

Abstract
Oral cancer is one of the most aggressive epithelial malignancies, whose incidence is on the rise. Previous studies have shown that in a subset of human oral squamous cell carcinoma (OSCC) tumor specimens, overexpression of the DPAGT1 gene, encoding the dolichol-P-dependent N-acetylglucoseamine-1-phosphate transferase, a key regulator of the metabolic pathway of protein N-glycosylation, drives tumor cell discohesion by inhibiting E-cadherin adhesive function. Recently, we reported that DPAGT1 was a target of the canonical Wnt signaling pathway. Here, we link overexpression of DPAGT1 in human OSCC tumor specimens to aberrant activation of canonical Wnt signaling. We report dramatic increases in β- and γ-catenins at the DPAGT1 promoter and correlate them with reduced expression of a Wnt inhibitor, Dickkopf-1 (Dkk-1). Using human squamous carcinoma cell lines of the head and neck, we show that partial inhibition of DPAGT1 reduces canonical Wnt signaling, indicating that DPAGT1 and canonical Wnt signaling function in a positive feedback loop. We provide evidence that E-cadherin inhibits DPAGT1, canonical Wnt signaling and the OSCC cancer phenotype by depleting nuclear β-and γ-catenins, with hypoglycosylated E-cadherin being the most effective.This suggests that in human OSCC, extensive N-glycosylation of E-cadherin compromises its ability to inhibit canonical Wnt signaling and DPAGT1 expression. Our studies reveal a novel interplay between DPAGT1/N-glycosylation and canonical Wnt signaling and suggest that dysregulation of this crosstalk is a key mechanism underlying OSCC. They also suggest that partial inhibition of DPAGT1 may represent an effective way to restore normal interactions among these essential pathways in oral cancer.
Keywords: Oral cancer, OSCC, canonical Wnt signaling, DPAGT1, N-glycosylation, Dkk-1, E-cadherin, β-catenin, γ-catenin
Introduction
Oral squamous cell carcinoma (OSCC) ranks among the most pernicious epithelial malignancies (1, 2). Recently, changes in the expression and mutational profiles of tumor suppressive and oncogenic pathways have contributed to identify the “mutational landscape” of this cancer (3, 4). Despite these advances,the key genes and pathways that drive early stages of development and progression of OSCC have remained elusive. In particular, little is known about the interplay between key metabolic and signaling pathways in oral tumorigenesis (1, 2, 4, 5).
Previously, we reported that a subset of OSCCs was associated with the overexpression of N-glycosylation gene, DPAGT1, leading to the loss of intercellular adhesion (6). DPAGT1 initiates protein N-glycosylation in the endoplasmic reticulum (ER) and is a key determinant of the quantity and quality of N-glycans on glycoproteins (7–9). One of the downstream targets of DPAGT1 is E-cadherin, an epithelial cell-cell adhesion receptor and a tumor suppressor (10–13). The N-glycosylation of E-cadherin affects its adhesive function by controlling its ability to organize dynamic multiprotein complexes at the plasma membrane known as adherens junctions (AJs) (10, 14, 15). High DPAGT1 expression leads to extensive modification of E-cadherin with complex N-glycans in unstable AJs, while low DPAGT1expression results in the hypoglycosylation of E-cadherin in mature AJs (10, 16).
Recently, we showed that DPAGT1 was a target of the canonical Wnt signaling pathway, with both β- and γ-catenins binding to the DPAGT1 promoter (17).Upregulation of DPAGT1 by canonical Wnt increased modification of E-cadherin with complex N-glycans, resembling the scenario in OSCC(17). Here, we show that overexpression of DPAGT1 in human OSCC specimens is associated with aberrant activation of canonical Wnt signaling due to diminished levels of a Wnt inhibitor, Dkk-1. Utilizing human head and neck cancer cell lines, we show that partial inhibition of DPAGT1 with siRNA downregulated canonical Wnt activity, indicating a positive feedback loop between Wnt signaling and DPAGT1. Also, hypoglycosylated E-cadherin effectively inhibited canonical Wnt and DPAGT1expression by depleting nuclear β- and γ-catenins. These results reveal novel interactions among the metabolic pathway of protein N-glycosylation, canonical Wnt signaling and E-cadherin adhesion and show that the dysregulation of their interplay promotes OSCC.
