

Abstract
It is now clear that there are a number of different forms or aspects of learning and memory that involve different brain systems. Broadly, memory phenomena have been categorized as explicit or implicit. Thus, explicit memories for experience involve the hippocampus–medial temporal lobe system and implicit basic associative learning and memory involves the cerebellum, amygdala, and other systems. Under normal conditions, however, many of these brain–memory systems are engaged to some degree in learning situations. But each of these brain systems is learning something different about the situation. The cerebellum is necessary for classical conditioning of discrete behavioral responses (eyeblink, limb flexion) under all conditions; however, in the “trace” procedure where a period of no stimuli intervenes between the conditioned stimulus and the unconditioned stimulus the hippocampus plays a critical role. Trace conditioning appears to provide a simple model of explicit memory where analysis of brain substrates is feasible. Analysis of the role of the cerebellum in basic delay conditioning (stimuli overlap) indicates that the memories are formed and stored in the cerebellum. The phenomenon of cerebellar long-term depression is considered as a putative mechanism of memory storage.
Current views recognize a number of different forms or aspects of learning and memory involving different neural systems in the brain (see Fig. 1, adapted from ref. 1). The papers in this Colloquium consider most of these memory systems. On the other hand it is likely that under normal conditions many or all of these brain–memory systems are engaged to some degree in most learning situations.
Figure 1.
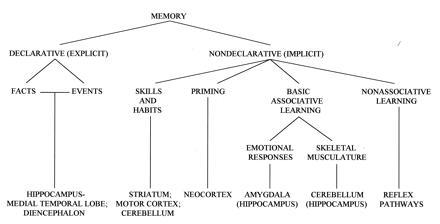
A tentative taxonomy of long-term memory and associated brain structures (adapted from ref. 1).
In this paper we will focus on basic associative learning and memory, “implicit memory,” using classical (Pavlovian) conditioning of discrete behavioral responses (e.g., eyeblink, limb flexion, etc.) as a model system. To the extent studied, the basic properties of learning and memory and the brain substrates for this form of learning are the same in all mammals, including humans. Even in this simple and basic paradigm, several brain–memory systems are engaged. Thus, the cerebellum is essential for this form of learning and memory under all conditions, as detailed below. However, the hippocampal system is also massively engaged in this simple form of learning and is necessary for learning and memory under some conditions (see below). Further, if the unconditioned or training stimulus is sufficiently aversive, learned fear will develop associated with the conditioned stimulus (CS) and involving the amygdalar and hippocampal systems. But we think that each of these brain systems is learning something rather different about the situation. The cerebellar system is learning to make specific behavioral responses that are most adaptive in dealing with the aversive event. The amygdalar system is learning fear and associated autonomic responses to deal with the situation (altered heart rate, blood pressure, etc.). The hippocampal system, we think, is learning what the situation is, “explicit memory,” forming declarative—i.e., experiential or episodic memories about the events and their relationships in the context of the organism’s ongoing experience.
At a more general level, all aspects of learning share a common thrust. As Rescorla (2) has stressed, basic associative learning is the way organisms, including humans, learn about causal relationships in the world. It results from exposure to relations among events in the world. For both modern Pavlovian and cognitive views of learning and memory, the individual learns a representation of the causal structure of the world and adjusts this representation through experience to bring it in tune with the real causal structure of the world, striving to reduce any discrepancies or errors between its internal representation and external reality (see also ref. 3).
Hippocampus and “Declarative” Memory
Interest in the critical role of the hippocampus in memory dates from the classic studies of patient HM (e.g., ref. 4). In 1978 Mishkin (5) published the first primate lesion study that appeared to mimic HM’s syndrome, using delayed nonmatching to sample. In the intervening years, a large number of studies on humans, monkeys, rabbits, rats, and mice have focused on animal models of human amnesia and on the presumed role(s) of the hippocampus and related structures in memory. The memory deficit following hippocampal lesions is not global but rather much more specific for one kind of memory, termed “declarative” (or explicit or relational) (6). Declarative memory is sometimes associated with consciousness or awareness, in contrast to many other forms of memory, including implicit (priming) memory in humans and a range of associative memory phenomena in humans and other mammals: motor and perceptual skills, classical conditioning, operant conditioning, habit formation, etc. (Fig. 1).
The lesions in Mishkin’s original study (5) were designed to reproduce HM’s lesions and included the hippocampus, amygdala, and adjacent cortical regions bilaterally. It now seems relatively clear that the amygdala, per se, is not critical, at least for declarative memory, but the hippocampus and related cortical structures are—e.g., perirhinal, parahippocampal, and entorhinal cortex (7). Lesions including all these structure produce the most profound amnesia and lesions including subsets produce substantial but less profound amnesia. Currently there is some question about the role of the hippocampus proper in this medial temporal lobe memory system (8).
A number of tasks in infraprimate mammals are sensitive to hippocampal damage—e.g., water maze (9), odor discriminations (10, 11), event timing (12), cue relationships (13), spatial memory (14), spatial alternation (15), radial arm maze (16), conditional learning (17), discrimination reversal (18), trace classical conditioning (19, 20), and contextual conditioning (21, 22). Although it is difficult to generalize, common threads in many of these tasks include relational memories—i.e., memories for relations among stimuli and events, and memories that utilize spatial–contextual information, both of which would seem reasonable analogs of primate declarative memory in lower mammals (see also ref. 23). For neurobiological analysis one would ideally wish to utilize preparations where much of the essential neuronal circuitry generating the behavior is known. Only a few of the animal models meet this modest requirement to some degree.
Hippocampus and Classical Conditioning
In eyeblink conditioning, neuronal unit cluster recordings in hippocampal fields CA1 and CA3 increase in discharge frequency in paired [tone CS–corneal airpuff unconditioned stimulus (US)] training trials very rapidly, shift forward in time as learning develops, and form a predictive “temporal model” of the learned behavioral response, both within trials and over the trials of training (24, 25). To summarize a large body of research, the growth of the hippocampal unit response is, under normal conditions, an invariable and strongly predictive concomitant of subsequent behavioral learning (see reviews in refs. 26, 27, 28). This increase in neuronal activity in the hippocampus becomes significant by the second or third trial of training, long before behavioral signs of learning develop, as would be expected of a declarative memory system. This initial hippocampal unit increase is in the US period; increases in the CS period appear at about the time point in training when behavioral conditioned responses (CRs) appear.
