

Abstract
The present study compared the selectivity of two homologous transport proteins, multidrug and toxin extruders 1 and 2-K (MATE1 and MATE2-K), and developed three-dimensional pharmacophores for inhibitory ligand interaction with human MATE1 (hMATE1). The human orthologs of MATE1 and MATE2-K were stably expressed in Chinese hamster ovary cells, and transport function was determined by measuring uptake of the prototypic organic cation (OC) substrate 1-methyl-4-phenylpyridinium (MPP). Both MATEs had similar apparent affinities for MPP, with Ktapp values of 4.4 and 3.7 μM for MATE1 and MATE2-K, respectively. Selectivity was assessed for both transporters from IC50 values for 59 structurally diverse compounds. Whereas the two transporters discriminated markedly between a few of the test compounds, the IC50 values for MATE1 and MATE2-K were within a factor of 3 for most of them. For hMATE1 there was little or no correlation between IC50 values and the individual molecular descriptors LogP, total polar surface area, or pKa. The IC50 values were used to generate a common-features pharmacophore, quantitative pharmacophores for hMATE1, and a Bayesian model suggesting molecular features favoring and not favoring the interaction of ligands with hMATE1. The models identified hydrophobic regions, hydrogen bond donor and hydrogen bond acceptor sites, and an ionizable (cationic) feature as key determinants for ligand binding to MATE1. In summary, using a combined in vitro and computational approach, MATE1 and MATE2-K were found to have markedly overlapping selectivities for a broad range of cationic compounds, including representatives from seven novel drug classes of Food and Drug Administration-approved drugs.
Introduction
A key physiological function of the kidneys is clearing the body of a structurally diverse array of organic compounds, the majority of which are exogenous, i.e., xenobiotic, in origin. These include plant-derived compounds found in typical diets and, increasingly, clinically relevant synthetic pharmaceuticals. So-called organic cations (OCs), molecules that carry a net positive charge at physiological pH, are a particularly significant subset of pharmaceuticals because they make up approximately 40% of all prescribed drugs (cimetidine, procainamide, pindolol, and metformin) (Neuhoff et al., 2003). The basic cellular model of renal OC secretion in renal proximal tubule (RPT) cells, described first by Holohan and Ross (1981), includes the sequential activity of 1) a basolateral “entry step,” from blood to cell, that involves an electrogenic organic cation transporter (OCT) and 2) an apical “exit step,” from cell to tubular filtrate (that is both the active and rate-limiting step in secretion; Wright and Dantzler, 2004), mediated by electroneutral OC/H+ exchange. After the cloning in 1994 of the first organic cation transporter, OCT1 (Gründemann et al., 1994), there is now a broad consensus that, in the human kidney, the basolateral step in this process is dominated by the activity of organic cation transporter 2 (OCT2) (Motohashi et al., 2002; Wright and Dantzler, 2004). However, establishing the molecular basis of the apical element in renal OC secretion, i.e., OC/H+ exchange, proved to be more elusive.
The cloning in 2005 of the first mammalian members of the multidrug and toxin extruder (MATE) family of transport proteins (Otsuka et al., 2005) provided the first viable candidates for the molecular identity of the apical OC/H+ exchanger. MATE1 and MATE2 proved to display the “physiological fingerprint” of the apical element of renal (and hepatic) OC secretion: 1) substantial expression in the luminal membrane of RPT cells (and, for MATE1, canilicular membrane of hepatocytes), 2) support of OC/H+ exchange, and 3) transport of structurally diverse OCs. The quantitative link between MATE activity and renal OC secretion was then firmly established by the observation that elimination of Mate1 in mice significantly reduces renal clearance of metformin (Tsuda et al., 2009) and cephalexin (Watanabe et al., 2010).
A primary focus of studies of MATE function has been establishing the interaction of MATE transporters (typically MATE1) with specific structural classes of drugs (Yokoo et al., 2007; Ohta et al., 2009; Watanabe et al., 2010; Cutler et al., 2012). However, lacking in these observations is an effort to identify the molecular determinants of ligand (substrate/inhibitor) interaction with MATE transporters, including establishing the differential selectivity of MATE1 versus MATE2 (Masuda et al., 2006; Komatsu et al., 2011) or with its kidney-specific isoform, MATE2-K. The multispecificity of the MATEs makes them important potential targets for unwanted drug-drug interactions (Yonezawa and Inui, 2011), so understanding the transport mechanisms that underlie the processes of renal and hepatic OC clearance, including the determinants of selectivity, is particularly relevant to efforts to predict and pre-empt the unwanted outcomes of drug exposure.
Previously, combining in vitro data with computational modeling of transporters enabled the development of pharmacophores and quantitative structure/activity relationships that have facilitated understanding the molecular basis of ligand interaction with transport proteins (Bednarczyk et al., 2003; Suhre et al., 2005; Zolk et al., 2008; Kido et al., 2011). In the current study we used sequential rounds of pharmacophore development and searching of a comprehensive set of FDA-approved drugs to: 1) characterize the relative selectivity of MATE1 and MATE2-K for a set of clinically important OCs, 2) identify novel inhibitors of these two transporters, and 3) develop initial predictive models of MATE1 selectivity by using an in vitro/in silico (IVIS) method that involves successive, iterative steps in the model-building process.
Materials and Methods
Reagents
Platinum high-fidelity DNA polymerase, phleomycin, hygromycin, Flp recombinase expression plasmid (pOG44), Chinese hamster ovary (CHO) cells containing a single integrated Flp recombination target (FRT) site (CHO Flp-In), and the mammalian expression vector pcDNA5/FRT/V5-His TOPO were obtained from Invitrogen (Carlsbad, CA). Ham's F12 Kaign's modification cell culture medium and test inhibitors of MATE transport activity were obtained from Sigma (St. Louis, MO). [3H]1-methyl-4-phenylpyridinium ([3H]MPP; 80 Ci/mmol) was synthesized at the Department of Chemistry and Biochemistry, University of Arizona, Tucson.
Cell Culture and Stable Expression of hMATE1 and hMATE2-K
The full-length human MATE1 sequence used in this study was generously provided by Dr. Kathleen Giacomini (University of California, San Francisco, CA) (Chen et al., 2007). The full-length human MATE2-K sequence used in this study was generously provided by Dr. Ken-ichi Inui (Kyoto University, Kyoto, Japan) (Masuda et al., 2006). CHO cells containing the Flp recombination target site were grown in Ham's F12 Kaighn's modification medium supplemented with 10% fetal calf serum and 100 μg/ml phleomycin. Cultures were split every 3 days. Aliquots of 5 × 106 cells were transfected by electroporation (BTX ECM 630; BTX, San Diego; 260 V and time constant of ∼25 ms) with 10 μg of salmon sperm, 18 μg of pOG44, and 2 μg of pcDNA5/FRT/V5-His TOPO containing the open reading frame of either hMATE1 or hMATE2-K construct. Cells were seeded in a T-75 flask after transfection and maintained under selection pressure with hygromycin (100 μg/ml) for at least 2 weeks before use in transport studies.