Materials and Methods
Tissue specimens and cell culture
Tissue specimens, representing incisional biopsies from resected primary tumors, were obtained at the Boston University Medical Center, Boston, MA and were approved for studies by the Institutional Review Board. Primary tumor samples were from 5 patients who underwent resection of squamous cell carcinoma of the oral cavity of the moderately differentiated grade. Paired samples, one representing the tumor and another from cytologically normal adjacent epithelia (AE) as defined by an on-site pathology analysis, were taken from each patient's specimen and snap-frozen at −80°C. One fraction of each was used for biochemical analyses, while another was fixed in 4% neutral buffered formalin for 24 h and processed for immunohistochemistry. Total tissue lysates (TTLs) from AE and OSCC were prepared as described (6). Protein concentrations were determined using the BCA assay (Pierce).
Head and neck carcinoma, CAL27 and A253, cells (ATCC) were maintained in DMEM and in McCoy's 5a medium, respectively, supplemented with 10% FBS, 1% penicillin and streptomycin. CAL27 cells were transfected with wild type E-cadherin (E-cad) or its hypoglycosylated variant, V13, and enriched for exogenous E-cadherins, as described before (16). Protein concentrations were determined using the BCA protein assay (Pierce).
RNA interference
SMART-pool™ of siRNAs targeting DPAGT1 (S) was obtained from Dharmacon. Non-silencing (NS) negative control siRNA was from Qiagen. A253 cells were transfected at 80% confluence with 200 nM of either NS or S using Lipofectamine 2000 and cultured for 72 h. The efficiency of transfections was determined using off-target Cy3-siRNA (Dharmacon). DPAGT1 expression was assessed by real-time PCR (RT-PCR), as described (6, 16).
Plasmids, antibodies and Western blot
TOP Flash and FOP Flash vectors were from Millipore. The FOP DPAGT1 vector, carrying three copies of the TCF sequence from the human DPAGT1 promoter in the FOP Flash reporter plasmid, and measurements of luciferase activity, including controls, were described (17). All antibodies were from commercial sources, with the exception of rabbit polyclonal antibody to hamster GPT (6, 17). Cell and tissue lysates and Western blots were according to standard procedures. Band intensities were quantified using Image J.
Microscopy, immunohistochemistry and immunofluorescence imaging
Scratch wound assays were examined using a Nikon Eclipse TE300 microscope. Immunohistochemistry was described before (6). Immunofluorescence analyses were carried out with a Zeiss LSM510 META confocal microscope; settings were fixed to the most highly stained sample and all other images were acquired at those settings (6). Negative controls lacked primary antibodies.
Cell growth and migration analyses
CAL27 cells were plated in 96-well plates at 1×103 cells/well in 100 μl of cell culture medium, grown and collected at 24 h intervals for 5 days and counted using a hemocytometer. For migration assays, cells were suspended in 100 μl of serum-free DMEM medium and added on top of Matrigel-coated Transwell (8.0 μM pore size; Corning Costar) chambers in triplicates. DMEM supplemented with 10% FBS was added to the lower chamber to serve as a chemoattractant. Cells that migrated through the Transwell filters were counted in 3 different fields under a light microscope.
Transepithelial resistance
Transepithelial resistance (TER) was measured using an epithelial voltohmmeter, as described (16). Values were calculated after subtracting background from blank Transwells cultured in parallel with the media. Statistical analysis was by ANOVA.
Scratch wound assay
Cells were seeded onto six-well tissue culture dishes, cultured to confluence, wounded using sterile pipette tips and photographed at 0, 24 and 48 h under a phase contrast microscope (10×). Three independent fields were photographed for each cell preparation, and each wounding assay was performed three times.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were carried out with chromatin cross-linked with 1% formaldehyde in E-cad- and V13-transfected cells, as described before (17). For tissue ChIP analyses, AE and OSCC were incubated in DMEM medium containing 1% formaldehyde, tissues were homogenized and sonicated at 4°C. The primers and the Taqman probe surrounding the Wnt responsive element of the human DPAGT1 promoter were described (17).
