

Abstract
Pituitary adenylate cyclase-activating polypeptide (PACAP) is an autocrine growth factor for some lung cancer cells. The activated PACAP receptor (PAC1) causes phosphatidylinositol turnover, elevates cAMP, and increases the proliferation of lung cancer cells. PAC1 and epidermal growth factor receptor (EGFR) are present in non–small-cell lung cancer (NSCLC) cells, and the growth of NSCLC cells is inhibited by the PAC1 antagonist PACAP(6–38) and the EGFR tyrosine kinase inhibitor gefitinib. Here, the ability of PACAP to transactivate the EGFR was investigated. Western blot analysis indicated that the addition of PACAP but not the structurally related vasoactive intestinal peptide increased EGFR tyrosine phosphorylation in NCI-H838 or H345 cells. PACAP-27, in a concentration-dependent manner, increased EGFR transactivation 4-fold 2 min after addition to NCI-H838 cells. The ability of 100 nM PACAP-27 to increase EGFR or extracellular signal-regulated kinase tyrosine phosphorylation in NCI-H838 cells was inhibited by PACAP(6–38), gefitinib, 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2; Src inhibitor), (R)-N4-hydroxy-N1-[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide (GM6001; matrix metalloprotease inhibitor), or antibody to transforming growth factor α (TGFα). By enzyme-linked immunosorbent assay, PACAP addition to NCI-H838 cells increased TGFα secretion into conditioned media. EGFR transactivation caused by the addition of PACAP to NCI-H838 cells was inhibited by N-acetyl-cysteine (antioxidant), tiron (superoxide scavenger), diphenylene iodonium (NADPH oxidase inhibitor), or 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122; phospholipase C inhibitor), but not N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide (H89; protein kinase A inhibitor). PACAP addition to NCI-H838 cells significantly increased reactive oxygen species, and the increase was inhibited by tiron. The results indicate that PACAP causes transactivation of the EGFR in NSCLC cells in an oxygen-dependent manner that involves phospholipase C but not protein kinase A.
Introduction
Pituitary adenylate cyclase-activating polypeptide (PACAP)- 27 has 67% sequence homology to vasoactive intestinal peptide (VIP) (Arimura, 1992). It is derived from a 176-amino acid precursor protein, and the gene for PACAP is located on chromosome 18p11 (Sherwood et al., 2000). The PACAP precursor contains an N-terminal 24-amino acid signal protein, a 29-amino acid PACAP-related peptide, and PACAP in the C-terminal domain. The precursor protein can be metabolized by prohormone convertase enzymes to biologically active PACAP-27 (preproPACAP131−158) or PACAP-38, which contains PACAP-27 plus an additional 11 amino acids at the C terminal (Miyata et al., 1989). Because PACAP(6–38) is an antagonist for PAC1, the N terminal of PACAP is essential for biological activity (Moody et al., 1994).
PACAP-27 binds with high affinity to the seven-transmembrane G protein-coupled receptors (GPCRs) VPAC1, VPAC2, and PAC1. PAC1 or type I binding site, however, binds VIP with low affinity, whereas VPAC1 and VPAC2 bind VIP with high affinity (Zia et al., 1995; Reubi et al., 2000). Addition of PACAP-27 to the human lung cancer cell line NCI-H345 increased cAMP and caused phosphatidylinositol (PI) turnover (Moody et al., 1993). These results suggest that PAC1 interacts with the guanine nucleotide binding proteins Gs and Gq. Elevated cAMP activates protein kinase A (Vaudry et al., 2009). The increase in PI 4,5-bisphosphate metabolism caused by phospholipase C results in diacylglycerol (activates protein kinase C) and inositol 1,4,5-trisphosphate (elevates cytosolic Ca2+). The addition of PACAP to NCI-H838 cells causes ERK phosphorylation, increases c-fos mRNA, and increases vascular endothelial growth factor expression (Draoui et al., 1996; Moody et al., 2002). PACAP stimulates the growth of lung cancer (NCI-H345 cells; Moody et al., 1993), pancreatic carcinoma (AR42-J cells; Buscail et al., 1992), and colon cancer (Bon cells; Germano et al., 2009). PACAP(6–38) is a PAC1 antagonist that inhibits the proliferation of lung, breast, and prostate cancer cells (Zia et al., 1995; Pisegna et al., 1997; Leyton et al., 1998). These results suggest that PAC1 regulates the proliferation of cancer cells.
Lung cancer causes the death of approximately 160,000 American citizens annually. Most of the patients die from non–small-cell lung cancer (NSCLC), which is characterized by p53 and k-ras mutations (Sekido et al., 2005). The epidermal growth factor receptor (EGFR) is overexpressed in approximately 30% of the patients with NSCLC examined. Activating mutations of the EGFR are associated with patient responses to EGFR tyrosine kinase inhibitors (gefitinib or erlotinib), including tumor reduction (Lynch et al., 2004; Paez et al., 2004; Helfrich et al., 2006). The lung cancer EGFR can be transactivated by GPCR for bombesin or neuromedin B (Lui et al., 2003, Moody et al., 2010). The GPCR regulates the release of endogenous transforming growth factor α (TGFα) from lung cancer cells in a Src- and matrix metalloprotease (MMP)-dependent manner. TGFα binds with high affinity to the EGFR, resulting in the tyrosine phosphorylation of the EGFR (Zhang et al., 2004).
