

Abstract
In the context of cancer, E-cadherin has traditionally been categorized as a tumor suppressor, given its essential role in the formation of proper intercellular junctions, and its downregulation in the process of epithelial-mesenchymal transition (EMT) in epithelial tumor progression. Germline or somatic mutations in the E-cadherin gene (CDH1) or downregulation by epigenetic mechanisms have been described in a small subset of epithelial cancers. However, recent evidence also points toward a promoting role of E-cadherin in several aspects of tumor progression. This includes preserved (or increased) E-cadherin expression in microemboli of inflammatory breast carcinoma, a possible “mesenchymal to epithelial transition” (MET) in ovarian carcinoma, collective cell invasion in some epithelial cancers, a recent association of E-cadherin expression with a more aggressive brain tumor subset, as well as the intriguing possibility of E-cadherin involvement in specific signaling networks in the cytoplasm and/or nucleus. In this review we address a lesser-known, positive role for E-cadherin in cancer.
Keywords: E-cadherin, epithelial-mesenchymal transition, mesenchymal-epithelial transition, oncogene, tumorigenesis, adherens junctions
1. Introduction
Cadherins are a family of cell surface glycoproteins that play a prominent role in cell-cell adhesion. Classical cadherins were the first members of the cadherin superfamily to be identified and are key structural components of the adherens junctions[1]. Cadherins mediate Ca2+ dependent, homophilic cell-cell adhesion via their extracellular domains[2]. In addition, they link the extracellular environment to the cytoskeleton and participate in cell signaling through the interaction of their conserved cytoplasmic domains with the catenins, in particular p120-, α- and β-catenin (reviewed in [3, 4]). E-cadherin, a type I classical cadherin, is a key component in the formation of cell-cell adherens-type junctions in epithelial tissues[3, 4].
Studies in a variety of cancers have documented E-cadherin’s role as a tumor suppressor; the evidence for this is covered briefly here, as it has been extensively reviewed elsewhere[5–8]. At the experimental level, suppression of E-cadherin function or expression leads to mesenchymal morphology and increased cell migration and invasion[9, 10] as well as metastasis[11–13]. Loss of E-cadherin facilitates the initial invasive behavior of certain epithelial-derived cancers[11, 14] which may allow basement membrane transgression and spread[10]. Conversely, re-introduction of E-cadherin in cell lines lacking expression leads to reversal of poorly differentiated carcinoma phenotypes (i.e. fibroblastic, highly invasive, poorly cell-cell adherent) back to a well-differentiated, minimally invasive epithelioid phenotype with well developed cell-cell junctions [15–18]. Loss of E-cadherin leads to activation of known oncogenic signaling pathways, including mitogen activated kinase (MAPK), rat sarcoma viral oncogene (Ras) and ras-related C3 botulinum toxin substrate (Rac1)[19, 20], as well as loss of Hippo signaling[21].
At the physiological level, the tumor suppressor role of E-cadherin has been particularly studied in breast cancer (reviewed in [7, 22]), where loss of heterozygosity in chromosome region 16q22.1, the gene region encoding E-cadherin (CDH1), is frequent[22]. In fact, loss of E-cadherin is one of the main phenotypic traits of lobular carcinoma, a breast carcinoma subtype characterized by prominent single cell infiltration, which has been colorfully referred to in classic pathology teaching as “indian filing”. A tumor suppressor role for E-cadherin has also been established or suggested in a variety of additional epithelial malignancies, including hepatocellular carcinoma[23], squamous cell carcinomas of the skin, head and neck[24, 25], and esophagus[26], as well as melanoma[27]. Many of these cancers do not show a high frequency of somatic mutations in CDH1; however E-cadherin expression can also be downregulated by promoter hypermethylation[28]. Inherited germline mutations in CDH1 are also a feature of hereditary gastric cancer[29].
The experimental and physiological observations regarding the tumor suppressor role of E-cadherin are often interpreted in the context of the epithelial-mesenchymal transition (EMT). This developmentally-important process includes a spectrum of changes that lead to the transient downregulation of epithelial cell traits such as apical-basal polarization and organized cell-cell adhesion and the acquisition of a mesenchymal cellular phenotype that is less adherent and more migratory (the process of EMT has been reviewed in [30, 31]). Together, these changes facilitate tissue reorganization and morphogenesis during development. It is increasingly apparent that facets of EMT underlie the progression of carcinomas (tumors of epithelial origin) toward increasingly malignant states (reviewed in [32–35]). One component of the EMT process is the cadherin “switch” whereby the expression of epithelial cadherins (e.g. E-cadherin) is downregulated and conversely mesenchymal cadherins (N-cadherin) are expressed (reviewed in [6, 36, 37]). Such changes in cadherin expression have been reported for a variety of cancers[37], and, as described above, are experimentally linked to increased cell motility and the ability to invade and/or metastasize. It is in this context that E-cadherin’s tumor suppressor role in cancer is most often viewed. In the most simplified model, E-cadherin’s presence prevents cell motility, invasion, and metastasis.
Although E-cadherin’s role as a tumor suppressor is established by agreed upon criteria with firm supporting experimental evidence, in recent years alternative roles for E-cadherin in tumor progression have become apparent. Particularly, in tumors from the ovary, a classical epithelial-mesenchymal transition does not occur, and to the contrary E-cadherin is consistently upregulated[38]. Similarly, although an EMT operates in breast carcinoma, evidence supports an important role for E-cadherin in tumor intravasation, which itself is a critical step required for metastasis([39], reviewed by [40]). For example, most breast ductal carcinomas, both primary and metastatic, consistently express E-cadherin[41], and E-cadherin plays an important role in maintaining intravasated microemboli in inflammatory breast carcinoma models, as described below. E-cadherin may also be important in stem cell biology, increasingly recognized as an important facet of tumor progression[42]; for example, in some studies E-cadherin upregulation increases the clonogenic activity of human embryonic stem cells in the absence of an effect on cell motility[43].
These observations suggest that E-cadherin not only acts as a tumor suppressor, but may also have a promoting role in tumor progression. A variety of proteins function as tumor suppressors or promoters, depending on the specific context; TGF-β is one example[44]. While compelling, the positive functions of E-cadherin in tumor progression have not yet been explored in a systematic fashion. In the remainder of this review we consider E-cadherin’s alternative, non-tumor suppressive roles, using specific tumor types as examples.
2. Positive role for E-cadherin in neoplastic progression
2.1 Support of intravasation and tumor cell survival in inflammatory breast carcinoma
Carcinoma of the breast is a tumor type in which the epithelial-mesenchymal transition with associated down regulation of E-cadherin has been studied extensively. Lobular carcinoma is a morphologic subtype, the hallmark of which is absence of E-cadherin expression[45], a diagnostically useful marker. However, the role of E-cadherin in breast cancer biology might be more complex, and a simple downregulation of E-cadherin may not accurately describe the whole process. In fact, invasive ductal carcinoma, the predominant histologic subtype of breast cancer, consistently expresses E-cadherin. A study of E-cadherin expression in primary breast carcinomas and their distant metastases demonstrated E-cadherin expression in all metastatic tumors of the ductal type (but not in the lobular tumors examined), with the same intensity or even stronger than their respective primaries[41].
Indeed, an unexpected role for E-cadherin in tumor progression has started to emerge in breast cancer. Intravasation, the process whereby tumor cells gain access to vascular channels, is rate-limiting in the formation of metastases[39], which is the predominant cause of death in breast and other epithelial cancers. This phenomenon is exaggerated in the clinical form of breast cancer known as inflammatory carcinoma, which usually displays extensive lymphovascular invasion and is associated with a dismal prognosis, i.e. a 2-fold greater mortality than for locally advanced breast cancer patients[46]. E-cadherin is overexpessed in this specific breast carcinoma subset at the protein level, both in cell lines and clinical tissues[47, 48], as is RhoC GTPase, another possible mediator of the inflammatory breast carcinoma phenotype[49, 50]. Functional in vitro experiments using introduction of dominant negative mutant E-cadherin into IBC cell lines leads to decreased invasion and downregulation of MAPK pathway signaling[51], suggesting a positive role for E-cadherin in IBC progression.
The histologic hallmark of inflammatory breast cancer is the production of numerous, cohesive tumor microemboli within the lymphatics. The biology of these microemboli is more complex than previously thought, as they seem to be curiously resistant to therapeutics, probably due in part to the cohesive nature of these emboli secondary to increased E-cadherin mediated homotypic interactions[52]. Experimental work using the “MARY-X” xenograft model of inflammatory breast carcinoma has demonstrated E-cadherin to be overexpressed in intravascular tumor emboli, resulting in compact, cohesive spheroid structures[47, 53–55]. These structures are unable to interact with endothelial cells due to the increased expression of sialyl-Lewis X/A-deficient-MUC1, which is unable to bind to the endothelial adhesion molecule E-selectin[55]. The combination of E-cadherin-mediated cell-cell adhesion and resistance to endothelial cell interaction facilitates the passive dissemination of tumor emboli in this model. Furthermore, expression of putative stem cell markers, such as stellar, rex-1, nestin, oct-4 and nanog, among others, has been observed[56]. In addition, IBC microemboli seem to have a molecular signature, at the mRNA and protein levels, that mediates adaptation to hypoxia and an increase in angiogenesis[57–60]; this occurs even during normoxic conditions. Taken together, these observations suggest that IBC microemboli are efficient structures for tumor cell survival. As increased E-cadherin levels promote microemboli formation, in this particular context E-cadherin plays a pro-survival, tumor-promoting role[61]. (Figure 1a)
Figure 1. Tumor-promoting functions of E-cadherin.

