

Abstract
The ER aminopeptidase associated with antigen processing, ERAAP, is essential for trimming peptides presented by MHC I molecules. ERAAP inhibition by cytomegalovirus causes immune evasion, and ERAAP polymorphisms are associated with autoimmune disorders. How normal ERAAP function is monitored is unknown. We found that ERAAP inhibition rapidly induced presentation of the FL9 peptide by the Qa-1b MHC Ib molecule. Antigen-experienced T cells specific for the Qa-1b-FL9 complex were frequent in naïve mice. Wild-type mice immunized with ERAAP-deficient cells mounted a potent CD8+ T cell response specific for the Qa-1b-FL9- complex. MHC Ib-restricted cytolytic effectors specifically eliminated ERAAP-deficient cells in vitro and in vivo. Thus, non-classical peptide-Qa-1b complexes direct cytotoxic T cells to targets with defective antigen processing in the ER.
Introduction
Cells generate peptide-MHC class I complexes (pMHC I) to display their intracellular protein milieu for immune surveillance1. Normal cells present “self” pMHC I, which are not recognized by self-tolerant cytotoxic CD8+ T lymphocytes (CTLs), while infected or transformed cells present “foreign” pMHC I that activate appropriate CTLs 2. The nature of the peptide repertoire presented by MHC I molecules, and rapidly responding antigen-specific CTLs, are key determinants of immune surveillance.
Peptide-MHC I complexes are generated by the antigen processing pathway in a series of concerted steps3. Generally, antigen processing begins with the cleavage of protein precursors by the proteasome, followed by transport of peptides into the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP). After transport, ERAAP, the ER aminopeptidase associated with antigen processing, trims N-terminally extended peptides to generate the final peptides presented by MHC I4. The peptides generated by this pathway can be presented by classical MHC I (MHC Ia) molecules as well as non-classical (MHC Ib) molecules5, although the role of ERAAP in generating peptides for presentation by MHC Ib molecules is unclear.
The MHC Ib molecules are closely related to the ubiquitous MHC Ia molecules H2-K, D and L (HLA-A, B and C in humans)5. An important difference is that MHC Ib molecules are not polymorphic—for example, there are only four known alleles of the mouse MHC Ib molecule Qa-1 (ref. 6) and only two of its human analog, HLA-E6. Both MHC Ia and MHC Ib molecules loaded with appropriate peptides in the ER go to the cell surface as potential ligands for circulating CD8+ T cells or natural killer (NK) cells1,3.
Immune evasion mechanisms frequently target key components of the antigen processing pathway to inhibit the formation and presentation of pMHC I 7,8. Counter measures have therefore evolved to detect the failure of various components of the antigen-processing pathway. For example, inhibition of peptide transport by TAP causes the severe loss of pMHC I from the cell surface 9. The absence of pMHC I complexes results in loss of CD8+ T cell recognition, but allows activation of NK cells that are released from pMHC I-mediated inhibition7,8. Similarly, immune responses can be inhibited or enhanced by changes in ERAAP activity. Expression of human ERAP1 is inhibited in tumor cells10, and human cytomegalovirus inhibits ERAP1 function by a microRNA-based mechanism11. Additionally, ERAP1 polymorphisms are associated with the autoimmune disorders ankylosing spondylitis and psoriasis12,13. It is not known if and how the immune system senses defects in ERAAP function.
The loss of ERAAP function does not generally cause a large change in pMHC I expression on the cell surface14-16. Nevertheless, we, and others, have found that loss of ERAAP substantially alters the repertoire of peptides presented by MHC I15-17. CTLs are elicited in wild-type mice immunized with ERAAP-deficient cells and vice versa17, suggesting that CTL-mediated recognition of changes in the pMHC I repertoire might indicate ERAAP dysfunction. However, the pMHC I ligands expressed by ERAAP-deficient cells that activate CTLs remain undefined.
Here, we used T cells as probes to analyze the immunogenic pMHC I ligands expressed uniquely by ERAAP-deficient cells. We found that cells lacking ERAAP elicited CD8+ T cells specific for both classical pMHC Ia and non-classical pMHC Ib complexes. We identified the highly conserved FL9 peptide that is presented by the MHC Ib molecule Qa-1b exclusively in ERAAP-deficient cells. Remarkably, we found that abundant antigen-experienced T cells, specific for the Qa-1b-FL9 complex, existed even in naïve wild-type mice, and expanded to yield cytotoxic effectors that rapidly eliminated cells lacking ERAAP in vitro as well as in vivo. Thus, non-classical MHC Ib molecules are sensors for defects in peptide processing in the ER.
Results
ERAAP-deficient cells present immunogenic MHC Ib ligands
The immune system efficiently detects differences between self- and non-self. Therefore, to identify immunologically significant changes caused by ERAAP deficiency, we immunized wild-type (C57BL/6J, H-2b) mice with MHC-matched spleen cells from ERAAP-deficient mice (14; referred to as ERAAP-KO in the text). We generated wild-type anti-ERAAP-KO CD8+ T cell lines by restimulating host spleen cells with ERAAP-KO antigen-presenting cells (APCs) in vitro. Similar to our earlier report17, CD8+ T cell lines generated from immunized mice responded strongly by producing interferon-γ (IFN-γ) and tumor necrosis factor (TNF) when cultured with ERAAP-deficient APCs compared to the background responses to self-APCs (Fig. 1a,b, Supplementary Fig. 1). Comparable fractions (30% versus 25% on average) of the same CD8+ T cells responded to ERAAP-deficient cells that also lacked any classical H2-Kb or H2-Db MHC Ia molecules (18; ERAAP-MHC Ia-TKO, Fig. 1a,b). MHC I molecules were nevertheless required for the IFN-γ response because APCs lacking ERAAP as well as β2-microglobulin (β2m; β2m-ERAAP-DKO), an essential structural subunit all MHC I molecules19,20, failed to stimulate the same wild-type anti-ERAAP-KO CD8+ T cells (Fig 1b). Likewise, APCs deficient in ERAAP as well as the TAP transporter (9; TAP-ERAAP-DKO), also failed to stimulate the same CD8+ T cells (Fig. 1b), demonstrating that their responses required peptide(s) transported into the ER. We therefore conclude that loss of ERAAP function results in the presentation of immunologically distinct peptides by MHC Ia and MHC Ib molecules to wild-type CD8+ T cells.
Figure 1. Wild-type CD8 T cells respond to pMHC Ia and pMHC Ib complexes expressed by ERAAP-deficient cells.

