

Abstract
Acute kidney injury is associated with a significant inflammatory response that has been the target of renoprotection strategies. Epoxyeicosatrienoic acids (EETs) are anti-inflammatory cytochrome P450-derived eicosanoids that are abundantly produced in the kidney and metabolized by soluble epoxide hydrolase (sEH; Ephx2) to less active dihydroxyeicosatrienoic acids. Genetic disruption of Ephx2 and chemical inhibition of sEH were used to test whether the anti-inflammatory effects of EETs, and other lipid epoxide substrates of sEH, afford protection against cisplatin-induced nephrotoxicity. EET hydrolysis was significantly reduced in Ephx2(−/−) mice and was associated with an attenuation of cisplatin-induced increases in serum urea nitrogen and creatinine levels. Histological evidence of renal tubular damage and neutrophil infiltration was also reduced in the Ephx2(−/−) mice. Likewise, cisplatin had no effect on renal function, neutrophil infiltration, or tubular structure and integrity in mice treated with the potent sEH inhibitor 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea) (AR9273). Consistent with the ability of EETs to interfere with nuclear factor-κB (NF-κB) signaling, the observed renoprotection was associated with attenuation of renal NF-κB activity and corresponding decreases in the expression of tumor necrosis factor (TNF) α, TNF receptor (TNFR) 1, TNFR2, and intercellular adhesive molecule-1 before the detection of tubular injury. These data suggest that EETs or other fatty acid epoxides can attenuate cisplatin-induced kidney injury and sEH inhibition is a novel renoprotective strategy.
Introduction
Acute kidney injury is a common disorder, and a complete understanding of the mechanisms responsible for its development and effective strategies for its prevention or treatment are still lacking. Cisplatin is a widely used broad-spectrum chemotherapeutic that has a dose-limiting renal toxicity associated with high morbidity and mortality (Pabla and Dong, 2008). Nephrotoxic doses of cisplatin lead to a robust induction of proinflammatory cytokines, although the exact mechanism by which cisplatin induces their release and how these cytokines, in turn, contribute to nephrotoxicity remain unknown (Ramesh and Reeves, 2002; Zhang et al., 2007).
Epoxyeicosatrienoic acids (EETs) are a major product of cytochrome P450 (P450) epoxygenase-catalyzed arachidonic acid metabolism (Kroetz and Zeldin, 2002). The metabolism of EETs to their corresponding dihydroxyeicosatrienoic acids (DHETs) is catalyzed by soluble epoxide hydrolase (sEH; encoded by EPHX2), and this serves as a critical regulatory point for the control of intracellular EET levels. EETs have numerous biological effects in the vasculature and are implicated in the regulation of blood pressure, inflammation, and atherosclerosis (Node et al., 1999; Yu et al., 2000b; Schmelzer et al., 2005; Wang et al., 2010). Clinical studies have also suggested that genetic variations in EPHX2 and P450 epoxygenase genes are associated with an increased risk of coronary heart disease and stroke (Fornage et al., 2004; Lee et al., 2006a, 2011; Burdon et al., 2008; Fava et al., 2010; Wang et al., 2010). In contrast to the increasing impact of EETs and EPHX2 in vascular biology and disease, little is known about their role in renal injury.
Given the abundance of EET production and degradation in isolated renal tissue and their recognition as vasoprotective and anti-inflammatory molecules (Node et al., 1999; Schmelzer et al., 2005), we hypothesize that renal EETs play a protective role during exposure to nephrotoxic stimuli. Consistent with this hypothesis, a selective sEH inhibitor has been reported to attenuate cisplatin-induced increases in biochemical markers of renal toxicity, but no mechanistic evidence for the protective effect was provided (Parrish et al., 2009). The objective of this study was to examine the renoprotective properties of lipid epoxides in a well characterized model of acute kidney injury. Ephx2(−/−) mice and a novel and selective inhibitor of sEH were used to determine the contribution of lipid epoxides to cisplatin-induced nephrotoxicity. We show that genetic or chemical disruption of sEH activity attenuates the renal damage induced by cisplatin. These effects are attributed to an attenuation of NF-κB signaling and resultant effects on cytokine secretion, adhesion molecule expression, and neutrophil infiltration that results from increased intracellular lipid epoxide levels.
Materials and Methods
Reagents.
Cisplatin was purchased from Aldrich Chemical Co. (Milwaukee, WI). The sEH chemical inhibitor 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea (AR9273) was synthesized and kindly provided by Arête Therapeutics (Hayward, CA). An antibody against neutrophils (NIMP-R14) was purchased from Abcam Inc. (Cambridge, MA), and goat anti-rat Alexa Fluor 488 antibody was purchased from Invitrogen (Carlsbad, CA).
Animal Experiments.
C57BL/6 mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA) and allowed to acclimate in the animal facility for at least 1 week before use. Ephx2(−/−) mice were originally derived in the Gonzalez laboratory at the National Cancer Institute (Bethesda, MD) and subsequently backcrossed onto a C57BL/6 genetic background for at least seven generations (Sinal et al., 2000). Ephx2(−/−) and Ephx2(+/+) breeder mice were kindly provided by the Gonzalez laboratory and subsequently bred in our laboratory at the University of California. Genotypes were determined in 3-week-old mice by using PCR (Sinal et al., 2000). A single 338-base pair PCR fragment is detected in Ephx2(+/+) mice, a single 295-base pair fragment is found in Ephx2(−/−) mice, and both fragments are detected in Ephx2(+/−) mice (Sinal et al., 2000).
In all studies, 8- to 10-week-old male mice weighing 20 to 25 g were used. Animal experiments were conducted in adherence to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and approved by the Animal Care and Use Committee of the University of California, San Francisco. Cisplatin and AR9273 were freshly prepared in sterile saline or 1% carboxymethylcellulose/0.1% Tween 80, respectively. C57BL/6 mice were given a daily 100 mg/kg dose of AR9273 or vehicle by oral gavage starting 24 h before and continuing for 24, 48, or 72 h after cisplatin treatment. Cisplatin was administered as a single intraperitoneal dose of 20 mg/kg, and an identical volume of sterile saline was administered to control mice. Mice were housed in metabolic cages for the collection of urine during the 24-h period before sacrifice. Mice were sacrificed at 24, 48, or 72 h after cisplatin treatment (immediately after the last dose of AR9273). Kidneys were removed and flash-frozen in liquid nitrogen. Blood samples were collected via cardiac puncture at the time of sacrifice. All tissue and fluid samples were stored at −80°C until analyzed. In studies involving Ephx2(−/−) and Ephx2(+/+) littermates, cisplatin treatment and sample collection were carried out exactly as described for the inhibitor studies, and samples were collected 72 h after cisplatin treatment.