Support of tumor cell survival, growth, and chemoresistance in the vasculature. a. In inflammatory breast carcinoma (IBC) models, E-cadherin is associated with the formation of intravascular tumor emboli. Experimentally, the increased cohesion mediated by E-cadherin leads to anchorage-independent survival of HSC-3 oral squamous cell carcinoma and nonepithelial Ewing tumor sarcoma cells and also promotes chemoresistance in Ewing tumor sarcoma and IBC cells. b. Experimental evidence from Silvera et al.[62] could explain the increased E-cadherin expression in IBC. Their data supports a model of IBC in which increased expression of the eIF4GI translation initiator leads to IRES-mediated translation of a specific mRNA molecular program that includes p120 catenin. The resulting increased expression of p120 catenin leads to stabilization of E-cadherin at cell junctions, formation and stabilization of tumor microemboli, and tumor dissemination in the lymphatics. H&E staining shows an IBC microembolus within the vasculature; E-cadherin staining shows its intense expression in the IBC microembolus.
Blue cells represent normal cells; pink cells represent tumor cells; orange cells represent endothelial cells; gray bars represent E-cadherin; white bars represent any other type of cadherin.
The upregulation of E-cadherin in tumor microemboli may be related to increased expression of p120 catenin[62] rather than changes in E-cadherin transcription[61](Figure 1b). In an elegant study, Silvera et al. demonstrated that the translation initiator eIF4GI is overexpressed in IBC, which leads to a pro-survival, protein synthesis program that favors emboli formation[62]. Silencing of eIF4GI resulted in a minor decrease in overall protein synthesis, but seemed to impair IBC growth in vivo, leading to small tumor size and reduced tumor emboli formation in xenograft models. The decreased tumor growth was not related to decreased viability, but rather to a destabilization of E-cadherin protein in the absence of changes to E-cadherin mRNA levels. Interestingly, the translation of p120 catenin, a known component of cell junctions that directly stabilizes E-cadherin, was impaired in tumors with silenced eIF4GI. Tumor emboli formation could be rescued by overexpression of p120 in eIF4GI deficient tumor cells.
Understanding the mechanisms of E-cadherin’s role in tumor microemboli genesis may prove useful in the future for understanding the effects of specific therapies. For example, in a recent in vitro study using IBC cell lines (SUM190, SUM149) and primary cultures, treatment with the histone deacetylase inhibitor SAHA was demonstrated to be an effective inhibitor of tumor spheroid self renewal[63]. SAHA treatment was also associated with a redistribution of E-cadherin from cell surface to cytoplasm, thus interfering with homotypic cell aggregation despite preserving total E-cadherin levels.
2.2 Ovarian cancer, mesenchymal-epithelial transition and AKT signaling
Among epithelial cancers, ovarian carcinoma is a prominent exception to the EMT rule: E-cadherin is expressed in the vast majority of ovarian carcinomas, irrespective of histologic type and degree of differentiation, but is low to absent in normal ovarian tissues[64–67]. (Figure 2a) High levels of E-cadherin are ubiquitous in primary ovarian carcinomas, and are also maintained when ovarian carcinoma metastasizes to the peritoneum and omentum[68], a hallmark of this tumor. Interestingly, E-cadherin expression in ovarian surface epithelium has been noted in patients with a family history of ovarian cancer, suggesting an additional possible role in tumor initiation and/or progression[69].
Figure 2. Tumor-promoting functions of E-cadherin.

Abnormal E-cadherin expression produces anomalous, E-cadherin-dependent activation of pro-survival, proliferative, and pro-migratory signaling. a. Aberrant expression of E-cadherin in cells that do not normally have it is a hallmark of ovarian carcinomas. This feature is also correlated with the increased proliferation and migration of a subset of glioblastoma tumor cells. b. Ligand-independent activation of the EGF receptor (EGFR) by E-cadherin has been demonstrated in ovarian and oral squamous cell carcinoma. Investigations with ovarian carcinoma indicate that the E-cadherin-catenin complex can coordinate the activation of PI3K, leading to localized pro-survival, pro-migratory AKT signaling. The E-cadherin-catenin complex can also coordinate the ErbB-family receptor tyrosine kinase-dependent activation of MAPK signaling, leading to proliferation. The precise molecular details of these interactions are not yet clear.
Blue cells represent normal cells; pink cells represent tumor cells; orange cells represent endothelial cells; gray bars represent E-cadherin; white bars represent any other type of cadherin.
Functional, in vitro experiments performed using ovarian surface epithelium cell lines have identified phenotypic changes associated with malignancy and increased proliferation after E-cadherin transfection[70], including features of a mesenchymal to epithelial transition[38]. Indeed ovarian carcinoma appears to be an excellent model of mesenchymal to epithelial transition [71]. Interestingly, although the EMT mediators Snail, Slug and SIP1 are upregulated at the mRNA level in malignant effusions of ovarian carcinoma[72], Snail and Slug protein levels are lower in these effusions compared to their primary tumors of origin[73], while membranous E-cadherin and α-, β- and γ-catenin proteins are upregulated[74]. Furthermore, Snail protein appears to localize to the cytoplasm in primary effusion cells, suggesting that it may not be active[73]. The variation in EMT proteins and E-cadherin levels in ovarian carcinoma appears to be a site-dependent and dynamic process. For example, as mentioned above, in carcinoma cells present in effusions Snail may be present in the cytoplasm, allowing increased transcription of E-cadherin, a process that may be mediated in part by lower Pak1 levels[71]. This increase in E-cadherin in free-floating effusion tumor aggregates may confer a survival advantage, reminiscent of the inflammatory breast carcinoma microemboli discussed above. Conversely, E-cadherin protein may be lower in solid metastases, with preservation of N-cadherin[75], and a more conventional EMT may be operative in this context.
Some investigators have suggested that these observations have mechanistic implications for ovarian carcinoma tumorigenesis. One study demonstrated an association between E-cadherin mediated intercellular junction formation and activation of the AKT and MAPK signaling pathways, a phenomenon related to ligand-independent activation of the EGF receptor (EGFR, also known as ErbB1 or HER1)[76]. Interestingly, adhesion-mediated ligand-independent activation of EGFR has also been implicated in oral squamous cell carcinoma[77]. Normally, E-cadherin interacts with EGFR[78, 79] and inhibits its function via the action of the neurofibromatosis type 2 (NF2) tumor suppressor merlin[80]. However, E-cadherin also associates with the p85 regulatory subunit of PI3K[81, 82], bringing it in close proximity to EGFR. When EGFR is activated, this close proximity contributes to PI3K/AKT activation[83]. It is therefore possible that under certain conditions and in the absence of a negative regulator, E-cadherin promotes EGFR-mediated PI3K activation instead of inhibiting it (Figure 2b).
Of course, cancer is a disease known for its heterogeneity of genotype and phenotype, and there appear to be subtypes of ovarian cancer that do not follow the altered pattern of E-cadherin and EMT mediator expression described above. For example, recent evidence suggests that changes associated with EMT and E-cadherin downregulation play a role in the progression and invasion of low grade serous carcinomas arising from borderline serous tumors[84, 85]. In particular, increased expression of key mediators of EMT (e.g. Snail, Slug, Twist1, SIP1), and decreased membranous E-cadherin in ovarian serous and clear cell carcinomas in particular, as opposed to surface epithelium and borderline tumors was reported by one group[86]. However, in the same study E-cadherin protein expression was increased, while Twist1 and Snail levels were decreased, in disseminated peritoneal lesions compared to the primary tumors, further supporting dynamic protein level alterations for these proteins in ovarian cancer as described above. In addition, some immunohistochemical studies have shown weak to absent E-cadherin staining in poorly differentiated subtypes compared to better differentiated/lower grade tumor types [87–89]. Finally, a recent study by Burkalter et al. suggests that integrin recruitment/activation caused by binding to collagen on the peritoneal surface and submesothelial matrix, leads to destabilization of cell junctions, and increased beta-catenin/WNT pathway signaling[90].