Many neurons that could be identified as pyramidal neurons in CA1 and CA3 (antidromic stimulation and collision) showed learning-related increases in discharge frequency in the trial period (many unidentified neurons showed decreases in the trial period) (26, 29). Typically, a given neuron modeled only some limited time period of the trial. Cumulating many such single pyramidal neuron responses produced the typical unit cluster model of the behavioral learned response. So the pyramidal neuron representation of the behavioral learned response is distributed over both space and time in the hippocampus. The high percentage of learning-influenced pyramidal neurons and their spatially distributed loci have been strikingly verified in studies by Disterhoft and associates (30, 31) using in vitro studies of hippocampal slices from trained versus control animals.
The work described above was all done using the basic delay paradigm, where hippocampal lesions do not impair simple acquisition (32). Similarly, humans with hippocampal–temporal lobe anterograde amnesia are able to learn simple acquisition of the eyeblink CR, but cannot describe it (33). However, hippocampal lesions severely impair discrimination reversal in the delay paradigm (rabbit eyeblink) (18). Studies by Daum, Grey, and associates (34, 35) on humans with brain damage show that hippocampal–medial temporal lobe lesion subjects are massively impaired on conditional discriminations in eyeblink conditioning (compared with frontal lesion or normal controls) but not on acquisition or simple discriminations (ruling out deficits in response inhibition). In a conditional task the subject must learn to blink to a CS (e.g., tone) only if it is preceded by another stimulus (e.g., a light). Ross et al. (17) had shown similar hippocampal lesion deficits in rats (locomotor response) in a conditional discrimination paradigm. Daum et al. (34, 35) make a strong case that the deficits seen in eyeblink conditional discriminations in humans and discrimination reversal in rabbits reflect deficits in declarative memory.
Trace conditioning was first described by Pavlov; the CS terminates and there is a period of no stimulation between CS offset and US onset (as Pavlov stressed, the organism must maintain a “trace” of the CS in the brain in order for the CS and the US to become associated). In eyeblink conditioning in animals, a typical trace interval is 500 ms. The trace CR is more difficult to learn than the standard “delay” procedure where the CS and US overlap in time. Bilateral removal of the dorsal plus some ventral hippocampus in rabbits markedly impaired subsequent acquisition of the 500-ms trace CR (19, 20). In recent work we explored the time-dependency of the role of the hippocampus in trace conditioning (36). In brief, animals were trained in the standard trace paradigm (250-ms tone CS, 500-ms trace interval of no stimuli, followed by a 100-ms corneal airpuff US) and then subjected to very large bilateral hippocampal lesions either 1 day or 1 month after reaching learning criterion. Results are shown in Fig. 2. The trace CR is essentially abolished in animals lesioned 1 day after learning but is unaffected by lesions 1 month after learning. Controls include both sham operates and cortical lesions; they do not differ from each other at 1 day or 1 month or from the 1 month hippocampal lesion animals. While hippocampectomy abolished immediate retention of trace CRs, the same lesions had no effect on immediate retention of delay CRs (Fig. 2). When these postlesion delay CR retention animals were then shifted immediately to the trace procedure, the short latency delay CR actually extinguished (Fig. 2).
Figure 2.
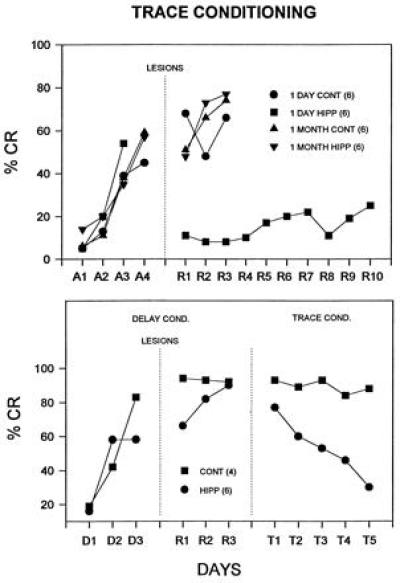
Effects of hippocampal lesions on retention of trace and delay CRs. Shown are the mean percentage of CRs during initial training and following postoperative training. (Upper) Trace procedure: 1 day cont, controls given cortical or sham lesions 1 day after training; 1 month cont, controls given lesions 1 month after training; 1 day hipp, bilateral hippocampal lesions made 1 day after training; 1 month hipp, hippocampal lesions made 1 month after training. Only the hippocampal lesions made immediately after training abolished the trace CR. (Lower) Animals initially trained on the delay procedure and lesioned 1 day after training (control lesions and hippocampal lesions). The hippocampal lesion had little effect on immediate retention of the delay CR. Animals were then shifted to the trace procedure. Control animals transferred, continuing to show asymptotic CRs, but the hippocampal lesioned animals actually showed extinction of the CR (from ref. 36; reprinted with permission from the American Psychological Association, Washington, DC).
To summarize, large bilateral lesions of the hippocampus made before training markedly impair learning of the trace CR. If the animals are first trained, lesions immediately after training abolish the trace CR but lesions made 1 month after training have no effect on memory of the trace CR. These results are strikingly consistent with the literature concerned with the declarative memory deficit following damage to the hippocampal system in humans and monkeys. These deficits have two key temporal characteristics: (i) profound and permanent anterograde amnesia, and (ii) profound but clearly time-limited retrograde amnesia. Subjects have great difficulty learning new declarative tasks/information and have substantial memory loss for events for some period just preceding brain damage (1 or more years in humans, 2–3 months for monkeys) but relatively intact memory for earlier events (37). Very similar results were found for classical conditioning of fear to context in rats (21). Analogous results have been found in McGaugh’s laboratory for instrumental avoidance learning and amygdalar lesions (38).