TaqMan Quantitative PCR.
Total RNA was isolated from kidney tissue by using the PARIS kit (Ambion, Austin, TX) according to the manufacturer's instructions. First-strand cDNA was synthesized by using SuperScript III reverse transcriptase (Invitrogen), and any remaining RNA was removed by incubating the samples with 20 U RNase H at 37°C for 15 min. All TaqMan probes and primers were purchased as Assays-on-Demand from Applied Biosystems (Foster City, CA). TaqMan analysis was performed by using an Applied Biosystems 7900HT real-time PCR system. Target gene expression was normalized to Gapdh, and change in expression was calculated as 2−ΔΔCT, where ΔΔCT = (ΔCttarget − ΔCtGapdh)treated − (ΔCttarget − ΔCtGapdh)control. PCR conditions were an initial hold at 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. An equal amount of template cDNA (100 ng) was used for each sample.
Renal Function and Histology.
Renal function was initially assessed by the measurement of serum creatinine and urea nitrogen in the clinical chemistry laboratory at San Francisco General Hospital. Paraformaldehyde (4%)-fixed and paraffin-embedded kidneys were sectioned at 3 μm and stained with hematoxylin and eosin or periodic acid-Schiff (PAS) by using standard methods. All renal histology evaluations were performed by a board-certified pathologist without knowledge of the experimental groups. Histological changes were evaluated in the cortex and outer stripe of the outer medulla and scored by counting the numbers of tubules that displayed apoptosis in 20 high-power fields (hpf) at 400× magnification. In addition, loss of brush border, cast formation, and tubule dilation were assessed. TUNEL staining was performed on paraffin-embedded kidney samples by using an in situ cell death detection kit, TMR Red (Roche Diagnostics, Mannheim, Germany), according to the manufacturer's directions.
Paraffin-embedded kidneys were sectioned at 3 μm. Neutrophils were immunostained with the rat monoclonal NIMP-R14 neutrophil antibody and detected with a goat anti-rat Alexa Fluor 488 antibody. Nuclei were counterstained with DAPI. Images were captured by using a Retiga charge-coupled device-cooled camera and associated QCapture Pro software (QImaging, Surrey, BC Canada). NIMP-R14-positive clusters were counted in 10 uniform fields in the cortex and outer medulla of each sample under high-power fields.
Ex Vivo Assay of sEH Activity.
The ethanol stock solution (1 mg/ml) of the substrate 14,15-EET (Omm Scientific, Dallas, TX), was evaporated to dryness under a stream of nitrogen and dissolved to 50 μM (5× the assay concentration) in assay buffer (0.1 mg/ml bovine serum albumin in 25 mM BisTris, pH 7.0) immediately before the initiation of reactions. Whole blood was diluted 1:10 with 0.1 mg/ml bovine serum albumin in 25 mM BisTris, pH 7.0 and equilibrated to 30°C. The reaction was initiated by the addition of 14,15-EET to a final substrate concentration of 10 μM. The reaction was quenched by the addition of two volumes of ice-cold methanol and stored at −80°C until analysis by LC/MS/MS. To determine background activity, a control incubation was performed with each sample containing 10 μM of the known sEH inhibitor, 12-(3-adamantyl-ureido)-dodecanoic acid. The rate of formation of the hydrolysis product, 14,15-DHET, was measured by LC/MS/MS. The sEH-specific activity was defined as the rate of product formation, corrected for the background activity.
Quantitation of AR9273 Plasma Levels.
Blood was collected into EDTA vacutainer tubes from C57BL/6 male mice after dosing with either vehicle or AR9273. Plasma was isolated by centrifugation, the samples were mixed with three volumes of 0.1% formic acid in acetonitrile, and the precipitates were removed by filtration. AR9273 was quantified in a single run by positive-mode electrospray ionization with tandem quadrupole mass spectroscopy.
Oxylipin Quantitation.
An API-4000 system (Applied Biosystems/MDS Sciex, Foster City, CA) was used to determine plasma lipid profiles. Plasma samples were mixed with four volumes of methanol, and the precipitates were removed via filtration. The filtrates were mixed with three plasma volumes of high-performance liquid chromatography water. Ten-microliter samples were injected onto a diphenyl column (2.1 × 3.3 mm, 2 μm) in a gradient run with mobile phase consisting of water and methanol containing 0.2% acetic acid. The lipids were quantified in a single run by negative mode electrospray ionization with tandem quadrupole mass spectroscopy.
NF-κB Activity Assay and Cytokine Quantitation.
NF-κB activity was measured by using a Quantitation NF-κB EIA kit (Oxford Biomedical Research, Oxford, MI) as described previously (Fife et al., 2008). This chemiluminescence assay uses an oligonucleotide containing the DNA binding NF-κB consensus sequence bound to a 96-well EIA plate. The level of TNFα in the kidney was quantified by using a Precoated Mouse TNFα ELISA kit (eBioscience, San Diego, CA), and soluble ICAM-1 (Thermo Fisher Scientific, Waltham, MA), TNRF1, and TNFR2 (R&D Systems, Minneapolis, MN) in serum were quantified by EIA kits. Assays were run in duplicate or triplicate exactly as described by the manufacturer. The amounts of NF-κB and cytokines were normalized to protein concentration.
Statistics.
Values are expressed as mean ± S.D. Data were analyzed by analysis of variance followed by Bonferroni post hoc multiple comparison testing using Prism 4.03 (GraphPad Software Inc., San Diego, CA). A p value of <0.05 was considered significant. All of the analyses were repeated in duplicate or triplicate by using samples from individual animals.
Results
Genetic Disruption of Ephx2 Attenuates Cisplatin-Induced Acute Kidney Injury and Cell Signaling.