Results
Overexpression of DPAGT1 is associated with increased canonical Wnt signaling in OSCC
Our previous work revealed that a subset of human OSCCs of the well to moderately differentiated histologic grade exhibited overexpression of DPAGT1 throughout the neoplasm, particularly at the tumor margins (6). Although these tumors displayed abundant expression of E-cadherin, its extensive modification with complex N-glycans prevented the formation of stable AJs and tight junctions (TJs) (6). Since DPAGT1 has been shown to be a target of canonical Wnt signaling in vitro (17), we asked whether its overexpression in OSCC tumor specimens was due to upregulation of canonical Wnt signaling. Total tissue lysates (TTLs) from 5 paired samples of human OSCC and cytologically normal adjacent epithelia (AE) (Fig. 1A) were analyzed for the abundance of the DPAGT1 protein, GPT, by Western blot (6). Consistently, OSCC exhibited 3–4 fold more GPT compared to AE (Fig. 1B). Also, immunofluorescence analysis showed increased GPT expression throughout OSCC (Fig. 1C).
Figure 1. Overexpression of DPAGT1 in OSCC is associated with increased promoter occupancy of β- and γ-catenins.

(A) Composite resection specimen of OSCC and AE. (B) Western blot of GPT expression in AE and OSCC total tissue lysates (TTLs). Bargraph, Fold change in GPT levels after normalization to actin. (*p<0.05).(C) Immunofluorescence localization of E-cadherin and GPT in AE and OSCC. Size bar: 10 μm. (D) ChIP analysis of β-catenin at the DPAGT1 promoter in AE and OSCC(**p< 0.01). (E) ChIP analysis of γ-catenin at the DPAGT1 promoter in AE and OSCC (**p< 0.01). All results are from three independent experiments.
Activation of DPAGT1 by canonical Wnt signaling has been shown to involve the binding of both β- and γ-catenins to the TCF sequence in the DPAGT1 promoter. Indeed, ChIP assays revealed a 10-fold increase in the amount of β-catenin and a 15-fold increase in the amount of γ-catenin bound to the DPAGT1 TCF sequence in OSCC (Fig. 1D & E). This correlated with 6- and 8-fold higher OSCC tissue levels of β-catenin and γ-catenin, respectively, compared to AE (Fig. 2A). Both β- and γ-catenins displayed diffuse, cytoplasmic staining in OSCC compared to a more localized cell-cell border pattern in AE (Fig. 2B).
Figure 2. OSCC displays elevated tissue levels of β- and γ-catenins.

(A) Western blot analysis of β-catenin and γ-catenin in AE and OSCC. Bargraph, Fold change in β-catenin and γ-catenin levels after normalization to actin. Results represent one of three experiments (*p<0.05). (B) Immunofluorescence localization of β- and γ-cateninsin AE and OSCC (arrows, insets).Size bar: 20 μm.
In addition, the OSCC tumor specimens had increased expression of a canonical Wnt ligand, Wnt3a, and decreased expression of Dkk-1, a canonical Wnt pathway inhibitor (18) (Figs. 3A and 3B). Immunofluorescence localization of Dkk-1 revealed robust expression in cytodifferentiated layers of AE and significantly diminished presence throughout the OSCC (Fig 3C). Thus, increased DPAGT1 expression in OSCC was associated with aberrant activation of the Wnt signaling pathway.
Figure 3. Upregulation of canonical Wnt in OSCC is associated with decreased Dkk-1 and elevated Wnt3a levels.
