising 6 patients with CD and 6 healthy controls observed a significantly higher urinary NGAL level in patients with CD which showed a nearly 60% reduction following administration of a single dose of Infliximab [190]. A larger study investigating the changes in the expression of genes encoding antimicrobial proteins in inflammatory bowel disease (IBD) following treatment with Infliximab noted that NGAL expression was strongly upregulated in the diseased colonic mucosa in untreated ulcerative colitis (UC, 18 fold) and in colonic Crohn’s disease patients (CDc,13 fold upregulation) compared to the normal colon mucosa [191]. However, there was no significant difference in expression between untreated UC and CDc mucosae. Patients who did not respond to the treatment (assessed by histopathological examination after 4–6 weeks) showed persistent elevation of NGAL mRNA in the diseased (11-fold and 13-fold higher in UC and CDc respectively compared to healthy colon tissue). In comparison, NGAL levels were significantly decreased in the affected areas of the colonic mucosa in patients who responded to infliximab therapy (5-fold and 2.5 fold downregulation in UC and CDc compared to pre-treatment expression levels). While the level of NGAL expression was not significantly different between ileal (CDi) and colonic (CDc) Crohn’s disease before starting Infliximab, they were nearly 22 fold higher in untreated CDi compared to the normal ileum. After treatment, NGAL decreased in CDi patients who responded to the therapy, although they were still nearly 11-fold higher in the affected ileum compared to healthy ileal tissue. CDi patients who did not respond to treatment (termed as non responders) had a nearly 16-fold higher NGAL expression in the affected ileal mucosa after Infliximab therapy compared to that prior to the start of therapy. There was a significant positive correlation between NGAL and IL8 expression in both the ileum and colon tissues affected by IBD (r=0.48 in both), suggesting that the two genes may be regulated similarly in this disease [191]. These results suggest that urine NGAL levels may be useful to follow the course of CD. Further, it suggests potential regulation of NGAL by TNF-α. Since CD is also accompanied by an increase in the levels of IL-1 and NF-κB, both potent inducers of NGAL, it is possible that the elevated levels of NGAL represent a general activation of pro-inflammatory pathways rather than a CD specific release from intestinal epithelial cells [190].
6.1.2. Infectious diseases
A study in HIV positive patients reported a significant positive correlation between serum NGAL levels and total neutrophil count in HIV-positive patients both at baseline and at 1 year following the initiation of highly active anti-retroviral therapy (Correlation coefficient 0.22 and 0.64 respectively). However, no such correlation was observed in healthy controls [41]. NGAL levels were also observed to be elevated in induced sputum from patients with chronic obstructive pulmonary disease (COPD) compared to that from patients with bronchial asthma [192]. This elevation in NGAL levels correlated with the higher concentration of neutrophils in the sputum of COPD patients and suggests that quantification of NGAL in induced sputum could be a potentially useful non-invasive marker to distinguish these two chronic inflammatory conditions.
6.1.3. Metabolic diseases
A study conducted in diabetic mice revealed that the circulating levels of Lcn2 are significantly elevated in diabetic mice compared to lean control mice [52]. Significantly, Lcn2 levels were noted to decrease significantly upon treatment with rosiglitazone for two weeks. Among human subjects, serum NGAL levels were significantly higher in obese compared to lean individuals. There was a significant positive correlation between NGAL levels and age of the patient, body-mass-index (BMI), waist-to-hip ratio, waist circumference, percentage of body fat and serum concentrations of insulin, triglycerides and glucose. A negative correlation was also observed between serum NGAL and the levels of HDL cholesterol. Serum NGAL remained an independent predictor of insulin resistance and hyperglycemia, suggesting a possible association with the pathogenesis of type-II diabetes mellitus. Significantly, NGAL levels were higher in females, both lean (BMI <23 kg/m2) and obese (BMI >30 kg/m2), and in older individuals suggesting a possible role for hormones and aging in regulating NGAL expression [52]. In a study conducted on obese Korean women however, baseline serum NGAL levels were marginally higher in obese (defined as individuals with BMI ≥25 kg/m2) women compared to age-matched non-obese women (mean serum NGAL levels being 49.2 ng/ml and 39.2 ng/ml respectively, p=0.054). Further, after a 3 month exercise regimen that included aerobic exercises (45 minutes per session or 300 kcal/day) and muscle strength exercises (20 minutes per session or 100 kcal/day) five times weekly, there was no significant change in the levels of serum NGAL in the obese women (mean serum NGAL 40.8 ng/ml, p=0.11) [193]. Several possibilities exist for the differences observed between these studies including differences between ethnic groups in the baseline production of NGAL and production of different transcript variant that may not be recognized by antibodies used in these assays. Whatever the underlying reason, these studies suggest that NGAL could be involved in lipid metabolism, possibly through paracrine signaling. With dyslipidemia taking on epidemic proportions throughout the globe, understanding the role of NGAL in modulating this process could constitute an important avenue for development of novel pharmacological interventions.
6.1.4. Cardiovascular disorders
Acute decompensated heart failure (ADHF) which is defined as “either a gradual or a rapid alteration in the signs and symptoms of heart failure that necessitate emergent institution of appropriate therapy” constitutes a major public health problem in the United States [194]. According to estimates, nearly 33 billion dollars were spent on the management of acute heart failure in the United States in 2007 alone, of which the majority were patients with ADHF [195] (acute heart failure syndromes have been excellently reviewed in [196]). An important contributor to the morbidity and mortality in ADHF is a decline in renal function (termed as worsening renal function or WRF), defined as a decline in serum creatinine by ≥0.3mg/dl from baseline levels [197]. A single center study involving 91 patients with ADHF observed that patients who developed WRF had significantly higher serum NGAL levels at the time of admission. Further, higher NGAL levels at admission were an associated with a greater risk of developing WRF (odds ratio 1.92, 95% C.I. 1.23–3.12). At a cut-off ≥ 140 ng/ml, serum NGAL was 86% sensitive and 54% specific for predicting the development of WRF in these patients [197]. Previous studies have identified an elevated serum creatinine (≥2.75 mg/dl) in patients with elevated blood urea nitrogen (BUN ≥43 mg/dl) as a significant predictor of in hospital mortality [198–200]. As serum NGAL is a more sensitive early marker of renal dysfunction than creatinine (discussed later), it could supplement or supplant creatinine in prognostication algorithms for patients with acute heart failure syndromes who generally have a high incidence of renal dysfunction (between 20%–30% in two large heart failure registries[198,200]).
Patients with chronic heart failure often suffer from chronically impaired renal function [201]. Estimated glomerular filtration rate (eGFR) is commonly elevated in patients with CKD and is used as a marker of renal impairment. However, it has been hypothesized that even patients with mildly elevated eGFR have an underlying renal dysfunction. In a recent study of 2, 130 patients with chronic heart failure and mildly impaired eGFR (68±21 ml/min/1.73 m2, normal <60 ml/min/1.73m2), urine NGAL levels were observed to be elevated nearly 35 fold (mean: 36 μg/gm of creatinine, normal being <1 μg/gm of creatinine) [202]. Following a mean follow-up period of 2.9 years, 655 (31%) patients reached the prognostic end point (either death from any cause or hospitalizations for heart failure). Urine NGAL (log10 transformed) showed a significant association with all cause mortality (but not hospitalizations for heart failure) both on univariate (HR 1.24, 1.08–1.42) and multivariate analysis (1.23, 1.07–1.41). The study showed that even in patients with mildly raised eGFR, NGAL was a highly sensitive marker of tubular injury although it did not show a significant correlation with other markers of renal tubular damage like KIM-1 (kidney injury molecule 1) and NAG (N-acetyl-beta-D-glucosaminidase). Interestingly, a study by Damman and co-workers demonstrated that unlike other markers of renal injury like KIM-1 and NAG, neither urine nor serum NGAL levels were significantly affected by diuretic therapy (with furosemide) in patients with chronic heart failure [203].
6.1.5. Renal disorders
Urinary NGAL level measurement has become the standard biomarker for both the early diagnosis and predicting the prognosis of patients with renal injury [204]. In one study, urinary NGAL levels were significantly elevated in patients with APKD who showed rapid disease progression compared to those with slower progression of the disease. Urine NGAL levels also correlated inversely with residual estimated glomerular filtration rate (eGFR) and microalbuminuria, two other indicators of adverse prognosis in patients with CKD [66]. Among 38 patients undergoing high risk cardiovascular surgery (thoracic aorta or aortic valve surgery), serum NGAL levels were significantly higher in patients with a higher IL-6 levels 1 hour after surgery. Further, 6 hours post surgery, serum NGAL levels were significantly higher in patients with high IL-10 levels [205]. These patients also had a significantly higher incidence of pulmonary failure (defined as >24 hours time on the ventilator), a longer ICU stay and a longer overall stay in the hospital. A mean arterial pressure (MAP) <50 mm Hg was found to be an independent predictor of a rise in both NGAL and IL-6 1 hour post-surgery suggesting that both cytokines may be released either from damaged endothelium or other extravascular sources in response to cardiovascular stress.
Urine NGAL was fairly accurate (AUC 0.59–0.65) in identifying patients with acute kidney injury (AKI) within 3 hours after cardiovascular surgery [206]. Interestingly, NGAL levels rose in all patients (even those without AKI) in the immediate post-operative period after cardiac surgery and progressively decreased thereafter. A larger cohort of 75 patients aimed at examining the performance of plasma NGAL as a marker of AKI following cardiopulmonary bypass (CPB) observed that although NGAL levels were higher in patients who developed AKI (vs. those who did not develop AKI), it had low sensitivity (38.7%), low positive predictive value (16.3%) but high specificity (81.5%) for predicting AKI [207]. At a cut-off >353.5 ng/ml plasma NGAL was an independent predictor of the risk of AKI (odds ratio 2.3, p=0.002).
In a recent study, the ability of NGAL levels in a single blood sample drawn immediately post-surgery to predict AKI was assessed in pediatric patients. Blood samples were drawn within 30 minutes of admission to the ICU, and within 2–4 hours after completion of cardiopulmonary bypass surgery for a congenital defect. NGAL levels were measured within 6 hours of blood draw using a fluorescence based ELISA using a commercial kit that has a sensitivity range between 60–1300 ng/ml. An analysis of the data showed no significant correlation between plasma NGAL levels and either age (range of age: 3 days-21.1 years), gender, BMI, duration of surgery or serum lactic acid concentration. Among newborns (age ≤10 days), NGAL levels showed a modest correlation with baseline serum creatinine (r=0.47, p=0.02) but not with peak post-operative creatinine levels. In older newborns (>10 days old) however, there was no correlation with baseline creatinine and a much weaker (r=0.21, p=0.003) correlation with peak creatinine levels. 122/218 (46%) patients developed renal failure (stage R or I) as measured by the RIFLE criteria. NGAL levels showed a weak but significant correlation with the severity of renal injury (r=0.15, p=0.03). Plasma NGAL levels were also slightly effective in predicting a rise in serum creatinine (AUC to predict a >50% rise in creatinine being about 0.54) in these patients. The study concluded that measuring plasma NGAL level in a single blood draw was not efficient way to predict renal complications following bypass surgery for a congenital heart defect [208].
The Acute Kidney Injury Network (AKIN) in 2007 introduced a modified system of diagnosing AKI with the aim of increasing the sensitivity of diagnosing early stage renal injury, in turn allowing earlier institution of therapy and decreasing morbidity and mortality [209]. In this system, AKI-3 constitutes severe AKI and is associated with a mortality of upto 50% [210]. One of the drawbacks of current markers of AKI (e.g. creatinine) is that they only measure glomerular damage. However, AKI is also associated with tubular damage, which can be measured by measuring the urinary levels of certain “low molecular weight (LMW) proteins” that appear in the urine due to loss of integrity of the tubular epithelial cell barrier. Some of the common LMW markers of tubular injury include cystatin C, α1-microglobulin, β2-microglobulin, N-acetyl glucosaminidase (NAG) and NGAL [211]. In a single center study among 50 adult patients undergoing elective cardiac surgery with cardiopulmonary bypass, the median urine NGAL levels (corrected for variation in degree of diuresis by multiplying with the ratio of serum to urine creatinine) were significantly higher in patients with AKI (AKIN stages 1–3) compared to those without renal injury within 6 hours of admission to the ICU (post-operatively) [15]. This was in comparison to the pre-operative period when there was no significant difference in urine NGAL (or creatinine) between those with or without AKI. In contrast, neither urinary α1-microglobulin nor cystatin-C levels differed significantly between the two groups of patients. Interestingly, the ratio of serum to urine creatinine appeared to parallel NGAL in its rise during the post-operative period [15]. A similar observation was also reported in a study comparing the levels of various potential AKI biomarkers among 123 patients undergoing cardiac surgery. An elevated urine NGAL (unadjusted for serum creatinine) 6 hours post-surgery was the best predictor of stage 3 AKI among patients undergoing cardiac surgery (AUC 0.88, 95% C.I. 0.73–0.99) [212]. Significantly, the baseline corrected urine NGAL level was a poor predictor of mild AKI (AKIN-1) but a better predictor of severe AKI (AKIN-3) (AUC being 0.49 and 0.71 respectively). Urine NGAL levels measured at the time of arrival to the ICU appeared to be better at predicting AKI than baseline levels (AUC 0.69 and 0.80 for predicting AKIN-1 and AKIN-3 respectively). A smaller study employing 29 patients (14 with AKI and 15 without AKI following cardiopulmonary bypass surgery) observed that urine NGAL (expressed as a ratio to urine creatinine levels) was significantly elevated at the time of admission to the ICU and again after 24 hours [213]. In comparison, serum creatinine levels rose progressively with time in patients who developed AKI (but not in control patients) when followed upto 72 hours after surgery. Retinol binding protein (RBP) and the cytokine IL-18 were also elevated at the time of ICU admission, suggesting that a combination of these markers may be useful in the early detection of AKI following cardiopulmonary bypass surgery [213]. While most studies till date have employed ELISA to assay the levels of urine NGAL, a comparison of ELISA (using a polyclonal-monoclonal antibody combination) with radioimmunoassay (RIA) using a polyclonal antibody revealed that the latter technique was more sensitive- 9.5-fold, 7.5-fold, 6.8-fold, 5-fold, 5-fold and 6-fold higher levels than that measured by ELISA among healthy controls and patients before and 2, 24, 48 and 72 hours following cardiac surgery respectively [214]. There was a significant positive correlation between the levels of urine NGAL measured by RIA and ELISA (average r2 being 0.72, p<0.00001) with the highest correlation observed in samples obtained from patients 24 hours post-cardiac surgery (r2 being 0.9). The study also confirmed that urine NGAL (corrected for creatinine) was a highly sensitive marker of AKI. Further, the levels of urine NGAL were positively correlated to the duration of extracorporeal circulation time, with patients with an extracorporeal circulation (ECC) time greater than 90 minutes having mean urine NGAL levels significantly higher than those with a shorter duration of ECC (7-fold and 4-fold higher urine NGAL levels in the former by RIA and ELISA respectively) [214]. During the process of ECC, the blood anticoagulated with heparin leaves the physiologically non-thrombogenic vascular system of the body and comes in contact with potentially thrombogenic synthetic surfaces of the extracorporeal circulation circuit. This has been demonstrated to promote significant qualitative and quantitative changes in platelets that in turn lead to the release of granulocyte granules. In vitro studies have revealed that passage of blood through the ECC leads to degranulation of neutrophils measured by a progressive rise in the levels of human neutrophil elastase (an enzyme present in azurophilic granules of neutrophils) [215]. This rise was observed as early as 2 minutes following start of ECC and began to show a decline after 24 hours (although it did not reach baseline levels). Significantly, the activation was specifically observed in blood samples exposed to the ECC circuit but not in heparinized blood left standing for the same amount of time. This process could be inhibited by prostaglandin E2 (PGE2) and by the local anesthetic lidocaine [215]. It is possible that a similar mechanism, distinct from renal injury contributes to the rise in urine NGAL levels in patients undergoing cardiopulmonary bypass, particularly those with prolonged ECC-time.
In an attempt to investigate the correlation of urine NGAL levels with clinical characteristics, 100 consecutive patients undergoing coronary angiography in an urban tertiary hospital had urine NGAL levels measured prior to and following the procedure. Pre-procedural urine NGAL levels in this cohort showed a weak but significant positive correlation with urine β2-microglobulin levels and a negative correlation with weight and urine sodium levels (correlation coefficient being 0.28 (p=0.004), −0.22 (p=0.03) and −0.24 (p=0.02)). Further, urine NGAL levels were significantly higher in females while there was no significant correlation with race, smoking status, positive history of diseases like hypertension, diabetes, hyperlipidemia, myocardial infarction or drugs (e.g. ACE inhibitors, Angiotensin-II receptor blockers, statins, aspirin, beta blockers and diuretics). Multiple regression analysis revealed that age and the urinary albumin to creatinine ratio but not gender, weight, serum or urine creatinine, urine sodium or beta β2-microglobulin levels or diuretics were significant predictors of pre-procedural levels of NGAL. Interestingly, there was no clear correlation between pre- and post-procedural urine NGAL levels [216]. These epidemiological studies on one hand suggest that urine NGAL excretion is modified by various patient related factors. At the same time they raise the possibility that several factors may be acting in concert or in contrasting manner to regulate NGAL in these patients. Some potential contributors include pre-procedural level of hydration, co-existing stress leading to induction of cytokines and effect of prior therapeutics. Careful in vitro and in vivo studies are needed to clarify the validity of such epidemiological correlations.
NGAL is not just a marker of renal injury in patients’ post-cardiac surgery but also in other critically ill adult patients. A study examined 301 patients admitted to the ICU for conditions arising primarily from the respiratory, cardiovascular, gastrointestinal or neurologic system or post-trauma. Of these 133 developed AKI, 90 within the first 24 hours and 43 after the first 24 hours, but all within the duration of their stay in the ICU [217]. A time course analysis revealed that plasma NGAL levels (uncorrected for creatinine) rose 24 hours prior to the diagnosis of AKI (by the RIFLE criteria), reached a peak 24 hours post-diagnosis, and began to decline thereafter. Plasma NGAL was a good predictor of the need for renal replacement therapy (RRT) during the stay of the patients in the ICU (AUC 0.82, 95% C.I. 0.70–0.95) and for the development of AKI within 48 hours from the time of NGAL measurement (AUC 0.78, 95% 0.45–0.92). However, it was a relatively poor predictor of AKI developing within the next 5 days and of ICU mortality in this heterogeneous adult population (AUC of 0.67 and 0.68 respectively). A plasma NGAL level >150 ng/ml was associated with a significant risk of AKI (odds ratio 11.8), advanced age, a greater likelihood of CKD, higher serum creatinine on admission to the ICU, higher median APACHE-II (Acute Physiology and Chronic Health Evaluation), SAPS-II (Simplified Acute Physiology Score) and SOFA (Sequential Organ Failure Assessment) scores and a greater likelihood of prior exposure to nephrotoxic agents. Significantly, this study found no difference in plasma NGAL levels between patients with or without sepsis or SIRS [217].