(a-b) Spleen cells from C57BL/6J (WT) mice, immunized with ERAAP-/- splenocytes, were restimulated in vitro to generate cytotoxic T lymphocyte (CTL) lines. IFN-γ production by the CTL lines was measured after incubation with the indicated splenic APCs. (a) Representative flow cytometry plots show IFN-γ produced by WT anti-ERAAP-/- CTLs. Numbers indicate % of IFN-γ+ cells among all the CD8+ cells in the sample. (b) The percentage of CD8+IFN-γ+ cells in cultures of individual immunized mice. Each symbol represents one mouse. One representative of three independent experiments is shown. (c-e) The lacZ response of BEko8Z hybridoma cells to the indicated LPS-stimulated spleen cells used as APCs. In (d) LPS-stimulated spleen cells were treated with the aminopeptidase inhibitor, leucinethiol and the BEko8Z hybridoma response measured. Data shown is representative of three independent experiments.
To further characterize those MHC Ib ligands expressed by ERAAP-deficient cells, we generated BEko8Z, a β-galactosidase (LacZ) inducible, monoclonal CD8+ T cell hybridoma, by fusing wild-type anti-ERAAP-KO CD8+ T cells with the BWZ.36/CD8α fusion partner21. The BEko8Z hybridoma produced lacZ when co-cultured with ERAAP-deficient, but not wild-type splenic APCs (Fig. 1c, left). Consistent with our earlier observations (Fig 1a), the BEko8Z ligand was expressed by ERAAP-deficient APCs and did not require expression of classical MHC Ia molecules (Fig. 1c, middle). BEko8Z did not respond to ERAAP-deficient APCs that lacked β2m or TAP (Fig. 1c). Thus, the BEko8Z hybridoma was specific for a TAP-transported peptide that was presented by a non-classical MHC Ib molecule exclusively on the surface of ERAAP-deficient cells.
Mice express a large number of non-classical MHC Ib molecules that serve as ligands for various innate as well as adaptive immune responses5. Among the three MHC Ib molecules known to present peptides, we considered H2-M3 the least likely because these molecules present N-formylated peptides that should be refractory to aminopeptidase activity of ERAAP. We tested the other two molecules, Qa-1 and Qa-2, as potential ligands for BEko8Z T cells. Splenocytes from Qa-2null (B6.K1, ref. 23) and Qa-1b-deficient (Qa-1-KO22) mouse strains were used as APCs, after treating them with leucinethiol, a potent inhibitor of aminopeptidases, including ERAAP (23; Fig. 1d). The BEko8Z hybridoma responded to leucinethiol-treated splenocytes from wild-type as well as Qa-2null B6.K1 mice, but failed to recognize untreated splenocytes, or those from Qa-1b-deficient mice (Fig. 1d). The requirement for Qa-1b was further confirmed when BEko8Z cells did not respond to APCs from mice with genetic deficiencies in both ERAAP and Qa-1b expression (ERAAP-Qa-1-DKO; Fig 1e). We conclude that BEko8Z T cells recognize a unique peptide presented by the Qa-1b MHC Ib molecule by ERAAP-deficient APCs.
The FL9 peptide is presented by Qa-1b to BEko8Z T cell
The Qa-1b molecule is best known for presenting the Qdm peptide, derived from the ER translocation signal sequence of classical MHC Ia molecules24,25. That the BEko8Z hybridoma recognizes ERAAP-MHC Ia-TKO APCs lacking H2-Db, which is the source of Qdm peptide in B6 mice, suggested that BEko8Z T cells were specific for a different peptide. Additionally, neither synthetic Qdm peptide (AMAPRTLLL) nor its N-terminally extended analogs were able to stimulate BEko8Z T cells (data not shown).
Next, we identified the naturally processed peptide presented by Qa-1b to BEko8Z by screening cDNA libraries prepared from ERAAP-deficient cells26. To increase the efficiency of the screen, we first determined that CD11c+B220- dendritic cells (DCs) from ERAAP-KO spleens were better APCs for stimulating BEko8Z than unfractionated spleen cells or other splenocyte subsets (Supplementary Fig. 2). A cDNA expression library was constructed using mRNA from splenic DCs isolated from ERAAP-KO mice treated with B16 cells expressing Flt3L27. The cDNA library was fractionated into small pools and transfected into Qa-1b-expressing L cells. Because the BEko8Z ligand was generated only in the absence of ERAAP, the transfected cells were treated briefly with leucinethiol to inhibit aminopeptidases, and then assessed for their ability to stimulate the BEko8Z T cell hybridoma. One of the 2880 pools screened stimulated the BEko8Z T cell hybridoma (Fig. 2a). After re-screening 24 individual cDNA clones from this cDNA pool, three stimulatory clones were identified, and clone 23D9.15 was chosen for further analysis (Supplementary Fig. 3). BEko8Z T cells responded strongly to APCs co-transfected with the 23D9.15 and Qa-1b cDNAs and treated with leucinethiol, compared to APCs treated with dithiothreitol (DTT) alone (Supplementary Fig. 4), suggesting that the 23D9.15 plasmid encoded the antigenic peptide. All the positive cDNAs encoded a “hypothetical protein” called Fam49b in the NCBI database (NM_144846).
Figure 2. The Fam49b gene is the source of a new antigen presented by Qa-1b in the absence of ERAAP function.

(a) cDNA pools were transfected into Qa-1b-expressing L cells, ERAAP function inhibited and BEko8Z hybridoma responses measured. One cDNA pool in plate 23 contained the antigenic activity. (b) A schematic of the clone 23D9.15 showing the Fam49b gene and the locations of the PCR primers used to make truncations. Truncations take the name of the reverse PCR primer used. The BEko8Z stimulatory activity was in region 13 (R13). The locations of PCR primers used to establish the C-terminus of the final peptide are shown on the amino acid sequence of R13. The indicated minigenes were tested as described for panel a. Data is graphed as Mean±SEM, of transfections done in duplicate. One representative of two experiments is shown.
The peptide-binding motif for Qa-1b is unknown. Therefore, to identify the minimal antigenic peptide within the Fam49b protein, we tested a series of deletion constructs of the 23D9.15 cDNA for their ability to generate the BEko8Z ligand (Fig. 2b). The antigenic activity was produced in cells transfected with the R1 and R4 fragments. In the next series of truncations of the R4 fragment, the C-terminal leucine in the R13 fragment was found to be absolutely essential for T cell stimulatory activity, as the R14 and R15 truncations lacking this leucine residue were unable to stimulate the T cell hybridoma (Fig. 2c). The antigenic activity thus mapped to the C-terminus of the R13 fragment.
FL9 is naturally processed and presented by Qa-1b
To establish the identity of the antigenic peptide and define its precise N-terminal boundary, we tested a panel of synthetic peptides of varying lengths that all terminated at the same essential leucine residue (Fig. 3a). The nine-residue FYAEATPML (FL9) or the ten residue LFYAEATPML (LL10) peptides were the most potent stimulators of BEko8Z T cells (Fig. 3a). In contrast, further addition to, or deletion of one or two residues from the N-terminus of the FL9 peptide (11,12 and 8, 7-mers) resulted in marked loss of activity. Thus, the FL9 nonapeptide defined the minimum core sequence for stimulating BEko8Z T cells.
Figure 3. The nonamer FL9 is the naturally processed peptide presented by Qa-1b molecules.