2.3 Growth and migration in glioblastoma
Although glial tumors (gliomas) are frequently viewed as “epithelial” in origin, the nervous tissue from which they arise is not classically epithelial in structure, and these tumors are more accurately described as “neuroepithelial”. The difference in terms can be partly understood by examination of cadherin expression in normal nervous tissue as compared to epithelial tissue. In the adult central nervous system cadherin expression is stable, mesenchymal cadherin (e.g. N-cadherin, cadherin 11) expression is the norm[92], and E-cadherin expression, in particular, is rare. A notable exception is expression of E-cadherin in arachnoid cells[93]. As with ovarian tumors, these observations make it conceptually difficult to apply the typical EMT cadherin-switching process to glial tumorigenesis. However the broader cadherin-switching concept, in which cells downregulate the cadherin they would normally express and upregulate an inappropriate cadherin, resulting in altered cell behavior, can still apply[37] (Figure 2a).
One of the main functional properties of gliomas is a marked tendency for infiltration of underlying parenchyma. This behavior provides a rationale to study cell-cell adhesion in glial tumorigenesis. Prior studies of cadherin expression in gliomas concluded that N-cadherin expression is frequent, while E-cadherin is rare to absent[94–100]. Studies of E-cadherin in astrocytomas in particular are scant and generally argue that E-cadherin is largely absent at the protein level[97]. Studies on the role of N-cadherin in glioma cell invasion have been inconclusive[94, 95, 97, 98], however, leading to the hypothesis that alterations in cell junction organization rather than differences in cadherin expression may be more important in the invasive properties of gliomas[96].
In this context, it is noteworthy that 73% of a subset of glioblastomas with epithelial and pseudoepithelial differentiation expresses E-cadherin, particularly in areas of epithelial differentiation[101]. Although this subset of tumors is rare (representing <2% of glioblastomas), E-cadherin expression was associated with decreased overall survival in these patients[102]. Further investigation revealed that while E-cadherin expression is rare in a panel of human high grade astrocytoma cell lines, it correlates with increased invasiveness in vivo, using orthotopic glioblastoma mouse xenografts[102]. Additionally, in a conventional glioma cell line (SF767), non-mutated endogenous E-cadherin was mislocalized throughout the cell membrane, rather than localizing predominantly at sites of cell-cell contact. Functional experiments using shRNA-mediated E-cadherin knockdown resulted in dose-dependent decreases in SF767 cell proliferation and migration. While the underlying mechanism needs to be established, these findings suggest that E-cadherin may have an important, positive role in the malignant progression of a subset of highly aggressive astrocytomas. The data lend additional support to the idea that E-cadherin is not limited to a tumor suppressor role in tumor progression.
3. Positive role of E-cadherin in tumor progression: possible mechanisms
As the previous discussion emphasizes, tumor conditions exist in which E-cadherin is present, and its presence makes tumor progression worse. Much remains to be learned about this positive role for E-cadherin in tumor-progression, in particular with respect to tumor context and specific mechanisms. In some cases, like the formation of tumor emboli that is seen in inflammatory breast carcinoma, cadherin-based cell-cell contacts may promote tumor cell survival, growth and invasion. In other cases, e.g. ovarian cancer, tumor-promoting signal cascades may be induced by dynamic E-cadherin complexes through mechanisms that need further exploration. The process of collective cell migration, which is also important in epithelial cancers, requires intact E-cadherin and proper intercellular junctions. Intact E-cadherin function may also be important in cell migration and motility, for example by providing anchoring points for cell movement and aiding in cytoskeletal contraction. These mechanisms are discussed here in more depth.
3.1 E-cadherin signal cascades that promote tumor cell survival
The ability of cells to survive and even proliferate in the absence of cell-extracellular matrix interactions is known as anchorage-independent growth, and is a feature of transformed cells. During this type of growth, signaling from cell-cell adhesions suppresses detachment-induced apoptosis (anoikis). Experimentally, E-cadherin has been shown to mediate the anchorage independent growth that occurs in multi-cellular spheres of epithelial HSC-3 oral squamous cell carcinoma cells[103] and nonepithelial Ewing tumor sarcoma cells[104]. Interestingly, in the latter example, E-cadherin expression was upregulated when the cells where grown under conditions of anchorage-independence. These experimental observations provide mechanistic insight into the importance of the previously discussed tumor microemboli seen in inflammatory breast carcinoma (Figure 1).
The molecular signaling that occurs downstream of E-cadherin engagement in the context of anchorage-independent growth could be pro-survival (anti-apoptotic), pro-growth (proliferation), or both. In the case of Ewing tumor sarcoma cells, investigation of anchorage-independent growth revealed reduced caspase-3 activation that was E-cadherin-dependent and downstream of ErbB4 receptor tyrosine kinase and PI3K activation[104]; thus, in these cells, pro-survival signaling results from E-cadherin engagement. Similarly, in the case of HSC-3 cells, anti-apoptotic mechanisms are clearly present, as E-cadherin engagement results in elevated levels of the anti-apoptotic proteins Bcl-2 and Bcl-xL downstream of EGFR and MAPK activation[77]. The MAPK path may also stimulate proliferative signaling in these cells, as HSC-3 cells growing under anchorage-independent conditions in which MAPK is activated do increase in number with time[77, 103].
In both of these experimental examples, E-cadherin is associated with receptor tyrosine kinase activation (Figure 2b). The E-cadherin protein itself does not have any intrinsic enzymatic activity[4]; therefore activation of the downstream effectors of receptor tyrosine kinases must be mediated by other proteins associated with the E-cadherin-catenin complex. It has been demonstrated that the E-cadherin-catenin complex can interact with EGFR in both tumor and non-tumorigenic cells, either by direct interaction of the E-cadherin extracellular domain with EGFR or possibly via β-catenin[76–79, 105, 106]. Depending on the cell context, this interaction can lead either to inhibition of EGFR signaling[79] or to ligand-independent EGFR activation and activation of MAPK signaling[76, 77, 105, 106], which is known to activate survival and proliferation mechanisms.
It is currently unclear why E-cadherin inhibits EGFR signaling in some cells, but activates ligand-independent EGFR signaling in others. Mechanisms related to the relative adhesion of normal vs. tumor cells can not completely explain the difference, as multi-cellular spheres growing under anchorage-independent conditions require cell-cell adhesion just as normal cells in an epithelial tissue do. However, unlike normal cells in a monolayer, cells growing as compact spheres exhibit loss of apical-basal polarity, a condition that may impact cadherin-mediated signaling events. In normal epithelial cells, E-cadherin-mediated cell-cell contact results in, among other mechanisms, NF2/Merlin-dependent inhibition of EGFR signaling[80]. It is possible that some E-cadherin positive tumors are NF2 mutated/deleted, allowing EGFR signaling to continue despite the formation of E-cadherin mediated cell-cell contacts. Another possibility is that NF2/Merlin function is impacted by changes in cell polarity. Importantly, ligand-stimulated and ligand-independent EGFR signaling have different outcomes. Ligand-independent EGFR signaling results in attenuated but sustained MAPK activation that is different from the high amplitude, transient MAPK signaling that results from EGF-induced EGFR activation([77], see also [107]). E-cadherin engagement in the context of tumor progression seems to more commonly induce ligand-independent EGFR activation. The resulting difference in MAPK signaling is likely important in interpreting the significance of E-cadherin-dependent EGFR activation.
In addition to EGFR, the E-cadherin-catenin complex can interact with the p85 subunit of PI3K[76, 81, 108], in both normal (MDCK) and tumor (OVCAR-3) cell lines, often leading to activation of PI3K and AKT-mediated pro-survival and pro-migratory signaling. As the E-cadherin-catenin complex is usually associated with decreased cell motility, mechanisms must exist to regulate pro-migratory PI3K signaling. In this respect, a report describing recruitment of the PI3K inhibitor PTEN to E-cadherin-catenin complexes by the β-catenin/MAGI-1b complex is important[109]. In the context of the MDCKts-src model of Src-induced invasiveness, recruitment of PTEN to cell-cell contacts by E-cadherin/β-catenin/MAGI-1b, and the resulting local down-regulation of PI3K/AKT signaling, is able to inhibit MDCKts-src cell invasion. These results suggest that, in the absence of locally recruited PTEN, the E-cadherin-catenin complex could facilitate PI3K/AKT activation, leading to pro-survival and pro-migratory signaling that could underlie tumor progression.
3.2 E-cadherin and cell migration
As the previous discussion suggests, the presence of E-cadherin does not preclude tumor cell migration/invasion during tumor progression. Rather, the molecular and signaling context within a cell determines its survival, proliferative, migratory, and invasive capabilities. Epithelial properties are not always lost in tumors, and invasion in the form of aggregates that remain closely together, i.e. “collective cell migration” [110], may enable spread of specific tumor types, including well-differentiated carcinomas, melanomas and rhabdomyosarcomas[110]. Preservation of cell-cell cohesion may, in fact, facilitate invasion of well-differentiated tumors by enabling passive migration and collective cell invasion of tumor cells (Figure 3).
Figure 3. Tumor-promoting functions of E-cadherin.