Transport Experiments
CHO cells expressing hMATE1 or hMATE2-K were grown to confluence in multiwell (typically 24-well) plates. Before transport experiments, the media were aspirated, and the cells were rinsed twice, briefly, with room-temperature Waymouth buffer (WB) containing 135 mM NaCl, 28 mM d-glucose, 5 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 0.8 mM MgSO4, and 13 mM HEPES-NaOH, pH 8.5. Transport was measured at room temperature and initiated by adding transport solution containing WB with 1 μCi/ml [3H]MPP (∼10–20 nM) and, in studies assessing the kinetics of transport, increasing concentrations of unlabeled substrate or inhibitor. To reduce the inhibitory effect of extracellular H+ on MATE transport activity the pH of the transport buffer in these studies was typically 8.5 (the impact of pH on the kinetics of MATE transport is discussed under Results). The solubility of some of the test agents required that stock solutions be prepared in dimethyl sulfoxide (DMSO), ethanol, or methanol, resulting in the presence of these solutes in some experimental solutions. Preliminary experiments revealed that 2% DMSO, ethanol, or methanol (the highest concentration required to maintain solubility of selected agents) reduced the rate transport of [3H]MPP by up to 40%. Consequently, for those agents that required the presence of DMSO, ethanol, or methanol in the test solutions, all solutions, including parallel control experiments (when no inhibitor was present), contained 2% DMSO, ethanol, or methanol in WB. Because initial experiments showed that uptake of [3H]MPP was linear for ∼10 min (Dangprapai and Wright, 2011), 5-min uptakes were used to approximate the initial rate of transport for use in kinetic analyses. After the transport period, the solution was aspirated, and the wells were rinsed three times with 1 ml of ice-cold WB. The cells were solubilized in 0.2 ml of 0.5 N NaOH with 1% SDS (v/v), and the resulting lysate was neutralized with 0.1 ml of 1 N HCl. Accumulated radioactivity was determined by liquid scintillation spectrometry (Beckman model LS3801; Beckman Coulter, Fullerton, CA). Mediated (i.e., inhibitable) accumulation of [3H]MPP into wild-type CHO cells is typically less than 2% of the mediated uptake into cells stably expressing MATE1 (Zhang and Wright, 2009), so it was ignored. Individual transport observations were typically performed in duplicate for each experiment, and observations were usually confirmed at least three times in separate experiments by using cells of a different passage.
Physicochemical Descriptors
LogP (log of the octanol-water partition coefficient) values were calculated with the ALOGPS 2.1 package (Tetko et al., 2005), which displays values calculated with ALOGPs, MLogP (Moriguchi octanol-water partition coefficient; Dragon 5.4; Talete, Milan, Italy), ALogP (Ghose–Crippen octanol-water partition coefficient; Dragon 5.4; Talete), and KowWin LogP (Syracuse Research Corporation, Syracuse, NY). The average value of these LogP calculations was used for our analysis. The pKa values were calculated with the SPARC On-Line Calculator (http://archemcalc.com/sparc) (Hilal et al., 1996). Topological polar surface area (TPSA) was calculated by using the Interactive PSA calculator (http://www.molinspiration.com/services/psa.html).
Computational Modeling
Common-Features hMATE1 Pharmacophore Development.
A common-features pharmacophore was developed by using Accelrys Discovery Studio version 2.5.5 (Accelrys, San Diego, CA) following the approach taken previously with other transporters (Diao et al., 2009, 2010). Template molecule structures were downloaded from ChemSpider (www.chemspider.com), and conformer generation was carried out by using the CAESAR algorithm (Conformer Algorithm based on Energy Screening and Recursive Buildup; Li et al., 2007) applied to the selected template molecules (maximum of 255 conformations per molecule and maximum energy of 20 kcal/mol).
3D-Quantitative Structure/Activity Relationship development used the Hypogen method in Discovery Studio. hMATE1 IC50 values were used as the indicator of biological activity. In the HypoGen approach (Ekins et al., 2002; Bednarczyk et al., 2003), 10 hypotheses were generated by using hydrophobic, hydrogen bond acceptor, hydrogen bond donor, and the positive and negative ionizable features, and the CAESAR conformer generation method was used. After assessing all generated hypotheses, the hypothesis with the lowest energy cost was selected for further analysis, because this model possessed features representative of all of the hypotheses and had the lowest total cost. The total energy cost of the generated pharmacophore was calculated from the deviation between the estimated activity and the observed activity, combined with the complexity of the hypothesis (i.e., the number of pharmacophore features). A null hypothesis, which asssumed that there was no relationship between chemical features and biological activity, was also calculated. Therefore, the greater the difference between the energy cost of the generated and null hypotheses, the less likely the generated hypothesis reflects a chance correlation. In addition, the quality of the structure-activity correlation between the predicted and observed activity values was estimated via correlation coefficient.
Quantitative Model Update with Variable Weights and Tolerances.
We selected hydrogen bond acceptor, hydrogen bond donor, hydrophobicity, and positive ionizable and negative ionizable features for model building (using CAESAR for conformation generation). Variable weights and tolerances were used, and a maximum of 10 pharmacophores were selected. The pharmacophore with the best correlation (lowest root mean square error) was used for further analysis.
Classification Bayesian Models.
Laplacian-corrected Bayesian classifier models were generated by using Discovery Studio. Molecular function class fingerprints of maximum diameter 6 (FCFP_6), ALogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area were calculated from input sdf files using the “calculate molecular properties” protocol. The “create Bayesian model” protocol was used for model generation (Diao et al., 2010).
Results
The Kinetics of MPP and H+ Interaction with hMATE1 and hMATE2-K
The kinetics of MATE1- and MATE2-K-mediated MPP transport (Fig. 1, A and B) were adequately described by the Michaelis-Menten equation for competitive interaction of labeled and unlabeled substrate (Malo and Berteloot, 1991).
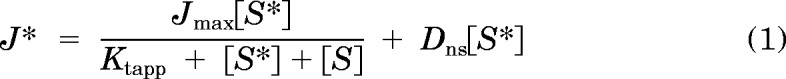 |
where J* is the rate of transport of the radiolabeled substrate (in this case, [3H]MPP) from a concentration of the labeled substrate equal to [S*]; Jmax is the maximal rate of mediated substrate transport; Ktapp is the apparent Michaelis constant of the transported substrate; [S] is the concentration of unlabeled substrate; and Dns is a first-order rate constant that describes the nonsaturable component of labeled substrate accumulation (reflecting the combined influence of diffusion, nonspecific binding, and incomplete rinsing of [3H]MPP from the cell culture well). In four separate experiments Ktapp and Jmax values for MATE1 and MATE2-K were 4.37 ± 0.32 and 3.72 ± 0.45 μM; and 2.14 ± 0.27 and 1.86 ± 0.28 pmol · cm−2 · min−1, respectively. Expressed per milligram of membrane protein, these Jmax values become 21.4 and 18.6 pmol · mg−1 · min−1 for MATE1 and MATE2-K, respectively.
Fig. 1.

Kinetic characteristics of transport mediated by hMATE1 and hMATE2-K expressed in CHO cells. A and B, kinetics of MPP transport mediated by hMATE1 (A) or hMATE2-K (B). C and D, effect of extracellular [H+] on MPP transport mediated by hMATE1 (C) or hMATE2-K (D). In all experiments, 5-min uptakes of [3H]MPP (∼13 nM) were measured in the presence of increasing concentrations of unlabeled MPP (A and B) at an external pH of 8.5 or increasing extracellular H+ concentrations (C and D). Each point is the mean (± S.E.M.) of uptakes measured in three wells of a 24-well plate from single representative experiments. Kinetic values shown represent the average of three to five experiments.
It is important to acknowledge that MATEs are sensitive to the intracellular and extracellular concentration of protons; therefore, we characterized the kinetics of H+ inhibition of [3H]MPP uptake. We previously showed that elevated concentrations of H+ in the extracellular solution inhibit transport mediated by hMATE1 (Dangprapai and Wright, 2011). Figure 1, C and D compares the pH sensitivity of MPP transport mediated by hMATE1 and hMATE2-K. As anticipated, transport activity of both proteins was inhibited by increasing concentrations of H+ in the extracellular solution and was described by the following relationship:
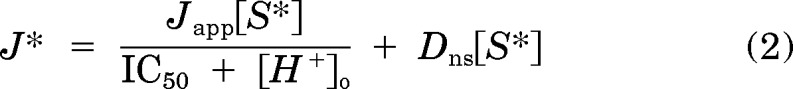 |
where Japp is the product of the maximum rate of S* (i.e., [3H]MPP) uptake (Jmax) and the ratio of the Ki (IC50) of H+ and Ktapp for MPP transport; and IC50 is the concentration of [H+]o (or other test inhibitor) that reduced mediated (i.e., blockable) [3H]MPP transport by 50%. In three experiments, the IC50 for H+-inhibitable hMATE1-mediated MPP uptake was 19.6 ± 0.7 nM, pH 7.73, similar to the value of 12.4 nM reported previously (Dangprapai and Wright, 2011). MATE2-K proved to be substantially more sensitive to H+, displaying an IC50 value of 3.5 ± 0.6 nM, pH 8.5 (n = 3).