In this article the effects of PACAP on EGFR transactivation were investigated by using lung cancer cells. The addition of PACAP, but not VIP, to human NCI-H838 NSCLC cells caused tyrosine phosphorylation of the EGFR. The transactivation of the EGFR caused by the addition of PACAP to NSCLC cells was inhibited by 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2; Src inhibitor), (R)-N4-hydroxy-N1-[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide (GM6001; MMP inhibitor), and anti-TGFα antibody. PACAP significantly increased TGFα release form NCI-H838 cells, which was inhibited by GM6001. In addition, N-acetyl-cysteine (NAC; antioxidant) and tiron (superoxide inhibitor) reduced the transactivation of the EGFR caused by the addition of PACAP to NCI-H838 cells. The results indicate that PACAP causes EGFR transactivation in lung cancer cells in a MMP-, Src-, and oxygen-dependent manner.
Materials and Methods
Cell Culture.
NCI-H838 cells, which are known to contain PAC1 and wild-type EGFR (Moody and Jensen, 2006), were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA). The cells were mycoplasma free and used when they were in exponential growth phase after incubation at 37°C in 5% CO2/95% air. NCI-H345 cells were cultured as floating aggregates in RPMI 1640 medium containing 10% FBS and were split weekly (1:1).
Receptor Binding.
Receptor binding studies were conducted by using NCI-H838 cells in 24-well plates. The plates were washed three times in SIT medium (RPMI 1640 medium containing 3 × 10−8 M sodium selenite, 5 μg/ml bovine insulin, and 10 μg/ml transferrin). The cells were incubated in SIT medium containing 2% bovine serum albumin and 2 mg/ml bacitracin with 0.2 nM [125I]PACAP-27 (2200 Ci/mmol) in the presence of varying concentrations of competitor. After incubation at 37°C for 30 min, free [125I]PACAP-27 was removed by washing three times in buffer, and the cells that contained bound [125I]PACAP-27 were dissolved in 0.2 N NaOH and counted in a gamma counter. The half-maximal inhibitory concentration (IC50) was calculated for each unlabeled competitor.
Western Blot.
The ability of PACAP to stimulate tyrosine phosphorylation of EGFR or ERK (p42/p44 mitogen-activated protein kinase) was investigated by Western blot. NCI-H838 cells were cultured in 10-cm dishes. When a monolayer of cells formed, they were placed in SIT media for 3 h. Routinely, NSCLC cells were treated with gefitinib, N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinamine (AG1478), PACAP(6–38), GM6001, PP2, NAC, tiron, or TGFα antibodies for 30 min before stimulation with PACAP-like peptides. Cells were treated with 100 nM PACAP-27 for 2 min, washed twice with phosphate-buffered saline, and lysed in buffer containing 50 mM Tris·HCl, pH 7.5, 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholate, 1% sodium azide, 1 mM EGTA, 0.4 M EDTA, 1.5 μg/ml aprotinin, 1.5 μg/ml leupeptin, 1 mM phenylmethylsulfonylfluoride, and 0.2 mM sodium vanadate (Sigma-Aldrich, St. Louis, MO). The lysate was sonicated for 5 s at 4°C and centrifuged at 10,000g for 15 min. Protein concentration was measured by using bicinchoninic acid reagent (Thermo Fisher Scientific, Waltham, MA), and 400 μg of protein was incubated with 4 μg of anti-phosphotyrosine (PY) monoclonal antibody, 4 μg of goat anti-mouse IgG, and 25 μl of immobilized protein G overnight at 4°C. The immunoprecipitates were washed three times with phosphate-buffered saline and analyzed by SDS/polyacrylamide gel electrophoresis and Western blotting. Immunoprecipitates were fractionated by using 4 to 20% polyacrylamide gels (Novex, San Diego, CA). Proteins were transferred to nitrocellulose membranes, and the membranes were blocked overnight at 4°C using Blotto (5% nonfat dried milk in solution containing 50 mM Tris·HCl, pH 8.0, 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20, and 0.02% sodium azide) and incubated for 16 h at 4°C with 1 μg/ml anti-EGFR antibody (Cell Signaling Technology, Danvers, MA) followed by anti-rabbit IgG-horseradish peroxidase conjugate (Millipore, Billerica, MA). The membrane was washed for 10 min with Blotto and twice for 10 min with washing solution (50 mM Tris·HCl, pH 8.0, 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20, and 0.02% sodium azide). The blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Kodak XAR film (Eastman Kodak, Rochester, NY). The intensity of the bands was determined by using a densitometer.