In terms of mechanisms of memory storage in the hippocampal system, the process of long-term potentiation (LTP) is widely favored (39, 40). Brief high frequency or appropriately patterned stimulation of axons induces long lasting monosynaptic increases in synaptic transmission in all three major subfields of the hippocampus—e.g., dentate gyrus, CA3, and CA1 (41). Considerable indirect evidence supports the view that a process like LTP may underlie processes of memory storage in the hippocampus (42). In the case of classical conditioning, there are a number of parallels between properties of LTP and the properties of the learning-induced increase in neuronal activity in the hippocampus (27). Both LTP and the learning-induced increase in hippocampal neuron activity are expressed by pyramidal neurons, both begin to develop after very brief periods (e.g., 100 Hz for 1 sec for LTP; 1–3 trials of training in eyeblink conditioning); both develop to asymptote over a period of many minutes; both show the same magnitude of increase; and both require very specific parameters of stimulation to develop. Further, there is a persisting increase in the monosynaptic population spike in the perforant path to dentate gyrus stimulation as a result of eyeblink conditioning (43) and following LTP induced by tetanus of the perforant path.
There are strikingly parallel and persisting increases in glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor binding on hippocampal membranes in the hippocampal subfields in both eyeblink conditioning (well-trained animals) and in in vivo expression of LTP by stimulation of the perforant path projection to hippocampal dentate gyrus. The pattern of increased binding is similar in both paradigms (39, 44, 45). Glutamate N-methyl-d-aspartate receptors play the critical role in induction of LTP (at least in dentate and CA1) (39) and also appear to be involved in acquisition of the trace eyeblink CR (46).
The most common current view of the memorial functions of the hippocampal–medial temporal lobe system is that declarative memories are stored there for some period of time (perhaps due to processes of synaptic plasticity like LTP) and then eventually transferred or consolidated to other brain regions for permanent storage, the cerebral cortex being the most commonly suggested site.
There are a number of key unsolved issues concerning the role of the hippocampal system in declarative memory. Are time-limited declarative memories actually stored in the hippocampal system? It seems very likely that they are, but conclusive evidence is lacking. What is the readout system from the hippocampal system to behavioral expression of learning in declarative memory? This key issue has received surprisingly little attention. Where are the long-term declarative memories stored after the hippocampal system is no longer necessary? Are they stored in the neocortex? What are the mechanisms of time-limited memory storage in hippocampus and storage of permanent memories in extra-hippocampal structures? Trace classical conditioning of discrete behavioral responses would seem a most valuable model system in which to explore these issues. As will be shown below, most is known about the brain circuitry essential for this basic form of associative learning and memory.
Cerebellar Substrate of Classical Conditioning of Discrete Responses
The literature concerned with the brain circuitry essential for delay classical conditioning of discrete behavioral responses has been reviewed in several recent publications and will be summarized only briefly here (see, for example, refs. 47, 48, 49, 50). Most of the work has used the conditioned eyeblink response as the model system for analysis. The highly simplified schematic block diagram of Fig. 3 can serve to summarize overall results to date and is a much simplified version of our current qualitative working model of the role of the cerebellum in basic delay classical conditioning of discrete responses. [Laterality is not shown; the critical region of the cerebellum is ipsilateral to the trained eye (or limb); the critical regions of the pontine nuclei, red nucleus, and inferior olive are contralateral.]
Figure 3.
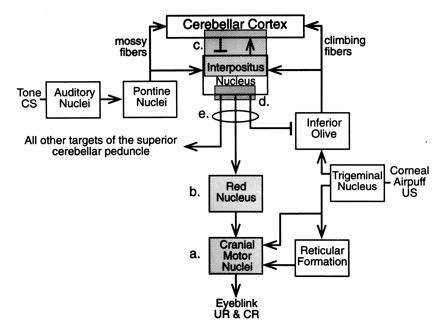
Simplified schematic of the essential brain circuitry involved in classical conditioning of discrete responses—e.g., eyeblink response. Shadowed boxes represent areas that have been reversibly inactivated during training. (a) Inactivation of motor nuclei including facial (7th) and accessory 6th. (b) Inactivation of magnocellular red nucleus. (c) Inactivation of dorsal aspect of the anterior interpositus nucleus and overlying cerebellar cortex. (d) Inactivation of ventral anterior interpositus nucleus and associated white matter. (e) Complete inactivation of the superior cerebellar peduncle (scp), essentially all output from the cerebellar hemisphere. See text for details. (Reprinted with permission from ref. 47, Annual Reviews, Inc., Palo Alto, CA.)
In brief, the reflex eyeblink response pathways activated by corneal airpuff (or periorbital shock) include the trigeminal nucleus, direct projections to the relevant motor nuclei (mostly the seventh and accessory sixth), and indirect projections to the motor nuclei via the brainstem reticular formation. Analysis of response latencies rules out any direct role of the cerebellum in the reflex response. The tone (and light) CS pathways project to the cerebellum as mossy fibers, mostly relaying through the pontine nuclei. The US pathway projects from the trigeminal nucleus to the inferior olive and from there to the cerebellum as climbing fibers. The CS-activated mossy fiber–parallel fiber pathway and the US activated climbing fiber pathway converge on Purkinje neurons in cerebellar cortex (parallel fiber–climbing fiber) and on neurons in the interpositus nucleus (mossy fiber–climbing fiber). The CR pathway projects from the interpositus nucleus of the cerebellum via the superior cerebellar peduncle to the red nucleus and from there to the motor nuclei (seventh and accessory sixth) controlling the eyeblink response.
This circuitry has been identified using lesions, electrophysiological, recordings, electrical microstimulation, anatomical characterization of projection pathways, etc. For example, neurons in the cerebellar cortex and interpositus nuclei respond to the CS and US before training and develop amplitude–time course models of the learned behavioral response that precede and predict the occurrence and form of the CR within trials and over the trials of training in animals (51) and by inference from positron-emission tomography analysis in humans (52). Appropriate lesions of the anterior interpositus nucleus completely and permanently abolish the CR with no effect on the unconditioned response (UR) in animals (53, 54); appropriate cerebellar lesions in humans similarly completely prevent learning of the CR with no effect on the UR (55). Appropriate lesions of the pontine nuclei can selectively abolish the CR to one modality of CS (56) and stimulation of the pontine nuclei serves as a supernormal CS yielding faster learning than peripheral CSs (57). Finally, lesions of the appropriate region of the inferior olive completely prevent learning if made before training and result in extinction of the CR if made after training [refs. 58 and 59 (limb flexion)]. Electrical microstimulation of this appropriate region of the inferior olive elicits discrete movements and the exact movements so elicited can be trained to occur to any neutral stimulus (60, 61). The inferior olive-climbing fiber system, incidentally, is the only system in the brain other than the reflex afferents where this can occur.