In the upcoming examination of the kinetics of ligand interaction with MATE transporters we elected to maximize control rates of MATE-mediated transport by running experiments at an external pH of 8.5, rather than use the “ammonia prepulse” method to acidify the cytoplasm and thereby create an outwardly directed pH gradient and a stimulation of OC uptake (Otsuka et al., 2005; Masuda et al., 2006; Kajiwara et al., 2007; Tanihara et al., 2007; Yasujima et al., 2010). The acidification of the cytoplasm after an ammonia pulse is generally short-lived and constantly changing (Kapus et al., 1994) during the several-minute time courses used to measure the rate of MATE-mediated transport, and these ill-defined conditions complicate the interpretation of kinetic measurements. It is noteworthy that we previously showed that cytoplasmic pH is effectively constant (at a internal pH of 7.5–7.6) during exposure of CHO cells to an external pH of 8.5 (Dangprapai and Wright, 2011), so transmembrane H+ gradients were both outwardly directed and unchanging during our transport measurements. The rank order of ligand selectivity at pH 8.5 and 7.4 is similar, if not identical, for the two transporters, as supported by the similar rank order of uptake ratios for transport of a structurally diverse set of organic cations into hMATE1 and hMATE2-K at these two pH values (Tanihara et al., 2007). However, given the apparent pKa values for the interaction of the two MATE transporters with H+ evident in Fig. 1B, the absolute IC50 values for inhibition of MATE1 and MATE2-K activity measured at pH 8.5 can be expected to underestimate the values anticipated at pH 7.4, by approximately 3- to 6-fold, respectively.
Inhibitory Selectivity of hMATE1 and hMATE2-K Test Set Selection
The test compounds (Table 1) were selected to represent a structurally diverse collection of drug and drug-like compounds, the intention being to interrogate the complex chemical space expected to influence interaction with the binding regions of multiselective organic cation transporters, i.e., MATE1 and MATE2-K. Weak bases and cations dominated the battery of test compounds; neutral compounds and those having a net negative charge at physiological pH were largely excluded. This bias toward cations reflected the existing database from the early literature on transport in isolated renal membranes (Holohan and Ross, 1980, 1981; Wright et al., 1995) and in intact renal tubules (McKinney, 1983; Dantzler et al., 1989; David et al., 1995), and from more recent work with MATE transporters (Tanihara et al., 2007), showing that cationic charge is a key criterion of ligand interaction with these processes (a conclusion supported by the present study, as documented below). Indeed, it was a specific goal of this study to identify molecular determinants of interaction of organic cations with MATE1 and MATE2-K. The final battery of organic compounds (Table 1) included 23 from the list of compounds generated by Ahlin et al. (2008) in their study of the selectivity of OCT1; 13 were selected because of previous evidence of their interaction with OC/H+ exchange activity in either native renal membranes, intact tubules, brush border membrane vesicles, or heterologous expression systems expressing MATE1 or MATE2-K (David et al., 1995; Wright et al., 1995; Ullrich and Rumrich, 1996; Wright and Wunz, 1998, 1999; Tanihara et al., 2007); and 23 were selected from lists of target hits from databases of compounds that were interrogated by the pharmacophore models developed during the course of this study.
TABLE 1.
IC50 values for inhibition of MPP transport into CHO cells stably expressed with either hMATE1 or hMATE2-K
Each value determined at pH 8.5 is a mean IC50 (in μM; ± S.E.M.) of two to four experiments. IC50 values listed for pH 7.4 were calculated. Underlined compounds indicate the compounds that comprised the initial set of 24 organic molecules used is in the first iteration of pharamacophore development; superscript a indicates the 15 compounds used to test the common features pharmacophore; and italicized compounds are those identified by the pharmacophores from the list of FDA-approved drugs during the course of model development.
| Compound | Drug Class | MATE1 IC50 pH 8.5 | MATE1 Calculated IC50 pH 7.4 | MATE2-K IC50 pH 8.5 | MATE2-K Calculated IC50 pH 7.4 |
|---|---|---|---|---|---|
| μM | μM | ||||
| Agmatine | NT | 53.9 ± 1.4 | 140 | 60.8 ± 9.1 | 400 |
| Allopurinol | XAI | NI | N.A. | NI | N.A. |
| Amantadine | AV | 7.53 ± 1.49 | 20 | 88.9 ± 9.0 | 580 |
| Amiloride | DI | 2.41 ± 0.18 | 6.3 | 3.06 ± 0.62 | 20 |
| APMI | PAL | 6.21 ± 0.26 | 16 | 0.501 ± 0.224 | 3.3 |
| Atropine | AC | 5.9 ± 1.3 | 15 | 52.8 ± 13.7 | 340 |
| Baclofen | AS, MR | NI | N.A. | NI | N.A. |
| Caffeine | S | 1096 ± 43 | 2900 | 451 ± 120 | 2900 |
| Chloramphenicol | AB | 1114 ± 156 | 2900 | 1951 ± 45 | 13,000 |
| Cinchonidinea | SC | 0.927 ± 0.154 | 2.4 | 4.38 ± 1.16 | 27 |
| Cinchoninea | SC | 1.86 ± 0.38 | 4.8 | 0.939 ± 0.043 | 5.8 |
| Cisplatin | CT | NI | N.A. | NI | N.A. |
| Clonidine | A2A | 8.09 ± 0.79 | 21 | 54.0 ± 1.3 | 350 |
| Creatinine | MBP | 195 ± 39 | 510 | 150 ± 26 | 980 |
| Ethohexadiola | IR | >2000 | N.A. | >1000 | N.A. |
| Famotidinea | H2RA | 2.16 ± 0.39 | 5.6 | 6.28 ± 0.45 | 39 |
| Guanfacine | A2AN | 3.5 ± 1.2 | 9.2 | 218 ± 64 | 1300 |
| Guanidine | MBP | >2100 | 5400 | >4000 | N.A. |
| H+ | EI | 0.020 ± 0.001 | N.A. | 0.004 ± 0.001 | N.A. |
| Histamine | IRR, NT | 761 ± 196 | 2000 | 775 ± 148 | 5000 |
| Imiquimoda | IMR | 13.9 ± 5.9 | 36 | 19.1 ± 5.7 | 120 |
| Ketoconazole | AF | 1.33 ± 0.17 | 3.4 | 9.33 ± 0.81 | 58 |
| Metformin | AD | 47.0 ± 2.2 | 120 | 89.3 ± 17.7 | 580 |
| Midodrinea | VP | 109 ± 18 | 280 | 87.1 ± 38.4 | 540 |
| MPP | NTX | 4.70 ± 0.55 | 12 | 3.3 ± 0.2 | 21 |
| Naloxonea | OAN | 24.1 ± 3.7 | 62 | 43.2 ± 12.9 | 280 |
| Nialamidea | MAOI | 212 ± 5 | 550 | 236 ± 31 | 1500 |
| Nicotine | S | 167 ± 37 | 440 | 134 ± 28 | 870 |
| Nicotinamide mononucleotide | POC | 147 ± 81 | 390 | 46.3 ± 5.3 | 500 |
| Paraquat | H | 50.5 ± 7.7 | 130 | 15.5 ± 1.3 | 100 |
| Phenformin | AD | 6.10 ± 0.19 | 16 | 11.2 ± 2.8 | 73 |
| Phentolaminea | AAA | 4.64 ± 0.37 | 12 | 5.3 ± 0.5 | 33 |
| Procainamide | AA | 24.0 ± 1.3 | 63 | 19.1 ± 3.4 | 120 |
| Proguanila | AM | 4.35 ± 1.70 | 11 | 1.39 ± 0.51 | 9.0 |
| Propranolola | BB | 7.81 ± 038 | 20 | 7.71 ± 0.10 | 48 |
| PYR | AM | 0.04 ± 0.01 | 0.11 | 0.059 ± 0.015 | 0.38 |
| PYR-2 | SC | 0.14 ± 0.01 | 0.35 | 0.068 ± 0.026 | 0.44 |
| PYR-3 | SC | 0.20 ± 0.01 | 0.51 | 0.058 ± 0.009 | 0.38 |
| Quinidine | AA | 1.57 ± 0.32 | 4.1 | 1.47 ± 0.27 | 9.6 |
| Quininea | AA | 1.90 ± 0.41 | 5.0 | 6.4 ± 1.7 | 42 |
| Ranitidine | H2RA | 5.40 ± 1.02 | 14 | 10.0 ± 1.9 | 65 |
| Scopolaminea | AC, AMS | 47.6 ± 4.8 | 120 | 272 ± 25 | 1700 |