Ephx2(−/−) mice were studied to evaluate the role of EETs or other lipid epoxides in cisplatin-induced renal injury. The Ephx2(−/−) mice completely lack immunoreactive sEH protein in their kidneys (Supplemental Fig. 1). Plasma EpOME/DiHOME ratios are validated biomarkers of in vivo sEH activity (Newman et al., 2002; Luria et al., 2007) and confirmed dramatic inhibition in the Ephx2(−/−) mice. EpOME hydrolysis is significantly impaired in the Ephx2(−/−) mice, as evident from an increase in EpOME/DiHOME ratios compared with their Ephx2(+/+) littermates (Fig. 1A). The increase in EpOME/DiHOME ratio in the saline-treated Ephx2(−/−) mice compared with Ephx2(+/+) mice was 2.9- and 78-fold for the 9,10-, and 12,13-EpOME/DiHOME ratios, respectively. Likewise, there was a 5.2- and 34-fold increase in the 9,10- and 12,13-EpOME/DiHOME ratios in the cisplatin-treated Ephx2(−/−) mice compared with the Ephx2(+/+) controls, respectively. Treatment of Ephx2(+/+) mice with cisplatin resulted in a significant increase in serum urea nitrogen and creatinine (Fig. 1, B and C). In contrast, identical treatment of Ephx2(−/−) mice had no effect on these serum markers of renal function. Histological analysis found only mild tubular injury characterized by mild spotty tubular dilation with rare apoptosis of tubular epithelial cells (35 ± 31 tubules/10 hpf) in cisplatin-treated Ephx2(−/−) mice (Fig. 1D). Tubular dilation was more widespread in the Ephx2(+/+) mice treated with cisplatin. Furthermore, frank tubular epithelial cell necrosis was identified with some sloughed cells within tubular lumina. Casts were also observed. Apoptosis was noted in 92 ± 77 tubules/10 hpf in cisplatin-treated Ephx2(+/+) mice [p = 0.064; Ephx2(+/+) versus Ephx2(−/−)]. TUNEL staining confirmed a reduction in cisplatin-induced apoptosis in Ephx2(−/−) mice relative to the Ephx2(+/+) mice (Fig. 2).
Fig. 1.

Genetic disruption of Ephx2 protects against cisplatin-induced acute kidney injury. Ephx2(+/+) and Ephx2(−/−) mice were treated with saline or cisplatin, and kidneys and blood were harvested 72 h later. A, plasma EpOME/DiHOME ratios are shown for the 9,10- (black bars) and 12,13- (hatched bars) regioisomers. Values shown are the mean ± S.D. from five or six mice per treatment group. Significant differences between Ephx2(+/+) and Ephx2(−/−) mice are indicated: *, p < 0.05 and **, p < 0.01. B and C, urea nitrogen (B) and creatinine (C) were measured in serum. Values shown are the mean ± S.D. from six mice per group. Significant differences between vehicle and cisplatin treatment groups are indicated for each strain: *, p < 0.05 and ***, p < 0.001. D, representative photomicrographs are shown from vehicle (Sal)- and cisplatin (Cis)-treated Ephx2(+/+) and Ephx2(−/−) mice. Note tubules containing casts (C) or sloughed tubular cells in cisplatin-treated Ephx2(+/+) kidneys. Apoptotic bodies (arrows) can also be seen in this group. The remaining groups show little damage. Tissue slices were stained with PAS. Photomicrographs are shown at 400× magnification. E, neutrophil staining is shown in representative slides from saline (Sal)- and cisplatin (Cis)-treated Ephx2(+/+) and Ephx2(−/−) mice. Kidney sections (3 μm) were immunostained with a rat monoclonal antibody against neutrophils (bright green), and nuclei were counterstained with DAPI (blue). Neutrophil clusters are marked with arrows. The bar indicates 50 μm; in the inset the scale bar corresponds to 5 μm.
Fig. 2.

Genetic disruption of Ephx2 protects against cisplatin (Cis)-induced apoptosis. Top, apoptotic cells were detected by TUNEL staining. Sal, saline. The bar indicates 100 μm. Bottom, the number of apoptotic cells were counted in 10 hpf, and the mean ± S.D. from three to four mice per group is expressed relative to control kidneys. Significant differences are indicated: ***, p < 0.001, between vehicle- and cisplatin-treated mice; †, p < 0.05 between cisplatin-treated Ephx2(+/+) and Ephx2(−/−) mice.
An early event in cisplatin-induced renal injury is infiltration of neutrophils into the kidney. Neutrophils were stained with an antibody and visualized by fluorescence microscopy (Fig. 1E). The degree of neutrophil infiltration into the cortex induced by cisplatin in Ephx2(−/−) mice (32 ± 2.9 cells/10 hpf) was attenuated in Ephx2(−/−) mice [21 ± 2.0 cells/10 hpf; p < 0.05; Ephx2(+/+) versus Ephx2(−/−)].
The effects of sEH inhibition on TNFα and ICAM-1 gene transcription were measured by using TaqMan real-time PCR. The induction of renal TNFα and ICAM-1 mRNA by cisplatin in Ephx2(+/+) mice was not evident in Ephx2(−/−) mice (Fig. 3, A and B). Consistent with the mRNA results, cisplatin induced renal TNFα and serum ICAM-1 protein levels in Ephx2(+/+) mice but not in the Ephx2(−/−) mice (Fig. 3, C and D).
Fig. 3.

Cisplatin-mediated renal inflammatory gene expression is prevented by genetic disruption of Ephx2. Ephx2(+/+) and Ephx2(−/−) mice were treated with saline or cisplatin, and renal and serum markers of inflammation were measured 72 h later. A and B, TNFα (A) and ICAM-1 (B) mRNA levels were measured in the kidneys of vehicle- and cisplatin-treated Ephx2(+/+) and Ephx2(−/−) mice by quantitative real-time PCR. RNA levels were normalized to Gapdh, and the data are presented as the relative change in expression between vehicle- and cisplatin-treated groups. C and D, renal TNFα (C) and serum ICAM-1 (D) protein levels were quantified by EIA. The values shown are the mean ± S.D. of five or six mice per treatment group. Significant differences between vehicle and cisplatin treatment in each strain are indicated: **, p < 0.01 and ***, p < 0.001.
Chemical Inhibition of sEH Attenuates Cisplatin-Induced Renal Injury and Cell Signaling.