In a large prospective study investigating the variation in plasma and urine NGAL levels among critically ill patients admitted to the ICU, all the patients who developed AKI (as defined by the RIFLE criteria) had significantly elevated plasma and urine NGAL levels compared to those who did not develop AKI [218]. Specifically, plasma NGAL concentration at the time of ICU admission correlated significantly with the severity of renal injury, the AUC for predicting the severity of AKI being 0.77 for RIFLE category R (risk), 0.80 for category I (injury) and 0.86 for category F (failure). A similar analysis for urine NGAL revealed an AUC of 0.80, 0.85 and 0.88 for RIFLE category R, I and F respectively. There was no significant difference between the AUCs of plasma and urine NGAL for predicting AKI. When comparing with the estimated glomerular filtration rate (eGFR), only plasma NGAL was significantly better than eGFR in predicting AKI. At a cut-off of >168 ng/ml, plasma NGAL was 91% sensitive (50% specific) while at a cut-off of >417 ng/ml, it was 90% specific (with sensitivity of 70%) for the prediction of severe renal injury (RIFLE group F). In comparison, urine NGAL was 98% sensitive (50% specific) for predicting RIFLE-F at a cut-off >98 ng/ml and 90% specific (at 55% sensitivity) at a cut-off >1,310 ng/ml. Both plasma and urine NGAL were superior to eGFR in predicting severe AKI (RIFLE-I and F) among patients with normal serum creatinine and eGFR. Adding plasma (but not urine) NGAL significantly improved the ability of eGFR, a diagnosis of sepsis, white blood count and temperature at admission to predict severe AKI (RIFLE F) within the first week of ICU stay. Significantly, patients diagnosed with sepsis had higher urine NGAL levels even if they did not meet the criteria for AKI. While both plasma and urine NGAL were significant predictors of the need for RRT, they did not perform better than either eGFR or serum creatinine. Unlike other studies, this study reported no advantage of serial NGAL measurement (in either plasma or urine) over a single measurement close to the time of maximal renal injury for predicting severe AKI (RIFLE-F) [218].
In a multicenter, prospective cohort study of patients with community acquired pneumonia and severe AKI (RIFLE-F), plasma NGAL was noted to be significantly lower in 93/181 patients who recovered (either survived or did not require renal replacement therapy) compared to those who did not recover (mean NGAL levels165 ng/ml vs. 371 ng/ml respectively). Plasma NGAL alone predicted failure to recover, with an area under the curve of 0.76 (95% C.I. 0.66–0.81). It was also a modest predictor of mortality, requirement for RRT and persistent severe AKI (AUC 0.71, 0.62 and 0.763 respectively). In a multivariate model, plasma NGAL was an independent predictor of the failure to recover normal kidney function (odds ratio: 2.0, 95% C.I. 1.03–3.3). Interestingly, adding plasma NGAL to a scoring system comprising age, serum creatinine, a pneumonia severity index and a Sequential Organ Failure Assessment score (SOFA score) did not significantly improve its ability to predict failure of pneumonia patients to recover from AKI [219].
NGAL mRNA expression in renal biopsies (within 1 hour post-transplant) and urinary NGAL levels (within 24 hours post-transplant) are both early predictor of delayed graft dysfunction (DGD defined as renal dysfunction severe enough to require dialysis 1 week post-transplant) which is observed in upto 50% of patients receiving cadaveric renal transplants [220,221]. Korbely and co-workers investigating the expression of four potential markers of DGD- NGAL, KIM-1, netrin-1 (NTN1) and cysteine rich angiogenic inducer 61 (CRAI-61) in microdissected needle biopsies from 34 deceased donors by quantitative real time PCR observed that NGAL and KIM-1 mRNA were significantly upregulated (nearly 3.8 and 3.5 fold respectively) in the cases where the recipients subsequently developed DGD. Specifically, this upregulation was observed in the tubulointerstitial portion of the kidney (microdissected from the glomeruli) [222]. Multiple regression analysis however revealed that neither marker was an independent predictor of DGD with advanced age being the only significant predictor of DGD (OR 1.11, 95% C.I. 1.005–1.221), P=0.04).
Plasma NGAL has also emerged as a highly sensitive and relatively specific marker of AKI among patients with suspected sepsis. In a multicenter prospective study, 24 patients (out of a total of 661 who met the inclusion criteria for suspected sepsis) developed AKI (defined as serum creatinine >0.5 mg/dl during 72 hours post-admission) within 72 hours after being brought to the Emergency department (ED) [223]. The median plasma NGAL level (in samples collected at the time of presentation to the ED) was significantly higher in patients who developed AKI (456 ng/ml) than in those who did not develop renal failure (134 ng/ml). At a cut-off of >150 ng/ml, plasma NGAL was 96% sensitive but only 51% specific for AKI (AUC: 0.82). In comparison, serum creatinine (at presentation) had similar sensitivity while being only 17% specific at a cut-off of 0.7 mg/dl. Interestingly, median plasma NGAL was highly elevated (431 ng/ml) in 52 patients who died from non-renal causes following admission to the hospital. Of these, 12 patients who died within 72 hours of admission from extra-renal causes had the highest plasma NGAL levels (median 876 ng/ml). Plasma NGAL (at a cut-off >150ng/ml) and serum creatinine (cut-off >0.7mg/dl) were significant predictors of in-hospital mortality from all causes within 72 hours of presentation to the ED (O.R. being 5.5 and 1.3 for NGAL and creatinine respectively) [223]. Renal dysfunction is a common observation in patients admitted to the hospital with heart failure (HF) and correlates with the severity of heart failure. Worsening renal function (WRF) defined as an increase in serum creatinine ≥0.3 mg/dl at any time during in hospital stay, measured relative to the level at the time of admission, complicates between 30%–50% of HF cases [224]. It is associated with a significantly worse prognosis (measured by in hospital and post-discharge risk of death and risk of rehospitalization). However, the late rise in serum creatinine relative to the time course of end-organ damage means that patients in the early stage of WRF may be missed. Serum NGAL measurement in 91 patients with Acute decompensated heart failure (ADHRF) revealed that those patients who developed WRF according to creatinine levels (38% of patients) had significantly higher median serum NGAL levels at the time of admission (194 ng/ml) compared to those who did not develop WRF (median serum NGAL at admission being 128 ng/ml) within 5 days of hospital admission. Multivariate analysis revealed that high serum NGAL at admission was associated with a greater likelihood of developing WRF (odds ratio 1.92, 95% confidence interval 1.23–3.12, p=0.004) [197]. Further, serum NGAL level ≥140 ng/ml could identify patients at risk of WRF with a sensitivity and specificity of 86% and 54% respectively (AUC 0.70, p=0.004). These results suggest that not only is NGAL an indicator of AKI in patients with presumed sepsis, but that plasma NGAL at admission to the ED may be very useful as a prognostic marker to predict early mortality in septic patients, both from renal and non-renal causes.
Sepsis is the most common cause for AKI in critically ill patients [225,226]. Distinguishing septic from non-septic AKI is important as non-septic AKI is associated with a significantly better survival [226,227]. On the contrary, septic AKI is associated with a greater chance of recovery of renal function. Comparison of plasma and urine NGAL levels in patients with AKI revealed that patients with septic AKI had significantly higher plasma and urine NGAL levels compared to those with non-septic AKI [228]. This difference in plasma NGAL levels was significant at 12 hours but not at 24 or 48 hours post-admission. Urine NGAL was significantly higher in patients with septic AKI at 12 and 24 hours but not after that. At a cut-off of ≥280 ng/ml, plasma NGAL was 75% sensitive and 76% specific for the diagnosis of septic AKI (Area under the curve 0.77, 95% C.I. 0.63–0.90). Urine NGAL was slightly inferior, being 69% sensitive and 60% specific for the diagnosis of septic AKI (AUC 0.70, 0.6–0.8). The peak plasma (but not urine) NGAL levels showed a significant positive correlation with the total white blood cell count only in septic (but not non-septic) AKI patients (r2=0.43, p=0.02). Patients in septic shock had higher levels of both plasma and urine NGAL compared to septic patients not in shock. It has been shown in other studies that NGAL released into the serum is filtered through the kidneys and thus appears in the urine over time. Consistent with that, the ratio of plasma to urine NGAL was observed to decrease over time in patients with septic AKI. Interestingly, in patients with non-septic AKI, the ratio showed a trend towards increase over time. Patients with sepsis as a result of a urinary tract infection (UTI) had higher (although non-significant) urine NGAL levels (821 ng/mg of creatinine vs. 201 ng/mg of creatinine in those with a non-UTI source). 20/83 (24%) patients in the study developed progressively worsening AKI at 48 hours. Peak plasma NGAL preceded the progression of AKI in 10 of them (50%). The RIFLE criterion was a better predictor of the need for renal replacement therapy (RRT) (AUC 0.95, 95% C.I. 0.91–0.99) compared to peak NGAL. Peak plasma and urine NGAL were less effective in predicting the need for RRT (AUC 0.71 and 0.70 respectively). Interestingly, urine NGAL at a cut-off ≥230 ng/μg of creatinine was nearly 78% sensitive and 81% specific for predicting the progression of AKI in non-septic AKI patients, but not in patients with septic AKI. Peak plasma and urine NGAL although significantly elevated in AKI patients who died, were not better discriminants in identifying in-hospital death among patients with AKI (AUC 0.70 (0.7–0.7) and 0.6 (0.5–0.8) for plasma and urine NGAL respectively) compared to the RIFLE criteria (AUC 0.70, 0.6–0.8) [228].
Pre-renal azotemia refers to renal injury that is completely reversible within 24–72 hours of onset. It is diagnosed primarily by the absence of features of tubular injury (i.e. casts) on microscopic examination of the urine, usually accompanied by a FENa (fractional excretion of sodium) less than 1% [64]. Reversibility distinguishes pre-renal azotemia from irreversible damage to the renal tubules (termed acute tubular necrosis or ATN). Separation of the two conditions is important given the observation in numerous epidemiological studies that significant differences exist in patient outcome (mortality) between those with transient vs. persistent renal injury [229]. Serum creatinine, the standard marker of renal injury does not distinguish between ATN and pre-renal azotemia with sufficient sensitivity and specificity [230]. In a study comparing the urinary level of NGAL, N-acetyl-β-D-glucosaminidase (NAG), α1-microglobulin and α1-acid glycoprotein in patients with either pre-renal azotemia (n=88), AKI (n=30), chronic kidney disease (n=106) or normal renal function (n=411) in an emergency department setting, urine NGAL was significantly elevated in patients with AKI compared to those with other renal disorders (mean ±SD being 416±387, 30±92, 22.5±41, 15.5±15 μg/g creatinine in patients with AKI, pre-renal azotemia, CKD and normal kidney function respectively) [231]. At a cut-off of >130μg/g creatinine, urine NGAL in a single urine specimen taken at admission was 90% sensitive (95% C.I. 73%–98%) and 99% specific (95% C.I. 99%–100%) in identifying AKI with a positive and negative likelihood ratio of 181.5 and 0.1 respectively. The performance of urinary NGAL was significantly better than other markers (including serum creatinine). Multiple logistic regression revealed that elevated urinary NGAL level (>130μg/g creatinine) was a significant predictor of poor outcome including nephrology consultation, requirement for dialysis, admission to the intensive care units and death (Odds ratio 24.7, 95% C.I. 7.7–79.5).
The results of this study was validated in an independent cohort of 32 patients with pre-renal, 75 with intrinsic renal (caused by ATN, nephrotoxins and acute glomerulonephritis) and 38 with unclassifiable AKI (did not meet criteria for either of the two sub-types) [232]. Urine NGAL levels were significantly higher (at the time of inclusion into the study) in patients with intrinsic AKI (based on RIFLE criteria) compared to those with pre-renal AKI (median urine levels being 36.5 ng/ml/g creatinine in pre-renal vs. 273.5 ng/ml/g creatinine in intrinsic renal AKI respectively, p<0.001). In comparison, serum creatinine but not fractional excretion of sodium (FENa) or urea (FEU) or the ratio of serum urea to creatinine was significantly higher in patients with intrinsic AKI (median serum creatinine being 175.5 μM in pre-renal vs. 233 μM in intrinsic AKI respectively). Significantly, urine NGAL levels (at inclusion) were significantly higher in patients who went on to develop an adverse outcome (n=40) as measured by either a step-up in severity of AKI, requirement for dialysis or death compared to those who did not develop these adverse outcomes (n=94) (median NGAL levels being 235.4 ng/ml/g of creatinine in the former vs. 71.8 ng/ml/g creatinine in the latter group, p<0.001). Urine NGAL excretion was also observed to increase with time in patients with intrinsic AKI but not in pre-renal AKI. The increase was particularly high (2-fold) in those with an adverse outcome during in-hospital stay. It appears that patients with intrinsic AKI who will go on to develop adverse outcomes have significantly elevated urine NGAL excretion at the time of admission which is maintained as complications develop In comparison, serum creatinine levels at inclusion could not predict who would develop an adverse outcome from those would have an uncomplicated clinical course. However, the peak serum creatinine levels measured within 7-days of inclusion into the study were significantly higher in patients who had an adverse outcome (median levels being 273 μM compared to 159 μM for those without these adverse events, p<0.001). Both uncorrected urine NGAL and that corrected for creatinine excretion (at the time of admission) were highly sensitive and specific in distinguishing between patients with pre-renal vs. those with intrinsic AKI (AUC being 0.87 and 0.89 respectively). In comparison, serum creatinine (AUC 0.74), RIFLE classification (AUC 0.72), FENa (AUC 0.54), FEU (AUC 0.59) and the serum urea to creatinine ratio (AUC 0.71) at admission were less accurate in discriminating these two categories of renal injury. Urine NGAL levels at the time of admission (both uncorrected and corrected for creatinine excretion) was also fairly accurate in predicting whether a patient would go on to have an adverse outcome (AUC 0.71 for both corrected and uncorrected urine NGAL). In comparison, serum creatinine (AUC 0.61), RIFLE class (AUC 0.56), FENa (AUC: 0.45), FEU (AUC: 0.49) and the serum urea to creatinine ratio (AUC 0.48) at admission were less effective at making this discrimination [232].
Not just in adults, but in children too, urine NGAL has emerged as a highly sensitive marker of AKI. In one study employing 252 children, 18 (7%) of whom developed AKI, urine NGAL was a fairly accurate marker of AKI (AUC 0.66). Further, the performance of NGAL was unaffected whether corrected or uncorrected for urine output (AUC 0.63 vs. 0.66 respectively) [233].
Children, particularly newborns represent a particular challenge when it comes to management of AKI. Incomplete development of nephrons in newborn results in higher baseline levels of serum creatinine than is normal in children. Further, mild to moderate glomerular damage (as happens during cardiopulmonary bypass surgery (CPB) may not produce significant elevations in serum creatinine in them. Thus, defining AKI in the neonatal period using serum creatinine has been quite challenging. Both plasma and urine NGAL were found to be significantly elevated in neonatal and non-neonatal pediatric patients as early as 2 hours after CPB and remained elevated upto 48 hours (maximum observation time) [234]. Higher NGAL levels (both in plasma and urine) correlated with younger age at surgery, a greater change in serum creatinine concentration (from baseline), longer CPB surgery time and a longer duration of AKI. Multivariate analysis revealed that both plasma and urine NGAL levels were independent predictors of AKI in neonatal as well as non-neonatal pediatric patients. Two hours after CPB, a urine NGAL >185 ng/ml or a plasma NGAL >95 ng/ml could identify AKI in the neonatal patients with sensitivity of 100% (88% for plasma) and specificity of 93% (for both urine and plasma). The corresponding cut-offs for non-neonatal patients were >45 ng/ml (urine), >48 ng/ml (plasma) with the sensitivity and specificity of 85% (90% for plasma) and 86% (88% for plasma) respectively [234].
While biomarkers may perform well in a homogenous patient population, their performance is often significantly worse in a heterogenous cohort. Part of the reason is that the level of a biomarker needs to be correlated keeping in mind the stage of the presumed disease process at which the biomarker level was measured. In a study aimed at investigating the performance of urine NGAL in predicting AKI in a patients population admitted to the ICU for various reasons, it was observed that the level of urine NGAL (corrected for creatinine excretion) at admission was poorly predictive of onset of AKI (AUC 0.66, 40% sensitive and 60% specific) or death within 7-days post-admission (AUC 0.66, 47% sensitive and 78% specific), but better at predicting the requirement for dialysis in the next seven days (AUC 0.79, 64% sensitive and 76% specific) [235]. Significantly, none of the potential biomarkers tested (NGAL, cystatin C, gamma glutamyl transpeptidase, alkaline phosphatase, IL-18 and kidney injury molecule-1) were able to predict AKI within 48 hours of admission in this heterogenous population. When patients were stratified according to the time elapsed after the renal insult however, urine NGAL was a better marker for predicting AKI between 12 hrs to 36 hrs following insult (AUC 0.71, 95% C.I. 0.61–0.82) than the other markers. When patients were stratified based on baseline renal function (at the time of entry into the ICU), urine NGAL was again the most significant predictor of AKI for patients with a baseline eGFR between 90–120 ml/minute (AUC 0.70, 95% C.I. 0.59–0.81). The conclusions from the study were that application of a biomarker in a heterogenous patient population is almost always associated with a poorer performance than in a homogenous patient cohort. Thus, instead of blindly applying a biomarker to all admissions with a particular condition, it is necessary to judiciously use markers (to diagnose or prognosticate) after accounting for the stage of the disease (at the time of admission) and the time elapsed since the presumed insult. Interestingly, the same study also observed that notwithstanding minor changes in AUC, the trend of variation in NGAL (and other biomarkers) remained similar irrespective of whether they were corrected for urinary creatinine excretion or not [235].