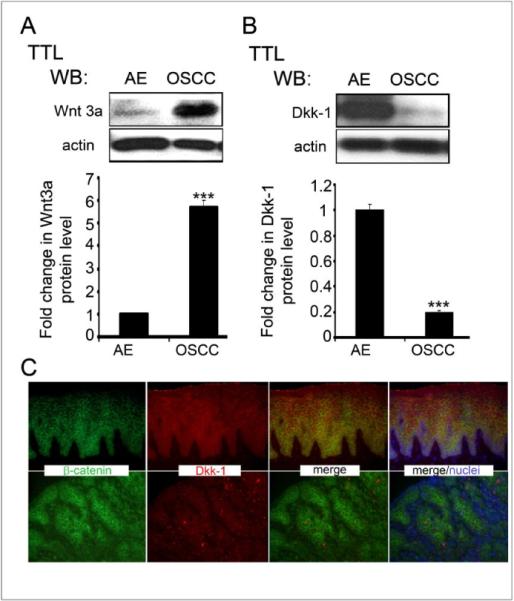
(A) Western blot of Dkk-1 expression in AE and OSCC. Bargraph, Fold change in Dkk-1 levels after normalization to actin (***p< 0.001). (B) Western blot of Wnt3a expression in AE and OSCC. Bargraph, Fold change in Wnt3a levels in OSCC in comparison to AE after normalization to actin (**p< 0.01). (C) Immunofluorescence staining of Dkk-1 and b-catenin in AE and OSCC. Size bar: 50 μm. All results represent one of three independent experiments
Partial silencing of DPAGT1 inhibits canonical Wnt activity
Partial inhibition of DPAGT1 with siRNA in human OSCC A253 cells reduced the N-glycosylation of E-cadherin with complex N-glycans and promoted AJ maturation via increased recruitment of γ-catenin (17). Therefore, we tested the consequence of partial silencing of DPAGT1 with siRNA on canonical Wnt activity in A253 cells using a TOP Flash vector. Treatment with 200 nM siRNA (S) resulted in downregulation of GPT protein levels by 50% when compared with non-silenced (NS) cells (Fig. 4A). This was associated with substantial inhibition of canonical Wnt and DPAGT1 promoter activities, as assessed by the luciferase reporter gene expression (Figs. 4B and 4C).
Figure 4. Partial silencing of DPAGT1 inhibits canonical Wnt activity in A253 cells.
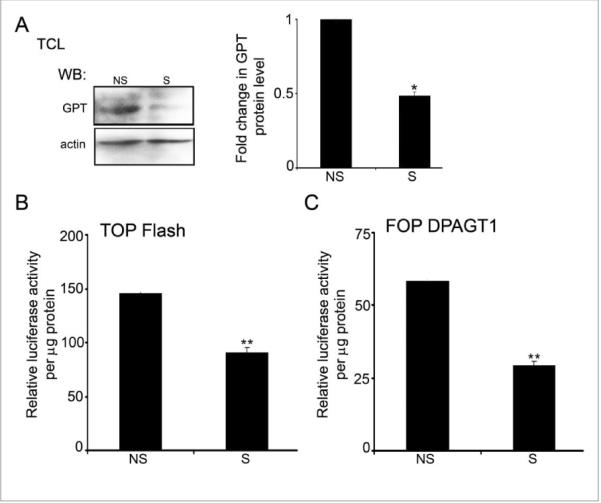
(A) Western blot of GPT expression after partial silencing of DPAGT1with siRNA. Bargraph, Fold change of GPT abundance in silenced (S) and non-silenced (NS) cells after normalization to actin (*p< 0.05). (B) Luciferase reporter activity from the TOP Flash vector in S and NS cells (**p< 0.01). (C) Luciferase activity from the FOP DPAGT1 vector in S and NS cells(***p< 0.001). All results are from two independent experiments with each experiment repeated twice.
Hypoglycosylated E-cadherin, V13, attenuates OSSC cancer phenotype
It has been documented that transfection of E-cadherin into normal and cancer cells strengthens cell-cell adhesion (12, 19–22). Also, reduction of the N-glycosylation status of E-cadherin further enhances intercellular adhesion (6, 23). Specifically, the hypoglycosylated E-cadherin variant, V13, lacking N-glycans at sites 1 and 3 in ectodomains 4 and 5, drove stabilization of AJs and increased intercellular adhesion in a manner similar to downregulation of DPAGT1 with siRNA (10, 16, 23). Thus, we examined the effects of V13 on CAL27 cells growth, migration, wound healing and TER. As expected, V13 cells produced hypoglycosylated E-cadherin that migrated with a reduced molecular size on a Western blot due to the lack of two N-glycan structures (Fig. 5A). Examination of growth rates revealed that untransfected cells grew faster than E-cad and V13 cells, with V13 cells displaying slowest growth (Fig. 5B). Therefore, the N-glycosylation status of E-cadherin impacted its ability to suppress OSCC proliferation in vitro.
Figure 5. Hypoglycosylated E-cadherin variant, V13, interferes with CAL27 cell growth and migration and enhances intercellular adhesion.