A second strategy to evaluate the renoprotective role of EETs or other lipid epoxides in cisplatin-induced acute kidney injury was to treat C57BL/6 mice with AR9273 to inhibit sEH-catalyzed epoxide hydrolysis. The plasma levels of AR9273 shortly after the fifth daily dose ranged from 2.39 to 13.4 μM, and all animals had levels that were many fold above the IC50 for mouse sEH of 2.3 nM (data not shown). No AR9273 was detected in vehicle-treated animals. Quantitation of EpOME and DiHOME plasma levels confirmed the inhibition of sEH in AR9273-treated mice (Fig. 4A). EpOME/DiHOME ratios increased 8- to 66-fold in mice treated with the sEH inhibitor. The 12,13-EpOME/DiHOME ratio increased to a greater extent (24- and 66-fold in vehicle- and cisplatin-treated mice, respectively) than the 9,10-EpOME/DiHOME ratio (8- and 9-fold in vehicle- and cisplatin-treated mice, respectively). Inhibition of sEH was also demonstrated with an ex vivo assay measuring EET hydrolysis in blood collected at 72 h after cisplatin treatment (data not shown). EET hydrolysis was readily measurable in the plasma of vehicle-treated mice but was barely detectable in plasma from AR9273-treated mice.
Fig. 4.

sEH inhibition prevents cisplatin-induced acute kidney injury in C57BL/6 mice. Mice were treated daily with 100 mg/kg AR9273 or vehicle for 5 days, and a single dose of 20 mg/kg cisplatin was administered on day 2. Kidneys and blood were harvested 24 to 72 h after cisplatin treatment and used for characterization of renal injury. A, plasma EpOME/DiHOME ratios are shown for the 9,10- (black bars) and 12,13- (hatched bars) regioisomers. The values represent the mean ± S.D. from five to eight samples per treatment group. Significant differences are indicated: ***, p < 0.001 between vehicle-and AR9273-treated mice; †††, p < 0.001 between mice treated with cisplatin alone and cisplatin with AR9273. B to D, urea nitrogen (B) and creatinine (C) were measured in serum, and Kim-1 mRNA levels (D) were measured in renal tissue. The values shown are the mean ± S.D. from 8 to 10 mice per group. The renal Kim-1 mRNA levels are expressed relative to vehicle-treated mice. Significant differences are indicated: **, p < 0.01 and ***, p < 0.001 between vehicle-and cisplatin-treated mice; ††, p < 0.01 and †††, p < 0.001 between mice treated with cisplatin alone and cisplatin with AR9273. E, kidney slices were stained with PAS, and representative photomicrographs are shown at 400× magnification. At 24 h little difference is seen between the mice treated with cisplatin (Cis) alone and cisplatin treated with AR9273 (AR). At 48 h an increase in the number of apoptotic bodies (arrows) is identified in the mice treated with cisplatin alone. At 72 h note the presence of casts and frank tubular necrosis in the groups treated with cisplatin alone. In addition, clusters of apoptotic bodies are seen compared with the single apoptotic cells seen in the other groups. Veh, vehicle. F, kidney sections (3 μm) were immunostained with a rat monoclonal antibody against neutrophils (bright green), and nuclei were stained with DAPI (blue). Neutrophil clusters are marked with arrows. Sal, saline; Cis, cisplatin; AR, AR9273. The bars indicate 50 μm; in the inset the scale bar corresponds to 5 μm.
The effects of 24- to 72-h treatment with AR9273 on cisplatin-induced renal injury were evaluated by measuring serum creatinine and urea nitrogen levels. AR9273 itself had no effect on serum urea nitrogen or creatinine, and elevation in these markers was not evident until 72 h after cisplatin treatment (Fig. 4, B and C). Elevation of renal Kim-1 mRNA levels was consistent with renal tubular damage as early as 24 h after cisplatin treatment (Fig. 4D). Histological examination of the kidneys showed no casts and only rare tubular epithelial cell apoptosis in any of the control animals at any time period (Fig. 4E). Cisplatin-treated mice showed focal mild tubular dilation with scattered casts and tubular epithelial cell apoptosis (Fig. 4E). The extent of tubular epithelial cell necrosis increased over the 3 days in response to cisplatin treatment, from 7.2 ± 2.9 apoptotic tubule cells/20 hpf at 24 h to 89 ± 21 apoptotic tubule cells/20 hpf at 72 h in the cortex (p < 0.001). At earlier times single-cell apoptosis was noted, but by day 3, clusters of apoptotic cells were seen. A similar trend was observed in the outer stripe of the outer medulla, although the number of affected tubules was less. It should also be noted that the apoptosis in the medulla was seen largely in collecting ducts, whereas proximal and distal tubules as well as collecting ducts showed injury in the cortex. These changes were significantly attenuated in AR9273-treated mice, affecting 34 ± 24 apoptotic tubules/20 hpf at 72 h (p < 0.001 compared with saline-treated mice at 72 h). Consistent with the histological analysis, TUNEL staining indicated a significant reduction in cisplatin-induced apoptosis in the presence of AR9273 (Fig. 5).
Fig. 5.

sEH inhibition protects against cisplatin-induced apoptosis. Top, apoptotic cells were detected by TUNEL staining. Sal, saline; AR, AR9273; Cis, cisplatin. The bar indicates 100 μm. Bottom, the number of apoptotic cells were counted in 10 hpf, and the mean ± S.D. from three to four mice per group is expressed relative to control kidneys. Significant differences are indicated: ***, p < 0.001, between vehicle- and cisplatin-treated mice; †, p < 0.05 between mice treated with cisplatin alone and cisplatin with AR9273.
Neutrophils were stained as an early measure of inflammation in response to cisplatin treatment. The majority of neutrophils were found in the glomeruli of cisplatin-treated mice (Fig. 4F; 3.6 ± 2.1, 9.4 ± 9.2, and 19.4 ± 10.6 cells/10 hpf at 24, 48, and 72 h after cisplatin treatment, respectively). Treatment with AR9273 reduced neutrophil infiltration at 48 and 72 h after treatment to 3.0 ± 2.2 and 9.1 ± 8.6 cells cells/10 hpf, respectively (p < 0.05 for difference at 72 h). In contrast, minimal neutrophil infiltration was detected in mice treated only with saline or inhibitor (<4.0 ± 1.0 cells/10 hpf 24–72 h after treatment).
Cisplatin treatment resulted in an increase in the renal mRNA levels of TNFα (Fig. 6A) and ICAM-1 (Fig. 6B) within 48 h of treatment. In both cases, AR9273 administration greatly attenuated this inflammatory response to cisplatin. Renal TNFα protein levels were not significantly elevated until 72 h after cisplatin treatment, and this increase was also prevented by AR9273 treatment (Fig. 6C). In contrast, a significant increase in soluble ICAM-1 levels in plasma was evident within 24 h of cisplatin treatment and remained elevated throughout the 72-h treatment period; this effect was attenuated by sEH inhibition (Fig. 6D).