While NGAL has emerged as a novel marker of AKI in critically ill patients, few studies have also demonstrated that NGAL can be an early diagnostic or prognostic marker in severely ill patients independent of the presence of an underlying AKI. In one study comparing plasma levels of several acute phase proteins in 27 patients without AKI but with systemic inflammatory response syndrome (SIRS), severe sepsis or septic shock with 18 patients who had concomitant AKI and septic shock, there was no significant difference in peak plasma NGAL levels (measured by RIA using a polyclonal antibody described in [214]) between septic shock patients with or without AKI (median (interquartile range) being 216 ng/ml (364) and 134 ng/ml (73) respectively). On the other hand, urine NGAL levels were significantly higher in patients with septic shock and a concomitant AKI compared to those patients in septic shock but without AKI (median (interquartile range) being 319 ng/mg of creatinine (1,933) and 63.5 ng/mg of creatinine (133) respectively, p<0.05). Both plasma and urine NGAL levels were also significantly higher in patients with septic shock compared to those with SIRS or severe sepsis (without signs of shock). A time-course of rise in plasma and urine NGAL levels among patients with AKI (according to the Risk-Injury-Failure-Loss-End stage renal disease (RIFLE) or AKIN criteria) revealed that NGAL levels both in the blood and urine were significantly elevated 12 hours prior to the diagnosis of AKI. At a cut-off of >120 ng/ml and >68 ng/mg of creatinine, plasma and urine NGAL were able to discriminate patients with AKI from those without AKI with high accuracy 12 hours after a diagnosis of AKI had been made with conventional criteria [AUC being 0.85 (83% sensitive and 86% specific) and 0.86 (71% sensitive and 100% specific) for plasma and urine NGAL respectively] [236]. Urine (but not plasma) NGAL level was also a significant predictor of AKI among patients with septic shock at 12 hours after the diagnosis of AKI by conventional criteria (AUC being 0.86 with a sensitivity and specificity of 71% and 100% respectively) [236].
Type-1 DM is a rapidly growing problem particularly in the developed world. Global estimates suggest a nearly 3% increase each year in its incidence in the last decade [237]. Diabetic nephropathy (DN) is a major long term complication of type-1 DM, its risk being related to the duration of diabetes (cumulative risk of 25%–40% after 25 years of either type-1 or type-2 DM), extent of hyperglycemia (measured by HbA1c levels), dyslipidemia and elevated blood pressure) [238–240]. DN involves both the glomeruli and the tubulointerstitial system. The gold standard for diagnosis of DN- renal biopsy is highly invasive, while microalbuminuria and is the most common clinically employed test. Given that NGAL is an early marker of kidney damage, particularly tubular damage, it is plausible that rise in NGAL levels may precede other clinically detectable changes such as microalbuminuria and decline in eGFR. In a study comparing urine and serum NGAL levels in 22 children (mean age 12.7±3.5 years) with type-1 DM and apparently normal renal function (normal eGFR and normal range urine albumin) treated for about 6 years on average, with 14 healthy children, both urine and serum NGAL concentrations were observed to be significantly higher in the children with type-1 DM compared to controls [241]. A significant positive correlation (r2=0.51, p<0.05) was also noted between both urine and serum NGAL levels and the albumin excretion rate. However, no correlation was observed between NGAL levels and either eGFR or glycosylated hemoglobin (HbA1c) levels.
Another study in 63 adult patients (mean age ±SD being 44±9 years) with DN (mean duration of diabetes 32±9 years) also observed that urine NGAL levels at baseline were significantly correlated with urinary albumin excretion (r=0.44) but not with the GFR. During a three year follow-up, patients who had higher baseline urine NGAL showed a faster decline in their GFR. However, when adjusted for other known factors that influence the progression of renal damage (including age, gender, HbA1c levels, systolic blood pressure and urine albumin excretion rate), NGAL was no longer a significant independent predictor of the progression of renal damage [242]. Thus, it appears that while NGAL measurement at baseline provides an indication of disease progression, in adult patients (unlike in children) it does not add to the prognostic power of urine albumin excretion, an established marker of renal damage.
Progression of DN involves a progressive increase in the amount of albumin excreted in the urine, measured as mg excreted per 24 hours. A study among 74 Chinese patients with DN revealed that the baseline serum and urine NGAL levels were inversely correlated with the degree of albumin excretion in the urine [stratified into normo-albuminuria (<30 mg/day), microalbuminuria (≥30 and <300 mg/24 hours) and macroalbuminuria (>300 mg/24 hrs)]. Thus, patients with normo-albuminuria had a higher serum and a lower urine NGAL level than those with macroalbuminuria. During a follow-up period of one year, serum NGAL levels increased in all three groups of patients, while urine NGAL showed a trend to decline, particularly in the patients with macroalbuminuria. While serum NGAL levels showed no correlation with GFR and only a weak correlation with other markers of renal dysfunction (cystatin C and BUN), urine NGAL showed a significant negative correlation with GFR and a positive correlation with both these markers. In addition, a positive correlation was found between urine NGAL levels and serum creatinine, an established marker of renal function both at baseline and at follow-up (r= 0.57 at baseline and 0.55 on follow-up, p<0.001). Interestingly, there was no significant correlation between serum and urine NGAL levels [173]. This study highlighted the potential of serum NGAL measurement as a tool to distinguish normo-albuminuric from micro-albuminuric (early stage) DN. Urine NGAL on the other hand, was more useful to identify late stage DN (macro-albuminuric patient) owing to its strong correlation with markers of renal damage.
NGAL levels, particularly urinary NGAL is significantly elevated not just in acute, but in chronic renal diseases as well. Among 85 pediatric patients with various chronic renal diseases, urinary NGAL levels (but not serum NGAL) were significantly higher in patients with chronic renal dysfunction (except in steroid responsive nephrotic syndrome in remission) [243]. Further, a sub-analysis revealed that tubular diseases were associated with a nearly 10–20 fold higher urinary NGAL level compared to glomerular diseases suggesting that the origin of urinary NGAL from the renal tubular epithelial cells. Both urine and serum NGAL levels showed a significant inverse correlation with the estimated glomerular filtration rate (eGFR) and a significant positive correlation with urine protein levels in patients with proteinuric renal diseases (e.g. proliferative glomerulonephritis, steroid-resistant nephrotic syndrome and relapse of steroid-sensitive nephrotic syndrome) [243]. Significantly, no significant correlation was found between serum NGAL levels and either serum or urine protein levels or patient age in this study.
AKI in the immediate post-operative period contributes significantly to the morbidity and mortality of patients receiving liver transplantation. De novo renal impairment is associated with the highest risk of mortality, with 90-day and 1 year mortality in patients requiring RRT being as high as 40% and 54% respectively [244]. Hence, there is an existing need to identify AKI early on so that appropriate management including RRT can be instituted. In one study, the difference in serum NGAL levels between two samples, one drawn at the time of surgery and the second within 2 hours after reperfusion of the liver was a fair marker of AKI among 59 adult patients undergoing liver transplantation (p=0.02) [245]. Among patients with a baseline serum creatinine <1.5 mg/dl, a single value of serum NGAL above a cutoff of >139ng/ml 2 hours post-reperfusion (of the liver) was a good predictor of AKI (AUC 0.79). In another study of 95 patients undergoing liver transplant, both plasma and urine NGAL were significant predictors of AKI in samples drawn immediately post-transplant [246]. The APACHE-II score and plasma NGAL levels were the strongest predictors of post-operative AKI on a multiple regression analysis (AUC being 0.87 [95% C.I. 0.77–0.97] and 0.87 [95% C.I. 0.77–0.92]. A score comprising of a combination of an APACHE-II score >13 and plasma NGAL >258 ng/ml was 100% sensitive and 76% specific in predicting severe AKI (defined as ≥stage 2 AKI according to AKIN criteria [209]) at a cut-off ≥1 (total maximum score being 2).
The above studies suggest that measurement of NGAL is not just an early marker for AKI, but could also be extremely useful for the follow-up of patients on anti-inflammatory therapy, for detecting recurrence of infection (or inflammation) and in identifying resistance to current therapy. Urine NGAL, particularly when corrected for the extent of diuresis appears to be more sensitive and specific than serum NGAL levels in its diagnostic efficacy. An explanation for the rise in urine NGAL levels following ischemia (e.g. aortic clamping during cardiac surgery) is hypoxia induced damaged to the medullary tubules of the kidney. These tubules, which are a part of the outer medulla, comprise the part of the kidney most sensitive to hypoxia owing to their dependence on oxidative metabolism for energy. A second mechanism for ischemia induced rise in NGAL is “ischemia-reperfusion” injury. Herein, the restoration of blood flow to previously ischemic tissues triggers an inflammatory response. Both endothelial cells and renal parenchymal cells release inflammatory cytokines and chemokines that lead to the recruitment of neutrophils to the site of reperfusion. There is a stimulation of free radical generation which also contributes to cell death. Inflammation induced neutrophil degranulation together with renal tubular necrosis probably contribute to the second peak in NGAL levels seen in many acute conditions during recovery from hypoperfusion. A third possibility is the release of NGAL from other extra-renal sources. How much this contributes to the elevated NGAL levels in ischemia reperfusion injury is however yet to be elucidated. An important consideration when interpreting the results of biomarker studies is the time elapsed as measured from the time of the initial insult. This is often difficult to establish and hence biomarker levels should preferably be used in combination and as an adjunct to other investigations both for diagnostic and prognostic applications. Given the late rise in serum creatinine levels following AKI, NGAL (both in plasma and urine) levels may be extremely useful to identify early AKI in those patients with low serum creatinine.
6.1.6. Hematologic disorders
Hematological disorders characterized by an increased tendency for spontaneous coagulation (coagulopathies) pose a serious threat to life. Apart from risk factors in the history like advanced age, coexisting vascular complications, inherited disorders in platelet function and a previous history of thrombotic events, there is no specific biomarker that can identify high-risk patients. A comparison of serum NGAL levels among patients with polycythemia vera (PV), essential thrombocythemia (ET), sepsis and healthy subjects revealed that serum NGAL levels were significantly elevated in patients with PV (mean±SEM 507.5 ng/ml+341 ng/ml), ET (521.3 ng/ml±363.4 ng/ml) and sepsis (272.5 ng/ml±212.4 ng/ml) compared to healthy controls (173 ng/ml±58 ng/ml) [247]. NGAL levels were significantly higher in patients with an elevated D-dimer (641 ng/ml±443 ng/ml vs. 404 ng/ml±202 ng/ml, p=0.01). There was a significant positive correlation between serum NGAL levels and both the total white blood cell count (r2=0.7) and neutrophil count (r2=0.8). However, there was no difference in NGAL levels between PV or ET patients with or without thrombosis. Further, there was no significant correlation of serum NGAL levels with CD11b (a marker of neutrophils), CD62 (marker of activated platelets), or the presence of JAK2 mutations (a marker of poor prognosis) [247]. ET and PV belong to the group of hematologic neoplasms termed as myeloproliferative neoplasms (MPNs) that also includes progressive myelofibrosis (PMF). PV is characterized by increase in the red cell mass (erythrocytosis) which is independent of erythropoietin (the main driver of erythropoiesis in the healthy bone marrow) while ET is a clonal stem cell disorder characterized by a tendency for an increase in platelet count and the risk of thrombotic events [248,249]. PV, like other MPNs carries a considerable risk of transformation into leukemia. A mutation in the Janus-associated kinase 2 (JAK2) gene identified by Kralovics and co-workers in 2005 [250] confers a proliferative advantage to the cells harboring this mutation. The mutation is present in PV, ET and PMF. Further, Data from multiple studies supports the idea that patients (ET) positive for the JAKV617F mutation have a greater risk of venous thrombosis (similar to that in patients with PV) than those without this mutation [251]. While the study did not find any differences in NGAL levels between patients with or without the JAK2 mutation, it does raise the possibility that NGAL is involved in the pathogenesis of MPNs and could be a potential serum based biomarker to diagnose MPNs early and possibly for follow-up for early detection of relapse.
Cancer is often associated with an increased tendency for the blood to clot (termed as pro-thrombotic state). This has been suggested to contribute in part to the mortality from cancer and formed the basis of treatment with anti-thrombotics, particularly low molecular weight heparins like enoxaparin in cancer patients [252]. A study of 110 patients with various cancers revealed that plasma NGAL levels were significantly increased in the blood at baseline measurement together with that of d-dimer, an established marker of pro-coagulant state. Importantly, both d-dimer and NGAL levels decreased after three months of treatment with enoxaparin [253]. This suggests that NGAL may have a role in cancer associated pro-coagulant state and may be a marker of decreased coagulability in multiple cancers. Whether it has a differential role in normal vs. cancer associated coagulation and whether its expression is directly regulated by enoxaparin (or other anticoagulants) remains to be investigated.
An important question that often faces blood based diagnostic studies is the influence of the storage method and anti-coagulant on the expression of the biomarker. Release (or degradation) of the protein over time can cause false elevations (or depression) in its levels and this can affect the reproducibility of the assay. As early as 1995, it was demonstrated that while storage of venous blood in tubes containing EDTA (ethylene diamine tetra-acetate) as the anticoagulant did not have any effect on blood NGAL levels, storage in presence of heparin as anticoagulant led to a progressive increase in NGAL levels in the blood over time [20].
6.2 MALIGNANT DISEASES
Several studies have investigated the level of circulating NGAL in the blood (plasma or serum) as a potential marker for the detection and prognostication of both solid tumor and hematologic malignancies.
6.2.1. Solid tumors
A comparison of serum NGAL levels among patients with benign, borderline and malignant ovarian tumors revealed that compared to healthy individuals (mean: 62 ng/ml) and those with benign ovarian tumors (mean: 67 ng/ml), serum NGAL levels were significantly elevated in patients with borderline and malignant ovarian neoplasms (mean being 79.6 ng/ml and 87.4 ng/ml respectively) [76]. Clinicopathologic correlation analysis revealed that patients with well-differentiated ovarian cancer and those with mucinous type of neoplasms had significantly higher serum NGAL levels (mean being 156 ng/ml and 167 ng/ml respectively) than either moderate-poorly differentiated tumors (mean: 78.5 ng/ml-79.3 ng/ml) or other histologic types of epithelial ovarian neoplasms. Although circulating NGAL levels were higher in patients with advanced stage (stage 3 and 4) ovarian cancer (mean level 90 ng/ml compared with 72.5 ng/ml in stage 1 or 2 diseases), this difference was not significant. In comparison, serum CA125, the current gold standard biomarker for the follow-up of patients with ovarian cancer was significantly higher in patients with borderline and malignant ovarian neoplasms (compared to healthy controls and benign ovarian tumors). Further, CA125 levels were higher in patients with advanced (stage 3 or 4) disease, in those with serous ovarian cancers and in well-differentiated tumors, although this difference did not reach statistical significance. Univariate regression analysis revealed that serum NGAL, serum CA125 and advanced stage were significantly associated with a lower survival (measured as the time from diagnosis to death) in treatment naïve ovarian cancer patients. On multivariate analysis however, no variable remained a significant independent predictor of overall survival. The area under a receiver operating characteristic curve (or ROC curve) is a good indicator of the ability of a biomarker to discriminate between two groups of patients. A comparison of the AUCs for serum NGAL and CA125 revealed that at a cut-off of 55.2 ng/ml NGAL was 72% sensitive but only 50% specific as a biomarker to distinguish ovarian cancer patients from cancer-free controls (AUC for NGAL being 0.62). CA125 on the other hand was 80% sensitive and 79% specific with an AUC of 0.91 suggesting that NGAL alone was not better than CA125 and thus not likely to be of significant use as a screening blood based marker for ovarian cancer [76].
Serum NGAL was a good marker to classify patients with either gastric cancer or healthy controls into “cancer” vs. “non-cancer” groups (AUC of 0.93). Further, at a cut-off of >14.31 ng/ml, an NGAL ELISA detected 43% of gastric cancer patients. Specifically, 38% of stage 1 and 58% of stage 2 patients were positive for NGAL. NGAL was significantly better in identifying patients with early stage gastric cancer (stage 1 and 2) than either CA19-9 (12.5% and 8% positivity at a cut-off of >37 U/ml) or carcinoembryonic antigen (CEA) (14.3% and 8% positivity at a cut-off of >5ng/ml) [82].
In a recently concluded prospective study to investigate novel diagnostic biomarkers in pancreatic juice, we observed that NGAL levels were significantly elevated in the pancreatic juice of both pancreatic cancer and chronic pancreatitis patients compared to controls (patients with epigastric pain who underwent endoscopy but did not have either pancreatitis or pancreatic cancer). NGAL was able to discriminate both pancreatic cancer and chronic patients from healthy controls with high accuracy (AUC 0.90 and 0.87 respectively). However, it was not able to distinguish between pancreatic cancer and chronic pancreatitis (unpublished results).
In another study, we have recently shown that plasma NGAL levels are significantly elevated in patients with pancreatic cancer and chronic pancreatitis patients compared to that in healthy individuals (mean being 103.5 ng/ml, 108.9 ng/ml and 67.4ng/ml respectively) (unpublished results). In patients with pancreatic cancer, plasma NGAL levels were significantly higher in those aged >60 years at the time of diagnosis and in those patients with a history of type-II diabetes. NGAL was highly specific in distinguishing pancreatic cancer or chronic pancreatitis patients from healthy controls (96% and 88% respectively). However, it was not very useful to distinguish between pancreatic cancer and chronic pancreatitis cases. When stratified by resectability, NGAL was highly specific (88%) in distinguishing resectable pancreatic cancer patients from healthy controls suggesting its potential utility as an adjunct to imaging in this malignancy.
NGAL has also emerged as a predictor of survival in several malignancies. Breast cancer patients whose tumors expressed NGAL had a significantly shorter disease specific (DSS-defined as the time from diagnosis to either death or loss of follow-up) and disease free (DFS-defined as the time of diagnosis to the date of first documented recurrence of the malignancy) survival, with NGAL positive and negative patients having a median DSS of 12.2 and 17.1 years respectively. Particularly, NGAL was a significant predictor of poor DFS (hazard ratio 1.98) in patients with ER positive tumors. Upon multivariate analysis however, NGAL positivity was a significant independent poor predictor of DFS (hazard ratio 1.85) but not DSS [74]. In another study involving 250 patients with breast cancer, Stoesz and Gould observed that NGAL expression in the malignant tissues correlated inversely with the expression of the estrogen and progesterone receptors (Spearman’s coefficient = −0.26 and −0.22 respectively, p<0.001) and positively with the percentage of cells in the S-phase (Spearman’s coefficient= 0.21, p=0.001) [88]. The inverse relationship between ER and PR expression and NGAL levels was also confirmed in breast cancer cell lines (MCF-7 and T47D) suggesting that the two hormone receptors may be negative regulators of NGAL expression in breast cancer. However, no significant difference was observed in NGAL expression among breast cancer cell lines sorted according to the phase of cell cycle. No significant correlation was also observed between NGAL expression in breast cancer tissues and either tumor size, nodal spread, age of the patient or type of therapy (chemotherapy, endocrine therapy vs. radiation). Further, NGAL expression did not have any significant correlation with either disease free or overall survival either alone or in combination with other factors in this study [88]. A recent large study comprising tissues from 855 breast cancer patients undergoing neoadjuvant chemotherapy demonstrated that NGAL is expressed in 42% of the cancer tissues [254]. Further, the intensity of NGAL expression correlated negatively with hormone receptor expression but positively with pathological complete response to neoadjuvant chemotherapy in patients with hormone receptor positive, node negative, grade 1 or 2 tumors <4cm in diameter. Univariate analysis revealed that higher intensity of NGAL expression correlated with decreased DFS (H.R.: 1.8, 95% C.I. 1.3–2.6) in breast cancer patients and this remained significant on multivariate analysis. The relationship of NGAL expression with prognosis in breast cancer remains to be elucidated in future studies.