(A) Western blot of V13 and E-cad. (B) Growth curves of untransfected, E-cad- and V13-transfected cells (*p<0.05). (C) Transepithelial resistance (TER) in untransfected, E-cad- and V13-transfected cells (**p< 0.01).(D) Transwell migration assays of untransfected, E-cad- and V13-transfected cells (**p< 0.01 and ***p< 0.001). (E) Scratch- wound assays of untransfected, E-cad- and V13-transfected cells. Size bar: 50 μm.
V13 cells also displayed higher TER than untransfected and E-cad cells (p< 0.01) (Fig. 5C), indicating that they assembled more functional TJs. In addition, Transwell migration assays showed that V13 cells migrated slower than E-cad cells, which were slower than untransfected cells (p< 0.01 and p< 0.001 for V13 and E-cad, respectively) (Fig. 5D). Lastly, scratch wound assays revealed that wound closure was significantly slower in V13 cells compared to untransfected and E-cad cells (Fig. 5E).Taken together, these data indicate that hypoglycosylated E-cadherin augments intercellular adhesion and slows the growth and migration of OSCC cells.
V13 inhibits canonical Wnt activity and DPAGT1 in CAL27 cells
Next, we examined DPAGT1 transcription and canonical Wnt activity in E-cad- and V13-transfectants. ChIP assays showed that E-cad, and to a greater extent V13, depleted nuclear β- and γ-catenins from the DPAGT1 promoter in OSCC cells (Fig. 6A). Also, V13 cells had significantly less TOP Flash and FOP DPAGT1 activities compared to E-cad cells (Figs. 6B and 6C). Collectively, these studies confirmed that the N-glycosylation status of E-cadherin affected its ability to sequester nuclear β- and γ-catenins and inhibit the canonical Wnt pathway.
Figure 6. Hypoglycosylated E-cadherin, V13, inhibits canonical Wnt activity and DPAGT1 transcription in CAL27 cells.
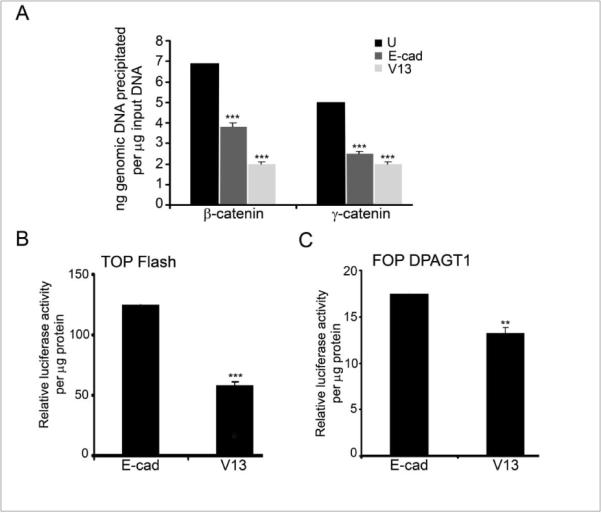
(A) ChIP analyses using antibodies against β- and γ-catenins from E-cad and V13 cells (***p< 0.001). (B) Luciferase activity from the TOP Flash vector in E-cad and V13 cells (***p< 0.001). (C) Luciferase activity from the FOP DPAGT1 vector in E-cad and V13 cells (**p< 0.01). All results are from two independent experiments with each experiment repeated twice.
Discussion
Studies presented here show that the overexpression of N-glycosylation gene, DPAGT1, in human OSCC specimens is associated with aberrant activation of canonical Wnt signaling. They also show that DPAGT1 and canonical Wnt signaling function in a positive feedback loop, while E-cadherin, and hypoglycosylated E-cadherin in particular, negatively regulates canonical Wnt and DPAGT1. OSCC cells fail to regulate the positive feedback loop between DPAGT1 and canonical Wnt signaling due to the loss of both Dkk-1 and mature AJs (Fig. 7). Loss of Dkk-1 leads to the activation of a positive feedback loop between canonical Wnt signaling and DPAGT1, which then usurps the ability of E-cadherin to downregulate their activities.
Figure 7. Interactions among canonical Wnt signaling, DPAGT1/N-glycosylation and E-cadherin in OSCC.