Fig. 6.

Cisplatin-mediated renal inflammatory gene expression is attenuated by inhibition of soluble epoxide hydrolase. Mice were treated daily with 100 mg/kg AR9273 or vehicle orally for 5 days, and a single dose of 20 mg/kg cisplatin was administered on day 2. Kidneys and blood were harvested 24 to 72 h after cisplatin treatment and used for measurement of inflammatory markers. A and B, renal TNFα (A) and ICAM-1 (B) mRNA levels were measured by quantitative real-time PCR. RNA levels were normalized to Gapdh, and the data are presented relative to the vehicle-treated group. C and D, renal TNFα (C) and serum ICAM-1 (D) protein levels were quantified by EIA. The values shown are the mean ± S.D. of 8 to 10 mice per treatment group. Significant differences are indicated: *, p < 0.05, **, p < 0.01, and ***, p < 0.001 between vehicle- and cisplatin-treated mice; †, p < 0.05, ††, p < 0.01, and †††, p < 0.001 between mice treated with cisplatin alone and cisplatin with AR9273.
TNF receptors are also regulated by cisplatin signaling, and serum levels were measured. As shown in Fig. 7, soluble TNFR1 and TNFR2 levels were dramatically elevated after treatment with cisplatin. In the case of TNFR1, this was evident within 48 h of treatment, whereas TNFR2 levels were already increased at 24 h after cisplatin. The levels of both TNF receptors were dramatically attenuated by AR9273 treatment. Likewise, genetic disruption of Ephx2 provided protection against cisplatin-induced TNFR elevations (Fig. 7).
Fig. 7.

Cisplatin-mediated induction of soluble TNFR1 and soluble TNFR2 is attenuated by genetic disruption of Ephx2 or inhibition of soluble epoxide hydrolase. A and B, Ephx2(+/+) and Ephx2(−/−) mice were treated with saline or cisplatin, and blood was collected 72 h later. C and D, in other studies, C57BL/6 mice were treated daily with 100 mg/kg AR9273 or vehicle orally for 5 days, and a single dose of 20 mg/kg cisplatin was administered on day 2. Blood was harvested 24 to 72 h after cisplatin treatment. sTNFR1 (A and C) and sTNFR2 (B and D) were measured by EIA. Values shown are the mean ± S.D. from five mice for each treatment group. Significant differences are indicated: *, p < 0.05, **, p < 0.01, and ***, p < 0.001 between vehicle- and cisplatin-treated mice; ††, p < 0.01 and †††, p < 0.001 between mice treated with cisplatin alone and cisplatin with AR9273.
Renoprotective Effects of sEH Inhibition Involve NF-κB Signaling.
Based on the effects of sEH inhibition on TNFα signaling and ICAM-1 expression and the known NF-κB inhibitory properties of EETs, we examined NF-κB activation in Ephx2(−/−) and AR9273-treated mice. Compared with Ephx2(+/+) mice, the basal level of active NF-κB was lower in Ephx2(−/−) mice but did not reach statistical significance (Fig. 8A). Cisplatin significantly increased NF-κB activity in Ephx2(+/+) mice but not in Ephx2(−/−) mice. Consistent with the findings in Ephx2(−/−) mice, chemical inhibition of sEH attenuated the cisplatin-induced increase in NF-κB activity at 72 h (Fig. 8B).
Fig. 8.
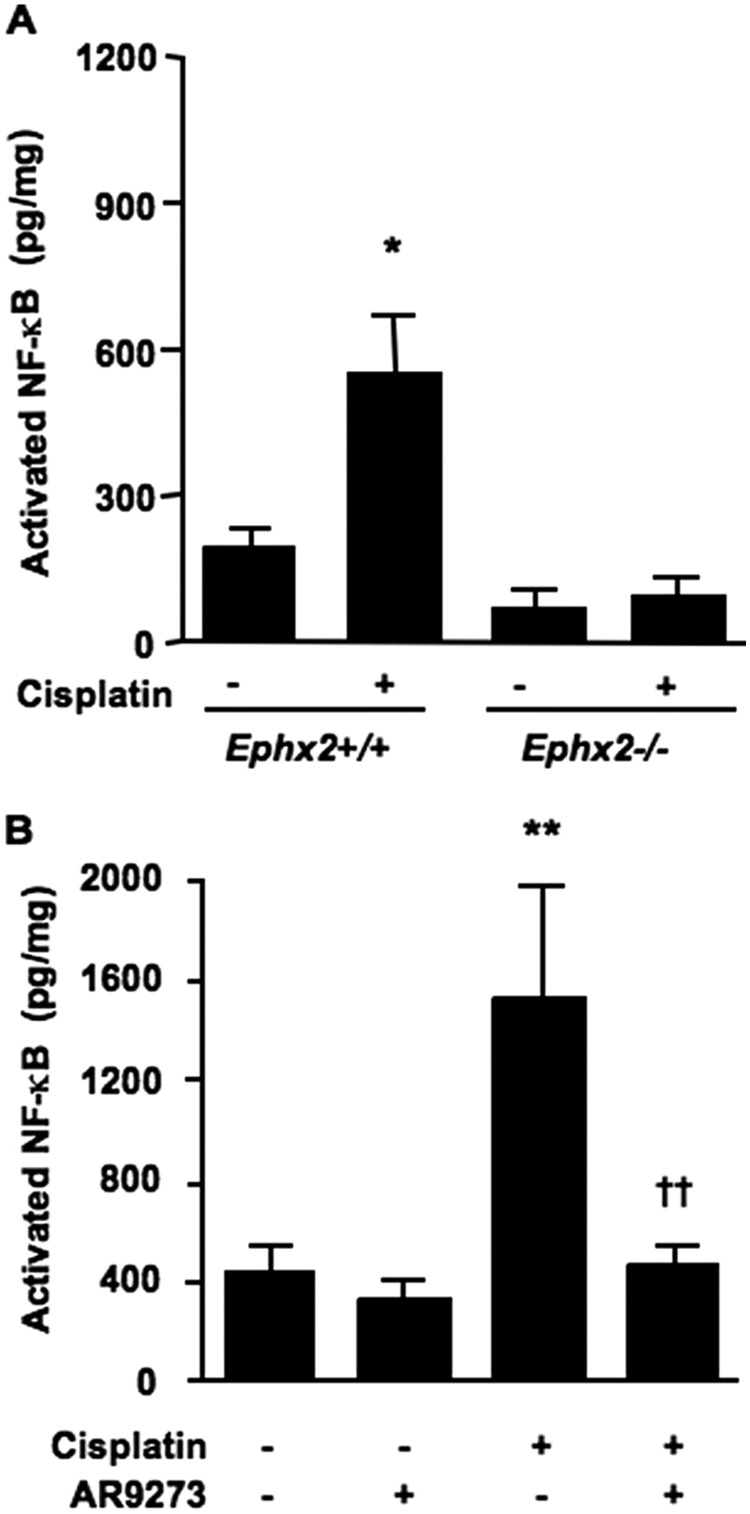
Activation of NF-κB is prevented by genetic disruption of Ephx2 or soluble epoxide hydrolase inhibition. Renal nuclear lysates collected 72 h after cisplatin treatment were used for measurement of activated NF-κB. The effect of cisplatin was compared between Ephx2(+/+) and Ephx2(−/−) mice (A) and in vehicle- and AR9273-treated mice (B). Values shown are the mean ± S.D. of five or six samples per treatment group. Significant differences are indicated: *, p < 0.05 and **, p < 0.01 for difference between vehicle and cisplatin treatment in Ephx2(+/+) and C57BL/6 mice, respectively; ††, p < 0.01 for difference between mice treated with cisplatin alone and cisplatin plus AR9273.