These results constituted an extraordinary verification of the much earlier theories of the cerebellum as a neuronal learning system developed initially in the classic papers of Marr (62) and Albus (63) and elaborated by Eccles (64) and Ito (65, 66) [see also Thach et al. (67)]. These theories proposed that mossy–parallel fibers convey information about stimuli and movement contexts (CSs here) and the climbing fibers convey information about specific movement errors and aversive events (USs here) and they converge on Purkinje neurons in cerebellar cortex to alter the synaptic efficacy of the parallel fiber synapses on their dendrites.
The Locus of the Long-Term Memory Trace
Overall, the results described to this point would seem to demonstrate conclusively that the cerebellum is necessary for learning, retention, and expression of classical conditioning of the eyeblink and other discrete responses. The next and more critical issue concerns the locus of the memory traces. Evidence summarized below would seem to demonstrate conclusively that the long-term memory traces for this type of learning are formed and stored in the cerebellum.
We and our associates have developed a new approach to the problem of localizing memory traces in the brain, namely the use of methods of reversible inactivation, together with recording of neuronal activity. Reversible inactivation methods (e.g., using drugs or cooling), per se, have existed for some time and have been used very effectively to produce temporary lesions (68). What we have done is to apply this method systematically to the major structures and pathways in the cerebellar–brain stem circuit we have identified as the essential (necessary and sufficient) circuit for classical conditioning of discrete responses (Fig. 3), during performance and during acquisition of the CR (see ref. 49 for a detailed discussion).
As noted above, the diagram of Fig. 3 shows in highly simplified schematic form the essential memory trace circuit for classical conditioning of discrete responses. Interneuron circuits are not shown, only net excitatory or inhibitory actions of projection pathways. Other pathways, known and unknown, may also of course be involved. Many uncertainties still exist—e.g., concerning details of sensory-specific patterns of projection to pontine nuclei and cerebellum (CS pathways), details of red nucleus projections to premotor and motor nuclei (CR pathway), and the possible roles of recurrent circuits.
Several parts of the circuit have been reversibly inactivated for the duration of training (eyeblink conditioning) in naive animals indicated by shadings labeled a, b, c, d, and encircled e in Fig. 3. The motor nuclei essential for generating the UR and CR (primarily 7th and accessory 6th and adjacent neural tissues) were inactivated by infusion of muscimol (6 days) or cooling (5 days) during standard tone-airpuff training (Fig. 3a) (69, 70). The animals showed no CRs and no URs during this inactivation training; indeed they showed no behavior at all—performance was completely abolished. However, the animals exhibited asymptotic CR performance and normal UR performance from the very beginning of post-inactivation training. Thus, performance of the CR and UR are completely unnecessary for normal learning, and the motor nuclei and adjacent inactivated tissue make no contribution at all to formation of the memory trace—they are completely efferent from the trace.
Inactivation of the magnocellular red nucleus is indicated in Fig. 3b. Inactivation by low doses of muscimol for 6 days of training or cooling for 5 days completely prevented the expression of the CR. Yet animals showed asymptotic learned performance of the CR from the beginning of postinactivation training (71, 72).
Inactivation of the dorsal anterior interpositus and overlying cortex (Fig. 3c) by low doses of muscimol (6 days), by lidocaine (3 days, 6 days), and by cooling (5 days) resulted in no expression of CRs during inactivation training and no evidence of any learning at all having occurred during inactivation training (71, 73, 74). In subsequent postinactivation training, animals learned normally as though completely naive; they showed no savings at all relative to noninactivated control animals. None of the methods of inactivation had any effect at all on performance of the UR on US alone trials. The loci in which [3H]muscimol was completely effective in preventing learning included the anterior dorsal interpositus and overlying cortex of lobule HVI, a volume ≈2% of the total volume of the cerebellum (71). The region of the cerebellum essential for learning this task is extremely localized.
Finally, essentially all of the output from the interpositus nucleus projecting to other regions of the brain, namely, the superior cerebellar peduncle (Fig. 3c), was inactivated using tetrodotoxin. Inactivation during training had no effect on performance of the UR and completely prevented performance of the CR. But when the inactivation was removed, the CR had been learned to asymptote (75).
Collectively, these data strongly support the hypothesis that the memory trace is formed and stored in a localized region of the cerebellum (anterior interpositus and overlying cortex). Indeed we can conceive of no rational alternative. Inactivation of this region (Fig. 3c) during training completely prevents learning but inactivation of the output pathway from the region (Fig. 3e) and its necessary (for the CR) efferent target, the red nucleus (Fig. 3b), do not prevent learning at all. In no case do the drug inactivations have any effect at all on performance of the reflex response on US alone trials. If even a part of the essential memory trace were formed prior to the cerebellum in the essential circuit, then following cerebellar inactivation training the animals would have to show savings and they show none at all. Similarly, if a part of the essential memory trace were formed in the red nucleus or other efferent targets of the interpositus (e.g., brainstem), then following red nucleus or superior cerebellar peduncle inactivation training, animals could not show asymptotic CR performance but they do.
Putative Mechanisms of Memory Storage in the Cerebellum
Classic theories of the cerebellum as a learning machine (see above) proposed that conjoint activation of Purkinje neurons by parallel fibers and climbing fibers would lead to alterations in synaptic efficacy of the parallel fiber synapses. Ito (76) discovered that such conjoint activation led to a long-lasting depression of parallel fiber synaptic efficacy on Purkinje neuron dendrites, the process of cerebellar long-term depression (LTD). He and his associates developed considerable evidence that such a process plays a key role in adaptation of the vestibulo-ocular reflex (66, 77, 78).
In eyeblink conditioning, many of the Purkinje neurons that exhibit learning-related changes show decreases in simple spike responses in the CS period (79), consistent with a mechanism of LTD (see also the discussion in ref. 80). Current evidence suggests that glutamate activation of AMPA and metabotropic receptors on Purkinje neuron dendrites together with increased intracellular calcium (normally by climbing fiber activation) yields the persisting decrease in AMPA receptor function at parallel fiber synapses on Purkinje neuron dendrites that produces LTD (see also ref. 77, 78, 81, 82).