(a) Progressively N-terminally extended synthetic peptides from the Fam49b R13 fragment were added to Qa-1b- L cells and tested for their ability to activate BEko8Z. (b) Synthetic FL9, and its N-terminally extended analogs were fractionated on a C18 RP-HPLC column and eluted using a 10-70% acetonitrile gradient. The presence of antigenic activity was assayed by BEko8Z activation. (c) Qa-1b-expressing COS-7 cells were transfected with the R13 minigene, or (d) a minigene encoding the FL9 peptide, and peptide extracts were fractionated as above. Arrows indicate the peak fractions for the indicated peptides. (e) COS-7 cells that did not express Qa-1b were transfected with the R13 minigene, and analyzed as above. One representative of three experiments is shown in all panels.
We analyzed cell extracts to define the naturally processed peptide recognized by the BEko8Z hybridoma. The extracts were fractionated by reverse-phase high performance liquid chromatography (RP-HPLC) and tested for the presence of antigenic activity. We took advantage of BEko8Z’s ability to detect FL9 as well as its N-terminally extended 10, 11 and 12-mer analogs (Fig. 3a). Each peptide eluted with a different retention time and could be readily distinguished from the others (Fig. 3b). We co-transfected Qa-1b expressing COS-7 cells with cDNA encoding the R13 fragment of Fam49b described in Fig. 2b above (Fig. 3c). Because generation of the final antigenic peptide from the Fam49b cDNA was greatly reduced in transfected cells when ERAAP function was not inhibited (Supplementary Fig. 4), we performed all subsequent experiments in the presence of ERAAP inhibition. Peptides were extracted from transfected cells after leucinethiol treatment to inhibit ERAAP, and fractionated by HPLC. The fractions were tested for their ability to stimulate BEko8Z in the presence of Qa-1b-expressing fibroblasts as APCs. A single peak of antigenic activity, corresponding to the synthetic FL9 peptide, was detected in HPLC fractionated R13-minigene expressing cells (Fig. 3c). A peak with the same retention time was detected when Qa-1b expressing COS-7 cells were transfected with a minigene encoding FL9 peptide alone (Fig. 3d). Furthermore the antigenic activity was detected only in cells that expressed the restricting Qa-1b MHC Ib molecule (Fig. 3e). The requirement for the restricting MHC molecule for detection of antigenic activity is strikingly similar to that observed with naturally processed peptides presented by classical MHC Ia molecules28,29. We conclude that the FL9 nonapeptide corresponds to the naturally processed peptide presented by Qa-1b in the absence of ERAAP function.
The Qa-1b-FL9 complex is an immunodominant T cell ligand
The Qa-1b-Qdm complex (referred to as Qdm) is recognized by the CD94-NKG2A receptor expressed on NK cells and some activated T cells. To assess whether the FL9 peptide presented by the same Qa-1b molecule (Qa-1b-FL9 complex: QFL) was also recognized by a subset of NK cells, we stained wild-type spleen cells with QFL tetramers obtained from the NIH tetramer core facility. In contrast to a distinct NKG2A+ population that bound the fluorescent Qdm tetramer, we did not detect binding of the QFL tetramer to either NKG2A+ or NKG2A- subsets of NK cells (Fig. 4a,b). We conclude that the QFL complex is a ligand for CD8+ T cells rather than NK cells.
Figure 4. The Qa-1b-FL9 complex is an immunodominant T cell ligand.
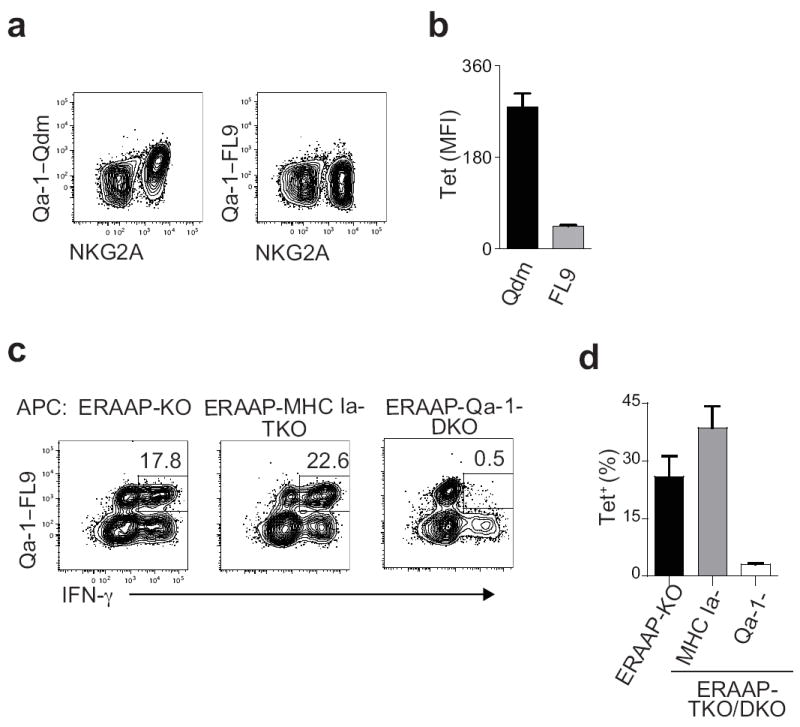
(a) Spleen cells from naïve mice were labeled with Qa-1b-Qdm or Qa-1b-FL9 tetramers together with markers for other spleen cell populations. Representative flow cytometry plots show tetramer labeling on gated B220-NK1.1+TCRβ- cells. (b) The mean fluorescent intensity of tetramer staining of NKG2A+NK1.1+TCRβ- cells of six individual mice. Data shown is the mean±SEM. (c) WT anti-ERAAP-/- CTL lines were generated, and IFN-γ production measured in response to the indicated T cell-depleted splenic APCs. Cells were labeled with Qa-1b-FL9 tetramers prior to intracellular IFN-γ staining. Representative flow cytometry plots of IFN-γ production by tetramer+ cells, gated on CD8+CD4- cells, are shown. The numbers indicate the % of tetramer+IFN-γ+ cells in the CD8+CD4- population. (d) The fraction of tetramer+ cells among CD4-CD8+IFN-γ+ cells in these experiments was measured. The mean±SEM shown were from two independent experiments with three mice per group.