Alternatively, 20 μg of cellular extract was loaded onto 15-well 4 to 20% polyacrylamide gels. After transfer to nitrocellulose, the blot was probed with anti-PY1068-EGFR, anti-EGFR, anti-PY-ERK, anti-ERK, or anti-tubulin (Cell Signaling Technology).
TGFα ELISA.
NCI-H838 cells in 24-well plates were washed in 0.25 ml of SIT media. The cells were incubated with inhibitors such as 10 μM GM6001 for 30 min, followed by the addition of 100 nM PACAP-27 for 5 min. The supernatant was collected and assayed for TGFα by ELISA (R&D Systems, Minneapolis, MN).
Reactive Oxygen Species.
NCI-H838 cells were placed in 96-well plates (30,000 cells/well) and cultured overnight. The cells were treated with 10 μM dichlorofluoresceindiacetate (H2DCF) for 1 h and washed three times with serum-free SIT medium. Some of the cells were treated with 5 mM tiron for 30 min and then stimuli, such as 100 nM PACAP-27 or 10 μM H2O2, was added. Fluorescence measurements were taken at the various times by using an excitation wavelength of 485 nm and emission wavelength of 585 nm.
Proliferation.
Growth studies in vitro were conducted by using clonogenic assays (Moody et al., 2003). In the clonogenic assay, the bottom layer of six-well plates contained 0.5% agarose in SIT medium containing 5% FBS in six-well plates. The top layer consisted of 3 ml of SIT medium in 0.3% agarose containing 5 × 104 NCI-H838 cells with PACAP-27, gefitinib, and/or PACAP(6–38). Triplicate wells were plated; after 2 weeks, 1 ml of 0.1% p-iodonitrotetrazolium violet was added; and after 16 h at 37°C, the plates were screened for colony formation. The number of colonies larger than 50 μm in diameter were counted by using an Omnicon image analysis system (BioLogics Inc., Manassas, VA).
Results
PACAP Agonists Increase EGF-R Tyrosine Phosphorylation.
Figure 1A shows that the addition of 100 nM PACAP-27 or PACAP-38, but not VIP, to NCI-H345 cells increased the phosphorylation of Tyr-1068 of the 170-kDa EGFR. In contrast, PACAP had little effect on total EGFR (Fig. 1A). PACAP-27 and PACAP-38, but not VIP, significantly increased EGFR transactivation 2-fold in NCI-H345 cells (Fig. 1B). Figure 1C shows that the addition of PACAP-27, but not VIP or PACAP(6–38), to NCI-H838 cells increased EGFR tyrosine phosphorylation. The addition of PACAP(6–38) to NCI-H838 cells blocked the increase in EGFR transactivation caused by PACAP-27 (Fig. 1C). In contrast, PACAP had little effect on tubulin (Fig. 1C). Figure 1D shows that the 3-fold increase in EGFR tyrosine phosphorylation caused by PACAP was significantly inhibited by PACAP(6–38). These results indicate that PACAP-27 and PACAP-38 are agonists for lung cancer PAC1, whereas PACAP(6–38) is an antagonist.
Fig. 1.

Specificity of EGFR transactivation by PACAP analogs. A, the ability of 100 nM VIP, PACAP (PC)-38, or PC-27 to stimulate EGFR tyrosine phosphorylation in NCI-H345 cells was determined by Western blot using antiphospho-Tyr-1068-EGFR antibody. B, the mean percentage of EGFR tyrosine phosphorylation ± S.D was indicated for four determinations. C, the ability of 100 nM PC-27 or VIP to stimulate EGFR tyrosine phosphorylation was determined by using NCI-H838 cells. Ten micrometers PC(6–38) had no effect on EGF-R tyrosine phosphorylation but antagonized the effects of 100 nM PC-27. D, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for four determinations. **, p < 0.01 by ANOVA.
The ability of PACAP analogs to bind to NCI-H838 cells was investigated. Table 1 shows that PACAP-27 inhibited specific [125I]PACAP-27 binding to NCI-H838 cells with an IC50 value of 3 ± 0.3 nM. Likewise, PACAP-38 and PACAP(6–38) inhibited specific [125I]PACAP-27 binding to NCI-H838 cells with IC50 values of 5.3 ± 0.6 and 31.4 ± 4.9 nM, respectively. In contrast, VIP inhibited [125I]PACAP-27 binding to NCI-H838 cells with an IC50 value of more than 1000 nM. Because PACAP-27, PACAP-38, and PACAP(6–38), but not VIP, inhibited specific [125I]PACAP-27 binding to NCI-H838 cells with high affinity, PAC1 is present on NSCLC cells.
TABLE 1.
Binding to lung cancer cells
The ability of the ligands to inhibit specific binding of [125I]PACAP-27 to NCI-H838 cells was determined at 37°C. The structures of PACAP-like peptides are shown below. Sequence homologies relative to PACAP-27 are underlined.
PACAP-27: His-Ser-Arg-Gly-Ile-Phe-Thr-Asp-Ser-Tyr-Ser-Arg-Tyr-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Ala-Ala-Val-Leu-NH2.