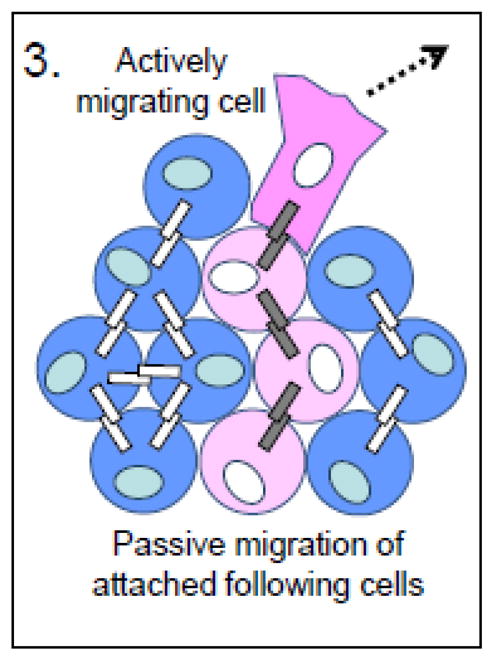
Facilitation of collective cell migration. Collective cell migration allows squamous cell carcinoma cells with intact E-cadherin-based junctions to move passively through a tissue by being pulled along by an actively migrating tumor cell.
Blue cells represent normal cells; pink cells represent tumor cells; orange cells represent endothelial cells; gray bars represent E-cadherin; white bars represent any other type of cadherin.
Collective cell migration can rely on E-cadherin in two ways. E-cadherin stabilizes interactions between cells such that traction forces generated by a leading cell or cells are able to pull adherent, following cells along (passive migration). Such a mechanism has been demonstrated experimentally: the collective invasion of vulvar squamous cell carcinoma A431 cells in 3D culture is dependent on intact E-cadherin/cell junctions and p120 catenin[111]. Furthermore, the creation of stromal “tracks” by non-neoplastic fibroblasts in the tumor microenvironment may facilitate collective cell invasion. It has been demonstrated that squamous cell carcinoma (SCC) cells alone are unable to invade a matrix composed of extracellular matrix protein, but can do so readily when co-cultured with stromal fibroblasts, or when exposed to matrix that had previously been cultured with stromal fibroblasts[112]. Such a mechanism obviates the need for an epithelial-mesenchymal transition that includes downregulated E-cadherin expression in the spread of certain tumor types.
Alternatively, E-cadherin could be directly responsible for generating the traction forces that allow tumor cells to move. At least one morphogenetic example of E-cadherin-mediated collective cell migration exists: in Drosophila oogenesis DE-cadherin is required for migration of clustered border cells on the surface of germline cells[113]. At the anterior end of the follicle, the border cells form a distinct group of two centrally-located, polarized cells surrounded by 6 additional, partially polarized cells; this group of cells migrates in between adjacent nurse cells, through the center of the follicle, until it reaches the oocyte. DE-cadherin is required for this process, specifically for the ability of the cells to migrate and not for adhesion between the border cells during migration. It is important to note that this migration occurs on the surface of other cells, as opposed to on/through extracellular matrix. The latter migration would depend on integrin signaling to generate the required traction.
In addition to E-cadherin-dependent collective cell migration, single cell migration can also occur in an E-cadherin dependent manner. In the developing zebrafish embryo, E-cadherin homophilic interactions provide crucial anchoring points that promote single cell migration of chemokine-guided germ cells[114]. These cells respond to the chemokine CXCL12a by forming protrusions (blebs) that are responsible for motility. F-actin brush structures form at the front of these protrusions in a Rac1-dependent manner, active RhoA controls the retrograde flow of actin, and the actin brushes are coupled to neighboring cells via E-cadherin, resulting in the generation of traction forces that move the cell forward. Interestingly, the level of E-cadherin in these cells is critical to their ability to move: cells that expressed abnormally high levels of E-cadherin migrated more slowly than normal germ cells, and cells that had been artificially knocked down for E-cadherin were unable to move[114]. Whether the mechanisms of E-cadherin-mediated motility described here occur in tumors in vivo remains to be demonstrated.
3.3 Functional significance of E-cadherin proteolytic cleavage fragments
In addition to roles for E-cadherin in intracellular signaling and migration, the effect on tumor progression of E-cadherin cleavage products resulting from proteolysis has also been explored. Recent evidence supports a role for soluble E-cadherin cleavage fragments in numerous cell roles, including junction disruption, migration and invasion, and initiation of signaling (reviewed in [115]). (Figure 4) It is important to note that in normal cells, degradation of E-cadherin does not occur via proteolytic cleavage at the cell membrane, but rather by endocytic internalization followed by targeting to the proteosome or lysosome (reviewed in [4]). In contrast, proteolytic cleavage of E-cadherin does occur in cells undergoing apoptosis[116]. There are two main cleavage sites adjacent to the E-cadherin transmembrane domain: proteolysis by metalloproteinases (e.g. ADAM10, ADAM15, others)[117, 118] (reviewed in [115]) or γ-secretases (e.g. Presenilin 1)[119] results in N-terminal (80-kDa) soluble (ecto-domain) or C-terminal intracellular fragments, respectively. The expression of metalloproteinases, in particular, is upregulated in a variety of cancers[120], making biological effects of the soluble and C-terminal E-cadherin fragments potentially important to tumor progression.
Figure 4. Tumor-promoting functions of E-cadherin.
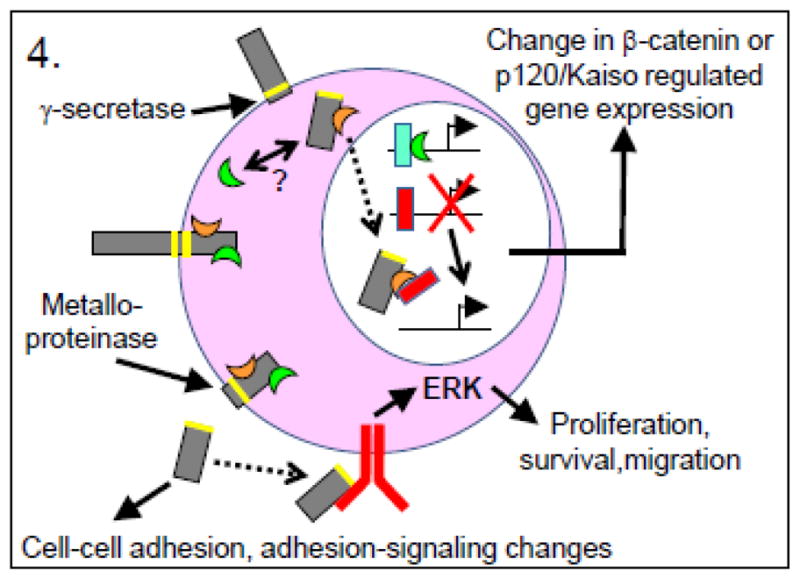
Aberrant signaling due to proteolytic cleavage fragments of E-cadherin. (Green crescent = β-catenin; orange crescent = p120 catenin; blue rectangle = Lef-1/Tcf transcription factors; red rectangle = Kaiso transcription regulator.) Proteolytic cleavage of E-cadherin by either γ-secretases or metalloproteinases produces cleavage fragments that influence cell-cell adhesion as well as, respectively, gene transcription events or adhesion-related or growth factor receptor signaling. For example, the cytoplasmic portion of E-cadherin may serve on the one hand as a source/sink for β-catenin, thereby regulating β-catenin/Lef-1/Tcf transcriptional events, or on the other hand as a co-regulator with p120 catenin of Kaiso-mediated transcriptional repression. Likewise, the soluble extracellular fragment of E-cadherin may alter existing adherens junctions, or may stimulate signaling through EGFR or other growth factor receptors.
Blue cells represent normal cells; pink cells represent tumor cells; orange cells represent endothelial cells; gray bars represent E-cadherin; white bars represent any other type of cadherin.
Extracellular N-terminal E-cadherin ectodomain “shedding” has been implicated in a variety of tumor-promoting activities, including loss of cell adhesion and facilitation of tumorigenic signaling, and its role in cancer and other disease has been recently comprehensively reviewed[115]. Purified E-cadherin fragments obtained from MCF-7 cells have been demonstrated to disrupt proper cell junctions in mice[121], while the E-cadherin ectodomain leads to disruption of cell junctions in ovarian carcinoma cells[122]. Furthermore, E-cadherin fragments may also interfere with re-formation of pancreatic carcinoma cell aggregates[123]. As mentioned previously, disruption of cell-cell junctions is widely viewed as a necessary step toward cell migration. That soluble E-cadherin fragments can actually stimulate migration was demonstrated in MCF7 cells[118], while stimulation of cell invasion was demonstrated with ovarian carcinoma[124], MCDK [125, 126], pancreatic cancer [123], and lung cancer cells[127]. Interestingly, the mechanism for stimulating MCF7 cell migration was shown to be dependent on ErbB receptor (Her2 and Her3) activation by the soluble E-cadherin fragment, leading to signaling via the Erk pathway[118]. This adds yet another dimension to the previous discussion of ligand-independent activation of EGFR by E-cadherin. Likewise, the stimulation of lung cancer cell invasion was shown to be due to soluble E-cadherin fragment-dependent upregulation of MMP-2, MMP-9, and MT1-MMP expression[127]. These MMPs are known for their ability to degrade basement membranes, leading to an interesting model in which tumor-derived metalloproteinases such as MMP-3, MMP-7, or ADAM15 could cleave E-cadherin, generating the soluble fragment, which then stimulates production of basement-membrane-degrading MMPs that facilitate tumor cell invasion[127]. How the soluble fragment of E-cadherin is able to regulate MMP gene expression is not known, but might involve signaling downstream of growth factor receptor activation as described above for EGFR.