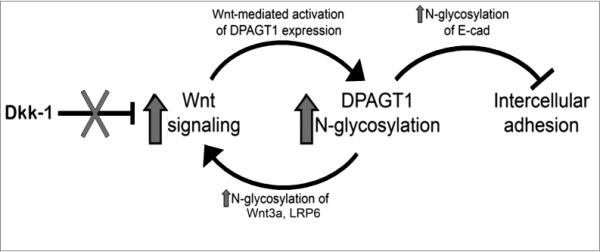
Wnt signaling activates DPAGT1 and protein N-glycosylation, leading to extensive N-glycosylation of E-cadherin and weak intercellular adhesion. In OSCC, this positive feedback loop between Wnt signaling and DPAGT1is amplified by diminished expression of Dkk-1, acanonical Wnt inhibitor. Furthermore, extensive N-glycosylation of E-cadherin prevents it from depleting nuclear β- and γ-catenins, allowing the postitive feedback between Wnt and DPAGT1 to operate without controls.
Aberrant increases in the activity of the canonical Wnt pathway have been reported in a number of human cancers. In OSCC, inappropriate activation of canonical Wnt signaling was documented by increased tissue levels of both β- and γ-catenins and by their greater abundance at the DPAGT1 promoter. Moreover, downregulation of Dkk-1 was, at least in part, responsible for the activation of canonical Wnt and DPAGT1 (24). Dkk-1 prevents the formation of Wnt-Frizzled-LRP5/6 receptor complexes and blocks canonical Wnt signaling (25). The DKK-1 gene promoter has Tcf/β-catenin binding sites, and β-catenin upregulates DKK-1 expression as part of a safety mechanism by which cells regulate canonical Wnt activity. We now show that OSCC has a defect in this autoregulatory loop, resembling colon carcinoma (24).
Partial silencing of DPAGT1 in A253 cells resulted in a significant reduction in canonical Wnt activity, indicating that DPAGT1 expression impacts this signaling pathway. This is substantiated by reports showing that N-glycosylation positively affects canonical Wnt signaling via modification of Wnt3a and LRP5/6 (26, 27). OSCC specimens with increased DPAGT1 expression displayed greater abundance of Wnt3a, suggesting that N-glycosylation affects its stability. Thus, an important finding of this study is that DPAGT1 and canonical Wnt signaling function in a positive feedback loop.
In OSCC cells, the hypoglycosylated E-cadherin variant, V13, significantly decreased the abundance β- and γ-catenins at the DPAGT1 promoter and attenuated Wnt activity more than wild type E-cad. This indicates that E-cadherin inhibits Wnt signaling and DPAGT1 expression by depleting β- and γ-catenins from Wnt target genes. While V13 preferentially recruits γ-catenin to AJs, the mechanism via which it reduces nuclear β-catenin is unknown.Thus, N-glycosylation of E-cadherin in OSCC interferes with its ability to downregulate canonical Wnt and DPAGT1, allowing them to operate without checks.
While loss of feedback mechanisms among key regulatory pathways represents a formidable challenge to cancer therapy, we also show that by downregulating DPAGT1 expression, or by transfecting cells with hypoglycosylated E-cadherin, it is possible to inhibit both canonical Wnt signaling and N-glycosylation. To date, many inhibitors of the canonical Wnt pathway have been studied, including those focused on β-catenin. The challenge has been to target nuclear β-catenin without affecting its structural role in cell-cell adhesion. By virtue of modulating canonical Wnt activity and E-cadherin adhesive function, DPAGT1 may represent an effective target for oral cancer therapy. Indeed, our studies suggest that it may be possible to partially inhibit DPAGT1 in human tumors to reestablish the correct interplay among these pathways, thus restoring normal control mechanisms that function in oral tissues,
Acknowledgments
This work was supported by NIDCR, National Institutes of Health grant RO1 DE015304 (to M.A.K.)