We then determined the frequency of QFL complex-specific T cells in wild-type mice that respond to ERAAP-deficient cells. Spleen cells from wild-type mice immunized with ERAAP-KO cells were re-stimulated with ERAAP-KO cells in vitro. The response of QFL tetramer+ cells towards APCs deficient in ERAAP alone, or deficient in both ERAAP and MHC Ia, or both ERAAP and Qa-1b was measured (Fig 4c,d). A substantial fraction of wild-type CD8+ T cells producing IFN-γ in response to ERAAP-KO APCs also bound the QFL tetramer (Fig. 4c,d). Likewise, a comparable fraction of CD8+ T cells producing IFN-γ in response to ERAAP-MHC Ia-TKO APCs were also tetramer+ (Fig 4c,d). Interestingly, a large fraction of T cells producing IFN-γ in response to ERAAP-MHC Ia-TKO APCs did not bind the tetramer (Fig 4c,d), suggesting that other pMHC Ib were also presented by these APCs. As expected, only background frequencies of IFN-γ-producing tetramer+ cells were detected when the T cells were stimulated with ERAAP-Qa-1-DKO APCs (Fig. 4c,d). We conclude that the QFL complex is an immunodominant ligand recognized by CD8+ T cells in wild-type mice immunized with ERAAP-deficient cells, although other immunogenic MHC Ia and MHC Ib ligands were also detected. We refer to this QFL-specific T cell population as QFL T cells.
QFL T cells are abundant and antigen-experienced in naïve mice
We assessed the abundance of QFL-specific cells in the spleens of naïve mice. Spleen cells were labeled with the QFL tetramers conjugated to two different fluorophores to increase specificity, followed by magnetic enrichment of tetramer+ cells, as described earlier for MHC Ia- and MHC II-restricted T cells (30-32; Fig. 5a,b). On average, 1030 QFL-specific T cells were detected in a naïve wild-type spleen, suggesting that QFL-specific T cells were present at a frequency of ~1:104 in naïve wild-type spleens. Enrichment of CD8+ T cells with the tetramer was specific, since the number of CD4+ tetramer+ cells enriched from wild-type mice were two orders of magnitude lower than the corresponding numbers of CD8+ tetramer+ cells (Supplementary Fig. 5 and Fig. 5a,b). On the other hand, the spleens of TAP-deficient mice contained less than 5% of the number of QFL-specific cells found in wild-type spleens (Fig 5a,b). Most remarkably, QFL-specific cells were also found in mice lacking Qa-1b expression, but only at about 30% of the numbers in wild-type spleens (Fig 5a,b). The frequency of QFL-specific cells was higher than that of typical pMHC Ia specific precursors, which range from 1:104 to 1:105 in combined spleens and lymph nodes of naïve mice31,33, compared to the numbers measured here in spleens alone. We conclude that QFL-specific CD8+ T cells that recognize the Qa-1b-FL9 complex are relatively abundant in normal wild-type mice. Furthermore, although TAP transport of antigenic peptides was crucial, the restricting Qa-1b molecule was not absolutely essential for the development of QFL cells, indicating that other MHC I molecules can also support development of QFL-specific cells.
Figure 5. Qa-1b-FL9-specific T cells are antigen experienced in naïve mice.

Spleen cells from naïve mice were stained with Qa-1b-FL9 tetramers labeled with two different fluorophores. Tetramer+ cells were enriched magnetically and counted during flow cytometry measurements. (a) Representative flow cytometry plots showing tetramer+ populations from WT, TAP-KO and Qa-1-KO mice before and after enrichment. The numbers indicate the average number of tetramer+ cells isolated from the indicated spleens. (b) The numbers of tetramer+ cells per spleen of the indicated mice. Each symbol represents one indivdual mouse. Data are pooled from six independent experiments. * P = 0.0043, **P = 0.0007. (c) Representative flow cytometry plots showing the percentage of enriched tetramer+ cells from WT and Qa-1-KO mice that express CD44. (d) The percentage of isolated tetramer+ cells that express CD44 compared to the percentage of total CD8+ cells in the same spleens, prior to enrichment, that also express CD44 are compared. Pooled data from six experiments are shown. Data shown as the mean±SEM
QFL-specific T cells in naïve wild-type mice expressed markers of antigen-experience. The majority of tetramer+ cells expressed the memory markers CD44 and the interleukin 2 receptor β chain CD122 (Fig. 5c, data not shown), compared to about 20% of total CD8+ T cells in the wild-type spleen that expressed CD44 and CD122 (Fig. 5c,d, data not shown). In contrast, the majority of tetramer+ cells in Qa-1-KO mice did not express the antigen-experienced CD44hiCD122+ phenotype seen in wild-type mice (Fig. 5c,d, data not shown). The fraction of tetramer+ cells in Qa-1-KO mice that were also CD44hi was comparable to the fraction of total CD8+ T cells in the same mice that were CD44hi at steady state (Fig. 5d). Thus, although the development of QFL-specific cells in Qa-1b-deficient mice did occur at a somewhat lower efficiency, the acquisition of the antigen-experienced CD44hi phenotype was absolutely dependent upon Qa-1b expression.
QFL T cells expand in response to ERAAP-deficient cells
MHC Ib-restricted T cells participate in primary immune responses in a variety of situations, but are not thought to generate strong secondary responses34-36. To determine how QFL-specific T cells respond to ERAAP-deficiency, wild-type mice were immunized with ERAAP-KO cells, and the frequency of QFL-specific T cells was measured ex vivo by tetramer staining. We analyzed animals either ten days after challenge, during the effector phase of the response, or 8 or 12 weeks post-challenge, during the memory phase. QFL-tetramer+ CD8+ T cells expanded during the effector phase in wild-type mice challenged with ERAAP-KO cells (Fig 6a,b). Importantly, memory QFL-specific T cells could also be clearly detected in wild-type mice 8-12 weeks after the initial priming with ERAAP-KO cells (Fig. 6a,b). Spleen cells from the same mice were restimulated with the FL9 peptide directly ex vivo, and intracellular cytokine staining for IFN-γ showed that both effector and memory QFL-specific T cells were readily activated (Fig. 6c,d). The activation and expansion of these cells was specific to challenge with ERAAP-KO cells, as they were not detected at comparable frequencies in naive mice (Fig. 6), or in mice challenged with wild-type cells (not shown). The results shown in Fig. 6 are from mice that were not re-challenged prior to measuring memory responses, and show that QFL-specific memory T cells had expanded and were maintained at high frequencies following a single encounter with ERAAP-KO cells. We conclude that, in contrast to other pMHC Ib-restricted T cells34-36, T cells specific for the QFL ligand form functional memory cells upon encountering ERAAP-KO cells.
Figure 6. Qa-1b-FL9-specific effector and memory T cells are generated in response to ERAAP-/- cells.

WT mice were immunized with ERAAP-KO cells and their spleens harvested 10 days later for the analysis of effector responses and either 8 or 12 weeks later for memory responses. (a) The frequency of tetramer+ cells was measured directly ex vivo either ten days or eight weeks post-immunization. Representative flow cytometry plots showing the frequency of PE- and APC-tetramer double-positive cells. Numbers indicate the percentage of double tetramer+ cells in the B220-CD4-CD8+ population (b) Pooled frequencies of tetramer+ cells from three mice per group. (c) IFN-γ production in response to the FL9 peptide was measured directly ex vivo 12 weeks after immunization. Representative flow cytometry plots showing IFN-γ production by B220- CD4-CD8+ cells are shown. The numbers indicate the percentage of IFN-γ+CD8+ cells in the B220-CD4- population. (d) Pooled frequencies of IFN-γ+ cells from three mice per group. Data shown as the mean±SEM. One representative of three experiments is shown.