In gastric cancer too, patients whose tumors expressed NGAL had a significantly shorter survival (35.6±1.2 months) compared to those where the primary tumor did not express NGAL (54.4±0.7 months). Upon multivariate analysis, the histologic type (diffuse type gastric cancer according to the Lauren classification system), presence of vascular invasion, TNM stage and NGAL expression were independent predictors of adverse outcome (hazards ratio being 2.33, 1.80, 1.66 and 1.84 respectively) [82]. NGAL expression was shown to correlate negatively with survival in patients with endometrial carcinoma (EC). Both a strong cytoplasmic and nuclear expression of NGAL were associated with a significant reduction in overall survival among EC patients suggesting the possibility that NGAL could be a clinically useful prognostic marker in EC [77].
Univariate analysis of colon cancer patients revealed that patients whose tumors had high NGAL expression had a significantly shorter overall survival than those whose tumors had low NGAL expression. In patients with stage II colon cancer, NGAL was also an independent predictor of poor prognosis. However, this was not significant predictor of OS in advanced stage (III/IV) colon cancer patients [94].
A comparison of bile samples from patients undergoing endoscopic retrograde cholangiopancreatography (ERCP) for either benign (n=36) or malignant (n=59) pancreatobiliary obstruction revealed that NGAL was one of the most differentially expressed proteins in those with malignant obstruction. Biliary NGAL levels (measured by ELISA) were significantly higher in patients in the malignant obstruction (1839 ng/ml) than in those with benign causes of obstruction (n=472 ng/ml). NGAL measurement in the bile was fairly accurate in discriminating benign from malignant pancreatobiliary obstruction with an area under the ROC curve of 0.76 (96% sensitive and 56% specific). In the same cohort of patients, no significant difference was found in both serum and urine NGAL levels between patients with benign vs. malignant biliary obstruction. Significantly, a combination of NGAL and serum CA19-9 was more accurate than CA19-9 alone in identifying patients with malignant pancreatobiliary obstruction (sensitivity, specificity, positive and negative predictive value of the combination being 85%, 82%, 79% and 87%) [255].
In a single center study of 85 patients with renal cell carcinoma (RCC) who received Sunitinib, a pan-receptor tyrosine kinase inhibitor, 17 patients (20%) had plasma NGAL levels higher than the normal range (42–177ng/ml)- the median NGAL level being 83.2ng/ml (range: 2.8–363.4ng/ml) [256]. The baseline plasma NGAL level was a predictor of progression free survival (PFS), with a 10ng/ml rise in NGAL levels associated with an increase in the relative risk (of progression) of 4%. Compared to patients who had a baseline plasma NGAL level ≤177 ng/ml, those with higher NGAL levels had a 1.86 higher risk of progression (p=0.03). The median PFS time for those with high (>177ng/ml) and low NGAL level was 3.35 months and 8.15 months respectively. Elevated NGAL levels were also a significant independent predictor of poor PFS in Sunitinib treated RCC patients (RR 1.91, 95% CI 1.39–2.42) [256].
With growing interest in NGAL as a biomarker has come the realization for the need to standardize the assay for implementation on an automated platform. Delanaye and Rozet in a recent study investigated the variation in NGAL levels in the urine of 20 healthy subjects (11 women) aged 33+10.3 years using a commercial assay platform (Abbott diagnostic assay on an Architect platform) found that the analytic coefficient of variation was 3.7% [257]. The biological CV for un-normalized NGAL was 84% for the first morning urine sample. When normalized to creatinine levels, the %CV was 81%. Further, the CV appeared to be similar in second urine sample, being 124% for un-normalized and 88% for normalized NGAL level. Given the variability in NGAL levels even in an automated platform, it has been suggested that a doubling of NGAL levels in at least 2 samples before making a conclusion that NGAL levels are indeed increasing. Another study comparing a traditional ELISA assay with a rapid fluorescent assay confirmed the robustness of the NGAL assay [258]. The study also supported the findings of the earlier study that two or more longitudinal NGAL measurements are required before we can make a diagnosis of increase in NGAL levels.
7. CONCLUSION AND PERSPECTIVE
Observations in animal models and human subjects suggest that lipocalin 2 (or NGAL) is required for the development and/or progression of both benign and malignant diseases. In BCR-ABL+ CML, Lcn2 is absolutely required for the malignant cells to establish in and produce features of leukemia [107,108]. On the other hand, NGAL appears to be an important tool in the armament of the innate immune system in combating bacterial infections and bacteria appear to have evolved ways to counteract its bacteriostatic effect including altered Glycosylation of siderophores (e.g. C5 glucosylation of enterobactin prevents binding of the siderophore to NGAL) and synthesizing siderophores that do not bind NGAL at all (e.g. petrobactin produced by Paracoccus denitrificans) [259–261].
While we are edging ever closer to understanding the complex role played by NGAL in health and disease, several questions remain unsolved. Why, for instance does it take 2 weeks for NGAL mRNA to be induced following stimulation (intimal injury)? Conversely, when NF-κB transcriptional activity is blocked in vivo by a dominant negative IKKβ, why does it take upto 14 days for a decrease in NGAL mRNA expression to occur? While the expression of the mono and multimeric forms of NGAL is well known, why in some diseases like CML, is a 21 kDa variant expressed in the blood? [107]. Glycosylation as a possible mechanism for the expression of this variant was ruled out, suggesting the possibility of alternative splicing of the mRNA. The function and incidence of expression of this shorter variant of NGAL remains a mystery. An interesting observation was the difference in NGAL expression in non-neoplastic pancreas adjacent to an area of adenocarcinoma compared to that in a specimen devoid of adjacent malignancy [86]. The expression of genes is known to be modulated in the vicinity of cancer cells primarily owing to the release of chemokines and other paracrine factors from the malignant cells (called the “field-effect” of cancer). Understanding the mechanisms by which cancer cells induce NGAL expression in adjacent normal cells would certainly offer us a deeper insight into the complex biology of cancer and potentially allow targeting of these pathways for therapeutic applications.
A measurement of NGAL levels in plasma and urine has been examined in several studies as both a diagnostic adjunct and in prognostication. For instance, plasma NGAL was one of the three markers (selected from nearly 150 potential markers) that could predict the development of organ dysfunction, septic shock and death among patients with presumptive sepsis admitted to the ED [188]. While these studies have investigated the expression of NGAL as a function of the systemic inflammatory state, the effect of therapy itself on NGAL expression has been largely neglected. For instance, while NGAL levels are elevated in patients with sepsis and AKI, these patients also receive large doses of various antibiotics and steroids among other medications. It is possible that one or more of these agents can independently influence NGAL expression and thus is a potential significant confounder, particularly in an acute care setting where medication doses are often rapidly altered. In the future, pointed studies are needed to address this issue to come up if possible with a correction factor to account for therapy induced rise in NGAL levels in various body fluids.
Another issue of significance for clinical applications of NGAL is the effect of techniques in general and antibodies in particular in affecting the sensitivity and specificity of NGAL detection in tissues and body fluids. Several studies have now been published that indicate that RIA may be more sensitive than ELISA in detecting NGAL levels [214,236]. However, ELISA continues to be more widely used. It is important to investigate on a common platform the sensitivity and specificity of NGAL RIA in comparison to ELISA using all available antibodies in order to come up with consensus guidelines on assay standardization and above all the optimum technique to identify alterations in the levels of NGAL at the earliest time point possible while maintaining specificity.
Several interesting findings have emerged from pre-clinical and clinical studies using NGAL as a biomarker which have however not reached statistical significance. For instance, patients with sepsis from a urinary infection have higher urine NGAL levels than those with a non-urinary source. Similarly, while in general, the ratio of plasma to urine NGAL decreases with time in septic AKI, the trend is reversed in non-septic AKI. These results may not be coincidental but reflect the difficulty with studies in human subjects where it is practically impossible to predict the exact onset of the underlying disease process. This could also be the reason why variations exist in the reported performance of NGAL between different studies among seemingly similar populations. Such observations should not however be looked upon as an indication of failure of a biomarker. Rather, we should use available resources including imaging together with a multi-marker biochemical panel to identify patients early on in the disease process. The differential expression of Lcn2 in the liver and kidneys is indicative of the influence of age on the expression of NGAL [38]. However, what mechanism (s) operates to affect this regulation remains unsolved. Pre-clinical models offer us the unique opportunity to understand the time-course of events underlying the rise and fall of potential biomarkers but the transition from the bench to the bedside is fraught with the dangers of false interpretations that should (when possible) be avoided by careful study design including patient selection and follow-up of individual patients.
As NGAL levels are demonstrated to be differentially altered in a number of diverse malignancies, a pertinent question that begs for an answer is the source of NGAL. While an obvious source is the tumor, the contribution of other factors including neutrophils cannot be ruled out. Further, it is possible that healthy organs release NGAL as a stress marker in response to the release of cytokines from the tumor cells. In a study on gastric cancer patients, it was observed that 20/26 (77%) patients with gastric cancer who had elevated levels of NGAL (defined as >14.3 ng/ml) also had intense NGAL staining in the gastric cancer tissues. In contrast, only 13/34 (38%) gastric cancer patients without NGAL elevation in serum had tumors that were positive for NGAL (correlation coefficient 0.38, p=0.002) [82]. Thus, there appears to be a positive correlation between the expression of NGAL by the tumor cells and its release into the bloodstream, at least for gastric cancer. It will be of interest in future studies to determine the sources of NGAL release into the blood and other body fluids in cancer patients in order to better understand the role of this cytokine in cancer progression.
The use of NGAL as a target for imaging currently suffers from the difficulties of specific targeting to the site of the lesion, adequate bioavailability and optimization of conditions for human clinical trials. In a recent study, we employed an antibody against human NGAL conjugated to a near infrared (NIR) dye (IRDye800CW) to demonstrate the feasibility of detecting NGAL expression in human pancreatic cancer cells both in vitro and in vivo [262]. In vivo imaging (at wavelength 785 nm) also revealed that a large proportion of the NIR-conjugated NGAL localized to the liver and urinary bladder (in addition to the tumor). Using micelles containing gadolinium conjugated to either NGAL specific or non-specific IgG antibodies, it was demonstrated that NGAL antibody conjugated micelles localized to the site of a carotid atherosclerotic plaque 72 hours post-injection (but not in the initial 24 hours) [46]. While the ability to image organs and tumors in vivo suggests that the antibody-dye complex is internalized into the cells, it also raises the potential problem of decreased bioavailability (owing to accumulation in off-target organs) that may be a practical problem in employing NGAL for in vivo imaging.
Its application in certain settings- e.g. as a surrogate marker of organ damage particularly AKI in patients consuming poisonous substances remains to be explored further [263]. The observation that Lcn2 is strongly induced in BMSCs induced to undergo osteoblastic differentiation suggests a possible role of Lcn2 in stem cell differentiation and possibly a therapeutic role as a target in stem cell based tissue engineering [111]. The observation that Lcn2 silencing decreases mitochondrial biogenesis [264] is highly significant from a therapeutic perspective. Studies have shown that in cancer cells, the mitochondria increase the rate of autophagy to remove aggregated proteins, and that inhibiting this process can cause cancer cells to die [181]. Thus, NGAL (LCN2) might be a therapeutic target to decrease autophagy and therefore increase sensitivity of cancer cells to chemotherapy and radiation. Further, Lcn2 downregulation also increased the expression of IRS-1, a potent inhibitor of tumorigenesis and EMT [264], further strengthening the premise of Lcn2 as a novel therapeutic target in cancer.
Given the significant role of NGAL in the pathogenesis of both benign and malignant human diseases, a pertinent question is – how can we target it therapeutically? Currently, there are no specific inhibitors (or inducers) of NGAL expression. However, recent research has identified several agents that can modulate NGAL expression. An siRNA targeting IKKβ for instance, significantly decreased ischemia reperfusion induced renal damage in rats and also decreased the expression of NGAL in the kidney [265]. From studies in mice, it is clear that NGAL is an important bacteriostatic agent that functions by binding to bacterial siderophores. It would be interesting to design NGAL mutants that have modifications in the ligand binding pocket that enhance its affinity for siderophores. Designing mutants specific for each bacterial species may be a potential strategy to alleviate the severity of bacterial infection. Recently, we have shown that the oil or ethanolic extracts from Ocimum sanctum (holy basil), a popular herbal remedy widely used in traditional systems of medicine can modulate NGAL expression in pancreatic cancer cells. The development of selective NGAL expression modulators will certainly be a focus of future research into the therapeutic targeting of this glycoprotein.
Recent analysis of tissues from patients with IBD has revealed a significant upregulation of NGAL in ulcerative colitis compared to the healthy colonic mucosa [80]. Given the availability of recombinant NGAL (both human and mouse) and specific antibodies, a theoretical possibility is to investigate whether treatment with antibodies can ameliorate the acute inflammatory process. On the other hand, recombinant NGAL could be potentially useful in diseases like HCC where it has been shown to inhibit the aggressive proliferation and invasion abilities of the cancer cells [154]. Potential hazards however remain including development of allergic reactions to the recombinant protein. However, the success of other recombinant protein based therapies, namely IL-2 for renal cell carcinoma [266] and insulin for diabetes suggests that protein engineering techniques can overcome these potential hurdles.
We have in recent years witnessed a spate of studies that have examined global expression profiles following in vitro or in vivo modulations (e.g. treatment with cytokines, gene knocking or knockouts). It has become increasingly clear that genes function as groups, and that a single gene (and by corollary its protein) will rarely function in isolation to mediate health or disease. As we pursue novel diagnostic and therapeutic targets like NGAL, identification of NGAL-associated functional groups of genes could give us a better “bird’s eye” view of the cellular processes than the gene by itself. Such a “systems biology” approach is likely to yield far reaching benefits and a faster implementation of bench side knowledge to bedside practice.
Acknowledgments
The authors on this work are supported, in part, by the grants from the National Institutes of Health (RO1 CA78590, RO1 CA133774, RO1 CA131944, RO1 CA139285, EDRN UO1CA111294, SPORE P50CA127297, TMEN U54CA163120) the Department of Defense (BC074639, BC083295, and BC09742).