Less is known about the effects of the E-cadherin C-terminus cleavage fragment on tumor progression. In a model proposed by Marambaud et al.[119], metalloproteinases cleave E-cadherin’s extracellular domain resulting in a 38kDa C-terminal E-cadherin domain (CTF1). This event is followed by cleavage of CTF1 by presenilin 1/γ-secretase activity at the membrane/cytosol junction, leading to a 33kDa cytoplasmic fragment (CTF2), which can still bind to β-catenin and may regulate Wnt-related β-catenin signaling[119]. Alternatively, CTF2 complexed with p120 catenin has been demonstrated to translocate to the nucleus in non-neoplastic cells, where it can regulate p120-Kaiso-dependent transcription[128]. Nuclear translocation of the C-terminal fragment has also been observed with N-cadherin[129] and γ-protocadherin[130] and associated with transcriptional activation in neural crest and cortical neurons, respectively. The transcriptional activation induced by the N-cadherin C-terminus may be mediated in part by increased β-catenin transcription[129]. Nuclear localization of E-cadherin has been reported in several tumor types, including Merkel cell carcinoma[131], stomach[132], colorectal[132, 133], renal[134], esophageal[135], and solid pseudopapillary tumor of the pancreas[136], and in some studies is also associated with increased nuclear β-catenin staining[132]. One study of esophageal squamous cell carcinoma demonstrated increased expression of the cytoplasmic C-terminus domain of E-cadherin, which was present in 60% of tumors examined[135]. Follow-up functional experiments using the cytoplasmic domain of E-cadherin demonstrated increased AP-1 and cyclin D1 promoter activity, but not β-catenin/Tcf transcription activity, consistent with transcriptional activation by E-cadherin fragments independent of β-catenin. It should be noted that most reports of nuclear localization of E-cadherin in cancer have utilized immunohistochemistry only, which must be interpreted with caution[137].
Whether biological effects of the soluble and CTF2 E-cadherin fragments occur in addition to or in place of the E-cadherin/junctional signaling described in the previous sections of this review is unclear. Some investigators have described a disruption of E-cadherin/junctional function due to the soluble E-cadherin fragment that results in increased cell motility/invasiveness[125]. Many others did not examine the state of the adherens junctions in the context of their studies (for example [118, 127]). It is important to note that, at least in some cell types, E-cadherin-dependent suppression of cell motility occurs due to signaling that is separate from its adhesive function[17, 18, 138]. The transcriptional effects of the CTF2 fragment are particularly poorly understood at this point. Although much work remains to be done to clarify these issues, it is clear that this is yet another mechanism by which E-cadherin expression in tumor cells can result in tumor progression rather than suppression.
4. Conclusion
Conventional interpretation of the key mediators of tumor promotion/progression has emphasized the presence of tumor suppressors and oncoproteins, roles that are discussed in sharply separated contexts. However, as we dissect the complex molecular mechanisms of cancer, it becomes evident that genes and proteins may have opposing roles depending on the specific context. E-cadherin has been well established as an important tumor suppressor in a variety of tumor types by a strong body of experimental evidence. A less well known, positive role in various aspects of tumor promotion/progression has also surfaced. The specific mechanisms operating in this context still need to be described, but may involve stabilization of cell contacts, or cooperative aberrant cytoplasmic and nuclear signaling. Therefore, aberrant as well as typical E-cadherin properties may play important roles in tumor progression according to tumor context, possibilities that still require testing in rigorous, experimental settings. Further studies focusing on specific tumor types may provide insights into these mechanisms, and may be of help in the identification of novel therapeutic strategies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 2.Nagafuchi A, Takeichi M. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J. 1988;7:3679–3684. doi: 10.1002/j.1460-2075.1988.tb03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 4.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 6.Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. 2004;1014:155–163. doi: 10.1196/annals.1294.016. [DOI] [PubMed] [Google Scholar]
- 7.Strumane K, Berx G, Van Roy F. Cadherins in cancer. Handb Exp Pharmacol. 2004:69–103. doi: 10.1007/978-3-540-68170-0_4. [DOI] [PubMed] [Google Scholar]
- 8.Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1:a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–119. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- 10.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 11.Mareel MM, Behrens J, Birchmeier W, De Bruyne GK, Vleminckx K, Hoogewijs A, Fiers WC, Van Roy FM. Down-regulation of E-cadherin expression in Madin Darby canine kidney (MDCK) cells inside tumors of nude mice. Int J Cancer. 1991;47:922–928. doi: 10.1002/ijc.2910470623. [DOI] [PubMed] [Google Scholar]
- 12.Osada T, Sakamoto M, Ino Y, Iwamatsu A, Matsuno Y, Muto T, Hirohashi S. E-cadherin is involved in the intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 1996;24:1460–1467. doi: 10.1053/jhep.1996.v24.pm0008938181. [DOI] [PubMed] [Google Scholar]
- 13.Luo J, Sharma N, Seftor EA, De Larco J, Heidger PM, Hendrix MJ, Lubaroff DM. Heterogeneous Expression of Invasive and Metastatic Properties in a Prostate Tumor Model. Pathol Oncol Res. 1997;3:264–271. doi: 10.1007/BF02904285. [DOI] [PubMed] [Google Scholar]
- 14.Behrens J, Mareel MM, Van Roy FM, Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol. 1989;108:2435–2447. doi: 10.1083/jcb.108.6.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–3556. [PubMed] [Google Scholar]
- 17.Wong AS, Gumbiner BM. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. J Cell Biol. 2003;161:1191–1203. doi: 10.1083/jcb.200212033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yanagisawa M, Anastasiadis PZ. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J Cell Biol. 2006;174:1087–1096. doi: 10.1083/jcb.200605022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wijnhoven BP, Dinjens WN, Pignatelli M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br J Surg. 2000;87:992–1005. doi: 10.1046/j.1365-2168.2000.01513.x. [DOI] [PubMed] [Google Scholar]
- 20.Soto E, Yanagisawa M, Marlow LA, Copland JA, Perez EA, Anastasiadis PZ. p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J Cell Biol. 2008;183:737–749. doi: 10.1083/jcb.200805113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108:11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berx G, Van Roy F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001;3:289–293. doi: 10.1186/bcr309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhai B, Yan HX, Liu SQ, Chen L, Wu MC, Wang HY. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J Gastroenterol. 2008;14:5665–5673. doi: 10.3748/wjg.14.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Field JK. Oncogenes and tumour-suppressor genes in squamous cell carcinoma of the head and neck. Eur J Cancer B Oral Oncol. 1992;28B:67–76. doi: 10.1016/0964-1955(92)90016-t. [DOI] [PubMed] [Google Scholar]
- 25.Ruggeri B, Caamano J, Slaga TJ, Conti CJ, Nelson WJ, Klein-Szanto AJ. Alterations in the expression of uvomorulin and Na+,K(+)-adenosine triphosphatase during mouse skin tumor progression. Am J Pathol. 1992;140:1179–1185. [PMC free article] [PubMed] [Google Scholar]
- 26.Ling ZQ, Li P, Ge MH, Zhao X, Hu FJ, Fang XH, Dong ZM, Mao WM. Hypermethylation-modulated down-regulation of CDH1 expression contributes to the progression of esophageal cancer. Int J Mol Med. 2011;27:625–635. doi: 10.3892/ijmm.2011.640. [DOI] [PubMed] [Google Scholar]