MHC Ib-specific T cells eliminate ERAAP-deficient cells
We hypothesized that ERAAP-/--specific CD8+ T cells elicited in wild-type mice may carry out immune surveillance of ERAAP deficiency. We tested whether CD8+ T cells from immunized wild-type mice were capable of eliminating ERAAP-deficient target cells. Indeed, wild-type anti-ERAAP-KO CTLs, generated as in Fig. 1a, were cytotoxic and efficiently killed both ERAAP-KO as well as ERAAP-MHC Ia-TKO target cells in vitro (Fig. 7a). Furthermore, wild-type anti-ERAAP-KO CTLs also killed RMA tumor cells that were treated for 5h with leucinethiol to inhibit ERAAP, but did not kill leucinethiol-treated TAP-deficient, RMA/s cells (Fig. 7b). Interestingly, B16 melanoma cells transfected with a minigene encoding the FL9 peptide, but not untransfected B16 cells, were also effectively killed by wild-type anti-ERAAP-KO CTLs (Fig. 7c), despite the absence of ERAAP inhibition. Thus, expression of FL9 is both necessary and sufficient for elimination of target cells. We conclude that wild-type CTLs, elicited by ERAAP-deficient cells, were capable of detecting and eliminating ERAAP-deficient or FL9-presenting cells.
Figure 7. WT CTLs specifically eliminate ERAAP-KO cells expressing novel pMHC Ib complexes.

(a) Spleen cells from the indicated mice were used as targets for WT anti-ERAAP-KO CTLs. Pooled data from three independent experiments and a total of 12 mice are shown. (b) RMA or TAP-deficient RMA/s cells were treated with leucinethiol to inhibit ERAAP function and used as targets for CTLs. (c) B16 melanoma cells, either stably expressing FL9 or not, were used as targets for WT anti-ERAAP-KO CTLs. One representative of three independent experiments is shown. (d) WT, and either ERAAP-KO or ERAAP-MHC Ia-TKO spleen cells were labeled with 0.25 μM and 2.5 μM, respectively, of the dye CFSE and injected into primed or naïve WT mice. Representative flow cytometry plots in the left panel show rejection of CFSEhi ERAAP-KO targets in primed mice. Pooled results of three independent experiments are shown in the right panel, *P < 0.0001 **P < 0.0005. Data shown as the mean±SEM (e) In experiments as described above, CD4+ or CD8+ cells were depleted 24 h prior to challenge. Pooled results from two independent experiments are shown. Data shown as the mean±SEM.
To assess whether ERAAP-deficient cells could also be eliminated in vivo, wild-type mice were injected with a mixture of self- and ERAAP-KO cells labeled with two different concentrations of the intracellular dye CFSE. The input cells could therefore be distinguished from each other and from unlabeled host cells by flow cytometry (Fig. 7d, left). Both self- and ERAAP-deficient fluorescent cells were recovered from naive mice in approximately the same 1:1 ratio as the input cell mixture. In contrast, ERAAP-KO cells were specifically rejected without affecting the recovery of self-cells by wild-type mice primed with ERAAP-deficient cells 7 days earlier (Fig. 7d). As expected, ERAAP-MHC Ia-TKO cells were also rejected with comparable efficiency in primed mice. Notably, the elimination or loss of target cells required CD8+ rather than CD4+ cells because mice depleted of CD8+ cells no longer rejected the ERAAP-KO targets, unlike mice depleted of CD4+ cells, which did (Fig. 7e). In contrast to another study that suggested a role for NK cells in rejecting ERAAP-deficient cells37, we did not detect a role for NK cells in our experiments with mice depleted of NK1.1+ cells (Fig. 7b). We conclude that ERAAP deficiency elicits a potent MHC Ib-restricted CD8+ T cell response in wild-type mice that effectively eliminates ERAAP-deficient cells in vitro as well as in vivo.
Discussion
The pMHC I repertoire is the immune system’s window into the state of the cellular proteome. Immune evasion mechanisms often target the antigen processing pathway to inhibit the presentation of pMHC I. Immune surveillance mechanisms in turn detect defects in pMHC I presentation. Here, we describe an MHC Ib-restricted CD8+ T cell mechanism that senses normal antigen processing in the ER.
Immune responses are influenced by alterations in ERAAP activity. A microRNA encoded by human cytomegalovirus specifically targets the predominant human ERAAP isoform (hERAP1) 11. Loss of hERAP1 function inhibits presentation of viral epitopes and likely contributes to the chronic infections established by HCMV. Likewise, changes in ERAAP expression in tumors might lead to the activation of NK cells 37. Finally, polymorphisms in hERAP1 have been found to be associated with two autoimmune disorders, ankylosing spondylitis and psoriasis 12,13. Thus, impaired ERAAP function can cause various immune disorders.
Analysis of the MHC I-bound peptide pool in ERAAP-deficient cells has shown a clear shift towards presentation of N-terminally extended as well as unique peptides by MHC Ia molecules 14-17,38. Here, we show that the changes in the pMHC I repertoire in ERAAP-KO cells also extend to MHC Ib molecules. Furthermore, the new pMHC Ib ligands on ERAAP-KO cells are immunodominant, inducing a large fraction of WT anti-ERAAP-KO CD8 T cells. An earlier study by our group 17 did not detect an MHC Ib-specific response of this magnitude; although, a small fraction of WT anti-ERAAP-KO CTLs in that study were activated by MHC Ia-deficient APCs. These differences in relative abundance of MHC Ia and MHC Ib restricted CTLs may arise from differences in the in vitro restimulation culture conditions. Also, in this study, the Qa-1-FL9 tetramer allows direct measurement of the ligand-specific CTLs.
We identified Fam49b as the source of the antigenic peptide presented by Qa-1b in ERAAP-deficient cells. Fam49b is highly conserved in vertebrates—from zebra fish to humans—and appears to be ubiquitously expressed (EBI gene atlas; http://www.ebi.ac.uk/gxa/gene/ENSMUSG00000022378), although its function is unknown. Intriguingly, expression of FAM49B transcripts has been detected at high levels in patients with relapsing multiple sclerosis, and in non-small cell lung cancer tissues 39,40. We suggest, therefore, that presentation of the FL9 peptide by Qa-1b could be a highly conserved mechanism for detecting ERAAP deficiency.
We used the QFL tetramer to characterize lymphocytes specific for this ligand. We were unable to detect NK cells specific for the QFL tetramer, in contrast to the readily detectable Qdm-specific subset of NKG2A+ NK cells. However, a large fraction of the CD8 T cells elicited by ERAAP-deficient cells were QFL tetramer+. Using QFL tetramers, we also enriched a sizeable number of QFL CD8 T cells from naïve WT mice, suggesting that these cells were present at a relatively high frequency. Intriguingly, even in naive mice, QFL T cells had an antigen-experienced, CD44hiCD122hi phenotype, similar to other innate-like MHC Ib-specific T cells, such as natural killer T and mucosa-associated invariant T MAIT cells 41. Whether QFL T cells share other developmental and functional characteristics with these innate-like effectors is not known.