Abbreviations
- Adp
Adult Periodontitis
- AKI
Acute Kidney Injury
- AMI
Acute Myocardial Infarction
- APACHE-II
Acute Physiology And Chronic Health Evaluation
- APKD
Adult Polycystic Kidney Disease
- BFGF
Basic Fibroblast Growth Factor
- BMI
Body-Mass-Index
- BMSCs
Bone Marrow Derived Mesenchymal Stem Cells
- CAD
Coronary Artery Disease
- CD
Crohn’s Disease
- CKD
Chronic Kidney Disease
- CMB
Carboxymycobactin
- CML
Chronic Myeloid Leukemia
- COPD
Chronic Obstructive Pulmonary Disease
- COX-2
Cytochrome c Oxidase-2
- CSC
Cancer Stem Cells
- CSF
Cerebrospinal Fluid
- DFO
Iron-Chelator Deferoxamine
- E2
17-B Estradiol
- EC
Endometrial Carcinoma
- EGF
Epidermal Growth Factor
- EGFR
Epidermal Growth Factor Receptor
- eGFR
Estimated Glomerular Filtration Rate
- ER
Estrogen Receptor
- ERCP
Endoscopic Retrograde Cholangiopancreatography
- ERE
Estrogen Response Element
- FAK
Focal Adhesion Kinase
- GFR
Glomerular Filtration Rate
- GRs
Glucocorticoid Receptors
- H.R
Hazards Ratio
- HCC
Hepatocellular Carcinoma
- HIF-1α
Hypoxia Inducible Factor-1
- Holo-Tf
Transferrin Loaded With Iron
- IGF-1
Insulin Like Growth Factor-1
- IHC
Immunohistochemistry
- IL
Interleukin
- IRI
Ischemia Reperfusion Injury
- JAK2
Janus-Associated Kinase 2
- Lcn2
Lipocalin 2
- Lcn2
Mouse Lipocalin 2
- LJP
Localized Juveline Periodontitis
- LMW
Low Molecular Weight
- LRP2
Lipoprotein Receptor Related Protein
- MEFs
Mouse Embryonic Fibroblasts
- MHC
Major Histocompatibility Antigen
- MMP-9
Matrix Metalloprotease-9
- Mouse Ngal
24p3
- MPO
Myloperoxidase Peroxidase
- NGAL
Neutrophil Gelatinase Associated Lipocalin
- NMR
Nuclear Magnetic Resonance
- NOD/SCID
Non-Obese Diabetic/Severe Combined Immunodeficient
- OSPC
Ovarian Serous Papillary Carcinoma
- PGC-1α
Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Alpha
- PHKs
Primary Human Keratinocytes
- RCC
Renal Cell Carcinoma
- ROS
Reactive Oxygen Species
- s.c
Subcutaneous
- SAPS-II
Simplified Acute Physiology Score
- SOFA
Sequential Organ Failure Assessment
- TGF-α
Transforming Growth Factor Alpha
- TICs
Tumor Initiating Cells
- TIMP-1
Tissue Inhibitor Of Metalloproteinase-1
- TLR
Toll-Like Receptor
- TNF- α
Tumor Necrosis Factor Alpha
- VEGF
Vascular Endothelial Growth Factor
- α2-MRP
α2-Microglobulin Related Protein
Footnotes
Financial and competing interest disclosure
Surinder K Batra, Subhankar Chakraborty and Sukhwinder Kaur are supported, in part, by grants from the NIH (RO1 CA78590, RO1 CA133774, RO1 CA131944, RO1 CA139285, EDRN UO1CA111294, SPORE P50CA127297, TMEN U54CA163120) the Department of Defense (BC074639, BC083295, and BC09742). Sushovan Guha is supported by grant from the NIH 2P30DK56338 PFS. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mussap M, Degrandi R, Fravega M, Fanos V. Acute kidney injury in critically ill infants: the role of urine Neutrophil Gelatinase-Associated Lipocalin (NGAL) J Matern Fetal Neonatal Med. 2010;23(Suppl 3):70–72. doi: 10.3109/14767058.2010.508217. [DOI] [PubMed] [Google Scholar]
- 2.Devarajan P. Review: neutrophil gelatinase-associated lipocalin: a troponin-like biomarker for human acute kidney injury. Nephrology (Carlton ) 2010;15:419–428. doi: 10.1111/j.1440-1797.2010.01317.x. [DOI] [PubMed] [Google Scholar]
- 3.Kunzendorf U, Haase M, Rolver L, Haase-Fielitz A. Novel aspects of pharmacological therapies for acute renal failure. Drugs. 2010;70:1099–1114. doi: 10.2165/11535890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Moore E, Bellomo R, Nichol A. Biomarkers of acute kidney injury in anesthesia, intensive care and major surgery: from the bench to clinical research to clinical practice. Minerva Anestesiol. 2010;76:425–440. [PubMed] [Google Scholar]
- 5.Chaudhary K, Phadke G, Nistala R, Weidmeyer CE, McFarlane SI, Whaley-Connell A. The emerging role of biomarkers in diabetic and hypertensive chronic kidney disease. Curr Diab Rep. 2010;10:37–42. doi: 10.1007/s11892-009-0080-z. [DOI] [PubMed] [Google Scholar]
- 6.Bolignano D, Coppolino G, Lacquaniti A, Buemi M. From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40:273–276. doi: 10.1111/j.1365-2362.2010.02258.x. [DOI] [PubMed] [Google Scholar]
- 7.Prabhu A, Sujatha DI, Ninan B, Vijayalakshmi MA. Neutrophil gelatinase associated lipocalin as a biomarker for acute kidney injury in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. Ann Vasc Surg. 2010;24:525–531. doi: 10.1016/j.avsg.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Rosner MH. Urinary biomarkers for the detection of renal injury. Adv Clin Chem. 2009;49:73–97. doi: 10.1016/s0065-2423(09)49004-8. [DOI] [PubMed] [Google Scholar]
- 9.Di GA, Giuffrida C, Carpinteri G, Narbone G, Pirrone G, Di MA, Calandra S, Noto P, Le MC, Alongi B, Nigro F. Neutrophil gelatinase-associated lipocalin: a novel biomarker for the early diagnosis of acute kidney injury in the emergency department. Eur Rev Med Pharmacol Sci. 2009;13:197–200. [PubMed] [Google Scholar]
- 10.Soni SS, Ronco C, Katz N, Cruz DN. Early diagnosis of acute kidney injury: the promise of novel biomarkers. Blood Purif. 2009;28:165–174. doi: 10.1159/000227785. [DOI] [PubMed] [Google Scholar]
- 11.Bolignano D, Coppolino G, Donato V, Lacquaniti A, Bono C, Buemi M. Neutrophil gelatinase-associated lipocalin (NGAL): a new piece of the anemia puzzle? Med Sci Monit. 2010;16:RA131–RA135. [PubMed] [Google Scholar]
- 12.Bolignano D, Donato V, Lacquaniti A, Fazio MR, Bono C, Coppolino G, Buemi M. Neutrophil gelatinase-associated lipocalin (NGAL) in human neoplasias: a new protein enters the scene. Cancer Lett. 2010;288:10–16. doi: 10.1016/j.canlet.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 13.Flower DR. Experimentally determined lipocalin structures. Biochim Biophys Acta. 2000;1482:46–56. doi: 10.1016/s0167-4838(00)00147-3. [DOI] [PubMed] [Google Scholar]
- 14.Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482:9–24. doi: 10.1016/s0167-4838(00)00148-5. [DOI] [PubMed] [Google Scholar]
- 15.Heise D, Rentsch K, Braeuer A, Friedrich M, Quintel M. Comparison of urinary neutrophil glucosaminidase-associated lipocalin, cystatin C, and alpha1-microglobulin for early detection of acute renal injury after cardiac surgery. Eur J Cardiothorac Surg. 2011;39:38–43. doi: 10.1016/j.ejcts.2010.05.044. [DOI] [PubMed] [Google Scholar]
- 16.Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure. 2005;13:29–41. doi: 10.1016/j.str.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim Biophys Acta. 2000;1482:272–283. doi: 10.1016/s0167-4838(00)00152-7. [DOI] [PubMed] [Google Scholar]
- 18.Hraba-Renevey S, Turler H, Kress M, Salomon C, Weil R. SV40-induced expression of mouse gene 24p3 involves a post-transcriptional mechanism. Oncogene. 1989;4:601–608. [PubMed] [Google Scholar]
- 19.Triebel S, Blaser J, Reinke H, Tschesche H. A 25 kDa alpha 2-microglobulin-related protein is a component of the 125 kDa form of human gelatinase. FEBS Lett. 1992;314:386–388. doi: 10.1016/0014-5793(92)81511-j. [DOI] [PubMed] [Google Scholar]
- 20.Axelsson L, Bergenfeldt M, Ohlsson K. Studies of the release and turnover of a human neutrophil lipocalin. Scand J Clin Lab Invest. 1995;55:577–588. doi: 10.3109/00365519509110257. [DOI] [PubMed] [Google Scholar]
- 21.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 22.Chu ST, Lin HJ, Huang HL, Chen YH. The hydrophobic pocket of 24p3 protein from mouse uterine luminal fluid: fatty acid and retinol binding activity and predicted structural similarity to lipocalins. J Pept Res. 1998;52:390–397. doi: 10.1111/j.1399-3011.1998.tb00663.x. [DOI] [PubMed] [Google Scholar]
- 23.Coles M, Diercks T, Muehlenweg B, Bartsch S, Zolzer V, Tschesche H, Kessler H. The solution structure and dynamics of human neutrophil gelatinase-associated lipocalin. J Mol Biol. 1999;289:139–157. doi: 10.1006/jmbi.1999.2755. [DOI] [PubMed] [Google Scholar]
- 24.Goetz DH, Willie ST, Armen RS, Bratt T, Borregaard N, Strong RK. Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin. Biochemistry. 2000;39:1935–1941. doi: 10.1021/bi992215v. [DOI] [PubMed] [Google Scholar]
- 25.Payne SM, Finkelstein RA. The critical role of iron in host-bacterial interactions. J Clin Invest. 1978;61:1428–1440. doi: 10.1172/JCI109062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neilands JB. Siderophores: structure and function of microbial iron transport compounds. J Biol Chem. 1995;270:26723–26726. doi: 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- 27.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 28.Le C, Calafat VJ, Borregaard N. Sorting of the specific granule protein, NGAL, during granulocytic maturation of HL-60 cells. Blood. 1997;89:2113–2121. [PubMed] [Google Scholar]
- 29.Van Dyke TE, Schweinebraten M, Cianciola LJ, Offenbacher S, Genco RJ. Neutrophil chemotaxis in families with localized juvenile periodontitis. J Periodontal Res. 1985;20:503–514. doi: 10.1111/j.1600-0765.1985.tb00834.x. [DOI] [PubMed] [Google Scholar]
- 30.Cowland JB, Sorensen OE, Sehested M, Borregaard N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol. 2003;171:6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 31.Le C, Cowland VJB, Calafat J, Borregaard N. Targeting of proteins to granule subsets is determined by timing and not by sorting: The specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proc Natl Acad Sci U S A. 1996;93:6454–6457. doi: 10.1073/pnas.93.13.6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borregaard N, Sorensen OE, Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28:340–345. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Askenazi DJ, Koralkar R, Levitan EB, Goldstein SL, Devarajan P, Khandrika S, Mehta RL, Ambalavanan N. Baseline values of candidate urine acute kidney injury biomarkers vary by gestational age in premature infants. Pediatr Res. 2011;70:302–306. doi: 10.1203/PDR.0b013e3182275164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mallbris L, O’Brien KP, Hulthen A, Sandstedt B, Cowland JB, Borregaard N, Stahle-Backdahl M. Neutrophil gelatinase-associated lipocalin is a marker for dysregulated keratinocyte differentiation in human skin. Exp Dermatol. 2002;11:584–591. doi: 10.1034/j.1600-0625.2002.110611.x. [DOI] [PubMed] [Google Scholar]
- 35.Owen HC, Roberts SJ, Ahmed SF, Farquharson C. Dexamethasone-induced expression of the glucocorticoid response gene lipocalin 2 in chondrocytes. Am J Physiol Endocrinol Metab. 2008;294:E1023–E1034. doi: 10.1152/ajpendo.00586.2007. [DOI] [PubMed] [Google Scholar]
- 36.Huang HL, Chu ST, Chen YH. Ovarian steroids regulate 24p3 expression in mouse uterus during the natural estrous cycle and the preimplantation period. J Endocrinol. 1999;162:11–19. doi: 10.1677/joe.0.1620011. [DOI] [PubMed] [Google Scholar]
- 37.Aigner F, Maier HT, Schwelberger HG, Wallnofer EA, Amberger A, Obrist P, Berger T, Mak TW, Maglione M, Margreiter R, Schneeberger S, Troppmair J. Lipocalin-2 regulates the inflammatory response during ischemia and reperfusion of the transplanted heart. Am J Transplant. 2007;7:779–788. doi: 10.1111/j.1600-6143.2006.01723.x. [DOI] [PubMed] [Google Scholar]
- 38.Garay-Rojas E, Harper M, Hraba-Renevey S, Kress M. An apparent autocrine mechanism amplifies the dexamethasone- and retinoic acid-induced expression of mouse lipocalin-encoding gene 24p3. Gene. 1996;170:173–180. doi: 10.1016/0378-1119(95)00896-9. [DOI] [PubMed] [Google Scholar]
- 39.Ding L, Hanawa H, Ota Y, Hasegawa G, Hao K, Asami F, Watanabe R, Yoshida T, Toba K, Yoshida K, Ogura M, Kodama M, Aizawa Y. Lipocalin-2/neutrophil gelatinase-B associated lipocalin is strongly induced in hearts of rats with autoimmune myocarditis and in human myocarditis. Circ J. 2010;74:523–530. doi: 10.1253/circj.cj-09-0485. [DOI] [PubMed] [Google Scholar]
- 40.Yndestad A, Landro L, Ueland T, Dahl CP, Flo TH, Vinge LE, Espevik T, Froland SS, Husberg C, Christensen G, Dickstein K, Kjekshus J, Oie E, Gullestad L, Aukrust P. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure 1. Eur Heart J. 2009;30:1229–1236. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- 41.Landro L, Damas JK, Flo TH, Heggelund L, Ueland T, Tjonnfjord GE, Espevik T, Aukrust P, Froland SS. Decreased serum lipocalin-2 levels in human immunodeficiency virus-infected patients: increase during highly active anti-retroviral therapy. Clin Exp Immunol. 2008;152:57–63. doi: 10.1111/j.1365-2249.2008.03592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lloyd-Jones D, Adams R, Carnethon M, De SG, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 43.Nagai Y, Kitagawa K, Sakaguchi M, Shimizu Y, Hashimoto H, Yamagami H, Narita M, Ohtsuki T, Hori M, Matsumoto M. Significance of earlier carotid atherosclerosis for stroke subtypes. Stroke. 2001;32:1780–1785. doi: 10.1161/01.str.32.8.1780. [DOI] [PubMed] [Google Scholar]
- 44.Anwaar I, Gottsater A, Hedblad B, Palmqvist B, Mattiasson I, Lindgarde F. Endothelial derived vasoactive factors and leukocyte derived inflammatory mediators in subjects with asymptomatic atherosclerosis. Angiology. 1998;49:957–966. doi: 10.1177/000331979804901201. [DOI] [PubMed] [Google Scholar]
- 45.Elneihoum AM, Falke P, Hedblad B, Lindgarde F, Ohlsson K. Leukocyte activation in atherosclerosis: correlation with risk factors. Atherosclerosis. 1997;131:79–84. doi: 10.1016/s0021-9150(96)06077-7. [DOI] [PubMed] [Google Scholar]
- 46.te Boekhorst BC, Bovens SM, Hellings WE, van der Kraak PH, van de Kolk KW, Vink A, Moll FL, van Oosterhout MF, de Vries JP, Doevendans PA, Goumans MJ, de Kleijn DP, van Echteld CJ, Pasterkamp G, Sluijter JP. Molecular MRI of murine atherosclerotic plaque targeting NGAL: a protein associated with unstable human plaque characteristics. Cardiovasc Res. 2011;89:680–688. doi: 10.1093/cvr/cvq340. [DOI] [PubMed] [Google Scholar]
- 47.Bu DX, Hemdahl AL, Gabrielsen A, Fuxe J, Zhu C, Eriksson P, Yan ZQ. Induction of neutrophil gelatinase-associated lipocalin in vascular injury via activation of nuclear factor-kappaB. Am J Pathol. 2006;169:2245–2253. doi: 10.2353/ajpath.2006.050706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, de SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Roger VL, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 49.Sahinarslan A, Kocaman SA, Bas D, Akyel A, Ercin U, Zengin O, Timurkaynak T. Plasma neutrophil gelatinase-associated lipocalin levels in acute myocardial infarction and stable coronary artery disease. Coron Artery Dis. 2011;22:333–338. doi: 10.1097/MCA.0b013e3283472a71. [DOI] [PubMed] [Google Scholar]
- 50.Pukstad BS, Ryan L, Flo TH, Stenvik J, Moseley R, Harding K, Thomas DW, Espevik T. Non-healing is associated with persistent stimulation of the innate immune response in chronic venous leg ulcers. J Dermatol Sci. 2010;59:115–122. doi: 10.1016/j.jdermsci.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Lee SY, Kim DH, Sung SA, Kim MG, Cho WY, Kim HK, Jo SK. Sphingosine-1-phosphate reduces hepatic ischaemia/reperfusion-induced acute kidney injury through attenuation of endothelial injury in mice. Nephrology (Carlton ) 2011;16:163–173. doi: 10.1111/j.1440-1797.2010.01386.x. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. doi: 10.1373/clinchem.2006.075614. [DOI] [PubMed] [Google Scholar]
- 53.Tan BK, Adya R, Shan X, Syed F, Lewandowski KC, O’Hare JP, Randeva HS. Ex vivo and in vivo regulation of lipocalin-2, a novel adipokine, by insulin. Diabetes Care. 2009;32:129–131. doi: 10.2337/dc08-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.D’Anna R, Baviera G, Corrado F, Giordano D, Recupero S, Di BA. First trimester serum neutrophil gelatinase-associated lipocalin in gestational diabetes. Diabet Med. 2009;26:1293–1295. doi: 10.1111/j.1464-5491.2009.02830.x. [DOI] [PubMed] [Google Scholar]
- 55.D’Anna R, Baviera G, Giordano D, Todarello G, Corrado F, Buemi M. Second trimester neutrophil gelatinase-associated lipocalin as a potential prediagnostic marker of preeclampsia. Acta Obstet Gynecol Scand. 2008;87:1370–1373. doi: 10.1080/00016340802464463. [DOI] [PubMed] [Google Scholar]
- 56.Bachorzewska-Gajewska H, Malyszko J, Sitniewska E, Malyszko JS, Pawlak K, Mysliwiec M, Lawnicki S, Szmitkowski M, Dobrzycki S. Could neutrophil-gelatinase-associated lipocalin and cystatin C predict the development of contrast-induced nephropathy after percutaneous coronary interventions in patients with stable angina and normal serum creatinine values? Kidney Blood Press Res. 2007;30:408–415. doi: 10.1159/000109102. [DOI] [PubMed] [Google Scholar]
- 57.Hirsch R, Dent C, Pfriem H, Allen J, Beekman RH, III, Ma Q, Dastrala S, Bennett M, Mitsnefes M, Devarajan P. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol. 2007;22:2089–2095. doi: 10.1007/s00467-007-0601-4. [DOI] [PubMed] [Google Scholar]
- 58.Ling W, Zhaohui N, Ben H, Leyi G, Jianping L, Huili D, Jiaqi Q. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clin Pract. 2008;108:c176–c181. doi: 10.1159/000117814. [DOI] [PubMed] [Google Scholar]