- 27.Molina-Ortiz I, Bartolome RA, Hernandez-Varas P, Colo GP, Teixido J. Overexpression of E-cadherin on melanoma cells inhibits chemokine-promoted invasion involving p190RhoGAP/p120ctn-dependent inactivation of RhoA. J Biol Chem. 2009;284:15147–15157. doi: 10.1074/jbc.M807834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winter JM, Ting AH, Vilardell F, Gallmeier E, Baylin SB, Hruban RH, Kern SE, Iacobuzio-Donahue CA. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res. 2008;14:412–418. doi: 10.1158/1078-0432.CCR-07-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinheiro H, Bordeira-Carrico R, Seixas S, Carvalho J, Senz J, Oliveira P, Inacio P, Gusmao L, Rocha J, Huntsman D, Seruca R, Oliveira C. Allele-specific CDH1 downregulation and hereditary diffuse gastric cancer. Hum Mol Genet. 2010;19:943–952. doi: 10.1093/hmg/ddp537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radisky DC. Epithelial-mesenchymal transition. J Cell Sci. 2005;118:4325–4326. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- 31.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 32.Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell. 2001;105:425–431. doi: 10.1016/s0092-8674(01)00365-8. [DOI] [PubMed] [Google Scholar]
- 33.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 34.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 35.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 36.Cavallaro U, Schaffhauser B, Christofori G. Cadherins and the tumour progression: is it all in a switch? Cancer Lett. 2002;176:123–128. doi: 10.1016/s0304-3835(01)00759-5. [DOI] [PubMed] [Google Scholar]
- 37.Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci. 2008;121:727–735. doi: 10.1242/jcs.000455. [DOI] [PubMed] [Google Scholar]
- 38.Auersperg N, Pan J, Grove BD, Peterson T, Fisher J, Maines-Bandiera S, Somasiri A, Roskelley CD. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc Natl Acad Sci U S A. 1999;96:6249–6254. doi: 10.1073/pnas.96.11.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J, Yu W, Kovalski K, Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- 40.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728:23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5:R217–222. doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Wang BH, Wang S, Moalim-Nour L, Mohib K, Lohnes D, Wang L. Individual cell movement, asymmetric colony expansion, rho-associated kinase, and E-cadherin impact the clonogenicity of human embryonic stem cells. Biophys J. 2010;98:2442–2451. doi: 10.1016/j.bpj.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 45.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995;14:6107–6115. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overmoyer BA. Inflammatory breast cancer: novel preoperative therapies. Clin Breast Cancer. 2010;10:27–32. doi: 10.3816/CBC.2010.n.003. [DOI] [PubMed] [Google Scholar]
- 47.Kleer CG, van Golen KL, Braun T, Merajver SD. Persistent E-cadherin expression in inflammatory breast cancer. Mod Pathol. 2001;14:458–464. doi: 10.1038/modpathol.3880334. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmeyer MR, Wall KM, Dharmawardhane SF. In vitro analysis of the invasive phenotype of SUM 149, an inflammatory breast cancer cell line. Cancer Cell Int. 2005;5:11. doi: 10.1186/1475-2867-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–2519. [PubMed] [Google Scholar]
- 50.Van den Eynden GG, Van der Auwera I, Van Laere S, Colpaert CG, van Dam P, Merajver S, Kleer CG, Harris AL, Van Marck EA, Dirix LY, Vermeulen PB. Validation of a tissue microarray to study differential protein expression in inflammatory and non-inflammatory breast cancer. Breast Cancer Res Treat. 2004;85:13–22. doi: 10.1023/B:BREA.0000021028.33926.a8. [DOI] [PubMed] [Google Scholar]
- 51.Dong HM, Liu G, Hou YF, Wu J, Lu JS, Luo JM, Shen ZZ, Shao ZM. Dominant-negative E-cadherin inhibits the invasiveness of inflammatory breast cancer cells in vitro. J Cancer Res Clin Oncol. 2007;133:83–92. doi: 10.1007/s00432-006-0140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crane K. Elucidating an uncommon disease: inflammatory breast cancer. J Natl Cancer Inst. 2011;103:1358–1360. doi: 10.1093/jnci/djr364. [DOI] [PubMed] [Google Scholar]
- 53.Alpaugh ML, Tomlinson JS, Shao ZM, Barsky SH. A novel human xenograft model of inflammatory breast cancer. Cancer Res. 1999;59:5079–5084. [PubMed] [Google Scholar]
- 54.Tomlinson JS, Alpaugh ML, Barsky SH. An intact overexpressed E-cadherin/alpha,beta-catenin axis characterizes the lymphovascular emboli of inflammatory breast carcinoma. Cancer Res. 2001;61:5231–5241. [PubMed] [Google Scholar]
- 55.Alpaugh ML, Tomlinson JS, Kasraeian S, Barsky SH. Cooperative role of E-cadherin and sialyl-Lewis X/A-deficient MUC1 in the passive dissemination of tumor emboli in inflammatory breast carcinoma. Oncogene. 2002;21:3631–3643. doi: 10.1038/sj.onc.1205389. [DOI] [PubMed] [Google Scholar]
- 56.Xiao Y, Ye Y, Yearsley K, Jones S, Barsky SH. The lymphovascular embolus of inflammatory breast cancer expresses a stem cell-like phenotype. Am J Pathol. 2008;173:561–574. doi: 10.2353/ajpath.2008.071214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colpaert CG, Vermeulen PB, Benoy I, Soubry A, van Roy F, van Beest P, Goovaerts G, Dirix LY, van Dam P, Fox SB, Harris AL, van Marck EA. Inflammatory breast cancer shows angiogenesis with high endothelial proliferation rate and strong E-cadherin expression. Br J Cancer. 2003;88:718–725. doi: 10.1038/sj.bjc.6600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bieche I, Lerebours F, Tozlu S, Espie M, Marty M, Lidereau R. Molecular profiling of inflammatory breast cancer: identification of a poor-prognosis gene expression signature. Clin Cancer Res. 2004;10:6789–6795. doi: 10.1158/1078-0432.CCR-04-0306. [DOI] [PubMed] [Google Scholar]
- 59.Van der Auwera I, Van Laere SJ, Van den Eynden GG, Benoy I, van Dam P, Colpaert CG, Fox SB, Turley H, Harris AL, Van Marck EA, Vermeulen PB, Dirix LY. Increased angiogenesis and lymphangiogenesis in inflammatory versus noninflammatory breast cancer by real-time reverse transcriptase-PCR gene expression quantification. Clin Cancer Res. 2004;10:7965–7971. doi: 10.1158/1078-0432.CCR-04-0063. [DOI] [PubMed] [Google Scholar]
- 60.Silvera D, Schneider RJ. Inflammatory breast cancer cells are constitutively adapted to hypoxia. Cell Cycle. 2009;8:3091–3096. doi: 10.4161/cc.8.19.9637. [DOI] [PubMed] [Google Scholar]
- 61.Ye Y, Tellez JD, Durazo M, Belcher M, Yearsley K, Barsky SH. E-cadherin accumulation within the lymphovascular embolus of inflammatory breast cancer is due to altered trafficking. Anticancer Res. 2010;30:3903–3910. [PubMed] [Google Scholar]
- 62.Silvera D, Arju R, Darvishian F, Levine PH, Zolfaghari L, Goldberg J, Hochman T, Formenti SC, Schneider RJ. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol. 2009;11:903–908. doi: 10.1038/ncb1900. [DOI] [PubMed] [Google Scholar]
- 63.Robertson FM, Woodward WA, Pickei R, Ye Z, Bornmann W, Pal A, Peng Z, Hall CS, Cristofanilli M. Suberoylanilide hydroxamic acid blocks self-renewal and homotypic aggregation of inflammatory breast cancer spheroids. Cancer. 2010;116:2760–2767. doi: 10.1002/cncr.25176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Inoue M, Ogawa H, Miyata M, Shiozaki H, Tanizawa O. Expression of E-cadherin in normal, benign, and malignant tissues of female genital organs. Am J Clin Pathol. 1992;98:76–80. doi: 10.1093/ajcp/98.1.76. [DOI] [PubMed] [Google Scholar]
- 65.Darai E, Scoazec JY, Walker-Combrouze F, Mlika-Cabanne N, Feldmann G, Madelenat P, Potet F. Expression of cadherins in benign, borderline, and malignant ovarian epithelial tumors: a clinicopathologic study of 60 cases. Hum Pathol. 1997;28:922–928. doi: 10.1016/s0046-8177(97)90007-1. [DOI] [PubMed] [Google Scholar]
- 66.Peralta Soler A, Knudsen KA, Tecson-Miguel A, McBrearty FX, Han AC, Salazar H. Expression of E-cadherin and N-cadherin in surface epithelial-stromal tumors of the ovary distinguishes mucinous from serous and endometrioid tumors. Hum Pathol. 1997;28:734–739. doi: 10.1016/s0046-8177(97)90184-2. [DOI] [PubMed] [Google Scholar]