| Serotonin | MA, NT | 28.8 ± 5.3 | 75 | 18.3 ± 1.4 | 120 |
| Sulfadimethoxine | AB | >>1000 | N.A. | >>1000 | N.A. |
| Sulfamerazine | AB | >>1000 | N.A. | >>1000 | N.A. |
| Tacrine | ACS | 0.51 ± 0.10 | 1.3 | 1.10 ± 0.21 | 7.2 |
| Tetrabutylammonium | SC | 11.6 ± 1.8 | 30 | N.A. | N.A. |
| TEA | POC | 40.6 ± 4.6 | 110 | 14.4 ± 8.8 | 94 |
| Triethylmethylammonium | SC | 51.6 ± 10.2 | 140 | 28.4 ± 0.7 | 180 |
| Ticlopidine | AP | >1000 | N.A. | >1000 | N.A. |
| Tetramethylammonium | SC | >5000 | N.A. | N.A. | N.A. |
| Topiramatea | ACT | ∼1000 | N.A. | NI | N.A. |
| TPeA | SC | 6.34 ± 0.23 | 16 | N.A. | N.A. |
| Tetrapropylammonium | SC | 14.8 ± 4.0 | 39 | N.A. | N.A. |
| Tramadol | OA | 17.8 ± 1.6 | 46 | 74.6 ± 17.6 | 490 |
| Trichlormethiazidea | DI | 249 ± 10 | 640 | 679 ± 10 | 4200 |
| Trimethoprim | AB | 9.70 ± 1.2 | 225 | 2.61 ± 1.12 | 17 |
| Tryptophan | AAM | NI | N.A. | NI | N.A. |
| Tyramine | MA | 86.6 ± 8.2 | 230 | 138 ± 11 | 900 |
| Verapamil | LTCB | 41.9 ± 6.8 | 110 | 37.9 ± 9.7 | 250 |
N.A., not available; A2A, α-2 adrenergic agonist; A2AN, α-2A norepinephrin receptor agonist; AA, antiarrhythmic; AAA, α adrenergic antagonist; AAC, amino acid; AB, antibiotic; AC, anticholinergic; ACS, anticholinesterase; ACT, anticonvulsant; AD, antidiabetic; AF, antifungal; AM, antimalarial; AMS, antimuscurinic; AP, antiplatelet; AS, antispastic; AV, antiviral; BB, β blocker; CT, chemotherapy drug; DI, diuretic; EI, endogenous ion; H, herbicide; H2RA, histamine H2-receptor antagonist; IMR, immune response modifier; IR, insect repellent; IRR, immune response regulator; LTCB, L-type calcium blocker; MA, monoamine; MAOI, monoamine oxidase inhibitor; MBP, metabolism by-product; MS, muscle relaxant; NT, neurotransmitter; NTX, neurotoxin; OA, opiate agonist; OAN, opiate antagonist; PAL, photoaffinity label; POC, prototypical organic cation; S, stimulant; SC, synthetic chemical; VP, vasopressor; XOI, xanthine oxidase inhibitor; NI, no interaction.
Figure 2 shows the range of inhibition of MATE-mediated transport activity produced by the 59 organic test compounds. At inhibitor concentrations of 10 μM, MPP transport was reduced by ≥50% by 20 (MATE1) or 14 (MATE2-K) of these compounds. Figure 3 shows inhibitory profiles against transport activity of MATE1 and MATE2-K produced by four compounds (quinidine, agmatine, nialamide, and allopurinol) selected to emphasize the spectrum of inhibitory effectiveness of this battery of test agents, with IC50 values (determined by using eq. 2) that ranged from low micromolar (quinidine), through near millimolar (nialamide), to no effective interaction at all (allopurinol). Table 1 lists the IC50 values (as measured at pH 8.5, and as calculated for pH 7.4 using the apparent IC50 values for H+ interaction with the transporters shown in Fig. 1, C and D) for all 59 organic molecules used to inhibit transport activity of one or both MATE transporters.
Fig. 2.

Range of inhibition of transport mediated by the human orthologs of MATE1 (A) and MATE2-K (B) produced by the battery of test compounds used in this study. The height of the gray bars indicates the degree of inhibition of mediated uptake (5 min) of [3H]MPP (∼13 nM) produced by 10 μM concentrations of 59 test compounds. The horizontal dashed lines indicate 50% inhibition of transport.
Fig. 3.

The effect on the rate of hMATE1- and hMATE2-K-mediated MPP transport of increasing concentration of representative test inhibitors. ● represent hMATE1, ○ represent hMATE2-K, and they show inhibition produced by quinidine (A), agmatine (B), nialamide (C), and allopurinol (D). Each point represents the mean value (± S.E.M.; determined in three separate experiments) of the 5-min uptake of [3H]MPP (∼13 nM) measured in the presence of increasing concentration of test inhibitor; uptakes were normalized to that measured in the absence of inhibitor. NI, no interaction.
As inferred from the results presented in Figs. 1 to 3, there was substantial overlap in the interaction of the test compounds with MATE1 and MATE2-K. The extent of this overlap is evident in the comparison of the MATE1 and MATE2-K IC50 values for the 59 organic molecules used to probe both transporters (Fig. 4A); 75% of these agents had IC50 values for the two transporters that differed by less than a factor of 3 at pH 8.5 (61% of the compounds, based on the calculated IC50 values at pH 7.4). MATE1 and MATE2-K did, however, markedly discriminate (ratio of IC50 values >5) between eight of the compounds examined in this study. For example, as shown in Fig. 4B, the apparent affinities of hMATE1 for atropine (IC50 of 5.90 ± 1.31 μM) and amantadine (7.50 ± 1.49 μM) were ∼10 times greater than those displayed by MATE2-K (52.8 ± 13.7 and 88.9 ± 9.0 μM, for atropine and amantadine, respectively); whereas the apparent affinity of hMATE2-K for azidoprocainamide (APMI) (Mol et al., 1989) was ∼10 times greater (IC50 of 0.50 ± 0.22 μM) than that displayed by hMATE1 (6.2 ± 0.3 μM; Fig. 4B). Despite these differences, the data in hand support the view that MATE1 and MATE2-K show far more similarities in selectivity than differences.
Fig. 4.

Comparison of hMATE1 and hMATE2-K IC50 values. A, hMATE1 IC50 values were graphed as a function of hMATE2-K IC50 values for the 59 test compounds measured at pH 8.5 (left) and as calculated for pH 7.4 (right). The solid lines represent equal IC50 values for the two transporters; the dashed lines indicate 3-fold ± differences in these values. B, inhibitory profiles for several test ligands (atropine, amantadine, and APMI, left to right) against MPP transport mediated by hMATE1 (●) and hMATE2-K (○). Each point is the mean of 5-min uptakes (± S.E.M.; normalized to uptake measured in the absence of inhibitor) determined in three separate experiments, each run in duplicate. IC50 values are average values from three separate experiments.
Modeling of MATE1 Selectivity
Influence of Selected Molecular Descriptors on MATE1-Mediated Transport Activity.
Figure 5 shows the log of the IC50 values for inhibition of hMATE1 activity graphed as a function of several commonly applied molecular descriptors for the test agents in this study. There was a significant, albeit weak, correlation between hMATE1 IC50 values and LogP (r value of 0.332; p < 0.05; Fig. 5A). It is noteworthy that when the IC50 values for inhibition of MATE1 were restricted to a structurally constrained subset of the test agents of the present study, i.e., an n-tetraalkylammonium series, the influence of LogP was more evident [r = 0.97 for tetraethylammonium (TEA) through tetrapentylammonium (TPeA); Fig. 5B]. There was no correlation between TPSA and hMATE1 IC50 (r value of 0.045; p > 0.05; Fig. 5C), and a modest, albeit significant, correlation between pKa and hMATE1 IC50 values (r = 0.423; p < 0.01; Fig. 5D).
Fig. 5.