- 59.Bennett M, Dent CL, Ma Q, Dastrala S, Grenier F, Workman R, Syed H, Ali S, Barasch J, Devarajan P. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol. 2008;3:665–673. doi: 10.2215/CJN.04010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumpers P, Hafer C, Lukasz A, Lichtinghagen R, Brand K, Fliser D, Faulhaber-Walter R, Kielstein JT. Serum neutrophil gelatinase-associated lipocalin at inception of renal replacement therapy predicts survival in critically ill patients with acute kidney injury. Crit Care. 2010;14:R9. doi: 10.1186/cc8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Geus HR, Betjes MG, Bakker J. Neutrophil gelatinase-associated lipocalin clearance during veno-venous continuous renal replacement therapy in critically ill patients. Intensive Care Med. 2010;36:2156–2157. doi: 10.1007/s00134-010-2015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14:2534–2543. doi: 10.1097/01.asn.0000088027.54400.c6. [DOI] [PubMed] [Google Scholar]
- 63.Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005;579:773–777. doi: 10.1016/j.febslet.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 64.Parikh CR, Coca SG. Acute kidney injury: defining prerenal azotemia in clinical practice and research. Nat Rev Nephrol. 2010;6:641–642. doi: 10.1038/nrneph.2010.128. [DOI] [PubMed] [Google Scholar]
- 65.Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. J Intern Med. 2010;268:456–467. doi: 10.1111/j.1365-2796.2010.02269.x. [DOI] [PubMed] [Google Scholar]
- 66.Viau A, El KK, Laouari D, Burtin M, Nguyen C, Mori K, Pillebout E, Berger T, Mak TW, Knebelmann B, Friedlander G, Barasch J, Terzi F. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. 2010;120:4065–4076. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 68.Bykov I, Junnikkala S, Pekna M, Lindros KO, Meri S. Effect of chronic ethanol consumption on the expression of complement components and acute-phase proteins in liver. Clin Immunol. 2007;124:213–220. doi: 10.1016/j.clim.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 69.Ouchi Y, Kubota Y, Ito C. Serial analysis of gene expression in methamphetamine- and phencyclidine-treated rodent cerebral cortices: are there common mechanisms? Ann N Y Acad Sci. 2004;1025:57–61. doi: 10.1196/annals.1316.007. [DOI] [PubMed] [Google Scholar]
- 70.Adler M, Hoffmann D, Ellinger-Ziegelbauer H, Hewitt P, Matheis K, Mulrane L, Gallagher WM, Callanan JJ, Suter L, Fountoulakis MM, Dekant W, Mally A. Assessment of candidate biomarkers of drug-induced hepatobiliary injury in preclinical toxicity studies. Toxicol Lett. 2010;196:1–11. doi: 10.1016/j.toxlet.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 71.Amberger A, Schneeberger S, Hernegger G, Brandacher G, Obrist P, Lackner P, Margreiter R, Mark W. Gene expression profiling of prolonged cold ischemia and reperfusion in murine heart transplants. Transplantation. 2002;74:1441–1449. doi: 10.1097/00007890-200211270-00017. [DOI] [PubMed] [Google Scholar]
- 72.Kusaka M, Kuroyanagi Y, Kowa H, Nagaoka K, Mori T, Yamada K, Shiroki R, Kurahashi H, Hoshinaga K. Genomewide expression profiles of rat model renal isografts from brain dead donors. Transplantation. 2007;83:62–70. doi: 10.1097/01.tp.0000250485.53865.b8. [DOI] [PubMed] [Google Scholar]
- 73.Nacht M, Ferguson AT, Zhang W, Petroziello JM, Cook BP, Gao YH, Maguire S, Riley D, Coppola G, Landes GM, Madden SL, Sukumar S. Combining serial analysis of gene expression and array technologies to identify genes differentially expressed in breast cancer. Cancer Res. 1999;59:5464–5470. [PubMed] [Google Scholar]
- 74.Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat. 2008;108:389–397. doi: 10.1007/s10549-007-9619-3. [DOI] [PubMed] [Google Scholar]
- 75.Santin AD, Zhan F, Bellone S, Palmieri M, Cane S, Bignotti E, Anfossi S, Gokden M, Dunn D, Roman JJ, O’Brien TJ, Tian E, Cannon MJ, Shaughnessy J, Jr, Pecorelli S. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int J Cancer. 2004;112:14–25. doi: 10.1002/ijc.20408. [DOI] [PubMed] [Google Scholar]
- 76.Cho H, Kim JH. Lipocalin2 expressions correlate significantly with tumor differentiation in epithelial ovarian cancer. J Histochem Cytochem. 2009;57:513–521. doi: 10.1369/jhc.2009.953257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miyamoto T, Kashima H, Suzuki A, Kikuchi N, Konishi I, Seki N, Shiozawa T. Laser-captured microdissection-microarray analysis of the genes involved in endometrial carcinogenesis: stepwise up-regulation of lipocalin2 expression in normal and neoplastic endometria and its functional relevance. Hum Pathol. 2011;42:1265–1274. doi: 10.1016/j.humpath.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 78.Bousserouel S, Kauntz H, Gosse F, Bouhadjar M, Soler L, Marescaux J, Raul F. Identification of gene expression profiles correlated to tumor progression in a preclinical model of colon carcinogenesis. Int J Oncol. 2010;36:1485–1490. doi: 10.3892/ijo_00000635. [DOI] [PubMed] [Google Scholar]
- 79.Furutani M, Arii S, Mizumoto M, Kato M, Imamura M. Identification of a neutrophil gelatinase-associated lipocalin mRNA in human pancreatic cancers using a modified signal sequence trap method. Cancer Lett. 1998;122:209–214. doi: 10.1016/s0304-3835(97)00391-1. [DOI] [PubMed] [Google Scholar]
- 80.Galamb O, Gyorffy B, Sipos F, Spisak S, Nemeth AM, Miheller P, Tulassay Z, Dinya E, Molnar B. Inflammation, adenoma and cancer: objective classification of colon biopsy specimens with gene expression signature. Dis Markers. 2008;25:1–16. doi: 10.1155/2008/586721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Linnerth NM, Sirbovan K, Moorehead RA. Use of a transgenic mouse model to identify markers of human lung tumors. Int J Cancer. 2005;114:977–982. doi: 10.1002/ijc.20814. [DOI] [PubMed] [Google Scholar]
- 82.Wang HJ, He XJ, Ma YY, Jiang XT, Xia YJ, Ye ZY, Zhao ZS, Tao HQ. Expressions of neutrophil gelatinase-associated lipocalin in gastric cancer: a potential biomarker for prognosis and an ancillary diagnostic test. Anat Rec (Hoboken ) 2010;293:1855–1863. doi: 10.1002/ar.21230. [DOI] [PubMed] [Google Scholar]
- 83.Alpizar YA, Ramirez BS, Fernandez DR, Capote AR, Hidalgo GG, Rodriguez RP, Molina LE. HER1-ECD vaccination dispenses with emulsification to elicit HER1-specific anti-proliferative effects. Hum Vaccin. 2009;5:158–165. doi: 10.4161/hv.5.3.7129. [DOI] [PubMed] [Google Scholar]
- 84.Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, Ryu B, Skinner HG, Goggins M, Jaffee EM, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4324. [PubMed] [Google Scholar]
- 85.Laurell H, Bouisson M, Berthelemy P, Rochaix P, Dejean S, Besse P, Susini C, Pradayrol L, Vaysse N, Buscail L. Identification of biomarkers of human pancreatic adenocarcinomas by expression profiling and validation with gene expression analysis in endoscopic ultrasound-guided fine needle aspiration samples. World J Gastroenterol. 2006;12:3344–3351. doi: 10.3748/wjg.v12.i21.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, Lele SM, Ouellette M, Pour PM, Sasson AR, Brand RE, Hollingsworth MA, Jain M, Batra SK. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540–1547. doi: 10.1038/sj.bjc.6604329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stoesz SP, Gould MN. Overexpression of neu-related lipocalin (NRL) in neu-initiated but not ras or chemically initiated rat mammary carcinomas. Oncogene. 1995;11:2233–2241. [PubMed] [Google Scholar]
- 88.Stoesz SP, Friedl A, Haag JD, Lindstrom MJ, Clark GM, Gould MN. Heterogeneous expression of the lipocalin NGAL in primary breast cancers. Int J Cancer. 1998;79:565–572. doi: 10.1002/(sici)1097-0215(19981218)79:6<565::aid-ijc3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 89.Lim R, Ahmed N, Borregaard N, Riley C, Wafai R, Thompson EW, Quinn MA, Rice GE. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. Int J Cancer. 2007;120:2426–2434. doi: 10.1002/ijc.22352. [DOI] [PubMed] [Google Scholar]
- 90.Cho H, Lim BJ, Kang ES, Choi JS, Kim JH. Molecular characterization of a new ovarian cancer cell line, YDOV-151, established from mucinous cystadenocarcinoma 1. Tohoku J Exp Med. 2009;218:129–139. doi: 10.1620/tjem.218.129. [DOI] [PubMed] [Google Scholar]
- 91.Emmanuel C, Gava N, Kennedy C, Balleine RL, Sharma R, Wain G, Brand A, Hogg R, Etemadmoghadam D, George J, Birrer MJ, Clarke CL, Chenevix-Trench G, Bowtell DD, Harnett PR, deFazio A. Comparison of expression profiles in ovarian epithelium in vivo and ovarian cancer identifies novel candidate genes involved in disease pathogenesis. PLoS One. 2011;6:e17617. doi: 10.1371/journal.pone.0017617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wong YF, Cheung TH, Lo KW, Yim SF, Siu NS, Chan SC, Ho TW, Wong KW, Yu MY, Wang VW, Li C, Gardner GJ, Bonome T, Johnson WB, Smith DI, Chung TK, Birrer MJ. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene. 2007;26:1971–1982. doi: 10.1038/sj.onc.1209986. [DOI] [PubMed] [Google Scholar]
- 93.Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Silva C, Rotellar F, Hernandez-Lizoain JL, Baixauli J, Valenti V, Pardo F, Salvador J, Fruhbeck G. Up-regulation of the novel proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and osteopontin as well as angiogenic-related factors in visceral adipose tissue of patients with colon cancer. J Nutr Biochem. 2011;22:634–641. doi: 10.1016/j.jnutbio.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 94.Sun Y, Yokoi K, Li H, Gao J, Hu L, Liu B, Chen K, Hamilton SR, Fan D, Sun B, Zhang W. NGAL expression is elevated in both colorectal adenoma-carcinoma sequence and cancer progression and enhances tumorigenesis in xenograft mouse models. Clin Cancer Res. 2011;17:4331–4340. doi: 10.1158/1078-0432.CCR-11-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chakraborty S, Baine MJ, Sasson AR, Batra SK. Current status of molecular markers for early detection of sporadic pancreatic cancer. Biochim Biophys Acta. 2011;1815:44–64. doi: 10.1016/j.bbcan.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tong Z, Kunnumakkara AB, Wang H, Matsuo Y, Diagaradjane P, Harikumar KB, Ramachandran V, Sung B, Chakraborty A, Bresalier RS, Logsdon C, Aggarwal BB, Krishnan S, Guha S. Neutrophil gelatinase-associated lipocalin: a novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 2008;68:6100–6108. doi: 10.1158/0008-5472.CAN-08-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 98.Hiukka A, Maranghi M, Matikainen N, Taskinen MR. PPARalpha: an emerging therapeutic target in diabetic microvascular damage. Nat Rev Endocrinol. 2010;6:454–463. doi: 10.1038/nrendo.2010.89. [DOI] [PubMed] [Google Scholar]
- 99.Reddy JK, Rao S, Moody DE. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Res. 1976;36:1211–1217. [PubMed] [Google Scholar]
- 100.Meyer K, Lee JS, Dyck PA, Cao WQ, Rao MS, Thorgeirsson SS, Reddy JK. Molecular profiling of hepatocellular carcinomas developing spontaneously in acyl-CoA oxidase deficient mice: comparison with liver tumors induced in wild-type mice by a peroxisome proliferator and a genotoxic carcinogen. Carcinogenesis. 2003;24:975–984. doi: 10.1093/carcin/bgg040. [DOI] [PubMed] [Google Scholar]
- 101.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 102.Smith ER, Zurakowski D, Saad A, Scott RM, Moses MA. Urinary biomarkers predict brain tumor presence and response to therapy. Clin Cancer Res. 2008;14:2378–2386. doi: 10.1158/1078-0432.CCR-07-1253. [DOI] [PubMed] [Google Scholar]
- 103.Barresi V, Tuccari G, Barresi G. NGAL immunohistochemical expression in brain primary and metastatic tumors. Clin Neuropathol. 2010;29:317–322. doi: 10.5414/npp29317. [DOI] [PubMed] [Google Scholar]
- 104.Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP, Barclay E, Calonje E, Manek S, Adams SJ, Bowers PW, Burrows NP, Charles-Holmes R, Cook LJ, Daly BM, Ford GP, Fuller LC, Hadfield-Jones SE, Hardwick N, Highet AS, Keefe M, MacDonald-Hull SP, Potts ED, Crone M, Wilkinson S, Camacho-Martinez F, Jablonska S, Ratnavel R, MacDonald A, Mann RJ, Grice K, Guillet G, Lewis-Jones MS, McGrath H, Seukeran DC, Morrison PJ, Fleming S, Rahman S, Kelsell D, Leigh I, Olpin S, Tomlinson IP. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12:1241–1252. doi: 10.1093/hmg/ddg148. [DOI] [PubMed] [Google Scholar]
- 105.Ashrafian H, O’Flaherty L, Adam J, Steeples V, Chung YL, East P, Vanharanta S, Lehtonen H, Nye E, Hatipoglu E, Miranda M, Howarth K, Shukla D, Troy H, Griffiths J, Spencer-Dene B, Yusuf M, Volpi E, Maxwell PH, Stamp G, Poulsom R, Pugh CW, Costa B, Bardella C, Di Renzo MF, Kotlikoff MI, Launonen V, Aaltonen L, El-Bahrawy M, Tomlinson I, Pollard PJ. Expression profiling in progressive stages of fumarate-hydratase deficiency: the contribution of metabolic changes to tumorigenesis. Cancer Res. 2010;70:9153–9165. doi: 10.1158/0008-5472.CAN-10-1949. [DOI] [PubMed] [Google Scholar]
- 106.Villalva C, Sorel N, Bonnet ML, Guilhot J, Mayeur-Rousse C, Guilhot F, Chomel JC, Turhan AG. Neutrophil gelatinase-associated lipocalin expression in chronic myeloid leukemia. Leuk Lymphoma. 2008;49:984–988. doi: 10.1080/10428190801942360. [DOI] [PubMed] [Google Scholar]
- 107.Leng X, Lin H, Ding T, Wang Y, Wu Y, Klumpp S, Sun T, Zhou Y, Monaco P, Belmont J, Aderem A, Akira S, Strong R, Arlinghaus R. Lipocalin 2 is required for BCR-ABL-induced tumorigenesis. Oncogene. 2008;27:6110–6119. doi: 10.1038/onc.2008.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arlinghaus R, Leng X. Requirement of lipocalin 2 for chronic myeloid leukemia. Leuk Lymphoma. 2008;49:600–603. doi: 10.1080/10428190701859664. [DOI] [PubMed] [Google Scholar]
- 109.Lin H, Monaco G, Sun T, Ling X, Stephens C, Xie S, Belmont J, Arlinghaus R. Bcr-Abl-mediated suppression of normal hematopoiesis in leukemia. Oncogene. 2005;24:3246–3256. doi: 10.1038/sj.onc.1208500. [DOI] [PubMed] [Google Scholar]
- 110.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 111.Xiao J, Wu Y, Chen R, Lin Y, Wu L, Tian W, Liu L. Expression of Pcp4 gene during osteogenic differentiation of bone marrow mesenchymal stem cells in vitro. Mol Cell Biochem. 2008;309:143–150. doi: 10.1007/s11010-007-9652-x. [DOI] [PubMed] [Google Scholar]
- 112.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 113.Geng S, Wang Q, Wang J, Hu Z, Liu C, Qiu J, Zeng W. Isolation and identification of a distinct side population cancer cells in the human epidermal squamous cancer cell line A431. Arch Dermatol Res. 2011;303:181–189. doi: 10.1007/s00403-010-1100-1. [DOI] [PubMed] [Google Scholar]
- 114.Tong Z, Wu X, Ovcharenko D, Zhu J, Chen CS, Kehrer JP. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochem J. 2005;391:441–448. doi: 10.1042/BJ20051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sengelov H, Boulay F, Kjeldsen L, Borregaard N. Subcellular localization and translocation of the receptor for N-formylmethionyl-leucyl-phenylalanine in human neutrophils. Biochem J. 1994;299(Pt 2):473–479. doi: 10.1042/bj2990473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carrano CJ, Raymond KN. Coordination chemistry of microbial iron transport compounds: rhodotorulic acid and iron uptake in Rhodotorula pilimanae. J Bacteriol. 1978;136:69–74. doi: 10.1128/jb.136.1.69-74.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miethke M, Skerra A. Neutrophil gelatinase-associated lipocalin expresses antimicrobial activity by interfering with L-norepinephrine-mediated bacterial iron acquisition. Antimicrob Agents Chemother. 2010;54:1580–1589. doi: 10.1128/AAC.01158-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin RB, Savory J, Brown S, Bertholf RL, Wills MR. Transferrin binding of Al3+ and Fe3+ Clin Chem. 1987;33:405–407. [PubMed] [Google Scholar]
- 119.Dertz EA, Xu J, Stintzi A, Raymond KN. Bacillibactin-mediated iron transport in Bacillus subtilis. J Am Chem Soc. 2006;128:22–23. doi: 10.1021/ja055898c. [DOI] [PubMed] [Google Scholar]
- 120.Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, Seifert M, Crouch ML, Hantke K, Akira S, Fang FC, Weiss G. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114:3642–3651. doi: 10.1182/blood-2009-05-223354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Miethke M, Klotz O, Linne U, May JJ, Beckering CL, Marahiel MA. Ferri-bacillibactin uptake and hydrolysis in Bacillus subtilis. Mol Microbiol. 2006;61:1413–1427. doi: 10.1111/j.1365-2958.2006.05321.x. [DOI] [PubMed] [Google Scholar]
- 122.Ollinger J, Song KB, Antelmann H, Hecker M, Helmann JD. Role of the Fur regulon in iron transport in Bacillus subtilis. J Bacteriol. 2006;188:3664–3673. doi: 10.1128/JB.188.10.3664-3673.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, Viltard M, Williams D, Paragas N, Leete T, Kulkarni R, Li X, Lee B, Kalandadze A, Ratner AJ, Pizarro JC, Schmidt-Ott KM, Landry DW, Raymond KN, Strong RK, Barasch J. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. 2010;6:602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 125.Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115:610–621. doi: 10.1172/JCI23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, Aucouturier P, Moskaug JO, Otto A, Christensen EI, Willnow TE. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol. 1999;155:1361–1370. doi: 10.1016/S0002-9440(10)65238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Demaurex N. pH Homeostasis of cellular organelles. News Physiol Sci. 2002;17:1–5. doi: 10.1152/physiologyonline.2002.17.1.1. [DOI] [PubMed] [Google Scholar]
- 128.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 129.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, Baumler AJ. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe. 2009;5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol. 2004;58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 132.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 133.Andre E, Stoeger T, Takenaka S, Bahnweg M, Ritter B, Karg E, Lentner B, Reinhard C, Schulz H, Wjst M. Inhalation of ultrafine carbon particles triggers biphasic pro-inflammatory response in the mouse lung. Eur Respir J. 2006;28:275–285. doi: 10.1183/09031936.06.00071205. [DOI] [PubMed] [Google Scholar]