The Cerebellar Cortex and LTD
Experimentally it has proved extremely difficult to determine the relative roles of the cerebellar cortex and interpositus nucleus in eyeblink conditioning using the lesion method. There is general argument that very large cortical lesions impair learning and memory of the eyeblink CR, but it is difficult to rule out damage to the interpositus nucleus; it lies immediately underneath the critical cortical tissue (50, 83, 84). A recent study made use of the mutant Purkinje cell degeneration (pcd) mouse strain (85). In this mutant, Purkinje neurons (and all other neurons studied) are normal throughout pre and perinatal development. At about 2–4 weeks postnatal, the Purkinje neurons in the cerebellar cortex degenerate and disappear (86). For a period of about two months after this time, other neuronal structures appear relatively normal (87). Thus, during this period of young adulthood, the animals have a complete functional decortication of the cerebellum.
Appropriate lesions of the interpositus nucleus in the wild-type control mice (normal cerebellum) completely prevented learning of the conditioned eyeblink response, as with all other mammals studied. So the cerebellum is completely necessary for learning in this species as well. The pcd mice learned very slowly, very poorly, and to a much lower level than wild-type controls, but showed extinction with subsequent training to the CS alone. Thus the cerebellar cortex plays a critically important role in normal learning (of discrete behavioral responses) but some degree of learning is possible without the cerebellar cortex.
Recent studies using “gene knockout” preparations have strengthened the argument for LTD as a key mechanism of memory storage in cerebellar cortex in classical conditioning. Thus, mice that lack the metabotropic glutamate receptor (mGluR1) show marked impairments in cerebellar cortical LTD and eyeblink conditioning (88). They also show generalized motor impairments—i.e., some degree of ataxia, as do the pcd mice (see above).
Interestingly, current studies present evidence supporting the view that LTD is more important for learning (e.g., eyeblink conditioning) than for motor coordination. Thus, using the protein kinase C γ (PKCγ) knockout mutant mouse, Kano et al. (89) showed that the Purkinje neurons in adult animals maintained the perinatal condition of more than one climbing fiber per neuron (wild-type adults have only one climbing fiber per Purkinje neuron). Chen et al. (90) showed that this mutant exhibited normal LTD but impaired motor coordination (due, presumably, to the multiple climbing fiber innervation of Purkinje neurons). In striking contrast, these animals learned the conditioned eyeblink response more rapidly than did the wild-type controls. This result is beautifully consistent with the evidence discussed earlier supporting the view that the climbing fiber system is the reinforcing or teaching pathway (see ref. 91).
Just the opposite result holds for a quite different mutant, namely the GFAP (glial fibrillary acidic protein) knockout mouse (92). Here, the cerebellar cortex appears to be anatomically normal. However, the animals are markedly deficient in cerebellar cortical LTD and in eyeblink conditioning (their performance is very similar to that of the pcd mice). In striking contrast, these animals do not show any impairments at all in motor coordination or general motor behavior!
The Shibuki et al. (92) study is important in another regard as well. GFAP is not present in neurons, only in glial cells. In the cerebellum it is normally present in substantial amounts in the Bergmann glia that surround the parallel fiber and climbing fiber–Purkinje neuron dendrite synapses. Although the Bergman glia appear morphologically normal in the GFAP knockout, they have no GFAP. The key point here is that an abnormality limited to glial cells markedly impairs a form of synaptic plasticity (LTD) and a form of basic associative learning and memory. To our knowledge, this may be the first direct evidence for a key role of glia in processes of learning and memory.
Thus, several lines of evidence support the hypothesis that a process of LTD in cerebellar cortex is a mechanism involved in memory storage in classical conditioning of discrete behavioral responses. Similarly, several lines of evidence support such a role for cerebellar cortical LTD in adaptation of the vestibulo-ocular reflex (see refs. 66 and 77 for detailed discussions). However, the fact that some degree of eyeblink learning occurs in the pcd mouse (see above), a preparation that functionally has a complete cerebellar decortication, argues that some degree of plasticity must occur in the interpositus nucleus. Similarly, some plasticity may also occur in a vestibular nucleus in adaptation of the vestibulo-ocular reflex (93). There is just one paper in the literature (94) reporting that tetanus of the white matter yields LTP in the interpositus nucleus.
Much work remains to be done exploring possible mechanisms of plasticity and memory storage in the cerebellum. The fact that γ-aminobutyric acid (GABA) agonists and antagonists infused in the cerebellum have such profound effects on the CR (71, 95) at least raises the possibility that GABAergic processes may be involved in cerebellar memory storage. It appears that GABAergic processes in cerebellar cortex may play a key role in CR timing (80). Insulin-like growth factor I released from the climbing fibers onto Purkinje neurons modulates glutamate induced GABA release by Purkinje neurons (96) and also plays a critical role in learning but not performance of the conditioned eyeblink response (97).
We have focused on the essential role of the cerebellum in classical conditioning of discrete behavioral responses, a basic form of associative learning and memory. This is perhaps the clearest and most decisive evidence for the localization of a memory trace to a particular brain region in mammals (cerebellum) that exists at present. A closely related and increasingly definitive literature supports the view that the cerebellum learns and stores complex, multijoint movements (67).
Actually, there is a growing evidence that the cerebellum may be critically involved in many other forms of learning and memory, including cardiovascular conditioning (98), discrete response instrumental avoidance learning (99), maze learning (100), spatial learning and memory (101, 102), adaptive timing (103). There is even a growing literature implicating the cerebellum in complex cognitive processes (104).
The message here is not that all learning occurs in the cerebellum; it does not. Certain structures, cerebellum, hippocampal system, and amygdala, play key roles in processes of learning and memory. But a “structure” like the hippocampus is not really a structure at all; evolution has simply resulted in it appearing so. The hippocampus is not an island unto itself; it is a set of interconnected neurons interconnected with other neurons in the brain. And so it is with all other brain “structures.”
Although molecular–genetic analysis may someday tell us the nature of the mechanisms of memory storage and perhaps even the exact loci of storage (e.g., in cerebellum or hippocampus or neocortex), such reductionistic analysis can never tell us what the memories are. Only a detailed characterization of the neural circuitries that code, store, and retrieve the memories can do this.