Surprisingly, we discovered tetramer+ QFL T cells in Qa-1b-deficient mice, albeit at a lower frequency than in wild-type mice. We did not, however, detect QFL tetramer+ cells in TAP- or β2m-deficient mice, suggesting that QFL T cells require the presentation of peptides by an MHC Ib molecule other than Qa-1b for development. Remarkably, acquisition of the antigen-experienced phenotype did require Qa-1b expression, because QFL T cells in naïve Qa-1b-deficient mice did not constitutively express CD44 or CD122. MHC class Ib-restricted T cells generally display an antigen-experienced phenotype in naïve mice, possibly because they are selected by APCs of hematopoietic origin 42,43. Our observations show that QFL T cells only express markers of antigen-experience on encountering Qa-1b presented peptides. An interesting implication of these findings is that QFL T cells may have encountered their ligand even in naïve mice. Because the FL9 peptide is exclusively produced in ERAAP-deficient cells, the Qa-1b-FL9 complex or a cross-reactive ligand might have been generated during a transient ERAAP deficiency in wild-type mice—caused, perhaps, by natural transformation events, endogenous viruses or commensal microbes.
CD8 T cells elicited by immunization with ERAAP-deficient cells eliminated both MHC Ia- and MHC Ib-expressing targets cells lacking ERAAP. WT anti-ERAAP-KO T cell lines also killed ERAAP-inhibited tumor cells and ERAAP-sufficient cells expressing FL9, suggesting that expression of this pMHC Ib complex is sufficient for the elimination of target cells. QFL T cells expanded robustly in response to a single challenge with ERAAP-KO cells, and unusually for a pMHC Ib-specific response, also formed stable memory T cells that retained their effector function without rechallenge. QFL T cells might therefore have the capacity to mediate long-term immune surveillance of ERAAP deficiencies. QFL complexes are rapidly presented, by APCs treated for five hours with an ERAAP inhibitor. QFL complexes are also a specific sign of ERAAP dysfunction, and are not presented by TAP-deficient cells, unlike the Qa-1-associated peptides isolated from TAP-deficient cells described in a recent study 44. Thus, the peptides presented by Qa-1b reflect the state of the antigen-processing pathway.
In conclusion, ERAAP deficiency results in the presentation of a highly conserved pMHC Ib complex, Qa-1b-FL9, on the cell surface. Abundant antigen-experienced Qa-1b-FL9 specific CD8+ T cells exist in naïve mice, expand and effectively eliminate ERAAP-deficient cells. This response appears to combine the best of both worlds—the exquisite specificity of adaptive immunity and the rapid responsiveness of the innate immune system.
Methods
Mice
ERAAP-KO mice (B6.129.Arts1tm1Ucb) have been previously described. ERAAP-MHC Ia-TKO, ERAAP-TAP-DKO, ERAAP-β2m-DKO and ERAAP-Qa-1-DKO mice were generated in our facility by crossing ERAAP-KO mice with H2-Kb-H-2Db-deficient, TAP1-deficient, β2m-deficient and Qa-1b-deficient mice, respectively. WT C57BL/6J, Qa-2-deficient B6.K1 and β2m-deficient mice were either purchased from the Jackson Laboratory or bred in our facility. H2-T23-deficient Qa-1-KO mice were generated in the laboratory of H. Cantor (Harvard University) and were the kind gift of E. Engleman (Stanford University). Mice were housed and all procedures carried out with the approval of, and in accordance with, the institutional animal care guidelines of the University of California Berkeley.
Antibodies, cell lines and peptides
Antibodies for flow cytometry were obtained from BD Biosciences (CD8α (53-6.7), CD4 (RM4-5), B220 (RA36B2), TCRβ (H57-597), CD44 (IM7), NK1.1 (PK136), Qa-1b (6A8.6F10.1A6), CD11c (HL3), H2-Kb (AF6-88.5), H2-Db (KH95), I-Ab (25-9-17), IFN-γ (XMG1.2) and TNF (MP6-XP22)) and eBioscience (NKG2A (20D5)). Antbodies used to deplete cells in vivo were from the UCSF Cell Culture Facility. All peptides were synthesized by D. King (UC Berkeley). Qa-1b-expressing L and COS cells were made by stably transducing Lmtk- and COS-7 cells respectively with lentiviral vectors expressing Qa-1b (kind gift of H. Cantor, Harvard University). B16.FL9 cells were made by transfecting B16.BL6 cells with a plasmid encoding the FL9 minigene and neomycin resistance. Stable cells lines were derived from single cells by limiting dilution in G418 selection.
CTL and T cell activation assays
WT CTL lines that recognize ERAAP-deficient cells were generated by immunizing C57BL/6J mice once with 2 × 107 ERAAP-KO spleen cells intraperitoneally. Spleens were harvested from immunized mice ten days after immunization. 5 × 106 responder spleen cells were restimulated in vitro with an equal number of irradiated ERAAP-/- spleen cells and 20 U/ml of recombinant human IL-2 (BD Biosciences) per well in 24-well plates. T cells were used for IFN-γ assays six days after restimulation, and for in vitro cytotoxicity assays five days after restimulation. To measure intracellular IFN-γ, CTL lines were harvested and stimulated for 5 h with CD4- and CD8-depleted LPS-stimulated spleen APCs of the indicated genotypes. Golgi-Plug (BD Biosciences) was added for the latter 4 h of the incubation period. Cells were then stained first with surface markers, or with tetramers followed by other surface markers, fixed, permeabilized and stained for intracellular IFN-γ. For ex vivo analysis of IFN-γ production, spleen cells were harvested from immunized mice and plated in 96-well plates with 1 μM of the indicated peptides for 5 h in the presence of Golgi-Plug. The cells were then stained as described.