- 67.Sundfeldt K, Piontkewitz Y, Ivarsson K, Nilsson O, Hellberg P, Brannstrom M, Janson PO, Enerback S, Hedin L. E-cadherin expression in human epithelial ovarian cancer and normal ovary. Int J Cancer. 1997;74:275–280. doi: 10.1002/(sici)1097-0215(19970620)74:3<275::aid-ijc7>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 68.Kobel M, Turbin D, Kalloger SE, Gao D, Huntsman DG, Gilks CB. Biomarker expression in pelvic high-grade serous carcinoma: comparison of ovarian and omental sites. Int J Gynecol Pathol. 2011;30:366–371. doi: 10.1097/PGP.0b013e31820d20ba. [DOI] [PubMed] [Google Scholar]
- 69.Wong AS, Maines-Bandiera SL, Rosen B, Wheelock MJ, Johnson KR, Leung PC, Roskelley CD, Auersperg N. Constitutive and conditional cadherin expression in cultured human ovarian surface epithelium: influence of family history of ovarian cancer. Int J Cancer. 1999;81:180–188. doi: 10.1002/(sici)1097-0215(19990412)81:2<180::aid-ijc3>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 70.Ong A, Maines-Bandiera SL, Roskelley CD, Auersperg N. An ovarian adenocarcinoma line derived from SV40/E-cadherin-transfected normal human ovarian surface epithelium. Int J Cancer. 2000;85:430–437. [PubMed] [Google Scholar]
- 71.Elloul S, Vaksman O, Stavnes HT, Trope CG, Davidson B, Reich R. Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin Exp Metastasis. 2010;27:161–172. doi: 10.1007/s10585-010-9315-2. [DOI] [PubMed] [Google Scholar]
- 72.Elloul S, Elstrand MB, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R, Davidson B. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103:1631–1643. doi: 10.1002/cncr.20946. [DOI] [PubMed] [Google Scholar]
- 73.Elloul S, Silins I, Trope CG, Benshushan A, Davidson B, Reich R. Expression of E-cadherin transcriptional regulators in ovarian carcinoma. Virchows Arch. 2006;449:520–528. doi: 10.1007/s00428-006-0274-6. [DOI] [PubMed] [Google Scholar]
- 74.Davidson B, Berner A, Nesland JM, Risberg B, Berner HS, Trope CG, Kristensen GB, Bryne M, Ann Florenes V. E-cadherin, alpha-, beta-, and gamma-catenin protein expression is up-regulated in ovarian carcinoma cells in serous effusions. J Pathol. 2000;192:460–469. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH726>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 75.Hudson LG, Zeineldin R, Stack MS. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin Exp Metastasis. 2008;25:643–655. doi: 10.1007/s10585-008-9171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reddy P, Liu L, Ren C, Lindgren P, Boman K, Shen Y, Lundin E, Ottander U, Rytinki M, Liu K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol Endocrinol. 2005;19:2564–2578. doi: 10.1210/me.2004-0342. [DOI] [PubMed] [Google Scholar]
- 77.Shen X, Kramer RH. Adhesion-mediated squamous cell carcinoma survival through ligand-independent activation of epidermal growth factor receptor. Am J Pathol. 2004;165:1315–1329. doi: 10.1016/S0002-9440(10)63390-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23:1739–1748. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woodfield RJ, Hodgkin MN, Akhtar N, Morse MA, Fuller KJ, Saqib K, Thompson NT, Wakelam MJ. The p85 subunit of phosphoinositide 3-kinase is associated with beta-catenin in the cadherin-based adhesion complex. Biochem J. 2001;360:335–344. doi: 10.1042/0264-6021:3600335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laprise P, Viel A, Rivard N. Human homolog of disc-large is required for adherens junction assembly and differentiation of human intestinal epithelial cells. J Biol Chem. 2004;279:10157–10166. doi: 10.1074/jbc.M309843200. [DOI] [PubMed] [Google Scholar]
- 83.De Santis G, Miotti S, Mazzi M, Canevari S, Tomassetti A. E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene. 2009;28:1206–1217. doi: 10.1038/onc.2008.470. [DOI] [PubMed] [Google Scholar]
- 84.Cheng JC, Auersperg N, Leung PC. Inhibition of p53 represses E-cadherin expression by increasing DNA methyltransferase-1 and promoter methylation in serous borderline ovarian tumor cells. Oncogene. 2011;30:3930–3942. doi: 10.1038/onc.2011.117. [DOI] [PubMed] [Google Scholar]
- 85.Cheng JC, Auersperg N, Leung PC. Inhibition of p53 induces invasion of serous borderline ovarian tumor cells by accentuating PI3K/Akt-mediated suppression of E-cadherin. Oncogene. 2011;30:1020–1031. doi: 10.1038/onc.2010.486. [DOI] [PubMed] [Google Scholar]
- 86.Yoshida J, Horiuchi A, Kikuchi N, Hayashi A, Osada R, Ohira S, Shiozawa T, Konishi I. Changes in the expression of E-cadherin repressors, Snail, Slug, SIP1, Twist, in the development and progression of ovarian carcinoma: the important role of Snail in ovarian tumorigenesis and progression. Med Mol Morphol. 2009;42:82–91. doi: 10.1007/s00795-008-0436-5. [DOI] [PubMed] [Google Scholar]
- 87.Davies BR, Worsley SD, Ponder BA. Expression of E-cadherin, alpha-catenin and beta-catenin in normal ovarian surface epithelium and epithelial ovarian cancers. Histopathology. 1998;32:69–80. doi: 10.1046/j.1365-2559.1998.00341.x. [DOI] [PubMed] [Google Scholar]
- 88.Cho EY, Choi Y, Chae SW, Sohn JH, Ahn GH. Immunohistochemical study of the expression of adhesion molecules in ovarian serous neoplasms. Pathol Int. 2006;56:62–70. doi: 10.1111/j.1440-1827.2006.01925.x. [DOI] [PubMed] [Google Scholar]
- 89.Voutilainen KA, Anttila MA, Sillanpaa SM, Ropponen KM, Saarikoski SV, Juhola MT, Kosma VM. Prognostic significance of E-cadherin-catenin complex in epithelial ovarian cancer. J Clin Pathol. 2006;59:460–467. doi: 10.1136/jcp.2005.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burkhalter RJ, Symowicz J, Hudson LG, Gottardi CJ, Stack MS. Integrin regulation of beta-catenin signaling in ovarian carcinoma. J Biol Chem. 2011;286:23467–23475. doi: 10.1074/jbc.M110.199539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohishi Y, Kurihara S, Takeuchi T, Aman M, Kaku T, Kobayashi H, Wake N, Oda Y. E-cadherin nuclear staining is useful for the diagnosis of ovarian adult granulosa cell tumor. Human pathology. 2011 doi: 10.1016/j.humpath.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 92.Redies C. Cadherins in the central nervous system. Prog Neurobiol. 2000;61:611–648. doi: 10.1016/s0301-0082(99)00070-2. [DOI] [PubMed] [Google Scholar]
- 93.Tohma Y, Yamashima T, Yamashita J. Immunohistochemical localization of cell adhesion molecule epithelial cadherin in human arachnoid villi and meningiomas. Cancer Res. 1992;52:1981–1987. [PubMed] [Google Scholar]
- 94.Shinoura N, Paradies NE, Warnick RE, Chen H, Larson JJ, Tew JJ, Simon M, Lynch RA, Kanai Y, Hirohashi S, et al. Expression of N-cadherin and alpha-catenin in astrocytomas and glioblastomas. Br J Cancer. 1995;72:627–633. doi: 10.1038/bjc.1995.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Asano K, Kubo O, Tajika Y, Huang MC, Takakura K, Ebina K, Suzuki S. Expression and role of cadherins in astrocytic tumors. Brain Tumor Pathol. 1997;14:27–33. doi: 10.1007/BF02478865. [DOI] [PubMed] [Google Scholar]
- 96.Perego C, Vanoni C, Massari S, Raimondi A, Pola S, Cattaneo MG, Francolini M, Vicentini LM, Pietrini G. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J Cell Sci. 2002;115:3331–3340. doi: 10.1242/jcs.115.16.3331. [DOI] [PubMed] [Google Scholar]
- 97.Utsuki S, Sato Y, Oka H, Tsuchiya B, Suzuki S, Fujii K. Relationship between the expression of E-, N-cadherins and beta-catenin and tumor grade in astrocytomas. J Neurooncol. 2002;57:187–192. doi: 10.1023/a:1015720220602. [DOI] [PubMed] [Google Scholar]
- 98.Asano K, Duntsch CD, Zhou Q, Weimar JD, Bordelon D, Robertson JH, Pourmotabbed T. Correlation of N-cadherin expression in high grade gliomas with tissue invasion. J Neurooncol. 2004;70:3–15. doi: 10.1023/b:neon.0000040811.14908.f2. [DOI] [PubMed] [Google Scholar]
- 99.Barami K, Lewis-Tuffin L, Anastasiadis PZ. The role of cadherins and catenins in gliomagenesis. Neurosurg Focus. 2006;21:e13. doi: 10.3171/foc.2006.21.4.14. [DOI] [PubMed] [Google Scholar]
- 100.Motta FJ, Valera ET, Lucio-Eterovic AK, Queiroz RG, Neder L, Scrideli CA, Machado HR, Carlotti-Junior CG, Marie SK, Tone LG. Differential expression of E-cadherin gene in human neuroepithelial tumors. Genet Mol Res. 2008;7:295–304. doi: 10.4238/vol7-2gmr424. [DOI] [PubMed] [Google Scholar]