Acknowledgments
The research reported here was supported in part by research grants from the National Science Foundation (IBN-9215069), the National Institute of Health (AG05142), the National Institute of Mental Health (MH52194), the Office of Naval Research (N00014-95-1-1152), and Sanyo Co., Ltd.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CS, conditioned stimulus; US, unconditioned stimulus; CR, conditioned response; UR, unconditioned response; LTP, long-term potentiation; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; LTD, long-term depression.
References
- 1.Squire L R, Knowlton B J. In: The Cognitive Neurosciences. Gazzaniga M, editor. Cambridge, MA: MIT Press; 1994. pp. 825–837. [Google Scholar]
- 2.Rescorla R A. Annu Rev Neurosci. 1988;11:329–352. doi: 10.1146/annurev.ne.11.030188.001553. [DOI] [PubMed] [Google Scholar]
- 3.Dudai Y. The Neurobiology Memory: Concepts, Findings, Trends. New York: Oxford Univ. Press; 1989. [Google Scholar]
- 4.Scoville W B, Milner B. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishkin M. Nature (London) 1978;273:297–298. doi: 10.1038/273297a0. [DOI] [PubMed] [Google Scholar]
- 6.Squire L R. Psychol Rev. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- 7.Squire L R, Zola-Morgan S. Science. 1991;253:1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- 8.Meunier, M., Hadfield, W., Bachevalier, J. & Murray, E. A. (1996) J. Neurophysiol., in press. [DOI] [PubMed]
- 9.Morris R G M, Anderson E, Lynch G S, Baudry M. Nature (London) 1986;319:774–775. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 10.Eichenbaum H, Fagan A, Cohen N J. J Neurosci. 1986;6:1876–1884. doi: 10.1523/JNEUROSCI.06-07-01876.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch G. Synapses, Circuits, and the Beginnings of Memory. Cambridge, MA: MIT Press; 1986. [Google Scholar]
- 12.Olton D S, Meck W H, Church R M. Brain Res. 1987;404:180–188. doi: 10.1016/0006-8993(87)91369-2. [DOI] [PubMed] [Google Scholar]
- 13.Sutherland R W, McDonald R J, Hill C R, Rudy J W. Behav Brain Res. 1989;52:331–356. doi: 10.1016/s0163-1047(89)90457-3. [DOI] [PubMed] [Google Scholar]
- 14.O’Keefe J, Nadel L. The Hippocampus as a Cognitive Map. London: Oxford Univ. Press; 1978. [Google Scholar]
- 15.Aggleton J P, Lindt H S, Rawlins J N P. Behav Neurosci. 1989;103:962–974. doi: 10.1037//0735-7044.103.5.962. [DOI] [PubMed] [Google Scholar]
- 16.Becker J T, Walker J A, Olton D S. Brain Res. 1980;200:307–320. doi: 10.1016/0006-8993(80)90922-1. [DOI] [PubMed] [Google Scholar]
- 17.Ross R T, Orr W B, Holland P C, Berger T W. Behav Neurosci. 1984;98:211–225. doi: 10.1037//0735-7044.98.2.211. [DOI] [PubMed] [Google Scholar]
- 18.Berger T W, Orr W B. Behav Brain Res. 1983;210:411–417. doi: 10.1016/0166-4328(83)90171-7. [DOI] [PubMed] [Google Scholar]
- 19.Moyer J R, Deyo R A, Disterhoft J F. Behav Neurosci. 1990;104:243–252. doi: 10.1037//0735-7044.104.2.243. [DOI] [PubMed] [Google Scholar]
- 20.Solomon P R, Vander Schaaf E R, Thompson R F, Weisz D J. Behav Neurosci. 1986;100:729–744. doi: 10.1037//0735-7044.100.5.729. [DOI] [PubMed] [Google Scholar]
- 21.Kim J J, Fanselow M S. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 22.Phillips R G, Ledoux J E. Behav Neurosci. 1992;106:294–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 23.Eichenbaum H, Otto T, Cohen N J. Brain Behav Sci. 1994;17:449–518. [Google Scholar]
- 24.Berger T W, Alger B E, Thompson R F. Science. 1976;192:483–485. doi: 10.1126/science.1257783. [DOI] [PubMed] [Google Scholar]
- 25.Berger T W, Thompson R F. Proc Natl Acad Sci USA. 1978;75:1572–1576. doi: 10.1073/pnas.75.3.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger T W, Rinaldi P C, Weisz D J, Thompson R F. J Neurophysiol. 1983;50:1197–1219. doi: 10.1152/jn.1983.50.5.1197. [DOI] [PubMed] [Google Scholar]
- 27.Berger T W, Berry S D, Thompson R F. In: The Hippocampus. Isaacson R L, Pribram K H, editors. Vol. 4. New York: Plenum; 1986. pp. 203–239. [Google Scholar]
- 28.Swanson L W, Teyler T J, Thompson R F. Neurosciences Research Program. Vol. 20. Boston, MA: MIT Press; 1982. [Google Scholar]
- 29.Berger T W, Thompson R F. Brain Res. 1978;145:323–346. doi: 10.1016/0006-8993(78)90866-1. [DOI] [PubMed] [Google Scholar]
- 30.Disterhoft J F, Coulter D A, Alkon D I. Proc Natl Acad Sci USA. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.deJonge M C, Black J, Deyo R A, Disterhoft J F. Exp Brain Res. 1990;80:450–462. doi: 10.1007/BF00227987. [DOI] [PubMed] [Google Scholar]
- 32.Solomon P R, Moore J W. J Comp Physiol Psychol. 1975;89:1192–1203. doi: 10.1037/h0077183. [DOI] [PubMed] [Google Scholar]
- 33.Weiskrantz L, Warrington E K. Neuropsychologia. 1979;17:187–194. doi: 10.1016/0028-3932(79)90009-5. [DOI] [PubMed] [Google Scholar]
- 34.Daum I, Channon S, Polkey C E, Gray J A. Behav Neurosci. 1991;105:396–408. doi: 10.1037//0735-7044.105.3.396. [DOI] [PubMed] [Google Scholar]