- 134.Kohl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol. 2006;586:71–94. doi: 10.1007/0-387-34134-X_6. [DOI] [PubMed] [Google Scholar]
- 135.Hawlisch H, Wills-Karp M, Karp CL, Kohl J. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol. 2004;41:123–131. doi: 10.1016/j.molimm.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 136.Haas KM, Hasegawa M, Steeber DA, Poe JC, Zabel MD, Bock CB, Karp DR, Briles DE, Weis JH, Tedder TF. Complement receptors CD21/35 link innate and protective immunity during Streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity. 2002;17:713–723. doi: 10.1016/s1074-7613(02)00483-1. [DOI] [PubMed] [Google Scholar]
- 137.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 138.Bahmani P, Halabian R, Rouhbakhsh M, Roushandeh AM, Masroori N, Ebrahimi M, Samadikuchaksaraei A, Shokrgozar MA, Roudkenar MH. Neutrophil gelatinase-associated lipocalin induces the expression of heme oxygenase-1 and superoxide dismutase 1, 2. Cell Stress Chaperones. 2010;15:395–403. doi: 10.1007/s12192-009-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roudkenar MH, Halabian R, Ghasemipour Z, Roushandeh AM, Rouhbakhsh M, Nekogoftar M, Kuwahara Y, Fukumoto M, Shokrgozar MA. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H(2)O(2) toxicity. Arch Med Res. 2008;39:560–566. doi: 10.1016/j.arcmed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 140.Roudkenar MH, Halabian R, Oodi A, Roushandeh AM, Yaghmai P, Najar MR, Amirizadeh N, Shokrgozar MA. Upregulation of neutrophil gelatinase-associated lipocalin, NGAL/Lcn2, in beta-thalassemia patients. Arch Med Res. 2008;39:402–407. doi: 10.1016/j.arcmed.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 141.Roudkenar MH, Kuwahara Y, Baba T, Roushandeh AM, Ebishima S, Abe S, Ohkubo Y, Fukumoto M. Oxidative stress induced lipocalin 2 gene expression: addressing its expression under the harmful conditions. J Radiat Res (Tokyo) 2007;48:39–44. doi: 10.1269/jrr.06057. [DOI] [PubMed] [Google Scholar]
- 142.Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C, Tell G, Salzano AM, Scaloni A, Vuttariello E, Chiappetta G, Formisano S, Leonardi A. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci U S A. 2008;105:14058–14063. doi: 10.1073/pnas.0710846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Seth P, Porter D, Lahti-Domenici J, Geng Y, Richardson A, Polyak K. Cellular and molecular targets of estrogen in normal human breast tissue. Cancer Res. 2002;62:4540–4544. [PubMed] [Google Scholar]
- 144.Shi H, Gu Y, Yang J, Xu L, Mi W, Yu W. Lipocalin 2 promotes lung metastasis of murine breast cancer cells. J Exp Clin Cancer Res. 2008;27:83. doi: 10.1186/1756-9966-27-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee HJ, Lee EK, Lee KJ, Hong SW, Yoon Y, Kim JS. Ectopic expression of neutrophil gelatinase-associated lipocalin suppresses the invasion and liver metastasis of colon cancer cells. Int J Cancer. 2006;118:2490–2497. doi: 10.1002/ijc.21657. [DOI] [PubMed] [Google Scholar]
- 146.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery. 2004;135:555–562. doi: 10.1016/j.surg.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 147.Baril P, Gangeswaran R, Mahon PC, Caulee K, Kocher HM, Harada T, Zhu M, Kalthoff H, Crnogorac-Jurcevic T, Lemoine NR. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene. 2007;26:2082–2094. doi: 10.1038/sj.onc.1210009. [DOI] [PubMed] [Google Scholar]
- 148.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 149.Venkatesha S. Lipocalin 2 antagonizes the proangiogenic action of ras in transformed cells. 2006 doi: 10.1158/1541-7786.MCR-06-0110. [DOI] [PubMed] [Google Scholar]
- 150.Sweeney SM, DiLullo G, Slater SJ, Martinez J, Iozzo RV, Lauer-Fields JL, Fields GB, San Antonio JD. Angiogenesis in collagen I requires alpha2beta1 ligation of a GFP*GER sequence and possibly p38 MAPK activation and focal adhesion disassembly. J Biol Chem. 2003;278:30516–30524. doi: 10.1074/jbc.M304237200. [DOI] [PubMed] [Google Scholar]
- 151.Philip PA. Targeting angiogenesis in pancreatic cancer. Lancet. 2008;371:2062–2064. doi: 10.1016/S0140-6736(08)60770-9. [DOI] [PubMed] [Google Scholar]
- 152.Hansen J. Increased breast cancer risk among women who work predominantly at night. Epidemiology. 2001;12:74–77. doi: 10.1097/00001648-200101000-00013. [DOI] [PubMed] [Google Scholar]
- 153.Sharman EH, Sharman KG, Bondy SC. Extended exposure to dietary melatonin reduces tumor number and size in aged male mice. Exp Gerontol. 2011;46:18–22. doi: 10.1016/j.exger.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee EK, Kim HJ, Lee KJ, Lee HJ, Lee JS, Kim DG, Hong SW, Yoon Y, Kim JS. Inhibition of the proliferation and invasion of hepatocellular carcinoma cells by lipocalin 2 through blockade of JNK and PI3K/Akt signaling. Int J Oncol. 2011;38:325–333. doi: 10.3892/ijo.2010.854. [DOI] [PubMed] [Google Scholar]
- 155.Tong Z, Chakraborty S, Sung B, Koolwal P, Kaur S, Aggarwal BB, Mani SA, Bresalier RS, Batra SK, Guha S. Epidermal growth factor down-regulates the expression of neutrophil gelatinase-associated lipocalin (NGAL) through E-cadherin in pancreatic cancer cells. Cancer. 2010 doi: 10.1002/cncr.25803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pacifico F, Crescenzi E, Mellone S, Iannetti A, Porrino N, Liguoro D, Moscato F, Grieco M, Formisano S, Leonardi A. Nuclear factor-{kappa}B contributes to anaplastic thyroid carcinomas through up-regulation of miR-146a. J Clin Endocrinol Metab. 2010;95:1421–1430. doi: 10.1210/jc.2009-1128. [DOI] [PubMed] [Google Scholar]
- 157.Bando M, Hiroshima Y, Kataoka M, Shinohara Y, Herzberg MC, Ross KF, Nagata T, Kido J. Interleukin-1alpha regulates antimicrobial peptide expression in human keratinocytes. Immunol Cell Biol. 2007;85:532–537. doi: 10.1038/sj.icb.7100078. [DOI] [PubMed] [Google Scholar]
- 158.Sorensen OE, Cowland JB, Theilgaard-Monch K, Liu L, Ganz T, Borregaard N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol. 2003;170:5583–5589. doi: 10.4049/jimmunol.170.11.5583. [DOI] [PubMed] [Google Scholar]
- 159.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 160.Underhill DM. Toll-like receptors: networking for success. Eur J Immunol. 2003;33:1767–1775. doi: 10.1002/eji.200324037. [DOI] [PubMed] [Google Scholar]
- 161.Zhang FX, Kirschning CJ, Mancinelli R, Xu XP, Jin Y, Faure E, Mantovani A, Rothe M, Muzio M, Arditi M. Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611–7614. doi: 10.1074/jbc.274.12.7611. [DOI] [PubMed] [Google Scholar]
- 162.Kirschning CJ, Wesche H, Merrill AT, Rothe M. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med. 1998;188:2091–2097. doi: 10.1084/jem.188.11.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 165.Zhu J, Mohan C. Toll-like receptor signaling pathways--therapeutic opportunities. Mediators Inflamm. 2010;2010:781235. doi: 10.1155/2010/781235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Whitmore MM, Iparraguirre A, Kubelka L, Weninger W, Hai T, Williams BR. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. doi: 10.4049/jimmunol.179.6.3622. [DOI] [PubMed] [Google Scholar]
- 167.Eller MS, Yaar M, Ostrom K, Harkness DD, Gilchrest BA. A role for interleukin-1 in epidermal differentiation: regulation by expression of functional versus decoy receptors. J Cell Sci. 1995;108(Pt 8):2741–2746. doi: 10.1242/jcs.108.8.2741. [DOI] [PubMed] [Google Scholar]
- 168.Watt FM, Mattey DL, Garrod DR. Calcium-induced reorganization of desmosomal components in cultured human keratinocytes. J Cell Biol. 1984;99:2211–2215. doi: 10.1083/jcb.99.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Seo EY, Lee WH, Piao YJ, Kim KH, Lee KM, Ahn KS, Yang JM, Seo YJ, Kim CD, Park JK, Lee JH. Identification of calcium-inducible genes in primary keratinocytes using suppression-subtractive hybridization. Exp Dermatol. 2004;13:163–169. doi: 10.1111/j.0906-6705.2004.0144.x. [DOI] [PubMed] [Google Scholar]
- 170.Seo SJ, Ahn JY, Hong CK, Seo EY, Kye KC, Lee WH, Lee SK, Lim JS, Hahn MJ, Kjeldsen L, Borregaard N, Kim CD, Park JK, Lee JH. Expression of neutrophil gelatinase-associated lipocalin in skin epidermis. J Invest Dermatol. 2006;126:510–512. doi: 10.1038/sj.jid.5700035. [DOI] [PubMed] [Google Scholar]
- 171.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 172.Florin L, Hummerich L, Dittrich BT, Kokocinski F, Wrobel G, Gack S, Schorpp-Kistner M, Werner S, Hahn M, Lichter P, Szabowski A, Angel P. Identification of novel AP-1 target genes in fibroblasts regulated during cutaneous wound healing. Oncogene. 2004;23:7005–7017. doi: 10.1038/sj.onc.1207938. [DOI] [PubMed] [Google Scholar]
- 173.Yang YH, He XJ, Chen SR, Wang L, Li EM, Xu LY. Changes of serum and urine neutrophil gelatinase-associated lipocalin in type-2 diabetic patients with nephropathy: one year observational follow-up study. Endocrine. 2009;36:45–51. doi: 10.1007/s12020-009-9187-x. [DOI] [PubMed] [Google Scholar]
- 174.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rong Z, Cheng L, Ren Y, Li Z, Li Y, Li X, Li H, Fu XY, Chang Z. Interleukin-17F signaling requires ubiquitination of interleukin-17 receptor via TRAF6. Cell Signal. 2007;19:1514–1520. doi: 10.1016/j.cellsig.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 176.Pillebout E, Burtin M, Yuan HT, Briand P, Woolf AS, Friedlander G, Terzi F. Proliferation and remodeling of the peritubular microcirculation after nephron reduction: association with the progression of renal lesions. Am J Pathol. 2001;159:547–560. doi: 10.1016/S0002-9440(10)61726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hynes NE, Macdonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 178.Mimeault M, Johansson SL, Vankatraman G, Moore E, Henichart JP, Depreux P, Lin MF, Batra SK. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol Cancer Ther. 2007;6:967–978. doi: 10.1158/1535-7163.MCT-06-0648. [DOI] [PubMed] [Google Scholar]
- 179.Gao H, Liang M, Bergdahl A, Hamren A, Lindholm MW, Dahlman-Wright K, Nilsson BO. Estrogen attenuates vascular expression of inflammation associated genes and adhesion of monocytes to endothelial cells. Inflamm Res. 2006;55:349–353. doi: 10.1007/s00011-006-5194-z. [DOI] [PubMed] [Google Scholar]
- 180.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 181.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 182.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 183.Schnarr B, Strunz K, Ohsam J, Benner A, Wacker J, Mayer D. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int J Cancer. 2000;89:506–513. doi: 10.1002/1097-0215(20001120)89:6<506::aid-ijc7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 184.Han CH, Cho JY, Moon JT, Kim HJ, Kim SK, Shin DH, Chang J, Ahn CM, Kim SK, Chang YS. Clinical significance of insulin receptor substrate-I down-regulation in non-small cell lung cancer. Oncol Rep. 2006;16:1205–1210. [PubMed] [Google Scholar]
- 185.Hibbs MS, Hasty KA, Seyer JM, Kang AH, Mainardi CL. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J Biol Chem. 1985;260:2493–2500. [PubMed] [Google Scholar]
- 186.Finlay GA, Russell KJ, McMahon KJ, D’Arcy EM, Masterson JB, FitzGerald MX, O’Connor CM. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax. 1997;52:502–506. doi: 10.1136/thx.52.6.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Van den Steen PE, Van A, Hvidberg IV, Piccard H, Fiten P, Jacobsen C, Moestrup SK, Fry S, Royle L, Wormald MR, Wallis R, Rudd PM, Dwek RA, Opdenakker G. The hemopexin and O-glycosylated domains tune gelatinase B/MMP-9 bioavailability via inhibition and binding to cargo receptors. J Biol Chem. 2006;281:18626–18637. doi: 10.1074/jbc.M512308200. [DOI] [PubMed] [Google Scholar]
- 188.Shapiro NI, Trzeciak S, Hollander JE, Birkhahn R, Otero R, Osborn TM, Moretti E, Nguyen HB, Gunnerson KJ, Milzman D, Gaieski DF, Goyal M, Cairns CB, Ngo L, Rivers EP. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med. 2009;37:96–104. doi: 10.1097/CCM.0b013e318192fd9d. [DOI] [PubMed] [Google Scholar]
- 189.Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, Lichtiger S, D’Haens G, Diamond RH, Broussard DL, Tang KL, van der Woude CJ, Rutgeerts P. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362:1383–1395. doi: 10.1056/NEJMoa0904492. [DOI] [PubMed] [Google Scholar]
- 190.Bolignano D, Della TA, Lacquaniti A, Costantino G, Fries W, Buemi M. Neutrophil gelatinase-associated lipocalin levels in patients with crohn disease undergoing treatment with infliximab. J Investig Med. 2010;58:569–571. doi: 10.231/JIM.0b013e3181ccc20c. [DOI] [PubMed] [Google Scholar]
- 191.Arijs I, De HG, Lemaire K, Quintens R, Van LL, Van SK, Leemans P, Cleynen I, Van AG, Vermeire S, Geboes K, Schuit F, Rutgeerts P. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One. 2009;4:e7984. doi: 10.1371/journal.pone.0007984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med. 1997;155:449–453. doi: 10.1164/ajrccm.155.2.9032177. [DOI] [PubMed] [Google Scholar]
- 193.Choi KM, Kim TN, Yoo HJ, Lee KW, Cho GJ, Hwang TG, Baik SH, Choi DS, Kim SM. Effect of exercise training on A-FABP, lipocalin-2 and RBP4 levels in obese women. Clin Endocrinol (Oxf) 2009;70:569–574. doi: 10.1111/j.1365-2265.2008.03374.x. [DOI] [PubMed] [Google Scholar]
- 194.Gheorghiade M, Zannad F, Sopko G, Klein L, Pina IL, Konstam MA, Massie BM, Roland E, Targum S, Collins SP, Filippatos G, Tavazzi L. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112:3958–3968. doi: 10.1161/CIRCULATIONAHA.105.590091. [DOI] [PubMed] [Google Scholar]
- 195.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart disease and stroke statistics--2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 196.Nieminen MS, Harjola VP. Definition and epidemiology of acute heart failure syndromes. Am J Cardiol. 2005;96:5G–10G. doi: 10.1016/j.amjcard.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 197.Aghel A, Shrestha K, Mullens W, Borowski A, Tang WH. Serum neutrophil gelatinase-associated lipocalin (NGAL) in predicting worsening renal function in acute decompensated heart failure. J Card Fail. 2010;16:49–54. doi: 10.1016/j.cardfail.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fonarow GC, Abraham WT, Albert NM, Gattis WA, Gheorghiade M, Greenberg B, O’Connor CM, Yancy CW, Young J. Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF): rationale and design. Am Heart J. 2004;148:43–51. doi: 10.1016/j.ahj.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 199.Fonarow GC, Adams KF, Jr, Abraham WT, Yancy CW, Boscardin WJ. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA. 2005;293:572–580. doi: 10.1001/jama.293.5.572. [DOI] [PubMed] [Google Scholar]
- 200.Fonarow GC, Abraham WT, Albert NM, Gattis SW, Gheorghiade M, Greenberg BH, O’Connor CM, Pieper K, Sun JL, Yancy CW, Young JB. Influence of a performance-improvement initiative on quality of care for patients hospitalized with heart failure: results of the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure (OPTIMIZE-HF) Arch Intern Med. 2007;167:1493–1502. doi: 10.1001/archinte.167.14.1493. [DOI] [PubMed] [Google Scholar]
- 201.Hillege HL, Girbes AR, de Kam PJ, Boomsma F, de ZD, Charlesworth A, Hampton JR, van Veldhuisen DJ. Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation. 2000;102:203–210. doi: 10.1161/01.cir.102.2.203. [DOI] [PubMed] [Google Scholar]
- 202.Damman K, Masson S, Hillege HL, Maggioni AP, Voors AA, Opasich C, van Veldhuisen DJ, Montagna L, Cosmi F, Tognoni G, Tavazzi L, Latini R. Clinical outcome of renal tubular damage in chronic heart failure. Eur Heart J. 2011;32:2705–2712. doi: 10.1093/eurheartj/ehr190. [DOI] [PubMed] [Google Scholar]
- 203.Damman K, Ng Kam Chuen MJ, MacFadyen RJ, Lip GY, Gaze D, Collinson PO, Hillege HL, van OW, Voors AA, van Veldhuisen DJ. Volume status and diuretic therapy in systolic heart failure and the detection of early abnormalities in renal and tubular function. J Am Coll Cardiol. 2011;57:2233–2241. doi: 10.1016/j.jacc.2010.10.065. [DOI] [PubMed] [Google Scholar]
- 204.Schmidt-Ott KM, Mori K, Kalandadze A, Li JY, Paragas N, Nicholas T, Devarajan P, Barasch J. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens. 2006;15:442–449. doi: 10.1097/01.mnh.0000232886.81142.58. [DOI] [PubMed] [Google Scholar]
- 205.Kim T, Arnaoutakis GJ, Bihorac A, Martin TD, Hess PJ, Jr, Klodell CT, Tribble CG, Ejaz AA, Moldawer LL, Beaver TM. Early blood biomarkers predict organ injury and resource utilization following complex cardiac surgery. J Surg Res. 2011;168:168–172. doi: 10.1016/j.jss.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Han WK, Wagener G, Zhu Y, Wang S, Lee HT. Urinary biomarkers in the early detection of acute kidney injury after cardiac surgery. Clin J Am Soc Nephrol. 2009;4:873–882. doi: 10.2215/CJN.04810908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Perry TE, Muehlschlegel JD, Liu KY, Fox AA, Collard CD, Shernan SK, Body SC. Plasma neutrophil gelatinase-associated lipocalin and acute postoperative kidney injury in adult cardiac surgical patients. Anesth Analg. 2010;110:1541–1547. doi: 10.1213/ANE.0b013e3181da938e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Koch AM, Dittrich S, Cesnjevar R, Ruffer A, Breuer C, Glockler M. Plasma neutrophil gelatinase-associated lipocalin measured in consecutive patients after congenital heart surgery using point-of-care technology. Interact Cardiovasc Thorac Surg. 2011;13:133–136. doi: 10.1510/icvts.2011.269647. [DOI] [PubMed] [Google Scholar]