Discussion
Cisplatin induces acute kidney injury in both animals and humans, and tissue injury is preceded by a robust inflammatory response (Kelly et al., 1999; Deng et al., 2001; Ramesh and Reeves, 2002, 2004). An early response to cisplatin treatment includes increases in NF-κB signaling and corresponding induction of TNFα and ICAM-1 expression. Recent studies of modalities that increase EETs, such as Ephx2(−/−) mice and inhibitors of sEH, suggest that EETs can antagonize inflammatory changes in endothelial cells and in systemic models of inflammation (Node et al., 1999; Fleming et al., 2001; Schmelzer et al., 2005; Smith et al., 2005; Luria et al., 2007; Manhiani et al., 2009; Liu et al., 2010). The anti-inflammatory effects of EETs are attributed, at least in part, to interruption of NF-κB signaling and decreased expression of inflammatory markers such as adhesion molecules and cytokines (Node et al., 1999; Fleming et al., 2001; Schmelzer et al., 2005; Manhiani et al., 2009; Liu et al., 2010). Based on our knowledge of EET action and the pattern of cisplatin nephrotoxicity, we hypothesized that increased intracellular EET levels would attenuate the nephrotoxic effects of cisplatin.
Potential strategies for increasing EET levels include induction of P450 epoxygenases, treatment with EETs or EET mimetics, or inhibition of EET hydrolysis. P450 epoxygenases are induced by dietary fatty acids and during hypertension, but there is no evidence for selective induction of these enzymes by small molecules, including cisplatin (Yu et al., 2000a, 2006). Induction of P450 epoxygenases as a viable approach for increasing EETs is therefore of limited use. Likewise, treatment with EETs is complicated by their rapid degradation (Spector et al., 2004). Although EET mimetics have been designed and are biologically active in vitro or in situ, their use in vivo has not been demonstrated (Yang et al., 2007b). Inhibition of EET hydrolysis is therefore the most attractive approach for increasing EET levels in vivo.
Ephx2(−/−) mice were used as one model of increased EET levels. Genetic disruption of sEH activity has previously been used to explore the role of EETs in blood pressure, cardioprotection, systemic inflammation, heart failure, diabetes, and vascular remodeling (Seubert et al., 2006; Luria et al., 2007; Monti et al., 2008; Luo et al., 2010; Simpkins et al., 2010). A series of potent and selective inhibitors of sEH have also been synthesized, and several have been tested in animal and cellular models (Yu et al., 2000b; Davis et al., 2002; Imig et al., 2002, 2005; Zhao et al., 2004; Schmelzer et al., 2005; Smith et al., 2005; Luria et al., 2007). The current study used a potent and selective inhibitor of lipid epoxide hydrolase activity, AR9273, selected for use in this study because of its oral bioavailability and the resulting changes in the plasma oxylipid ratios it affords in mice. Daily oral treatment of mice with AR9273 results in almost complete inhibition of sEH, as reflected by an ex vivo assay of EET hydrolysis and plasma EpOME/DiHOME ratios. The level of EETs and DHETs in plasma samples from individual mice is close to the limit of detection of the LC/MS/MS assay, but it has previously been shown that the corresponding levels of the C18 fatty acid epoxides and diols, EpOMEs and DiHOMEs, reflect the levels of EETs and DHETs (Newman et al., 2002; Luria et al., 2007). In addition to epoxide hydrolase activity, sEH has phosphatase activity, which is not affected by AR9273. The inhibitor therefore allows us to focus specifically on the role of EETs and other lipid epoxides, while the Ephx2(−/−) mice informs us about both the epoxide hydrolase and phosphatase activities of sEH. Relatively little is known about the biological effects of other fatty acid epoxide substrates of sEH, and it is possible that multiple oxylipins have renoprotective properties similar to those attributed to the EETs.
Genetic disruption of Ephx2 or the treatment of mice with a sEH inhibitor was associated with an almost complete attenuation of cisplatin-induced renal damage, measured either as elevations in plasma or renal biomarkers of renal injury, histological changes in renal tubular structure, or neutrophil infiltration. The combined strategy of chemical and genetic disruption of sEH activity provides strong evidence for a renoprotective role for fatty acid epoxides during cisplatin exposure. It is noteworthy that renoprotective effects of the potent sEH inhibitor AR9273 were evident within 24 h of cisplatin treatment, as measured by the early renal injury biomarker Kim-1, changes in tubular structure, neutrophil infiltration, and the number of apoptotic cells. Consistent with these findings, the effects of sEH inhibition on cytokine and adhesion molecule expression were also observed 24 to 48 h before the effects of cisplatin on serum creatinine and urea nitrogen were apparent at 72 h after treatment. While these data were being analyzed, a report was published showing that the sEH inhibitor 12-(3-adamantan-1-yl-ureido)-dodecanoic acid prevented cisplatin-induced increases in blood urea nitrogen and tubular necrosis (Parrish et al., 2009). Our results confirm and extend those findings by using a novel sEH inhibitor with Ephx2(−/−) mice. We also provide evidence for an effect of inhibition of sEH hydrolase activity or genetic disruption of Ephx2 on NF-κB signaling in response to cisplatin. AR9273 had no significant effect on the function of renal transporters involved in cisplatin uptake (data not shown), providing further support that the observed renoprotective effects are a result of changes in EET degradation by sEH.