Hybridomas and expression cloning
WT anti-ERAAP-KO CTL lines were harvested 4 d after in vitro restimulation and fused to the CD8+TCRαβ− cell line BWZ.36 that expresses a TCR-inducible lacZ gene. Putative positive clones were screened using LPS-stimulated ERAAP-/- spleen cells as APCs, and positive wells were subcloned by limiting dilution to obtain clones derived from a single cell. Once a stable hybridoma BEko8Z was established, it was used to screen cDNA libraries, as described in Sanderson et al45. ERAAP-/- mice were immunized subcutaneously with 3 × 106 Flt3L-secreting B16 melanoma cells, as described by Mach et al27. Two weeks later, their spleens were harvested and blasted overnight with 200ng/ml LPS (Sigma Aldrich). CD11c+ cells were enriched using CD11c microbeads followed by magnetic enrichment using LS columns (Miltenyi Biotec). The enriched cells were pelleted, frozen, then homogenized using a rotor-stator homogenizer. mRNA was isolated using the Oligotex mRNA isolation kit from Qiagen. cDNA was synthesized and cloned into pCMV-SPORT6 using the SuperScript Plasmid kit from Invitrogen, and transformed into bacteria. Plasmid DNA was isolated from bacterial cultures grown in 96-well plates and used to transfect Qa-1b-expressing L cells. 15 μM leucinethiol in 500 μM DTT was added 42 h later, and BEko8Z hybridoma cells added 6 h after that and incubated overnight. T cell activation was detected by the colorimetric cleavage of chlorophenylred-β-D-galactopyranoside (CPRG; Roche) by TCR-induced β-galactosidase. When a positive well was detected, 1 μl of cDNA from the well was transformed in bacteria and plasmid DNA isolated from multiple individual colonies. Each of the individual cDNAs was transfected into Qa-1b-expressing L cells. The positive cDNAs were sequenced and identified. Progressive truncations were made in the coding region of Fam49b using nested PCR primers, cloned into pcDNA1, and their activity tested by transfection into Qa-1b-expressing L cells. Once the candidate region was narrowed down to region 13, and the final leucine was determined to be essential, N-terminally extended peptides were synthesized and tested. The minigenes were cloned in the pcDNA1 expression vector, and were designed to encode the desired amino acid sequence with an ATG initiation codon. The FL9 minigene, for example, encodes Met-FL9 (MFYAEATPML) amino acid sequence.
Enrichment of tetramer+ cells
Qa-1b-FL9 tetramers were synthesized by the NIH tetramer core facility. Tetramer+ cells were enriched as previously described by Moon et al32. Briefly, spleens from the indicated mice were harvested and homogenized using nylon filters. RBCs were lysed, and the cells resuspended in 200 μl of sorter buffer (PBS+0.1%sodium azide+5% FCS). PE- and APC-labeled Qa-1b-FL9 tetramers were added at a final dilution of 1:200. The cells were incubated for 30 min at 23°C, washed, resuspended in 0.45ml of sorter buffer. 50 μl of anti-PE microbeads (Miltenyi Biotec) were added to each sample and incubated for 15min at 4-8°C. Cells were washed and PE-labeled cells isolated by passing the cells over an LS magnetic column (Miltenyi Biotec). The entire positively selected fraction was collected and stained with antibodies to B220, CD4, CD8α, TCRβ and CD44. Tetramer+ cells were gated as B220-CD4-CD8α+TCRβ+PE Tet+ and APC Tet+. A fixed number of CountBright beads (Invitrogen) were added to each sample to allow the measurement of cell numbers, and the samples acquired on a BD LSR II.
RP-HPLC
COS-7 or Qa-1b-expressing COS cells were transfected with cDNA encoding either Fam49b region 13, or a minigene encoding the FL9 peptide. 15 μM leucinethiol in 500 μM DTT was added 42 h later. After 5 h of leucinethiol treatment, the cells were harvested and peptides extracted using 10% acetic acid. Extracts were filtered through a 10 kDa exclusion filter, and all the material <10 kDa was fractionated using a C18 reversed-phase HPLC column (Grace Vydac). The fractionated material was collected in 96-well plates and lyophilized. 5 × 104 Qa-1b-expressing L cells and 1 × 105 BEko8Z cells were added to each well and incubated overnight at 37°C. The presence of activating peptide was detected the following day using colorimetric conversion of CPRG. 1 pmole of the indicated synthetic peptides were also run during the same experiment and detected in the same way.
Killing assays
For in vitro cytotoxicity assays, the cells used as targets were labeled with the dye PKH26 (Sigma Aldrich), counted, and plated. Effector CTL lines were harvested and added at the indicated effector:target ratios and incubated at 37 °C for 1 h 45 min, after which the dye YoPRO-1 (Invitrogen) was added to a final concentration of 1 μM, to visualize apoptotic cells. The cells were washed, and visualized on a flow cytometer. Apoptotic targets were measured as those cells that were both PKH26+ and YoPRO-1+. The percentage of apoptotic targets were calculated as 100*(%PKH26+YoPRO-1+E:T=X—%PKH26+YoPRO-1+E:T=0)/(100—%PKH26+YoPRO-1+E:T=0).
For in vivo cytotoxicity assays, WT B6 and ERAAP-/- spleen cells were harvested and labeled with 0.25 μM and 2.5 μM of carboxyfluorescein-succinimidyl ester (CFSE, Invitrogen) respectively. 5 × 106 each of WT (CFSElow) and ERAAP-/- (CFSEhigh) cells were mixed and the relative ratio of each measured immediately prior to injection. This ratio is called the input ratio. Targets were injected intravenously into either naïve mice, or mice that had been primed 7 days prior with ERAAP-/- spleen cells intraperitoneally. Spleens from injected mice were harvested 24 h later and the ratios of CFSElo to CFSEhi cells measured by flow cytometry. The “Targets lost (%)” in a sample was calculated as 100—100*(Fraction CFSEhi/Input Fraction CFSEhi), where fraction CFSEhi was calculated as %CFSEhi/(%CFSEhi+%CFSElo). Mice that received ERAAP-MHC Ia-TKO cells as targets were depleted of NK cells with 200 μg of anti-NK1.1 (PK136) antibody 36 h prior to challenge. In experiments where the role of CD4+ and CD8+ cells were measured, the indicated cells were depleted using 200 μg of anti-CD8 (YTS169.4) or anti-CD4 (GK1.5) 24 h prior to challenge.
Statistcal Analyses
All statistical analyses were carried out using GraphPad Prism. Non-parametric Mann-Whitney U tests were used to estimate statistical significance.
Supplementary Material
Acknowledgments
We thank the NIH Tetramer Core Facility for tetramer reagents; K. Söderstrom and E. Engleman from Stanford University for Qa-1b-deficient mice generated by H. Cantor and his colleagues at Harvard University; D. King for peptide synthesis; H. Nolla for assistance with cell sorting; K. C. Lind and A. H. Bakker for discussions and comments on the manuscript; and H. Dani for technical assistance. N.A.N. was supported in part by the Irvington Institute Fellowship program of the Cancer Research Institute. This research was supported by grants from the NIH to N.S.
Footnotes
Author Contributions
N.A.N and N.S. designed the study, N.A.N and F.G. carried out the experiments, and N.A.N and N.S. wrote the manuscript.
The authors declare that they have no competing financial interests.