- 209.Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11:R31. doi: 10.1186/cc5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med. 1998;104:343–348. doi: 10.1016/s0002-9343(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 211.Vaidya VS, Waikar SS, Ferguson MA, Collings FB, Sunderland K, Gioules C, Bradwin G, Matsouaka R, Betensky RA, Curhan GC, Bonventre JV. Urinary biomarkers for sensitive and specific detection of acute kidney injury in humans. Clin Transl Sci. 2008;1:200–208. doi: 10.1111/j.1752-8062.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Koyner JL, Vaidya VS, Bennett MR, Ma Q, Worcester E, Akhter SA, Raman J, Jeevanandam V, O’Connor MF, Devarajan P, Bonventre JV, Murray PT. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol. 2010;5:2154–2165. doi: 10.2215/CJN.00740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Che M, Xie B, Xue S, Dai H, Qian J, Ni Z, Axelsson J, Yan Y. Clinical usefulness of novel biomarkers for the detection of acute kidney injury following elective cardiac surgery. Nephron Clin Pract. 2010;115:c66–c72. doi: 10.1159/000286352. [DOI] [PubMed] [Google Scholar]
- 214.Cai L, Borowiec J, Xu S, Han W, Venge P. Assays of urine levels of HNL/NGAL in patients undergoing cardiac surgery and the impact of antibody configuration on their clinical performances. Clin Chim Acta. 2009;403:121–125. doi: 10.1016/j.cca.2009.01.030. [DOI] [PubMed] [Google Scholar]
- 215.Wachtfogel YT, Kucich U, Greenplate J, Gluszko P, Abrams W, Weinbaum G, Wenger RK, Rucinski B, Niewiarowski S, Edmunds LH., Jr Human neutrophil degranulation during extracorporeal circulation. Blood. 1987;69:324–330. [PubMed] [Google Scholar]
- 216.Weber CL, Bennett M, Er L, Bennett MT, Levin A. Urinary NGAL levels before and after coronary angiography: a complex story. Nephrol Dial Transplant. 2011;26:3207–3211. doi: 10.1093/ndt/gfr033. [DOI] [PubMed] [Google Scholar]
- 217.Cruz DN, de CM, Garzotto F, Perazella MA, Lentini P, Corradi V, Piccinni P, Ronco C. Plasma neutrophil gelatinase-associated lipocalin is an early biomarker for acute kidney injury in an adult ICU population. Intensive Care Med. 2010;36:444–451. doi: 10.1007/s00134-009-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.de Geus HR, Bakker J, Lesaffre EM, le Noble JL. Neutrophil gelatinase-associated lipocalin at ICU admission predicts for acute kidney injury in adult patients. Am J Respir Crit Care Med. 2011;183:907–914. doi: 10.1164/rccm.200908-1214OC. [DOI] [PubMed] [Google Scholar]
- 219.Srisawat N, Murugan R, Lee M, Kong L, Carter M, Angus DC, Kellum JA. Plasma neutrophil gelatinase-associated lipocalin predicts recovery from acute kidney injury following community-acquired pneumonia. Kidney Int. 2011 doi: 10.1038/ki.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Parikh CR, Jani A, Mishra J, Ma Q, Kelly C, Barasch J, Edelstein CL, Devarajan P. Urine NGAL and IL-18 are predictive biomarkers for delayed graft function following kidney transplantation. Am J Transplant. 2006;6:1639–1645. doi: 10.1111/j.1600-6143.2006.01352.x. [DOI] [PubMed] [Google Scholar]
- 221.Mishra J, Ma Q, Kelly C, Mitsnefes M, Mori K, Barasch J, Devarajan P. Kidney NGAL is a novel early marker of acute injury following transplantation. Pediatr Nephrol. 2006;21:856–863. doi: 10.1007/s00467-006-0055-0. [DOI] [PubMed] [Google Scholar]
- 222.Korbely R, Wilflingseder J, Perco P, Kainz A, Langer RM, Mayer B, Oberbauer R. Molecular biomarker candidates of acute kidney injury in zero-hour renal transplant needle biopsies. Transpl Int. 2011;24:143–149. doi: 10.1111/j.1432-2277.2010.01162.x. [DOI] [PubMed] [Google Scholar]
- 223.Shapiro NI, Trzeciak S, Hollander JE, Birkhahn R, Otero R, Osborn TM, Moretti E, Nguyen HB, Gunnerson K, Milzman D, Gaieski DF, Goyal M, Cairns CB, Kupfer K, Lee SW, Rivers EP. The diagnostic accuracy of plasma neutrophil gelatinase-associated lipocalin in the prediction of acute kidney injury in emergency department patients with suspected sepsis. Ann Emerg Med. 2010;56:52–59. doi: 10.1016/j.annemergmed.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 224.Metra M, Nodari S, Parrinello G, Bordonali T, Bugatti S, Danesi R, Fontanella B, Lombardi C, Milani P, Verzura G, Cotter G, Dittrich H, Massie BM, Dei CL. Worsening renal function in patients hospitalised for acute heart failure: clinical implications and prognostic significance. Eur J Heart Fail. 2008;10:188–195. doi: 10.1016/j.ejheart.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 225.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 226.Bagshaw SM, George C, Bellomo R. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12:R47. doi: 10.1186/cc6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bagshaw SM, Uchino S, Bellomo R, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Oudemans-van Straaten HM, Ronco C, Kellum JA. Septic acute kidney injury in critically ill patients: clinical characteristics and outcomes. Clin J Am Soc Nephrol. 2007;2:431–439. doi: 10.2215/CJN.03681106. [DOI] [PubMed] [Google Scholar]
- 228.Bagshaw SM, Bennett M, Haase M, Haase-Fielitz A, Egi M, Morimatsu H, D’amico G, Goldsmith D, Devarajan P, Bellomo R. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med. 2010;36:452–461. doi: 10.1007/s00134-009-1724-9. [DOI] [PubMed] [Google Scholar]
- 229.Uchino S, Bellomo R, Bagshaw SM, Goldsmith D. Transient azotaemia is associated with a high risk of death in hospitalized patients. Nephrol Dial Transplant. 2010;25:1833–1839. doi: 10.1093/ndt/gfp624. [DOI] [PubMed] [Google Scholar]
- 230.Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–2210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- 231.Nickolas TL, O’Rourke MJ, Yang J, Sise ME, Canetta PA, Barasch N, Buchen C, Khan F, Mori K, Giglio J, Devarajan P, Barasch J. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med. 2008;148:810–819. doi: 10.7326/0003-4819-148-11-200806030-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Singer E, Elger A, Elitok S, Kettritz R, Nickolas TL, Barasch J, Luft FC, Schmidt-Ott KM. Urinary neutrophil gelatinase-associated lipocalin distinguishes pre-renal from intrinsic renal failure and predicts outcomes. Kidney Int. 2011;80:405–414. doi: 10.1038/ki.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Du Y, Zappitelli M, Mian A, Bennett M, Ma Q, Devarajan P, Mehta R, Goldstein SL. Urinary biomarkers to detect acute kidney injury in the pediatric emergency center. Pediatr Nephrol. 2011;26:267–274. doi: 10.1007/s00467-010-1673-0. [DOI] [PubMed] [Google Scholar]
- 234.Krajcovicova-Kudlackova M, Dusinska M. Oxidative DNA damage in relation to nutrition. Neoplasma. 2004;51:30–33. [PubMed] [Google Scholar]
- 235.Endre ZH, Pickering JW, Walker RJ, Devarajan P, Edelstein CL, Bonventre JV, Frampton CM, Bennett MR, Ma Q, Sabbisetti VS, Vaidya VS, Walcher AM, Shaw GM, Henderson SJ, Nejat M, Schollum JB, George PM. Improved performance of urinary biomarkers of acute kidney injury in the critically ill by stratification for injury duration and baseline renal function. Kidney Int. 2011;79:1119–1130. doi: 10.1038/ki.2010.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Martensson J, Bell M, Oldner A, Xu S, Venge P, Martling CR. Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med. 2010;36:1333–1340. doi: 10.1007/s00134-010-1887-4. [DOI] [PubMed] [Google Scholar]
- 237.Incidence and trends of childhood Type 1 diabetes worldwide 1990–1999. Diabet Med. 2006;23:857–866. doi: 10.1111/j.1464-5491.2006.01925.x. [DOI] [PubMed] [Google Scholar]
- 238.Andersen AR, Christiansen JS, Andersen JK, Kreiner S, Deckert T. Diabetic nephropathy in Type 1 (insulin-dependent) diabetes: an epidemiological study. Diabetologia. 1983;25:496–501. doi: 10.1007/BF00284458. [DOI] [PubMed] [Google Scholar]
- 239.Ballard DJ, Humphrey LL, Melton LJ, III, Frohnert PP, Chu PC, O’Fallon WM, Palumbo PJ. Epidemiology of persistent proteinuria in type II diabetes mellitus. Population-based study in Rochester, Minnesota. Diabetes. 1988;37:405–412. doi: 10.2337/diab.37.4.405. [DOI] [PubMed] [Google Scholar]
- 240.Raile K, Galler A, Hofer S, Herbst A, Dunstheimer D, Busch P, Holl RW. Diabetic nephropathy in 27,805 children, adolescents, and adults with type 1 diabetes: effect of diabetes duration, A1C, hypertension, dyslipidemia, diabetes onset, and sex. Diabetes Care. 2007;30:2523–2528. doi: 10.2337/dc07-0282. [DOI] [PubMed] [Google Scholar]
- 241.Zachwieja J, Soltysiak J, Fichna P, Lipkowska K, Stankiewicz W, Skowronska B, Kroll P, Lewandowska-Stachowiak M. Normal-range albuminuria does not exclude nephropathy in diabetic children. Pediatr Nephrol. 2010;25:1445–1451. doi: 10.1007/s00467-010-1443-z. [DOI] [PubMed] [Google Scholar]
- 242.Nielsen SE, Andersen S, Zdunek D, Hess G, Parving HH, Rossing P. Tubular markers do not predict the decline in glomerular filtration rate in type 1 diabetic patients with overt nephropathy. Kidney Int. 2011;79:1113–1118. doi: 10.1038/ki.2010.554. [DOI] [PubMed] [Google Scholar]
- 243.Nishida M, Kawakatsu H, Okumura Y, Hamaoka K. Serum and urinary neutrophil gelatinase-associated lipocalin levels in children with chronic renal diseases. Pediatr Int. 2010;52:563–568. doi: 10.1111/j.1442-200X.2010.03067.x. [DOI] [PubMed] [Google Scholar]
- 244.Gonwa TA, Mai ML, Melton LB, Hays SR, Goldstein RM, Levy MF, Klintmalm GB. Renal replacement therapy and orthotopic liver transplantation: the role of continuous veno-venous hemodialysis. Transplantation. 2001;71:1424–1428. doi: 10.1097/00007890-200105270-00012. [DOI] [PubMed] [Google Scholar]
- 245.Niemann CU, Walia A, Waldman J, Davio M, Roberts JP, Hirose R, Feiner J. Acute kidney injury during liver transplantation as determined by neutrophil gelatinase-associated lipocalin. Liver Transpl. 2009;15:1852–1860. doi: 10.1002/lt.21938. [DOI] [PubMed] [Google Scholar]
- 246.Portal AJ, McPhail MJ, Bruce M, Coltart I, Slack A, Sherwood R, Heaton ND, Shawcross D, Wendon JA, Heneghan MA. Neutrophil gelatinase--associated lipocalin predicts acute kidney injury in patients undergoing liver transplantation. Liver Transpl. 2010;16:1257–1266. doi: 10.1002/lt.22158. [DOI] [PubMed] [Google Scholar]
- 247.Allegra A, Alonci A, Bellomo G, Campo S, Cannavo A, Penna G, Russo S, Centorrino R, Gerace D, Petrungaro A, Musolino C. Increased serum levels of neutrophil gelatinase-associated lipocalin in patients with essential thrombocythemia and polycythemia vera. Leuk Lymphoma. 2011;52:101–107. doi: 10.3109/10428194.2010.531413. [DOI] [PubMed] [Google Scholar]
- 248.Means RT. JAK2 V617F and the evolving paradigm of polycythemia vera. Korean J Hematol. 2010;45:90–94. doi: 10.5045/kjh.2010.45.2.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Beer PA, Green AR. Pathogenesis and management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2009:621–628. doi: 10.1182/asheducation-2009.1.621. [DOI] [PubMed] [Google Scholar]
- 250.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 251.Harrison C. Rethinking disease definitions and therapeutic strategies in essential thrombocythemia and polycythemia vera. Hematology Am Soc Hematol Educ Program. 2010;2010:129–134. doi: 10.1182/asheducation-2010.1.129. [DOI] [PubMed] [Google Scholar]
- 252.Lip GY, Chin BS, Blann AD. Cancer and the prothrombotic state. Lancet Oncol. 2002;3:27–34. doi: 10.1016/s1470-2045(01)00619-2. [DOI] [PubMed] [Google Scholar]
- 253.Hoppensteadt D, Litinas E, Khan H, Cunanan J, Thethi I, Fareed J. The Effect of Enoxaparin on Inflammatory and Thrombotic Mediators in Cancer Patients As Studied Using Protein and Biochip Array Approaches. 2011:2547. [Google Scholar]
- 254.Wenners AS, Mehta K, Loibl S, Park H, Arnold N, Hamann S, Weimer J, Ataseven B, Schem C, Khandan F, Thomssen W, Jonat W, Holzhausen H, Von Minckwitz G, Denkert C, Bauer M. Evaluation of Neutrophil Gelatinase-Associated Lipocalin (NGAL) As Predictor of Response to Neoadjuvant Chemotherapy (NACT) in Primary Breast Cancer. 2011:595. [Google Scholar]
- 255.Zabron AA, Horneffer-van der Sluis VM, Wadsworth CA, Laird F, Gierula M, Thillainayagam AV, Vlavianos P, Westaby D, Taylor-Robinson SD, Edwards RJ, Khan SA. Elevated levels of neutrophil gelatinase-associated lipocalin in bile from patients with malignant pancreatobiliary disease. Am J Gastroenterol. 2011;106:1711–1717. doi: 10.1038/ajg.2011.187. [DOI] [PubMed] [Google Scholar]
- 256.Porta C, Paglino C, De AM, Quaglini S, Sacchi L, Imarisio I, Canipari C. Predictive value of baseline serum vascular endothelial growth factor and neutrophil gelatinase-associated lipocalin in advanced kidney cancer patients receiving sunitinib. Kidney Int. 2010;77:809–815. doi: 10.1038/ki.2009.552. [DOI] [PubMed] [Google Scholar]
- 257.Delanaye P, Rozet E, Krzesinski JM, Cavalier E. Urinary NGAL measurement: biological variation and ratio to creatinine. Clin Chim Acta. 2011;412:390. doi: 10.1016/j.cca.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 258.Cavalier E, Bekaert AC, Carlisi A, Legrand D, Krzesinski JM, Delanaye P. Neutrophil gelatinase-associated lipocalin (NGAL) determined in urine with the Abbott Architect or in plasma with the Biosite Triage? The laboratory’s point of view. Clin Chem Lab Med. 2011;49:339–341. doi: 10.1515/CCLM.2011.044. [DOI] [PubMed] [Google Scholar]
- 259.Abergel RJ, Wilson MK, Arceneaux JE, Hoette TM, Strong RK, Byers BR, Raymond KN. Anthrax pathogen evades the mammalian immune system through stealth siderophore production. Proc Natl Acad Sci U S A. 2006;103:18499–18503. doi: 10.1073/pnas.0607055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Fischbach MA, Lin H, Liu DR, Walsh CT. How pathogenic bacteria evade mammalian sabotage in the battle for iron. Nat Chem Biol. 2006;2:132–138. doi: 10.1038/nchembio771. [DOI] [PubMed] [Google Scholar]
- 261.Hantke K, Nicholson G, Rabsch W, Winkelmann G. Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci U S A. 2003;100:3677–3682. doi: 10.1073/pnas.0737682100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wang W, Lin J, Guha S, Tong Z, Cameron AG, Zhang F, Qiu X, Zou C, Gao X, Mawad ME, Ke S. Target-Specific Agents Imaging Ectopic and Orthotopic Human Pancreatic Cancer Xenografts. Pancreas. 2011;40:689–694. doi: 10.1097/MPA.0b013e31821f6b14. [DOI] [PubMed] [Google Scholar]
- 263.Roberts DM, Wilks MF, Roberts MS, Swaminathan R, Mohamed F, Dawson AH, Buckley NA. Changes in the concentrations of creatinine, cystatin C and NGAL in patients with acute paraquat self-poisoning. Toxicol Lett. 2011;202:69–74. doi: 10.1016/j.toxlet.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Jin D, Zhang Y, Chen X. Lipocalin 2 deficiency inhibits cell proliferation, autophagy, and mitochondrial biogenesis in mouse embryonic cells. Mol Cell Biochem. 2011;351:165–172. doi: 10.1007/s11010-011-0724-6. [DOI] [PubMed] [Google Scholar]
- 265.Wan X, Fan L, Hu B, Yang J, Li X, Chen X, Cao C. Small interfering RNA targeting IKKbeta prevents renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol. 2011;300:F857–F863. doi: 10.1152/ajprenal.00547.2010. [DOI] [PubMed] [Google Scholar]
- 266.Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, Sherry RM, Royal RE, Steinberg SM, Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma : a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113:293–301. doi: 10.1002/cncr.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17–23. doi: 10.1006/geno.1997.4896. [DOI] [PubMed] [Google Scholar]
- 268.Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E, Vendrell J, Izquierdo M, Martinez C, Nolfe G, Ricart W, Mingrone G, Tinahones F, Fernandez-Real JM. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int J Obes (Lond) 2010;34:240–249. doi: 10.1038/ijo.2009.242. [DOI] [PubMed] [Google Scholar]
- 269.Sethi H, Srinivasan P, Marangoni G, Prachalias A, Rela M, Heaton N. Pancreaticoduodenectomy with radical lymphadenectomy is not contraindicated for patients with established chronic liver disease and portal hypertension. Hepatobiliary Pancreat Dis Int. 2008;7:82–85. [PubMed] [Google Scholar]
- 270.Nomura I, Gao B, Boguniewicz M, Darst MA, Travers JB, Leung DY. Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J Allergy Clin Immunol. 2003;112:1195–1202. doi: 10.1016/j.jaci.2003.08.049. [DOI] [PubMed] [Google Scholar]
- 271.Friedl A, Stoesz SP, Buckley P, Gould MN. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem J. 1999;31:433–441. doi: 10.1023/a:1003708808934. [DOI] [PubMed] [Google Scholar]
- 272.Zhang XF, Zhang Y, Zhang XH, Zhou SM, Yang GG, Wang OC, Guo GL, Yang GY, Hu XQ. Clinical significance of Neutrophil gelatinase-associated lipocalin(NGAL) expression in primary rectal cancer. BMC Cancer. 2009;9:134. doi: 10.1186/1471-2407-9-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhang H, Xu L, Xiao D, Xie J, Zeng H, Wang Z, Zhang X, Niu Y, Shen Z, Shen J, Wu X, Li E. Upregulation of neutrophil gelatinase-associated lipocalin in oesophageal squamous cell carcinoma: significant correlation with cell differentiation and tumour invasion. J Clin Pathol. 2007;60:555–561. doi: 10.1136/jcp.2006.039297. [DOI] [PMC free article] [PubMed] [Google Scholar]