Our findings are consistent with the widely accepted major role for TNFα in mediating a robust inflammatory response to cisplatin treatment (Deng et al., 2001; Ramesh and Reeves, 2002, 2004; Li et al., 2005; Lee et al., 2006b; Zhang et al., 2007). Increased TNFα levels were an early response to cisplatin. Both TNFR1 and TNFR2 have been implicated in cisplatin-induced TNFα signaling. Studies in TNFR1-deficient mice have shown that signaling through TNFR1 is involved in tubular cell apoptosis after cisplatin treatment (Tsuruya et al., 2003). In contrast, others have shown that TNFR2 plays a more important role than TNFR1 in mediating the inflammatory and apoptotic effects of cisplatin, including ICAM-1 induction (Ramesh and Reeves, 2003). The fact that sEH inhibition can inhibit both TNFR2 and ICAM-1 induction to a similar degree and time dependence is consistent with EETs interfering with this signaling pathway. In the current study, cisplatin induced a more dramatic increase in circulating TNFR1 levels compared with TNFR2. It is noteworthy that the induction of both TNFR1 and TNFR2 was an early response to cisplatin treatment and evident within 24 h of treatment. The more dramatic effect on cisplatin-induced apoptosis compared with neutrophil infiltration is consistent with the significant changes in TNFR1 expression. The ability of sEH inhibition to attenuate these early increases in TNFα and soluble TNFR expression suggests that this could be an effective strategy for ameliorating both the inflammatory and apoptotic effects of cisplatin. This is significant because previous studies focused on the inhibition of inflammation alone did not prevent cisplatin-induced renal injury (Faubel et al., 2007).
The induction of renal NF-κB activity by cisplatin was also prevented by the inhibition of sEH activity and is consistent with the ability of EETs to disrupt NF-κB signaling (Node et al., 1999). In endothelial cells, EETs interfere with NF-κB signaling by disrupting IκB kinase activity, resulting in decreased expression of proinflammatory proteins (Node et al., 1999). Consistent with these findings in isolated cells, the induction in mice of the proinflammatory proteins cyclooxygenase-2 and inducible nitric-oxide synthase by endotoxin treatment is attenuated by sEH inhibition (Schmelzer et al., 2005). Likewise, acute exposure to tobacco smoke results in inflammatory cell infiltration into bronchial lavage fluid, a response that is greatly attenuated by treatment with a sEH inhibitor (Smith et al., 2005). The effect of sEH inhibition on cisplatin-induced changes in renal NF-κB activity is similar to the effect of salicylate, fibrate, or rosiglitazone treatment in this model (Ramesh and Reeves, 2004; Li et al., 2005; Lee et al., 2006b). In all cases, the attenuation of NF-κB activity is almost complete, supporting a critical role for these molecules in cisplatin nephrotoxicity.
NF-κB signaling is implicated in the regulation of both TNFα and TNFR2. TNFα itself can activate IκB kinase, leading to NF-κB activity (Kelliher et al., 1998). Both TNFR2 and TNFα have NF-κB binding sites, which could account for their induction in response to cisplatin treatment (Santee and Owen-Schaub, 1996; Yao et al., 1997). Further downstream effects of NF-κB activation include induction of ICAM-1 and subsequent neutrophil infiltration (Ramesh and Reeves, 2002, 2003; Francescato et al., 2007). The ability of EETs to interfere with NF-κB signaling is consistent with the findings that sEH inhibition attenuates the effect of cisplatin on TNFα, TNFR, and ICAM-1 expression and provides a plausible mechanism for the observed renoprotection afforded by sEH inhibition.
Collectively, these data provide convincing evidence that sEH inhibition protects against cisplatin-induced renal injury, at least in part by attenuation of NF-κB signaling. In addition to their interaction with the NF-κB signaling pathway, EETs interfere with other signaling pathways implicated in cisplatin toxicity, including peroxisome proliferator-activated receptor α and mitogen-activated protein kinase (Ng et al., 2007; Yang et al., 2007a). Whether these signaling pathways are involved in the renoprotective effects of sEH inhibition require further study. It will also be of interest to explore whether renoprotection by EETs is afforded in other models of acute kidney injury, such as ischemia reperfusion and mechanical injury. sEH inhibition does protect against renal injury associated with salt-sensitive hypertension, but in this case it is hard to determine the contribution of lowering blood pressure to the observed effects (Imig et al., 2005; Manhiani et al., 2009). Indeed, the combined effects of EETs on inflammatory signaling and vasoactivity may enhance the therapeutic potential of sEH inhibition for renoprotection.
Supplementary Material
Acknowledgments
We thank Julie Siegenthaller for assistance with neutrophil image capture and useful discussions.
This work was supported by the University of California Discovery Program [Grant Bio06-1-576]; the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK084147]; and Arête Therapeutics. J.M. was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Training Grant T32-GM007175].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- EET
- epoxyeicosatrienoic acid
- DHET
- dihydroxyeicosatrienoic acid
- P450
- cytochrome P450
- sEH
- soluble epoxide hydrolase
- NF-κB
- nuclear factor-κB
- TNFα
- tumor necrosis factor α
- TNFR
- TNFα receptor
- ICAM-1
- intercellular adhesive molecule-1
- AR9273
- 1-adamantan-1-yl-3-(1-methylsulfonyl-piperidin-4-yl-urea)
- EpOME
- epoxyoctadecenoic acid
- DiHOME
- dihydroxyoctadecenoic acid
- Kim-1
- kidney injury molecule-1
- PAS
- periodic acid-Schiff
- LC/MS/MS
- liquid chromatography-tandem mass spectrometry
- Gapdh
- glyceraldehyde-3-phosphate dehydrogenase
- hpf
- high-power fields
- EIA
- enzyme immunoassay
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
- PCR
- polymerase chain reaction
- DAPI
- 4,6-diamidino-2-phenylindole.
Authorship Contributions
Participated in research design: Liu, Webb, Olson, and Kroetz.
Conducted experiments: Liu, Fukushima, Micheli, and Markova.
Performed data analysis: Liu, Fukushima, and Kroetz.
Wrote or contributed to the writing of the manuscript: Liu, Webb, Fukushima, Micheli, Markova, Olson, and Kroetz.