PACAP(6–38): Phe-Thr-Asp-Ser-Tyr-Ser-Arg-Tyr-Arg-Lys-Gln-Met-Ala-Val-Lys- Lys-Tyr-Leu-Ala-Ala-Val-Leu-Gly-Lys-Arg-Tyr-Lys-Gln-Arg-Val-Lys-Asn-Lys-NH2.
VIP: His-Ser-Asp-Ala-Val-Phe-Thr-Asp-Asn-Tyr-Thr-Arg-Leu-Arg-Lys-Gln-Met-Ala-Val-Lys-Lys-Tyr-Leu-Asn-Ser-Ile-Leu-Asn-NH2.
| Ligand | IC50 |
|---|---|
| nM | |
| PACAP-27 | 3.0 ± 0.3 |
| PACAP-38 | 5.3 ± 0.6 |
| PACAP(6–38) | 31.4 ± 4.9 |
| VIP | >1000 |
| Gefitinib | >1000 |
PACAP-27 Causes EGFR and ERK Tyrosine Phosphorylation.
The ability of PACAP-27 to cause EGFR and ERK tyrosine phosphorylation was investigated with NCI-H838 cells. Figure 2A shows that 100 nM PACAP-27 increased EGFR tyrosine phosphorylation at 1, 2, or 3 min after addition to NCI-H838 cells. Figure 2B shows that the addition of PACAP-27 to NCI-H838 cells increased EGFR transactivation significantly at 1, 2, or 3 min by 2.2-, 2.8- or 3.3-fold, respectively. In contrast, phosphorylation of the 42-and 44-kDa ERK was increased 2 or 3 min after the addition of 100 nM PACAP-27 to NCI-H838 cells (Fig. 2C). Figure 2D shows that ERK tyrosine phosphorylation was significantly increased 2.5-fold 2 or 3 min, but not 1 min, after the addition of PACAP-27 to NCI-H838 cells. Figure 2E shows that the addition of 1, 10, or 100 nM PACAP-27 to NCI-H838 cells caused phosphorylation of Tyr-1068 of the 170-kDa EGFR. Figure 2F shows that the addition of 1, 10, or 100 nM PACAP-27 significantly increased EGFR transactivation 3.0-, 3.3-, and 4.2-fold, respectively. Likewise, ERK tyrosine phosphorylation was significantly increased by 1, 10, or 100 but not 0.1 nM PACAP-27 (data not shown). These results indicate that the addition of PACAP to NCI-H838 cells increases EGFR and ERK tyrosine phosphorylation in a time- and concentration-dependent manner.
Fig. 2.

PACAP-27 increases EGFR and ERK tyrosine phosphorylation in a time- and dose-dependent manner. A, the ability of 100 nM PACAP-27 to increase EGFR tyrosine phosphorylation was investigated as a function of time by using NCI-H838 cells and antiphospho-Tyr-1068-EGFR antibody. B, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for three determinations. C, the ability of 100 nM PACAP-27 to increase ERK tyrosine phosphorylation is indicated by using NCI-H838 cells. D, the mean percentage of ERK tyrosine phosphorylation ± S.D is indicated for four determinations. E, the ability of PACAP-27 to increase EGFR tyrosine phosphorylation is indicated as a function of dose by using NCI-H838 cells. F, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for four determinations. **, p < 0.01 by ANOVA.
Gefitinib Blocks EGFR and ERK Tyrosine Phosphorylation.
The ability of gefitinib to inhibit EGFR and ERK tyrosine phosphorylation was investigated. Figure 3A shows that the addition of 100 nM PACAP-27 to NCI-H838 cells increased EGFR tyrosine phosphorylation, which was blocked by 10 μg/ml gefitinib. PACAP significantly increased EGFR transactivation 4.1-fold, and the increase was inhibited by 10 but not 0.1 μg/ml gefitinib (Fig. 3B). Figure 3C shows that the addition of 100 nM PACAP-27 to NCI-H838 cells increased ERK tyrosine phosphorylation, which was inhibited by 10 μg/ml gefitinib. PACAP significantly increased ERK tyrosine phosphorylation 2.5-fold, and the increase was inhibited by 10 but not 0.1 μg/ml gefitinib (Fig. 3D). These results suggest that the EGFR and ERK tyrosine phosphorylation caused by the addition of 100 nM PACAP-27 to NCI-H838 cells depends on EGFR tyrosine kinase activity.
Fig. 3.