- 35.Daum I, Channon S, Gray J A. Behav Brain Res. 1992;52:159–165. doi: 10.1016/s0166-4328(05)80226-8. [DOI] [PubMed] [Google Scholar]
- 36.Kim J J, Clark R E, Thompson R F. Behav Neurosci. 1995;109:195–203. doi: 10.1037//0735-7044.109.2.195. [DOI] [PubMed] [Google Scholar]
- 37.Zola-Morgan S M, Squire L R. Science. 1990;250:288–290. doi: 10.1126/science.2218534. [DOI] [PubMed] [Google Scholar]
- 38.McGaugh J. Annu Rev Neurosci. 1989;12:255–287. doi: 10.1146/annurev.ne.12.030189.001351. [DOI] [PubMed] [Google Scholar]
- 39.Baudry M, Davis J L, editors. Long-Term Potentiation: A Debate of Current Issues. Cambridge, MA: MIT Press; 1991. [Google Scholar]
- 40.Lynch G, Baudry M. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- 41.Landfield P W, Deadwyler S A, editors. Long-Term Potentiation: A Debate of Current Issues. Cambridge, MA: MIT Press; 1988. [Google Scholar]
- 42.Baudry M, Thompson R F, Davis J L, editors. Synaptic Plasticity, Molecular, Cellular, and Functional Aspects. Cambridge, MA: MIT Press; 1993. [Google Scholar]
- 43.Weisz D J, Clark G A, Thompson R F. Behav Brain Res. 1984;12:145–154. doi: 10.1016/0166-4328(84)90037-8. [DOI] [PubMed] [Google Scholar]
- 44.Maren S, Tocco G, Standley S, Baudry M, Thompson R F. Proc Natl Acad Sci USA. 1993;90:9654–9658. doi: 10.1073/pnas.90.20.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tocco G, Maren S, Shors T, Baudry M, Thompson R F. Brain Res. 1992;573:228–234. doi: 10.1016/0006-8993(92)90767-4. [DOI] [PubMed] [Google Scholar]
- 46.Thompson L T, Deyo R A, Disterhoft J F. Nature (London) 1992;359:838–841. [Google Scholar]
- 47.Lavond D G, Kim J J, Thompson R F. Annu Rev Psychol. 1993;44:317–342. doi: 10.1146/annurev.ps.44.020193.001533. [DOI] [PubMed] [Google Scholar]
- 48.Thompson R F, Bao S, Berg M S, Chen L, Cipriano B D, Kim J J, Thompson J K, Tracy J, Krupa D J. In: The Cerebellum and Cognition. Schmahmann J, editor. San Diego: Academic; 1997. in press. [Google Scholar]
- 49.Thompson R F, Krupa D J. Annu Rev Neurosci. 1994;17:519–549. doi: 10.1146/annurev.ne.17.030194.002511. [DOI] [PubMed] [Google Scholar]
- 50.Yeo C H. Ann NY Acad Sci. 1991;627:292–304. doi: 10.1111/j.1749-6632.1991.tb25933.x. [DOI] [PubMed] [Google Scholar]
- 51.McCormick D A, Clark G A, Lavond D G, Thompson R F. Proc Natl Acad Sci USA. 1982;79:2731–2742. doi: 10.1073/pnas.79.8.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Logan C G, Grafton S T. Proc Natl Acad Sci USA. 1995;92:7500–7504. doi: 10.1073/pnas.92.16.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clark G A, McCormick D A, Lavond D G, Thompson R F. Brain Res. 1984;291:125–136. doi: 10.1016/0006-8993(84)90658-9. [DOI] [PubMed] [Google Scholar]
- 54.Yeo C H, Hardiman M J, Glickstein M. Exp Brain Res. 1985;60:87–98. doi: 10.1007/BF00237022. [DOI] [PubMed] [Google Scholar]
- 55.Daum I, Schugens M M, Ackermann H, Lutzenberger W, Dichgans J, Birbaumer N. Behav Neurosci. 1993;106:877–888. doi: 10.1037//0735-7044.107.5.748. [DOI] [PubMed] [Google Scholar]
- 56.Steinmetz J E, Logan C G, Rosen D J, Thompson J K, Lavond D G, Thompson R F. Proc Natl Acad Sci USA. 1987;84:3531–3535. doi: 10.1073/pnas.84.10.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steinmetz J E, Rosen D J, Chapman P F, Lavond D G, Thompson R F. Behav Neurosci. 1986;100:878–887. doi: 10.1037//0735-7044.100.6.878. [DOI] [PubMed] [Google Scholar]
- 58.McCormick D A, Steinmetz J E, Thompson R F. Brain Res. 1985;359:120–130. doi: 10.1016/0006-8993(85)91419-2. [DOI] [PubMed] [Google Scholar]
- 59.Voneida T, Christie D, Boganski R, Chopko B. J Neurosci. 1990;10:3583–3593. doi: 10.1523/JNEUROSCI.10-11-03583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mauk M D, Steinmetz J E, Thompson R F. Proc Natl Acad Sci USA. 1986;83:5349–5353. doi: 10.1073/pnas.83.14.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steinmetz J E, Lavond D G, Thompson R F. Synapse. 1989;3:225–232. doi: 10.1002/syn.890030308. [DOI] [PubMed] [Google Scholar]
- 62.Marr D. J Physiol (London) 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Albus J S. Math Biosci. 1971;10:25–61. [Google Scholar]
- 64.Eccles J C. Brain Res. 1977;127:327–352. doi: 10.1016/0006-8993(77)90550-9. [DOI] [PubMed] [Google Scholar]
- 65.Ito M. In: Control of Gaze by Brain Stem Neurons. Baker R, Berthoz A, editors. Elsevier: Amsterdam; 1977. pp. 391–398. [Google Scholar]
- 66.Ito M. The Cerebellum and Neural Control. New York: Appleton Century-Crofts; 1984. [Google Scholar]
- 67.Thach W T, Goodkin H G, Keating J G. Annu Rev Neurosci. 1992;15:403–442. doi: 10.1146/annurev.ne.15.030192.002155. [DOI] [PubMed] [Google Scholar]
- 68.Mink J W, Thach W T. J Neurophysiol. 1991;65:330–351. doi: 10.1152/jn.1991.65.2.330. [DOI] [PubMed] [Google Scholar]
- 69.Krupa D J, Weng J, Thompson R F. Behav Neurosci. 1996;110:219–227. doi: 10.1037//0735-7044.110.2.219. [DOI] [PubMed] [Google Scholar]
- 70.Zhang, A. A. (1992) Dissertation (Univ. of Southern California, Los Angeles).