- 101.Rodriguez FJ, Scheithauer BW, Giannini C, Bryant SC, Jenkins RB. Epithelial and pseudoepithelial differentiation in glioblastoma and gliosarcoma: a comparative morphologic and molecular genetic study. Cancer. 2008;113:2779–2789. doi: 10.1002/cncr.23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN, Anastasiadis PZ. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS One. 2010;5:e13665. doi: 10.1371/journal.pone.0013665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kantak SS, Kramer RH. E-cadherin regulates anchorage-independent growth and survival in oral squamous cell carcinoma cells. J Biol Chem. 1998;273:16953–16961. doi: 10.1074/jbc.273.27.16953. [DOI] [PubMed] [Google Scholar]
- 104.Kang HG, Jenabi JM, Zhang J, Keshelava N, Shimada H, May WA, Ng T, Reynolds CP, Triche TJ, Sorensen PH. E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res. 2007;67:3094–3105. doi: 10.1158/0008-5472.CAN-06-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pece S, Gutkind JS. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem. 2000;275:41227–41233. doi: 10.1074/jbc.M006578200. [DOI] [PubMed] [Google Scholar]
- 106.Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS. E-cadherin binding modulates EGF receptor activation. Cell Commun Adhes. 2003;10:105–118. [PubMed] [Google Scholar]
- 107.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 108.Pece S, Chiariello M, Murga C, Gutkind JS. Activation of the protein kinase Akt/PKB by the formation of E-cadherin-mediated cell-cell junctions. Evidence for the association of phosphatidylinositol 3-kinase with the E-cadherin adhesion complex. J Biol Chem. 1999;274:19347–19351. doi: 10.1074/jbc.274.27.19347. [DOI] [PubMed] [Google Scholar]
- 109.Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 2005;19:115–117. doi: 10.1096/fj.04-1942fje. [DOI] [PubMed] [Google Scholar]
- 110.Friedl P, Hegerfeldt Y, Tusch M. Collective cell migration in morphogenesis and cancer. Int J Dev Biol. 2004;48:441–449. doi: 10.1387/ijdb.041821pf. [DOI] [PubMed] [Google Scholar]
- 111.Macpherson IR, Hooper S, Serrels A, McGarry L, Ozanne BW, Harrington K, Frame MC, Sahai E, Brunton VG. p120-catenin is required for the collective invasion of squamous cell carcinoma cells via a phosphorylation-independent mechanism. Oncogene. 2007;26:5214–5228. doi: 10.1038/sj.onc.1210334. [DOI] [PubMed] [Google Scholar]
- 112.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 113.Niewiadomska P, Godt D, Tepass U. DE-Cadherin is required for intercellular motility during Drosophila oogenesis. J Cell Biol. 1999;144:533–547. doi: 10.1083/jcb.144.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kardash E, Reichman-Fried M, Maitre JL, Boldajipour B, Papusheva E, Messerschmidt EM, Heisenberg CP, Raz E. A role for Rho GTPases and cell-cell adhesion in single-cell motility in vivo. Nat Cell Biol. 2010;12:47–53. doi: 10.1038/ncb2003. sup pp 41–11. [DOI] [PubMed] [Google Scholar]
- 115.Grabowska MM, Day ML. Soluble E-cadherin: more than a symptom of disease. Front Biosci. 2012;17:1948–1964. doi: 10.2741/4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Steinhusen U, Weiske J, Badock V, Tauber R, Bommert K, Huber O. Cleavage and shedding of E-cadherin after induction of apoptosis. J Biol Chem. 2001;276:4972–4980. doi: 10.1074/jbc.M006102200. [DOI] [PubMed] [Google Scholar]
- 117.Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P. ADAM10 mediates E-cadherin shedding, regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2005;102:9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Najy AJ, Day KC, Day ML. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem. 2008;283:18393–18401. doi: 10.1074/jbc.M801329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68:3853–3868. doi: 10.1007/s00018-011-0763-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wheelock MJ, Buck CA, Bechtol KB, Damsky CH. Soluble 80-kd fragment of cell-CAM 120/80 disrupts cell-cell adhesion. J Cell Biochem. 1987;34:187–202. doi: 10.1002/jcb.240340305. [DOI] [PubMed] [Google Scholar]
- 122.Symowicz J, Adley BP, Gleason KJ, Johnson JJ, Ghosh S, Fishman DA, Hudson LG, Stack MS. Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Res. 2007;67:2030–2039. doi: 10.1158/0008-5472.CAN-06-2808. [DOI] [PubMed] [Google Scholar]
- 123.Johnson SK, Ramani VC, Hennings L, Haun RS. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer. 2007;109:1811–1820. doi: 10.1002/cncr.22606. [DOI] [PubMed] [Google Scholar]
- 124.Gil OD, Lee C, Ariztia EV, Wang FQ, Smith PJ, Hope JM, Fishman DA. Lysophosphatidic acid (LPA) promotes E-cadherin ectodomain shedding and OVCA429 cell invasion in an uPA-dependent manner. Gynecol Oncol. 2008;108:361–369. doi: 10.1016/j.ygyno.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 125.Noe V, Willems J, Vandekerckhove J, Roy FV, Bruyneel E, Mareel M. Inhibition of adhesion and induction of epithelial cell invasion by HAV-containing E-cadherin-specific peptides. J Cell Sci. 1999;112(Pt 1):127–135. doi: 10.1242/jcs.112.1.127. [DOI] [PubMed] [Google Scholar]
- 126.Ryniers F, Stove C, Goethals M, Brackenier L, Noe V, Bracke M, Vandekerckhove J, Mareel M, Bruyneel E. Plasmin produces an E-cadherin fragment that stimulates cancer cell invasion. Biol Chem. 2002;383:159–165. doi: 10.1515/BC.2002.016. [DOI] [PubMed] [Google Scholar]
- 127.Nawrocki-Raby B, Gilles C, Polette M, Bruyneel E, Laronze JY, Bonnet N, Foidart JM, Mareel M, Birembaut P. Upregulation of MMPs by soluble E-cadherin in human lung tumor cells. Int J Cancer. 2003;105:790–795. doi: 10.1002/ijc.11168. [DOI] [PubMed] [Google Scholar]
- 128.Ferber EC, Kajita M, Wadlow A, Tobiansky L, Niessen C, Ariga H, Daniel J, Fujita Y. A role for the cleaved cytoplasmic domain of E-cadherin in the nucleus. J Biol Chem. 2008;283:12691–12700. doi: 10.1074/jbc.M708887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shoval I, Ludwig A, Kalcheim C. Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development. 2007;134:491–501. doi: 10.1242/dev.02742. [DOI] [PubMed] [Google Scholar]
- 130.Hambsch B, Grinevich V, Seeburg PH, Schwarz MK. {gamma}-Protocadherins, presenilin-mediated release of C-terminal fragment promotes locus expression. J Biol Chem. 2005;280:15888–15897. doi: 10.1074/jbc.M414359200. [DOI] [PubMed] [Google Scholar]
- 131.Han AC, Soler AP, Tang CK, Knudsen KA, Salazar H. Nuclear localization of E-cadherin expression in Merkel cell carcinoma. Arch Pathol Lab Med. 2000;124:1147–1151. doi: 10.5858/2000-124-1147-NLOECE. [DOI] [PubMed] [Google Scholar]
- 132.Moon KC, Cho SY, Lee HS, Jeon YK, Chung JH, Jung KC, Chung DH. Distinct expression patterns of E-cadherin and beta-catenin in signet ring cell carcinoma components of primary pulmonary adenocarcinoma. Arch Pathol Lab Med. 2006;130:1320–1325. doi: 10.5858/2006-130-1320-DEPOEA. [DOI] [PubMed] [Google Scholar]
- 133.Cespedes MV, Larriba MJ, Pavon MA, Alamo P, Casanova I, Parreno M, Feliu A, Sancho FJ, Munoz A, Mangues R. Site-dependent E-cadherin cleavage and nuclear translocation in a metastatic colorectal cancer model. Am J Pathol. 2010;177:2067–2079. doi: 10.2353/ajpath.2010.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gervais ML, Henry PC, Saravanan A, Burry TN, Gallie BL, Jewett MA, Hill RP, Evans AJ, Ohh M. Nuclear E-cadherin and VHL immunoreactivity are prognostic indicators of clear-cell renal cell carcinoma. Lab Invest. 2007;87:1252–1264. doi: 10.1038/labinvest.3700684. [DOI] [PubMed] [Google Scholar]
- 135.Salahshor S, Naidoo R, Serra S, Shih W, Tsao MS, Chetty R, Woodgett JR. Frequent accumulation of nuclear E-cadherin and alterations in the Wnt signaling pathway in esophageal squamous cell carcinomas. Mod Pathol. 2008;21:271–281. doi: 10.1038/modpathol.3800990. [DOI] [PubMed] [Google Scholar]
- 136.Serra S, Salahshor S, Fagih M, Niakosari F, Radhi JM, Chetty R. Nuclear expression of E-cadherin in solid pseudopapillary tumors of the pancreas. JOP. 2007;8:296–303. [PubMed] [Google Scholar]
- 137.Ordonez NG. Value of E-cadherin and N-cadherin immunostaining in the diagnosis of mesothelioma. Hum Pathol. 2003;34:749–755. doi: 10.1016/s0046-8177(03)00285-5. [DOI] [PubMed] [Google Scholar]
- 138.Chen H, Paradies NE, Fedor-Chaiken M, Brackenbury R. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J Cell Sci. 1997;110(Pt 3):345–356. doi: 10.1242/jcs.110.3.345. [DOI] [PubMed] [Google Scholar]