Gefitinib inhibits EGFR and ERK tyrosine phosphorylation caused by PACAP. A, gefitinib in a dose-dependent manner inhibits EGFR transactivation caused by PC. B, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for four determinations. **, p < 0.01 relative to control by ANOVA. a, p < 0.05 relative to PC + 0.1 μg/ml gefitinib. aa, p < 0.01 relative to PC + 0.1 μg/ml gefitinib. C, gefitinib is a dose-dependent manner inhibits ERK tyrosine phosphorylation caused by 100 nM PC. D, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for four determinations. **, p < 0.01 relative to control by ANOVA. aa, p < 0.01 relative to PC + 0.1 μg/ml gefitinib. E, the EGFR tyrosine phosphorylation was determined by using antiphospho-Tyr-1068-EGFR antibody after adding 100 nM PC to NCI-H838 cells in the absence or presence anti-HB-EGF, antiamphiregulin (Amph), or anti-TGFα. F, the mean percentage of EGFR tyrosine phosphorylation ± S.D is indicated for four determinations. **, p < 0.01 relative to control. *, p < 0.05 by ANOVA. a, p < 0.05 relative to PC. aa, p < 0.01 relative to PC.
TGFα Is a Ligand for EGFR Transactivation.
The ability of antibodies to alter EGFR transactivation was investigated. Figure 3E shows that the addition of 100 nM PACAP-27 to NCI-H838 cells increased EGFR tyrosine phosphorylation. Anti-heparin binding (HB)-EGF or antiamphiregulin had little effect on EGFR transactivation; however, anti-TGFα reduced EGFR tyrosine phosphorylation caused by the addition of 100 nM PACAP-27 to NCI-H838 cells. Figure 3F shows that PACAP significantly increased EGFR transactivation 3.5-fold, and anti-TGFα, but not anti-HB-EGF or antiamphiregulin, reversed the increase in EGFR transactivation caused by PACAP. Table 2 shows that PACAP-27 significantly increased TGFα release from NCI-H838 cells 5.5-fold. The MMP inhibitor GM6001 reversed the increased release of TGFα caused by the addition of PACAP-27 to NCI-H838 cells. These results suggest that MMP may catalyze the conversion of pro-TGFα to TGFα in NCI-H838 cells.
TABLE 2.
TGFα release from lung cancer cells
NCI-H838 cells were treated with or without 10 μM GM6001 for 30 min in SIT medium; 100 nM PACAP-27 was added for 5 min, and the supernatant was removed and assayed for TGFα by ELISA. The mean value ± S.D. of three experiments, each repeated in quadruplicate, is indicated.
| Addition | TGFα Release |
|---|---|
| pg/ml | |
| None | 8 ± 2 |
| PACAP-27 | 44 ± 7** |
| PACAP + GM6001 | 9 ± 3 |
P < 0.01 by ANOVA.
Inhibitors of EGFR Transactivation.
The ability of various agents to inhibit EGFR transactivation caused by the addition of PACAP to NCI-H838 cells was investigated. Figure 4A shows that GM6001 inhibited the ability of PACAP to cause EGFR transactivation; similar results were obtained for the Src inhibitor PP2 (data not shown). Figure 4B shows that 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122; PLC inhibitor) but not N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide (H89; PKA inhibitor) reduced the ability of PACAP to cause EGFR transactivation in lung cancer cells. These results suggest that PI turnover, but not adenylyl cyclase stimulation, is important for EGFR transactivation. Figure 4C shows that BAPTA (calcium chelator) and GF109203X (PKC inhibitor) inhibited the ability of PACAP to cause EGFR transactivation. These results suggest that PI metabolites such as IP3 (elevates cytosolic Ca2+) and diacylglycerol (activates protein kinase C) are important in the transactivation of the EGFR. Figure 4D shows that NAC or tiron inhibited the increase in EGFR tyrosine caused by the addition of PACAP-27 to NCI-H838 cells. Similar results were obtained with diphenylene iodonium, an NADPH oxidase inhibitor (data not shown). Table 3 shows that the addition of PACAP to NCI-H838 cells significantly increased the production of reactive oxygen species (ROS). The increase caused by PACAP was reversed significantly by tiron, a superoxide scavenger. These results suggest that PACAP causes EGFR transactivation in an oxygen-dependent manner.
Fig. 4.
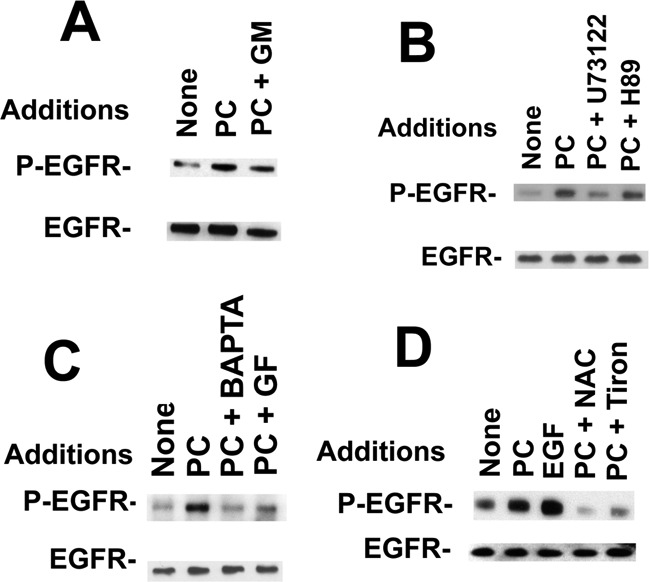
Inhibitors of EGFR transactivation. A, the ability of 10 μM GM6001 (GM) to inhibit the EGFR transactivation caused by the addition of 100 nM PC to NCI-H838 cells was investigated by using antiphospho-Tyr-1068-EGFR antibody. B, the ability of 50 μM U73122 or H89 to inhibit the EGFR transactivation caused by the addition of 100 nM PACAP-27 to NCI-H838 cells was investigated. C, the ability of 10 μM BAPTA and 1 mM EGTA as well as 10 μM GF109203X (GF) to inhibit EGFR transactivation caused by the addition of 100 nM PACAP-27 (PC) to NCI-H838 cells was investigated. D, the ability of 5 mM NAC or 5 mM tiron to inhibit the EGFR transactivation caused by 100 nM PACAP-27 to NCI-H838 cells was investigated. These experiments are representative of three others.