Grant sponsor: NIH; Grant number: DE 015304
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res. 2008;87:14–32. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Ordonez B, Beauchemin M, Jordan RC. Molecular biology of squamous cell carcinoma of the head and neck. J Clin Pathol. 2006;59:445–53. doi: 10.1136/jcp.2003.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuo J-H, Zhu W, Li M-Y, Li X-H, Yi H, Zeng G-Q, et al. Activation of EGFR Promotes Squamous Carcinoma SCC10A Cell Migration and Invasion via Inducins EMT-like PhenotypeCHange and MMP-9-Mediated Degradation of E-cadherin. J Cell Biochem. 2011;112:2508–17. doi: 10.1002/jcb.23175. [DOI] [PubMed] [Google Scholar]
- 6.Nita-Lazar M, Noonan V, Rebustini I, Walker J, Menko AS, Kukuruzinska MA. Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res. 2009;69:5673–80. doi: 10.1158/0008-5472.CAN-08-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kukuruzinska MA, Lennon K. Protein N-glycosylation: Molecular genetics and functional significance. Critical Reviews in Oral Biology & Medicine. 1998;9:415–48. doi: 10.1177/10454411980090040301. [DOI] [PubMed] [Google Scholar]
- 8.Mendelsohn RD, Helmerhorst EJ, Cipollo JF, Kukuruzinska MA. A hypomorphic allele of the first N-glycosylation gene, ALG7, causes mitochondrial defects in yeast. Biochim Biophys Acta. 2005;1723:33–44. doi: 10.1016/j.bbagen.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Rush JS, Karaoglu D, Krasnewich D, Lubinsky MS, Waechter CJ, et al. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Hum Mutat. 2003;22:144–50. doi: 10.1002/humu.10239. [DOI] [PubMed] [Google Scholar]
- 10.Liwosz A, Lei T, Kukuruzinska MA. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J Biol Chem. 2006;281:23138–49. doi: 10.1074/jbc.M512621200. [DOI] [PubMed] [Google Scholar]
- 11.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 12.Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasineness of human carcinoma cells. J Cell Biol. 1991;113:173–85. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Margulis A, Andriani F, Fusenig N, Hashimoto K, Hanakawa Y, Garlick JA. Abrogation of E-cadherin-mediated adhesion induces tumor cell invasion in human skin-like organotypic culture. J Invest Dermatol. 2003;121:1182–90. doi: 10.1046/j.1523-1747.2003.12523.x. [DOI] [PubMed] [Google Scholar]
- 14.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–48. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 15.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nita-Lazar M, Rebustini I, Walker J, Kukuruzinska MA. Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp Cell Res. 2010;316:1871–84. doi: 10.1016/j.yexcr.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengupta PK, Bouchie MP, Kukuruzinska MA. N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J Biol Chem. 2010;285:31164–73. doi: 10.1074/jbc.M110.149195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–14. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Paradies NE, Fedor-Chaliken M, Brackenbury R. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J Cell Sci. 1997;110:345–56. doi: 10.1242/jcs.110.3.345. [DOI] [PubMed] [Google Scholar]
- 20.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;21:1739–48. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo J, Lubaroff DM, Hendrix MJ. Suppression of prostate cancer invasive potential and matrix metalloproteinase activity by E-cadherin transfection. Cancer Res. 1999;59:3552–6. [PubMed] [Google Scholar]
- 22.Semb H, Christofori G. The tumor-suppressor function of E-cadherin. Am J Hum Genet. 1998;63:1588–93. doi: 10.1086/302173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jamal BT, Nita-Lazar M, Gao Z, Amin B, Walker J, Kukuruzinska MA. N-glycosylation status of E-cadherin controls cytoskeletal dynamics through the organization of distinct beta-catenin- and gamma-catenin-containing AJs. Cell Health Cytoskelet. 2009;2009:67–80. doi: 10.2147/chc.s5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguilera O, Fraga MF, Ballestar E, Paz MF, Herranz M, Espada J, et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25:4116–21. doi: 10.1038/sj.onc.1209439. [DOI] [PubMed] [Google Scholar]
- 25.Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429–33. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 26.Khan Z, Vijayakumar S, de la Torre TV, Rotolo S, Bafico A. Analysis of endogenous LRP6 function reveals a novel feedback mechanism by which Wnt negatively regulates its receptor. Mol Cell Biol. 2007;27:7291–301. doi: 10.1128/MCB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komekado H, Yamamoto H, Chiba T, Kikuchi A. Glycosylation and palmitoylation of Wnt-3a are coupled to produce an active form of Wnt-3a. Genes Cells. 2007;12:521–34. doi: 10.1111/j.1365-2443.2007.01068.x. [DOI] [PubMed] [Google Scholar]