- 71.Krupa D J, Thompson J K, Thompson R F. Science. 1993;260:989–991. doi: 10.1126/science.8493536. [DOI] [PubMed] [Google Scholar]
- 72.Clark R E, Lavond D G. Behav Neurosci. 1993;107:264–270. doi: 10.1037//0735-7044.107.2.264. [DOI] [PubMed] [Google Scholar]
- 73.Clark R E, Zhang A A, Lavond D G. Behav Neurosci. 1992;106:879–888. doi: 10.1037//0735-7044.106.6.879. [DOI] [PubMed] [Google Scholar]
- 74.Nordholm A F, Thompson J K, Dersarkissian C, Thompson R F. Behav Neurosci. 1993;107:882–886. doi: 10.1037//0735-7044.107.5.882. [DOI] [PubMed] [Google Scholar]
- 75.Krupa D J, Thompson R F. Proc Natl Acad Sci USA. 1995;92:5097–5101. doi: 10.1073/pnas.92.11.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ito M, Sakurai M, Tongroach P. J Physiol (London) 1982;324:113–134. doi: 10.1113/jphysiol.1982.sp014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ito M. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- 78.Ito M. In: Synaptic Plasticity: Molecular and Functional Aspects. Baudry M, Davis J L, Thompson R F, editors. Cambridge, MA: MIT Press; 1994. pp. 117–128. [Google Scholar]
- 79.Thompson R F. Philos Trans R Soc London B. 1990;329:161–170. doi: 10.1098/rstb.1990.0161. [DOI] [PubMed] [Google Scholar]
- 80.Chen C, Thompson R F. Learn Mem. 1995;2:185–198. doi: 10.1101/lm.2.3-4.185. [DOI] [PubMed] [Google Scholar]
- 81.Ito M, Karachot L. NeuroReport. 1990;1:129–132. doi: 10.1097/00001756-199010000-00012. [DOI] [PubMed] [Google Scholar]
- 82.Linden D J, Connor J A. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- 83.Lavond D G, Steinmetz J E, Yokaitis M H, Thompson R F. Exp Brain Res. 1987;67:569–593. doi: 10.1007/BF00247289. [DOI] [PubMed] [Google Scholar]
- 84.Logan, C. G. (1991) Dissertation (Stanford Univ., Palo Alto, CA).
- 85.Chen L, Bao S, Lockard J M, Kim J J, Thompson R F. J Neurosci. 1996;16:2829–2838. doi: 10.1523/JNEUROSCI.16-08-02829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Landis S C, Mullen R J. J Comp Neurol. 1978;177:125–144. doi: 10.1002/cne.901770109. [DOI] [PubMed] [Google Scholar]
- 87.Goldowitz D, Eisenman L M. In: Genetically Defined Animal Models of Neurobehavioral Dysfunctions. Driscoll P, editor. Boston: Birkhauser; 1992. pp. 66–88. [Google Scholar]
- 88.Aiba A, Kano M, Chen C, Stanton M E, Fox G D, Herrup K, Zwingman T A, Tonegawa S. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 89.Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 90.Chen C, Masanobu K, Abeliovich A, Chen L, Bao S, Kim J J, Hashimoto K, Thompson R F, Tonegawa S. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- 91.Thompson R F. In: The Olivocerebellar System in Motor Control. Strata P, editor. New York: Springer; 1989. pp. 347–362. [Google Scholar]
- 92.Shibuki K, Gomi H, Chen C, Bao S, Kim J J, Wakatsuki H, Fujisaki T, Fujimoto K, Ikeda T, Chen C, Thompson R F, Itohara S. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 93.Lisberger S G. Science. 1988;242:728–735. doi: 10.1126/science.3055293. [DOI] [PubMed] [Google Scholar]
- 94.Racine R J, Wilson D A, Gingell R, Sutherland D. Exp Brain Res. 1986;63:158–162. doi: 10.1007/BF00235658. [DOI] [PubMed] [Google Scholar]
- 95.Mamounas L A, Thompson R F, Madden J IV. Proc Natl Acad Sci USA. 1987;84:2101–2105. doi: 10.1073/pnas.84.7.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Castro-Alamancos M A, Torres-Aleman I. Proc Natl Acad Sci USA. 1993;90:7386–7390. doi: 10.1073/pnas.90.15.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Castro-Almancos M A, Torres-Aleman I. Proc Natl Acad Sci USA. 1994;91:10203–10207. doi: 10.1073/pnas.91.21.10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Supple W F, Jr, Leaton R N. Behav Neurosci. 1990;104:934–947. doi: 10.1037//0735-7044.104.6.934. [DOI] [PubMed] [Google Scholar]
- 99.Steinmetz J E, Logue S F, Miller D P. Behav Neurosci. 1993;107:941–954. doi: 10.1037//0735-7044.107.6.941. [DOI] [PubMed] [Google Scholar]
- 100.Pellegrino L J, Altman J. J Comp Physiol Psychol. 1979;93:1–33. doi: 10.1037/h0077589. [DOI] [PubMed] [Google Scholar]
- 101.Goodlett C R, Hamre K M, West J R. Behav Brain Res. 1992;47:129–141. doi: 10.1016/s0166-4328(05)80119-6. [DOI] [PubMed] [Google Scholar]
- 102.Lalonde R, Botez M I. Brain Res Rev. 1990;15:325–332. doi: 10.1016/0165-0173(90)90006-a. [DOI] [PubMed] [Google Scholar]
- 103.Keele S W, Ivry R B. In: The Development and Neural Bases of Higher Cognitive Functions. Diamond A, editor. New York: New York Acad. of Sci. Press; 1990. pp. 179–211. [DOI] [PubMed] [Google Scholar]
- 104.Schmahmann J, editor. The Cerebellum and Cognition. New York: Academic; 1997. [Google Scholar]