TABLE 3.
Reactive oxygen species
The relative fluorescence intensity was determined 0.5 h after the addition of PACAP-27 or H2O2 to NCI-H838 cells. Tiron was added 0.5 h before the addition of PACAP. The mean ± S.D. of six determinations is indicated.
| Addition | Fluorescence Intensity |
|---|---|
| % | |
| None | 100 ± 3 |
| PACAP-27, 1 μM | 151 ± 19* |
| PACAP + tiron, 5 mM | 103 ± 11 |
| Tiron | 93 ± 5 |
| H2O2, 100 uM | 367 ± 27** |
P < 0.05;
P < 0.01 by ANOVA.
Proliferation.
The effects of PACAP-like peptides and gefitinib on NCI-H838 proliferation were investigated. PACAP-27 significantly increased NCI-H838 colony formation, whereas the increase caused by PACAP-27 was reversed by PACAP(6–38) or gefitinib (Table 4). PACAP(6–38) or gefitinib significantly reduced NCI-H838 colony number, whereas the addition of PACAP(6–38) plus gefitinib strongly reduced NCI-H838 colony number. The results indicate that PACAP(6–38) or gefitinib reduce basal lung cancer proliferation as well as that induced by PACAP-27.
TABLE 4.
Colony number
The mean value ± S.D. of three determinations is indicated using NCI-H838 cells. This experiment is representative of three others.
| Addition | Colony Number |
|---|---|
| None | 15 ± 2 |
| PACAP-27, 10 nM | 27 ± 3** |
| PACAP(6–38), 10 μM | 9.5 ± 0.7* |
| PACAP + PACAP(6–38) | 17 ± 2.2 |
| Gefitinib 1 μg/ml | 7.7 ± 1.7** |
| PACAP + gefitinib | 10.7 ± 1.4* |
| PACAP(6–38) + gefitinib | 2.7 ± 0.3** |
P < 0.05;
P < 0.01, by ANOVA.
Discussion
Mutations of the EGFR occur in approximately 16% of patients with lung cancer (Landi and Cappuzzo, 2011). The most common mutations, which include deletions in exon 19 or the L858R substitution in exon 21, make the patients more responsive to tyrosine kinase inhibitors such as erlotinib and gefitinib (Rosell et al., 2009). The complete or partial response rate to erlotinib was 70% in patients with lung cancer with EGFR mutations. The progression-free survival was 13 months in patients treated with erlotinib but only 4.6 months in patients treated with platinum-based chemotherapy. It is important to increase the response rate of patients with wild-type EGFR. Studies using human cancer cells indicate that activation of GPCR can influence EGFR tyrosine phosphorylation by transactivation. In this article we studied the effects of PACAP on lung cancer cells with wild-type EGFR.
PAC1 has splice variants (SVs) in intracellular loop 3 (IC3). The regular (Reg) receptor contains 467 amino acids (Pisegna and Wank, 1993; Spengler et al., 1993). SV-1 or the hip receptor has a 28-amino acid insert in IC3. SV-2 or the hop receptor (HOP1) has a different 28-amino acid insert in IC3. SV-3 or the hip-hop receptor has both 28-amino acid inserts in IC3. PACAP addition to cells transfected with PAC1-R SV-2 strongly increased PI turnover, whereas cells transfected with Reg, SV-1, or SV-3 had a weaker PI response (Pisegna and Wank, 1996). In addition, extracellular loop 1 (EC1) deletions or SV insertions exist (Lutz et al., 2006). Ushiyama et al. (2007, 2010) expressed variants of the EC1 domain [normal (N) or short (S) form] and variants of IC3 (Reg or HOP1). The resulting four PAC1 isoforms (N/Reg, N/HOP1, S/Reg, or S/HOP1) bound PACAP-27 with high affinity. N/Reg, S/Reg, or S/HOP1, but not N/HOP1, strongly increased cAMP. These results indicate that alterations in EC1 or IC3 do not affect PAC1 binding, but do affect second-messenger production. Preliminary data (T. W. Moody, unpublished work) indicate that NCI-H345 and H838 lung cancer cell lines have PAC1 SV2 and wild-type EGFR.