References
- Burdon KP, Lehtinen AB, Langefeld CD, Carr JJ, Rich SS, Freedman BI, Herrington D, Bowden DW. (2008) Genetic analysis of the soluble epoxide hydrolase gene, EPHX2, in subclinical cardiovascular disease in the Diabetes Heart Study. Diab Vasc Dis Res 5:128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. (2002) Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A 99:2222–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Kohda Y, Chiao H, Wang Y, Hu X, Hewitt SM, Miyaji T, McLeroy P, Nibhanupudy B, Li S, et al. (2001) Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int 60:2118–2128 [DOI] [PubMed] [Google Scholar]
- Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, et al. (2007) Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther 322:8–15 [DOI] [PubMed] [Google Scholar]
- Fava C, Montagnana M, Danese E, Almgren P, Hedblad B, Engström G, Berglund G, Minuz P, Melander O. (2010) Homozygosity for the EPHX2 K55R polymorphism increases the long-term risk of ischemic stroke in men: a study in Swedes. Pharmacogenet Genomics 20:94–103 [DOI] [PubMed] [Google Scholar]
- Fife KL, Liu Y, Schmelzer KR, Tsai HJ, Kim IH, Morisseau C, Hammock BD, Kroetz DL. (2008) Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. J Pharmacol Exp Ther 327:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Michaelis UR, Bredenkötter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. (2001) Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 88:44–51 [DOI] [PubMed] [Google Scholar]
- Fornage M, Boerwinkle E, Doris PA, Jacobs D, Liu K, Wong ND. (2004) Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 109:335–339 [DOI] [PubMed] [Google Scholar]
- Francescato HD, Costa RS, Scavone C, Coimbra TM. (2007) Parthenolide reduces cisplatin-induced renal damage. Toxicology 230:64–75 [DOI] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. (2002) Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39:690–694 [DOI] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. (2005) An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 46:975–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. (1998) The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8:297–303 [DOI] [PubMed] [Google Scholar]
- Kelly KJ, Meehan SM, Colvin RB, Williams WW, Bonventre JV. (1999) Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int 56:922–931 [DOI] [PubMed] [Google Scholar]
- Kroetz DL, Zeldin DC. (2002) Cytochrome P450 pathways of arachidonic acid metabolism. Curr Opin Lipidol 13:273–283 [DOI] [PubMed] [Google Scholar]
- Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, Hammock BD, Couper DJ, Heiss G, Zeldin DC. (2006a) Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet 15:1640–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Pretorius M, Schuck RN, Burch LH, Bartlett J, Williams SM, Zeldin DC, Brown NJ. (2011) Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with forearm vasodilator responses in humans. Hypertension 57:116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim W, Moon SO, Sung MJ, Kim DH, Kang KP, Jang YB, Lee JE, Jang KY, Park SK. (2006b) Rosiglitazone ameliorates cisplatin-induced renal injury in mice. Nephrol Dial Transplant 21:2096–2105 [DOI] [PubMed] [Google Scholar]
- Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. (2005) Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol 289:F469–F480 [DOI] [PubMed] [Google Scholar]
- Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. (2010) Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol 79:880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. (2010) Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther 334:430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. (2007) Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem 282:2891–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. (2009) Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol 297:F740–F748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gösele C, Heuser A, Fischer R, Schmidt C, Schirdewan A, et al. (2008) Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 40:529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JW, Watanabe T, Hammock BD. (2002) The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J Lipid Res 43:1563–1578 [DOI] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. (2007) Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor α. Drug Metab Dispos 35:1126–1134 [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285:1276–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla N, Dong Z. (2008) Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73:994–1007 [DOI] [PubMed] [Google Scholar]
- Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD. (2009) Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol 25:217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh G, Reeves WB. (2002) TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110:835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh G, Reeves WB. (2003) TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285:F610–F618 [DOI] [PubMed] [Google Scholar]
- Ramesh G, Reeves WB. (2004) Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-α. Kidney Int 65:490–499 [DOI] [PubMed] [Google Scholar]
- Santee SM, Owen-Schaub LB. (1996) Human tumor necrosis factor receptor p75/80 (CD120b) gene structure and promoter characterization. J Biol Chem 271:21151–21159 [DOI] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. (2005) Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A 102:9772–9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, et al. (2006) Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res 99:442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD. (2010) Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol 298:H795–H806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. (2000) Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem 275:40504–40510 [DOI] [PubMed] [Google Scholar]
- Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. (2005) Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc Natl Acad Sci U S A 102:2186–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. (2004) Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43:55–90 [DOI] [PubMed] [Google Scholar]
- Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, Fukuda K, Kanai H, Kishihara K, Hirakata H, et al. (2003) Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int 63:72–82 [DOI] [PubMed] [Google Scholar]
- Wang YX, Ulu A, Zhang LN, Hammock B. (2010) Soluble epoxide hydrolase in atherosclerosis. Curr Atheroscl Rep 12:174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Lin L, Chen JX, Lee CR, Seubert JM, Wang Y, Wang H, Chao ZR, Tao DD, Gong JP, et al. (2007a) Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-α via MAPK and PI3K/Akt signaling pathways. Am J Physiol Heart Circ Physiol 293:H142–H151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Holmes BB, Gopal VR, Kishore RV, Sangras B, Yi XY, Falck JR, Campbell WB. (2007b) Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J Pharmacol Exp Ther 321:1023–1031 [DOI] [PubMed] [Google Scholar]
- Yao J, Mackman N, Edgington TS, Fan ST. (1997) Lipopolysaccharide induction of the tumor necrosis factor-α promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-κB transcription factors. J Biol Chem 272:17795–17801 [DOI] [PubMed] [Google Scholar]
- Yu Z, Huse LM, Adler P, Graham L, Ma J, Zeldin DC, Kroetz DL. (2000a) Increased CYP2J expression and epoxyeicosatrienoic acid formation in spontaneously hypertensive rat kidney. Mol Pharmacol 57:1011–1020 [PubMed] [Google Scholar]
- Yu Z, Ng VY, Su P, Engler MM, Engler MB, Huang Y, Lin E, Kroetz DL. (2006) Induction of renal cytochrome P450 arachidonic acid epoxygenase activity by dietary γ-linolenic acid. J Pharmacol Exp Ther 317:732–738 [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, et al. (2000b) Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87:992–998 [DOI] [PubMed] [Google Scholar]
- Zhang B, Ramesh G, Norbury CC, Reeves WB. (2007) Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int 72:37–44 [DOI] [PubMed] [Google Scholar]
- Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. (2004) Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol 15:1244–1253 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.