A, C, and D, relationship between hMATE1 IC50 values and the molecular descriptors LogP (A), TPSA (C), and pKa (D). B, the relationship between LogP and the IC50 values for a structurally constrained n-tetraalkylammonium series (TMA, tetramethylammonium; TEA; TPrA, tetrapropylammonium; TBA, tetrabutylammonium; and TPeA).
Computational Analysis of hMATE1 Inhibition.
The value of the approach offered by computational assessment of structure/activity relationships is that it may enable more insight into the molecular basis of ligand interaction than a view restricted to the influence of single physicochemical parameters alone. Following previous in silico modeling efforts on drug transporters (Suhre et al., 2005; Diao et al., 2009, 2010) we extended our IVIS strategy in this study to the use of multiple iterations of model development. The IVIS approach uses a comparatively small amount of in vitro data for development of an initial model that is then used to screen a database of potential additional compounds for testing. The results obtained from these initial tests feed into a further round of model building and database searching, and so on. The advantage of such an approach is the model is codeveloped with data acquisition, and it is validated and tuned with each additional set of test compounds. The approach does not require a large library of compounds to be tested and can save reagents and money (associated with testing many inactive compounds). Using this pharmacophore approach may suggest nonintuitive compounds as inhibitors because, although they include one or more of the initially mapped features, they may still prove to be low-affinity ligands (owing to the absence of what proves to be a missing critical feature). This in particular may be another potentially valuable side effect of the approach, enabling us to find novel compounds that may have a dissimilar two-dimensional structure, but similar 3D shape to known inhibitors or different mapping to pharmacophore features.
Initial Round: hMATE1 Common-Features Pharmacophore.
There were 26 compounds in the initial round of inhibitors studied (Table 1, underlined), from which five were selected to generate a common features pharmacophore: two high-affinity compounds, pyrimethamine (PYR; IC50 of 0.04 μM) and quinidine (IC50 of 1.57 μM), and three low-affinity compounds, histamine (IC50 of 761 μM), caffeine (IC50 of 1096 μM), and chloramphenicol (IC50 of 1115 μM) (Fig. 6A). By restricting the initial set of test compounds to these extremes (very high affinity versus very low affinity), the intent was to identify key features that may influence effective interaction with the transporter. In other words, common molecular and chemical features of the high-affinity substrates were included in the pharmacophore, whereas the molecular features of the low-affinity substrates were excluded from the pharmacophore. The resulting common-features pharmacophore had two hydrophobic regions, one H-bond donor, and one H-bond acceptor. The pharmacophore is depicted as an overlay of the structure of PYR (the highest-affinity substrate; Fig. 6B).
Fig. 6.

Inhibitory profiles for the five test ligands (A) used to generate a common-features pharmacophore (B). Each point is the mean of 5-min uptake (normalized to the transport measured in the absence of inhibitor) determined in single representative experiments with PYR (▵), quinidine (▴), histamine (♦), caffeine (■), and chloramphenicol (●). The common-features pharmacophore (displayed with the structure of PYR) includes two hydrophobic regions (cyan), one H-bond donor (magenta), and one H-bond acceptor (green).
First Iteration: hMATE1 Common-Features Pharmacophore Testing.
The common-features pharmacophore, with the van der Waals surface of PYR to provide a shape restriction (the two-dimensional molecule structures and Discovery Studio pharmacophores are available on request from the authors), was used to search a 3D database of 2690 FDA-approved compounds (www.collaborativedrug.com) and identified 126 molecules as potential inhibitors (see Supplemental Table 1). Fifteen commercially available compounds were selected from this list and tested as inhibitors of MATE1 (and MATE2-K), and the resulting IC50 values are presented in Table 1 (identified with the superscript a). Nine compounds in this test set proved to be comparatively high-affinity inhibitors of MATE1 (IC50 values of 0.9–25 μM), whereas four displayed modest affinity (IC50 values of 26–300 μM), two had weak interactions (<80% inhibition at 1000 μM), and one exerted no inhibition of either transporter at a concentration of 1 mM.
The inhibitory profiles produced by two of the 15 compounds in this test set, cinchonidine and ethohexadiol (Fig. 7, A and B), provided particular insight into the molecular determinants associated with ligand interaction with MATE1. Although both molecules were good fits for the common-features pharmacophore (Fig. 7, C and D; fit values of 3.1 and 2.7, respectively; data not shown), cinchonidine was an effective, high-affinity inhibitor of hMATE1 (IC50 = 0.93 μM; Fig. 7A), whereas ethohexadiol (Fig. 7B) had a weak interaction with hMATE1 (∼20% inhibition at a concentration of 1 mM). Thus, the presence in ethohexadiol of the common structural features of the initial pharmacophore, the two hydrophobic regions, one H-bond donor site, and one H-bond acceptor site, was not sufficient for an effective inhibitory interaction with MATE1. It is noteworthy that ethohexadiol is not an organic cation; its presence in the hit list reflected the absence of a cationic feature in the common-features pharmacophore that, in turn, reflected the presence of such a feature in all three of the weak inhibitors of MATE1 activity used to generate the pharmacophore. In other words, whereas a cationic feature was not sufficient to insure a strong interaction with MATE1, these data argue that charge does exert a strong, permissive influence on the binding interaction.
Fig. 7.

A and B, kinetics of inhibition of hMATE1-mediated MPP transport produced by cinchonidine (A) and ethohexadiol (B). Each point is mean (± S.E.M.) of 5-min uptake measured in triplicate in single representative experiments. IC50 values are average values from three separate experiments. C and D, common-features pharmacophores are shown for cinchonidine (C) and ethohexadiol (D).
First Iteration: Quantitative Pharmacophore Development for hMATE1.
We generated a quantitative pharmacophore in parallel to the common-features hMATE1 pharmacophore, taking advantage of the broad range of activities (IC50 values from 40 nM to ≫1 mM) displayed by the initial round of inhibitors. Twenty-four of the initial 26 compounds (H+, because of its restricted size, and verapamil, because it was a racemic mixture, were not included) were used in an analysis that resulted in a model containing two hydrophobic features, one hydrogen-bond donor, and, unlike the common features pharmacophore, one positive ionizable feature (Fig. 8A). The model had a small cost difference as total cost = 125.97 and null cost = 137.57, suggesting a modest quality model. Nevertheless, unlike the modeling efforts based on single physical descriptors (Fig. 5), the correlation between observed and predicted IC50 values resulted in r = 0.68 (p < 0.0001; Fig. 8B).
Fig. 8.

A, quantitative pharmacophore generated from analysis of data obtained by using the first round of hMATE1 inhibitors (see “First Iteration: Quantitative Pharmacophore Development for hMATE1” in Results). Twenty four of the initial 26 compounds were used in an analysis that resulted in a model containing two hydrophobic features (cyan), one hydrogen bond donor (magenta), and one positive ionizable feature (red), shown here with the structure of cinchonidine. B, the relationship between measured and predicted IC50 values based on the model shown in A (r = 0.68; p < 0.0001). C, quantitative pharmacophore generated from analysis of the data that incorporated the second round of hMATE1 inhibitors. Analysis of 43 compounds (including the initial 24 plus the test set of 15 compounds that probed the common features model) resulted in a model that included two hydrophobes (cyan), two hydrogen bond acceptors (green), and an ionizable feature (red). D, the relationship between measured and predicted IC50 values based on the model shown in C (r = 0.71; p < 0.0001). E, quantitative pharmacophore generated from analysis of 46 of the 59 test ligands (see “Final Iteration: Quantitative Pharmacophore Development for hMATE1” in Results for inclusion criteria). The model included two hydrophobes (cyan), a hydrogen bond acceptor (magenta), and an ionizable feature (red). F, the relationship between measured and predicted IC50 values based on the N46 model (r = 0.73; p < 0.0001). Quinidine is mapped to all pharmacophores.
Second Iteration: Quantitative Pharmacophore Development for hMATE1.