PACAP-27 binds with high affinity to VPAC1, VPAC2, and PAC1. NCI-H838 cells have VPAC1 and PAC1, but not VPAC2, mRNA (Pisegna and Wank, 1996). PACAP-27 and PACAP-38, but not VIP, inhibit specific [125I]PACAP-27 binding to NCI-H838 cells with high affinity. Likewise, PACAP-27 and PACAP-38, but not VIP, cause transactivation of the EGFR. Furthermore, PACAP(6–38) inhibited specific [125I]PACAP-27 binding to NCI-H838 cells with high affinity. PACAP(6–38), which is a PAC1 antagonist, blocked the ability of PACAP-27 to cause tyrosine phosphorylation of the EGFR. These results suggest that PAC1 mediates the transactivation of the EGFR in lung cancer cells.
In contrast, VPAC1 mediated the transactivation of the EGFR and HER2 in breast cancer cells (Valdehita et al., 2009, 2012). HER2 has an inactive ligand binding domain but functional tyrosine kinase domain and hence it can be phosphorylated when it forms heterodimers with the EGFR (Bhola and Grandis, 2008). The addition of 100 nM VIP to T47D breast cancer cells rapidly increased HER2 tyrosine phosphorylation after 1 min; however, it took 15 min to maximally increase tyrosine phosphorylation of the EGFR (Valdehita et al., 2009). In contrast, using lung cancer cells PAC1 maximally increased EGFR tyrosine phosphorylation after 1 min, whereas VIP had no effect. These results suggest that the EGFR transactivation in breast and lung cancer cells is different.
Because lung cancer cells have PAC1 SV2, the addition of PACAP to NCI-H838 cells elevates cytosolic cAMP and causes PI turnover. Because the increase in lung cancer EGFR transactivation caused by PACAP-27 was blocked by U73122 but not H89, phospholipase C but not PKA is important. Phospholipase C metabolizes phosphatidylinositol bisphosphate to IP3, which can cause release of Ca2+ from intracellular pools. Ca2+ is increased within seconds after the addition of PACAP-27 to NCI-H838 cells, and the increase in cytosolic Ca2+ is blocked by U73122, but not H89 (T. W. Moody, unpublished work). In addition, BAPTA reduces the ability of PACAP-27 to cause increased cytosolic Ca2+ and EGFR transactivation in NCI-H838 cells. These results suggest that phospholipase C, but not PKA, is important for PACAP-27 to stimulate EGFR transactivation in lung cancer cells. In contrast, the ability of VIP to cause transactivation of the EGR and HER2 in breast cancer cells was blocked by H89, the PKA inhibitor (Valdehita et al., 2009). These results indicate that PKA is important in the transactivation of the EGFR in breast but not lung cancer cells.
PACAP induces multiple neurotrophic signaling pathways. PACAP activation of PAC1 HOP1 caused ERK1/2 and ERK5 activation, but abrogated stress-activated protein/c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase signaling (May et al., 2010). PACAP caused ERK1/2 and ERK5 phosphorylation of primary sympathetic neurons within minutes and phosphorylation of AKT within hours (May et al., 2010). We are investigating whether PACAP causes AKT phosphorylation in NSCLC cells, but we recently found that PACAP causes focal adhesion kinase and paxillin tyrosine phosphorylation within minutes after the addition to NCI-H838 cells (Moody et al., 2012). Figure 5 shows that PACAP may alter gene expression, motility, and/or migration NSCLC cells.
Fig. 5.

Signal transduction mechanisms for lung cancer PAC1. Activated PAC1 interacts with Gα-stimulating adenylyl cyclase (AC), leading to PKA activation, cAMP response element-binding protein (Creb) phosphorylation, and altered gene expression. PKC activates MMP and/or ADAM, releasing TGFα from its precursor protein in the plasma membrane causing EGFR tyrosine phosphorylation. The phosphorylated EGFR causes Ras and Raf activation, resulting in phosphorylation of mitogen-activated protein kinase kinase (MEK) and Erk1/2, leading to proliferation. The phosphorylated EGFR causes tyrosine phosphorylation of phosphatidylinositol 3-kinase (PI3K) resulting in the activation of pyruvate dehydrogenase lipoamide kinase isozyme 1 (PDK-1), Akt, and mammalian target of rapamycin (mTor) phosphorylation, leading to cancer cellular survival. Activated PAC1 interacts with Gαq, leading to phospholipase C (PLC) stimulation and PI turnover. The IP3 and diacylglycerol (DAG) released cause elevation of cytosolic Ca2+ and PKC activation, respectively. The PKC or Ca2+ causes Src tyrosine phosphorylation, leading to phosphorylation of focal adhesion kinase (FAK), pyruvate kinase 2 (PYK2), and paxillin, affecting cellular motility, secretion and migration. ER, endoplasmic reticulum; PIP2, phosphatidylinositol bisphosphate; RTK, receptor tyrosine kinase.