References
- 1.Shastri N, Schwab S, Serwold T. Producing nature’s gene-chips: the generation of peptides for display by MHC class I molecules. Annu Rev Immunol. 2002;20:463–493. doi: 10.1146/annurev.immunol.20.100301.064819. [DOI] [PubMed] [Google Scholar]
- 2.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 3.Peaper DR, Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu Rev Cell Dev Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 4.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 5.Rodgers JR, Cook RG. MHC class Ib molecules bridge innate and acquired immunity. Nat Rev Immunol. 2005;5:459–471. doi: 10.1038/nri1635. [DOI] [PubMed] [Google Scholar]
- 6.Strong RK, et al. HLA-E allelic variants. Correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J Biol Chem. 2003;278:5082–5090. doi: 10.1074/jbc.M208268200. [DOI] [PubMed] [Google Scholar]
- 7.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 8.Tortorella D, Gewurz BE, Furman MH, Schust DJ, Ploegh HL. Viral subversion of the immune system. Annu Rev Immunol. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- 9.Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4-8+ T cells. Cell. 1992;71:1205–1214. doi: 10.1016/s0092-8674(05)80068-6. [DOI] [PubMed] [Google Scholar]
- 10.Fruci D, et al. Altered expression of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in transformed non-lymphoid human tissues. J Cell Physiol. 2008;216:742–749. doi: 10.1002/jcp.21454. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, et al. Human cytomegalovirus microRNA miR-US4-1 inhibits CD8(+) T cell responses by targeting the aminopeptidase ERAP1. Nat Immunol. 2011 doi: 10.1038/ni.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans DM, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43:761–767. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strange A, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. The aminopeptidase ERAAP shapes the peptide repertoire displayed by major histocompatibility complex class I molecules. Nat Immunol. 2006;7:103–112. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- 15.Yan J, et al. In vivo role of ER-associated peptidase activity in tailoring peptides for presentation by MHC class Ia and class Ib molecules. J Exp Med. 2006;203:647–659. doi: 10.1084/jem.20052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci U S A. 2006;103:9202–9207. doi: 10.1073/pnas.0603095103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammer GE, Gonzalez F, James E, Nolla H, Shastri N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat Immunol. 2007;8:101–108. doi: 10.1038/ni1409. [DOI] [PubMed] [Google Scholar]
- 18.Vugmeyster Y, et al. Major histocompatibility complex (MHC) class I KbDb -/- deficient mice possess functional CD8+ T cells and natural killer cells. Proc Natl Acad Sci U S A. 1998;95:12492–12497. doi: 10.1073/pnas.95.21.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science. 1990;248:1227–1230. doi: 10.1126/science.2112266. [DOI] [PubMed] [Google Scholar]
- 20.Zijlstra M, et al. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
- 21.Sanderson S, Shastri N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
- 22.Hu D, et al. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat Immunol. 2004;5:516–523. doi: 10.1038/ni1063. [DOI] [PubMed] [Google Scholar]
- 23.Serwold T, Gaw S, Shastri N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol. 2001;2:644–651. doi: 10.1038/89800. [DOI] [PubMed] [Google Scholar]
- 24.Lu L, Werneck MB, Cantor H. The immunoregulatory effects of Qa-1. Immunol Rev. 2006;212:51–59. doi: 10.1111/j.0105-2896.2006.00418.x. [DOI] [PubMed] [Google Scholar]
- 25.Aldrich CJ, et al. Identification of a TAP-dependent leader peptide recognized by alloreactive T cells specific for a class 1b antigen. Cell. 1994;79:649–658. doi: 10.1016/0092-8674(94)90550-9. [DOI] [PubMed] [Google Scholar]
- 26.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proc Natl Acad Sci U S A. 1992;89:6020–6024. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mach N, et al. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 2000;60:3239–3246. [PubMed] [Google Scholar]
- 28.Falk K, Rötzschke O, Rammensee HG. Cellular peptide composition governed by major histocompatibility complex class I molecules. Nature. 1990;348:248–251. doi: 10.1038/348248a0. [DOI] [PubMed] [Google Scholar]
- 29.Malarkannan S, Goth S, Buchholz DR, Shastri N. The role of MHC class I molecules in the generation of endogenous peptide/MHC complexes. J Immunol. 1995;154:585–598. [PubMed] [Google Scholar]
- 30.Moon JJ, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28:859–869. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moon JJ, et al. Tracking epitope-specific T cells. Nat Protoc. 2009;4:565–581. doi: 10.1038/nprot.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haluszczak C, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. 2009;206:435–448. doi: 10.1084/jem.20081829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerksiek KM, Busch DH, Pilip IM, Allen SE, Pamer EG. H2-M3-restricted T cells in bacterial infection: rapid primary but diminished memory responses. J Exp Med. 1999;190:195–204. doi: 10.1084/jem.190.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cho H, Choi HJ, Xu H, Felio K, Wang CR. Nonconventional CD8+ T cell responses to Listeria infection in mice lacking MHC class Ia and H2-M3. J Immunol. 2011;186:489–498. doi: 10.4049/jimmunol.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouwer HG, Barry RA, Hinrichs DJ. Lack of expansion of major histocompatibility complex class Ib-restricted effector cells following recovery from secondary infection with the intracellular pathogen Listeria monocytogenes. Infect Immun. 2001;69:2286–2292. doi: 10.1128/IAI.69.4.2286-2292.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cifaldi L, et al. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res. 2011;71:1597–1606. doi: 10.1158/0008-5472.CAN-10-3326. [DOI] [PubMed] [Google Scholar]
- 38.Blanchard N, et al. Endoplasmic reticulum aminopeptidase associated with antigen processing defines the composition and structure of MHC class I peptide repertoire in normal and virus-infected cells. J Immunol. 2010;184:3033–3042. doi: 10.4049/jimmunol.0903712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petroziello J, et al. Suppression subtractive hybridization and expression profiling identifies a unique set of genes overexpressed in non-small-cell lung cancer. Oncogene. 2004;23:7734–7745. doi: 10.1038/sj.onc.1207921. [DOI] [PubMed] [Google Scholar]
- 40.Gilli F, et al. Learning from nature: pregnancy changes the expression of inflammation-related genes in patients with multiple sclerosis. PLoS One. 2010;5:e8962. doi: 10.1371/journal.pone.0008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamagata T, Benoist C, Mathis D. A shared gene-expression signature in innate-like lymphocytes. Immunol Rev. 2006;210:52–66. doi: 10.1111/j.0105-2896.2006.00371.x. [DOI] [PubMed] [Google Scholar]
- 42.Urdahl KB, Sun JC, Bevan MJ. Positive selection of MHC class Ib-restricted CD8(+) T cells on hematopoietic cells. Nat Immunol. 2002;3:772–779. doi: 10.1038/ni814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho H, Bediako Y, Xu H, Choi HJ, Wang CR. Positive selecting cell type determines the phenotype of MHC class Ib-restricted CD8+ T cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13241–13246. doi: 10.1073/pnas.1105118108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oliveira CC, et al. The nonpolymorphic MHC Qa-1b mediates CD8+ T cell surveillance of antigen-processing defects. J Exp Med. 2010;207:207–221. doi: 10.1084/jem.20091429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanderson S, Shastri N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int Immunol. 1994;6:369–376. doi: 10.1093/intimm/6.3.369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.