Of the 39 compounds (the initial 24 and the test set of additional compounds derived from searching the database of FDA-approved compounds) used to generate and validate the two pharmacophores, PYR was the most potent inhibitor of hMATE1 (and MATE2-K). Consequently, we chose to probe two structural analogs of PYR: (5-(4-chlorophenyl)-6-ethyl-2,4-pyrimidinediamine):1-(2-chlorophenyl)-6,6-dimethyl-1,6-dihydro-1,3,5-triazine-2,4-diamine (PYR-2) and 1-(3-chlorophenyl)-6,6-dimethyl-1,6-dihydro-1,3,5-triazine-2,4-diamine (PYR-3). The IC50 values of 0.04, 0.14, and 0.20 μM for PYR, PYR-2, and PYR-3, respectively (Table 1), showed that the modest differences in structure between these three compounds had comparatively little impact on their inhibitory interactions with MATE1 and suggested that the structural features of this series of compounds may provide insight into molecular characteristics that optimize ligand interactions with the binding site/surface transport of the protein. A second quantitative pharmacophore model for hMATE1 reflecting these data (a total of 43 compounds) was generated (Fig. 8C) and also included two hydrophobes, two hydrogen-bond acceptors, and an ionizable feature (Fig. 8C). The correlation between observed and predicted IC50 values resulted in an r value of 0.71 (p < 0.0001; Fig. 8D).
Final Iteration: Quantitative Pharmacophore Development for hMATE1.
We ultimately screened 59 compounds, adding several novel structural groups including the n-tetraalkylammonium series mentioned earlier. To minimize interpretational issues associated with compounds that were weakly ionized at the experimental pH of 8.5 we chose to eliminate for modeling analysis all compounds with pKa values below 8.0 (a total of 13). The result was a final pharmacophore based on 46 structures and depicted in Fig. 8E. This N46 model had several features in common with the previous iteration models, in that it included two hydrophobes, a hydrogen bond-acceptor, and an ionizable feature, although the spatial distribution of these elements differed somewhat from the previous models. Figure 8F displays the relationship between measured and predicted IC50 values based on the N46 model (r value of 0.73; p < 0.0001).
Bayesian Model.
A Bayesian model for MATE 1 (at pH 8.5) was generated by using the N46 set of molecules; the receiver operator characteristic was 0.88. After leave-out 50% × 100 this value is 0.82 (concordance = 82.6 ± 4.7%; specificity = 83.9 ± 5.5%; selectivity = 66 ± 7.5%). These results suggest the model is stable.
Use of the molecular “function class fingerprints of maximum diameter 6” (FCFP_6) descriptors allowed the identification of molecular features that favored inhibition (Fig. 9A), as well as features that did not promote inhibition (Fig. 9B). Particularly noteworthy were the inclusion of nitrogen-containing six-membered rings as Bayesian good features and the exclusion of nitrogen-containing five-membered rings (pyrrole). The probable distinguishing characteristic between the two groups is the typically low basicity of the pyrrole moiety. In other words, pyrrole-containing structures generally are not cationic at physiological pH, again underscoring the importance of charge for increasing the effectiveness of ligand interaction with MATE transporters.
Fig. 9.

A, FCFP_6 features associated with hMATE1 inhibitors–pH 8.5 N46 model. B, FCFP_6 features associated with hMATE1 noninhibitors–pH 8.5 N46 model. Each panel shows the naming convention for one fragment, the numbers of compounds containing the fragment, and the Bayesian score for the fragment.
Discussion
The current study is the most complete to date on the selectivity of the human orthologs of MATE1 and MATE2-K, both of which are likely to play key roles in the renal secretion of organic cations. Previous data on the selectivity of human MATEs are sufficiently sparse that it is difficult to compare our observations with those in the literature. Nevertheless, a few comparisons are noteworthy for their agreement with the observations reported here. The antimalarial drug PYR, the highest-affinity inhibitor in the present study (IC50 of 42 nM; Table 1), was reported to have an IC50 of 93 nM against hMATE1-mediated metformin transport (Kusuhara et al., 2011), very similar to the IC50 value we predicted for inhibition at pH 7.4 of MATE1-mediated MPP transport (IC50 at pH 7.4 of 109 nM). In addition, both hMATE1 and hMATE2-K are known to transport the antidiabetic drug metformin with Ktapp values of 238 μM (Meyer zu Schwabedissen et al., 2010) and 1.1 mM (Masuda et al., 2006), respectively, not unlike the calculated IC50 at pH 7.4 values we measured here (123 and 581 μM). But not all of the comparisons of the present observations corresponded so closely to those observed in previous studies. For example, whereas tacrine had an IC50 value of 0.6 μM for inhibition of hMATE1-mediated MPP transport, which agreed well with the 1.1 μM value recently reported (Kido et al., 2011) for inhibition of hMATE1-mediated transport of 4–4-dimethlaminostyryl)-N-methyl-pyridinium (ASP), the IC50 of 1.1 μM for tacrine inhibition of hMATE2-K-mediated MPP transport contrasted sharply with the value of >100 μM for the inhibition of ASP transport by MATE2-K (Kido et al., 2011). The experimental conditions used in the aforementioned study (i.e., human embryonic kidney 293 cells at pH 7.4) differed from those used here and could account for some of the difference. However, we suggest it is more plausible that these differences are caused by the use of MPP as the transported substrate versus ASP (Kido et al., 2011), and we provide support for this suggestion later in this article.
The present observations support several conclusions concerning the molecular basis of selectivity of the mammalian MATEs. First, they support the hypothesis that no single physicochemical parameter of ligand structure is likely to provide an adequate predictor of interaction with MATE1 or MATE2-K. Previous studies of inhibition of OC/H+ exchange activity in isolated renal brush border membrane vesicles (Wright et al., 1995; Wright and Wunz, 1998, 1999) and intact microperfused renal proximal tubules (Ullrich et al., 1991, 1992; David et al., 1995; Somogyi et al., 1996; Ullrich and Rumrich, 1996) sought to correlate inhibitor effectiveness with selected, single physicochemical characteristics of the test agents included in these studies, such as LogP (hydrophobicity) and pKa (basicity). In fact, the IC50 for inhibition of OC/H+ exchange activity in rabbit renal brush border membrane vesicles (Wright and Wunz, 1999) and intact rat RPT (David et al., 1995) was shown to be strongly correlated with LogP, but the inhibitors used in those studies were more structurally constrained than those used in the present work and that may have masked the influence of steric (and other physicochemical) parameters on ligand interaction with the transporters. In support of this interpretation, the correlation between IC50 and LogP for the n-tetraalkylammonium series was clearly evident (Fig. 5B). We were not surprised that single molecular descriptors were not particularly effective predictors of inhibitory interaction with the MATEs. Given the broad structural diversity of compounds that interact effectively with the MATEs, it is probable that binding is a more complex process requiring multiple molecular interactions and therefore single physicochemical properties alone are likely to be of limited predictive value.
The second set of conclusions arising from the present work stems from the application of computational methods to identify several physicochemical parameters that influence ligand binding to MATE1, namely, the presence and location of multiple hydrophobic moieties, hydrogen donors, and an ionizable (i.e., cationic) feature. With respect to these two latter points, a recent study by Kido et al. (2011) that screened some 900+ compounds for inhibitory interaction with human OCT2 noted that inhibitory effectiveness was particularly influenced by 1) ligand liphophilicity, 2) average charge, 3) molecular volume, 4) TPSA, and 5) the number of hydrogen bond donors and acceptors. In our study the several iterations of pharmacophore development led to the identification and subsequent verification of several novel clinical classes of compounds as MATE ligands. Table 2 identifies 12 drug classes not previously shown to interact with the human MATEs; seven of them displayed IC50 values less than 25 μM, three of which (ketoconazole, proguanil, and imiquimod) represented a ratio of maximum plasma concentration (Cmax) versus IC50 of <0.1.
TABLE 2.
MATE1 inhibitors from novel drug classes identified during the course of pharmacophore development
| Inhibitor | Class | IC50 |
|---|---|---|
| μM | ||
| Ketoconazole | AF | 1.3 |
| Proguanil | PAM | 4.4 |
| Phentolamine | AAA | 4.6 |
| Propranolol | BB | 7.8 |
| lmiquimod | IMR | 13.9 |
| Tramadol | OA | 17.8 |
| Naloxone | OAN | 24.1 |
| Midodrine | VP | 109 |
| Nialamide | MAOI | 212 |
| Topiramate | ACT | 1000 |
| Ticlopidine | AP | 1000 |
| Ethohexadiol | IR | 2000 |
AF, antifungal; PAM, prophylactic antimalarial; AAA, α adrenergic antagonist; BB, β blocker; IMR, immune response modifier; OA, opiate agonist; OAN, opiate antagonist; VP, vasopressor; MAOI, monoamine oxidase inhibitor; ACT, anticonvulsant; AP, antiplatelet; IR, insect repellant.
The final pharmacophore (Fig. 8, E and F) provides insight concerning the molecular basis of ligand interaction with the MATEs. Whereas the agreement between “predicted” and “measured” IC50 values for 65% of the test compounds was within a factor of 5, for others the model displayed substantially less predictive capability. For example, the prototypic substrates of MATE1, MPP and TEA, were predicted to have IC50 values some 10 to 15 times greater than their measured values of 5 and 50 μM, respectively. These “misses” may reflect an underlying assumption of pharamcophore analysis, namely, that there is a unique most effective structure for interaction of ligand with a binding site, with the pharmacophore representing both the location and physicochemical character of chemical features important for interaction with that singular site. However, the physiological role of MATE transporters requires that they interact effectively with a multitude of structurally diverse compounds, a characteristic that is, arguably, inconsistent with the existence of a single site for substrate/inhibitor interaction. Instead, we suggest our data are consistent with inhibitory ligand interactions at several structurally distinct sites that overlap with the areas most favored for interaction with MPP. In this view, the pharmacophores represent a statistical average of the influence of selected structural features of test ligands on the inhibition of transport of a specific test probe. Thus, the inhibitory effectiveness of these ligands is likely to be influenced by the structural features of several distinct and potentially very different sites, a situation that would be difficult to describe with a single pharmacophore. A corollary to this suggestion is the hypothesis that the profiles of inhibition of structurally distinct substrates could result in distinct pharmacophores. Evidence in support of this idea was found in a preliminary analysis of data reported in the recent study by Kido et al. (2011) that included IC50 values for the inhibition of MATE1-mediated transport of the fluorescent OC, ASP. We used these IC50 values (six compounds, spanning 2.5 orders of magnitude) to generate for hMATE1 a quantitative pharmacophore that proved to have a fundamentally different structure than those we generated based on inhibition of [3H]MPP transport (Fig. 10). The quantitative pharmacophore based on inhibition of ASP transport had three hydrophobes, two hydrogen bond acceptors, and three excluded volumes (Fig. 10A) arranged in a spatial configuration that differed substantially from that of our model (Fig. 10B). These data suggest that a comprehensive assessment of selectivity of MATEs (and other multidrug transporters) may require the use of several structurally distinct substrates. This situation is analogous to what has been observed for the enzyme CYP3A4 (Kenworthy et al., 1999).
Fig. 10.

hMATE1 quantitative pharmacophores based on the inhibition of MATE1-mediated transport of ASP (A) or MPP (B). The ASP-derived pharmacophore (with ondansetron) had an r = 0.95 and was generated by using a data set obtained from Kido et al. (2011) that consisted of six compounds with IC50 values ranging from 0.15 to 66 μM. The MPP-derived N46 pharmacophore (with quinidine) was developed as described in “Computational Modeling” in Materials and Methods, and in Fig. 8E for the other pharmacophores and in Fig. 8E. Pharmacophore features are as described in Fig. 8 (with the addition of gray features indicating excluded volumes).
In summary, we have generated the first relatively large database for ligand inhibition of hMATE1 and hMATE2-K. Applying these data over the course of several computational modeling iterations using the IVIS approach resulted in a series of pharmacophores for hMATE1. The hMATE1 pharmacophores identified key structural features strongly correlated with ligand binding to hMATE1. The observations also supported the view that inhibitory profiles derived from the use of structurally distinct transported substrates can result in distinct pharmacophores, consistent with the contention that hMATE1 may have a complex binding surface for ligand interaction, rather than a single binding site.
Supplementary Material
Acknowledgments
S.E. thanks Accelrys for providing access to Discovery Studio.
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant 1R01DK080801; National Research Service Award DK752422]; the National Institutes of Health National Institute of Environmental Health Sciences [Grant 5P30ES006694]; and the National Institutes of Health National Heart, Lung, and Blood Institute [Grant 5T32HL07249].
S.E. consults for Collaborative Drug Discovery, Inc.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- OC
- organic cation
- OCT
- OC transporter
- MATE
- multidrug and toxin extruder
- hMATE
- human MATE
- RPT
- renal proximal tubule
- MPP
- 1-methyl-4-phenylpyridinium
- FDA
- Food and Drug Administration
- CHO
- Chinese hamster ovary
- FRT
- Flp recombination target
- WB
- Waymouth buffer
- DMSO
- dimethyl sulfoxide
- IVIS
- in vitro/in silico
- ASP
- 4–4-dimethlaminostyryl)-N-methyl-pyridinium
- PYR
- pyrimethamine
- PYR-2
- (5-(4-chlorophenyl)-6-ethyl-2,4-pyrimidinediamine):1-(2-chlorophenyl)-6,6-dimethyl-1,6-dihydro-1,3,5-triazine-2,4-diamine
- PYR-3
- 1-(3-chlorophenyl)-6,6-dimethyl-1,6-dihydro-1,3,5-triazine-2,4-diamine
- APMI
- azidoprocainamide
- TPSA
- topological polar surface area
- 3D
- three-dimensional
- TEA
- tetraethylammonium
- TPeA
- tetrapentylammonium
- CAESAR
- Conformer Algorithm based on Energy Screening and Recursive Buildup.
Authorship Contributions
Participated in research design: Astorga, Ekins, and Wright.
Conducted experiments: Astorga and Morales.
Performed data analysis: Astorga, Ekins, Morales, and Wright.
Wrote or contributed to the writing of the manuscript: Astorga, Ekins, and Wright.
References
- Ahlin G, Karlsson J, Pedersen JM, Gustavsson L, Larsson R, Matsson P, Norinder U, Bergström CA, Artursson P. (2008) Structural requirements for drug inhibition of the liver specific human organic cation transport protein. J Med Chem 51:5932–5942 [DOI] [PubMed] [Google Scholar]
- Bednarczyk D, Ekins S, Wikel JH, Wright SH. (2003) Influence of molecular structure on substrate binding to the human organic cation transporter, hOCT1. Mol Pharmacol 63:489–498 [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang S, Sorani M, Giacomini KM. (2007) Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol Exp Ther 322:695–700 [DOI] [PubMed] [Google Scholar]
- Cutler MJ, Urquhart BL, Velenosi TJ, Meyer zu Schwabedissen HE, Dresser GK, Leake BF, Tirona RG, Kim RB, Freeman DJ. (2012) In vitro and in vivo assessment of renal drug transporters in the disposition of mesna and dimesna. J Clin Pharmacol 52: 530–542 [DOI] [PubMed] [Google Scholar]
- Dangprapai Y, Wright SH. (2011) Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am J Physiol Renal Physiol 301:F520–F528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzler WH, Brokl OH, Wright SH. (1989) Brush-border TEA transport in intact proximal tubules and isolated membrane vesicles. Am J Physiol Renal Physiol 256:F290–F297 [DOI] [PubMed] [Google Scholar]
- David C, Rumrich G, Ullrich KJ. (1995) Luminal transport system for H+/organic cations in the rat proximal tubule. Kinetics, dependence on pH; specificity as compared with the contraluminal organic cation-transport system. Pflugers Arch 430:477–492 [DOI] [PubMed] [Google Scholar]
- Diao L, Ekins S, Polli JE. (2009) Novel inhibitors of human organic cation/carnitine transporter (hOCTN2) via computational modeling and in vitro testing. Pharm Res 26:1890–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao L, Ekins S, Polli JE. (2010) Quantitative structure activity relationship for inhibition of human organic cation/carnitine transporter. Mol Pharm 7:2120–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S, Kim RB, Leake BF, Dantzig AH, Schuetz EG, Lan LB, Yasuda K, Shepard RL, Winter MA, Schuetz JD, et al. (2002) Application of three-dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Mol Pharmacol 61:974–981 [DOI] [PubMed] [Google Scholar]
- Gründemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. (1994) Drug excretion mediated by a new prototype of polyspecific transporter. Nature 372:549–552 [DOI] [PubMed] [Google Scholar]
- Hilal SH, El-Shabrawy Y, Carreira LA, Karickhoff SW, Toubar SS, Rizk M. (1996) Estimation of the ionization pKa of pharmaceutical substances using the computer program Sparc. Talanta 43:607–619 [DOI] [PubMed] [Google Scholar]
- Holohan PD, Ross CR. (1980) Mechanisms of organic cation transport in kidney plasma membrane vesicles: 1. Countertransport studies. J Pharmacol Exp Ther 215:191–197 [PubMed] [Google Scholar]
- Holohan PD, Ross CR. (1981) Mechanisms of organic cation transport in kidney plasma membrane vesicles: 2. ΔpH studies. J Pharmacol Exp Ther 216:294–298 [PubMed] [Google Scholar]
- Kajiwara M, Terada T, Asaka J, Ogasawara K, Katsura T, Ogawa O, Fukatsu A, Doi T, Inui K. (2007) Critical roles of Sp1 in gene expression of human and rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol 293:F1564–F1570 [DOI] [PubMed] [Google Scholar]
- Kapus A, Grinstein S, Wasan S, Kandasamy R, Orlowski J. (1994) Functional characterization of three isoforms of the Na+/H+ exchanger stably expressed in Chinese hamster ovary cells. ATP dependence, osmotic sensitivity, and role in cell proliferation. J Biol Chem 269:23544–23552 [PubMed] [Google Scholar]
- Kenworthy KE, Bloomer JC, Clarke SE, Houston JB. (1999) CYP3A4 drug interactions: correlation of 10 in vitro probe substrates. Br J Clin Pharmacol 48:716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido Y, Matsson P, Giacomini KM. (2011) Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem 54:4548–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Hiasa M, Miyaji T, Kanamoto T, Matsumoto T, Otsuka M, Moriyama Y, Omote H. (2011) Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int J Biochem Cell Biol 43:913–918 [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, Inoue K, Yuasa H, Sugiyama Y. (2011) Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther 89:837–844 [DOI] [PubMed] [Google Scholar]
- Li J, Ehlers T, Sutter J, Varma-O'brien S, Kirchmair J. (2007) CAESAR: a new conformer generation algorithm based on recursive buildup and local rotational symmetry consideration. J Chem Inf Model 47:1923–1932 [DOI] [PubMed] [Google Scholar]
- Malo C, Berteloot A. (1991) Analysis of kinetic data in transport studies: new insights from kinetic studies of Na+-d-glucose cotransport in human intestinal brush-border membrane vesicles using a fast sampling, rapid filtration apparatus. J Membr Biol 122:127–141 [DOI] [PubMed] [Google Scholar]
- Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui K. (2006) Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol 17:2127–2135 [DOI] [PubMed] [Google Scholar]
- McKinney TD. (1983) Procainamide uptake by rabbit proximal tubules. J Pharmacol Exp Ther 224:302–306 [PubMed] [Google Scholar]
- Meyer zu Schwabedissen HE, Verstuyft C, Kroemer HK, Becquemont L, Kim RB. (2010) Human multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter: functional characterization, interaction with OCT2 (SLC22A2), and single nucleotide polymorphisms. Am J Physiol Renal Physiol 298:F997–F1005 [DOI] [PubMed] [Google Scholar]
- Mol WEM, Müller M, Kurz G, Meijer DKF. (1989) Synthesis of 4-azido-N-[2-(diethylmethylammonium)ethyl]benzamide iodide: a photolabile derivative of procainamidethobromide. Arch Pharm 322:613–615 [Google Scholar]
- Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, Inui K. (2002) Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol 13:866–874 [DOI] [PubMed] [Google Scholar]
- Neuhoff S, Ungell AL, Zamora I, Artursson P. (2003) pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res 20:1141–1148 [DOI] [PubMed] [Google Scholar]
- Ohta KY, Imamura Y, Okudaira N, Atsumi R, Inoue K, Yuasa H. (2009) Functional characterization of multidrug and toxin extrusion protein 1 as a facilitative transporter for fluoroquinolones. J Pharmacol Exp Ther 328:628–634 [DOI] [PubMed] [Google Scholar]
- Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci U S A 102:17923–17928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi AA, Rumrich G, Fritzsch G, Ullrich KJ. (1996) Stereospecificity in contraluminal and luminal transporters of organic cations in the rat renal proximal tubule. J Pharmacol Exp Ther 278:31–36 [PubMed] [Google Scholar]
- Suhre WM, Ekins S, Chang C, Swaan PW, Wright SH. (2005) Molecular determinants of substrate/inhibitor binding to the human and rabbit renal organic cation transporters hOCT2 and rbOCT2. Mol Pharmacol 67:1067–1077 [DOI] [PubMed] [Google Scholar]
- Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. (2007) Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol 74:359–371 [DOI] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, Palyulin VA, Radchenko EV, Zefirov NS, Makarenko AS, et al. (2005) Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des 19:453–463 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K. (2009) Targeted disruption of the multidrug and toxin extrusion 1 (mate1) gene in mice reduces renal secretion of metformin. Mol Pharmacol 75:1280–1286 [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Papavassiliou F, David C, Rumrich G, Fritzsch G. (1991) Contraluminal transport of organic cations in the proximal tubule of the rat kidney. I. Kinetics of N1-methylnicotinamide and tetraethylammonium, influence of K+, HCO3, pH; inhibition by aliphatic primary, secondary and tertiary amines, and mono- and bisquaternary compounds. Pflugers Arch 419:84–92 [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G. (1996) Luminal transport system for choline+ in relation to the other organic cation transport systems in the rat proximal tubule. Kinetics, specificity: alkyl/arylamines, alkylamines with OH, O, SH, NH2, ROCO, RSCO and H2PO4-groups, methylaminostyryl, rhodamine, acridine, phenanthrene and cyanine compounds. Pflugers Arch 432:471–485 [DOI] [PubMed] [Google Scholar]
- Ullrich KJ, Rumrich G, Neiteler K, Fritzsch G. (1992) Contraluminal transport of organic cations in the proximal tubule of the rat kidney. II. Specificity: anilines, phenylalkylamines (catecholamines), heterocyclic compounds (pyridines, quinolines, acridines). Pflugers Arch 420:29–38 [DOI] [PubMed] [Google Scholar]
- Watanabe S, Tsuda M, Terada T, Katsura T, Inui K. (2010) Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J Pharmacol Exp Ther 334:651–656 [DOI] [PubMed] [Google Scholar]
- Wright SH, Dantzler WH. (2004) Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev 84:987–1049 [DOI] [PubMed] [Google Scholar]
- Wright SH, Wunz TM. (1998) Influence of substrate structure on turnover of the renal organic cation/H+ exchanger of the renal luminal membrane. Pflugers Arch 436:469–477 [DOI] [PubMed] [Google Scholar]
- Wright SH, Wunz TM. (1999) Influence of substrate structure on substrate binding to the renal organic cation/H+ exchanger. Pflugers Arch 437:603–610 [DOI] [PubMed] [Google Scholar]
- Wright SH, Wunz TM, Wunz TP. (1995) Structure and interaction of inhibitors with the TEA/H+ exchanger of rabbit renal brush border membranes. Pflugers Arch 429:313–324 [DOI] [PubMed] [Google Scholar]
- Yasujima T, Ohta KY, Inoue K, Ishimaru M, Yuasa H. (2010) Evaluation of 4′,6-diamidino-2-phenylindole as a fluorescent probe substrate for rapid assays of the functionality of human multidrug and toxin extrusion proteins. Drug Metab Dispos 38:715–721 [DOI] [PubMed] [Google Scholar]
- Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. (2007) Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol 74:477–487 [DOI] [PubMed] [Google Scholar]
- Yonezawa A, Inui K. (2011) Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol 164:1817–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wright SH. (2009) MATE1 has an external COOH terminus, consistent with a 13-helix topology. Am J Physiol Renal Physiol 297:F263–F271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF. (2008) Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379:337–348 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.