GPCR-mediated transactivation of the EGFR results, because EGFR proligands are metabolized in a MMP-dependent manner (Bhola and Grandis, 2008). A disintegrin and metalloprotease (ADAM) family of metalloproteases are responsible for the cleavage of membrane-bound precursors to TGFα, amphiregulin, and HB-EGF, which bind with high affinity to the EGFR (Bhola and Grandis, 2008). We found that the MMP inhibitor GM6001 and the Src inhibitor PP2 reversed the ability of PACAP to cause EGFR transactivation in lung cancer cells. Likewise, PP2 and the MMP inhibitor TNF-α protease inhibitor-1 inhibited the ability of VIP to cause EGFR and HER2 transactivation in breast cancer cells (Valdehita et al., 2009). The ability of PACAP to cause EGFR transactivation in lung cancer cells was inhibited by the addition of TGFα antibodies. Further PACAP addition to NCI-H838 cells significantly increased the release of immunoreactive TGFα, which was reduced by the addition of GM6001. Figure 5 shows that TGFα is a ligand that mediates EGFR transactivation in lung cancer cells.
ROS, which reduce the activity of phosphatases, result in an increase of protein tyrosine phosphorylation. In Calu-6 cells, the GPCR formyl peptide receptor-like 1 activation resulted in EGFR tyrosine phosphorylation, p47phox phosphorylation, NADPH-oxidase-dependent superoxide generation, and c-Src kinase activity (Cattaneo et al., 2011). Here, PACAP addition to NCI-H838 cells significantly increased ROS, which was reversed by tiron. Tiron (superoxide scavenger), NAC (antioxidant), and diphenylene iodonium (NADPH oxidase inhibitor I) reversed the ability of PACAP to cause EGFR transactivation. The results indicate that ROS are important in the transactivation of the EGFR in lung cancer cells.
The transactivation of EGFR caused by GPCR is blocked by tyrosine kinase inhibitors and GPCR antagonists. For lung cancer cells with PAC1, PACAP(6–38) blocked the EGFR transactivation. For breast cancer cells with VPAC1, JV-1-53 (VPAC1 antagonist) blocked EGFR and HER2 transactivation (Valdehita et al., 2009). The tyrosine kinase inhibitors AG1478 (EGFR) and 3-[3-[(2-benzothiazolylthio)methyl]-4-hydroxy-5-methoxyphenyl]-2-cyano-2-propenamide (AG825; HER2) blocked the transactivation of the EGFR and HER2 caused by the addition of VIP to breast cancer cells (Valdehita et al., 2012). In lung cancer cells gefitinib blocked the increase in EGFR and ERK tyrosine phosphorylation caused by the addition of PACAP to NCI-H838 cells. It remains to be determined whether gefitinib blocks additional signal transduction caused by PACAP such as increased c-fos and vascular endothelial growth factor mRNA (Moody et al., 2002).
PACAP is an autocrine growth factor for lung cancer cells. PACAP and PAC1 mRNA are present in many lung cancer cells (Moody and Jensen, 2006). PACAP(6–38) inhibited the increase in cAMP, cytosolic Ca2+, and the proliferation of lung cancer cells caused by the addition of PACAP-27. In addition, PACAP(6–38) inhibited basal proliferation of NCI-H838 cells in vitro and tumors in vivo (Zia et al., 1995). Although clinical studies with PACAP(6–38) have not been done, gefitinib is used to treat patients with lung cancer with EGFR mutations. Gefitinib is cytotoxic for lung cancer cells with EGFR mutations, whereas cells with wild-type EGFR have reduced sensitivity. Because only approximately 16% of patients with lung cancer have activating mutations of the EGFR, they are resistant to gefitinib in vivo. Strategies to improve the sensitivity of gefitinib for patients with NSCLC who have wild-type EGFR would be beneficial. One such strategy is to combine GPCR antagonists with tyrosine kinase inhibitors. Previously, we showed that the neuromedin B receptor antagonist 2S-3-(1H-indol-3-yl)-2-methyl-2-[(4-nitrophenyl) carbamoylamino]-N[(1-pyridin)-2-ylcyclohexyl)methyl] propanamide (PD168368) was synergistic with gefitinib at inhibiting lung cancer growth in vitro (Moody et al., 2010). Here, we found that the addition of gefitinib significantly reduced NCI-H838 colony number by 50%, whereas gefitinib plus PACAP(6–38) strongly reduced colony number by 80%. Figure 5 shows that PACAP may increase the survival and proliferation of lung cancer cells.
In summary, PAC1 regulates EGFR transactivation in lung cancer cells. PACAP functions as an autocrine growth factor in some lung cancer cells, and PACAP(6–38) inhibits the growth of NSCLC cells in vitro and in vivo. It remains to be determined whether PACAP(6–38) will increase the potency of gefitinib in vivo.
Acknowledgments
We thank Samuel Mantey for helpful discussions.
This research was supported by the Intramural Research Program of the National Institutes of Health (National Cancer Institute and National Institute of Diabetes, Digestive, and Kidney Disease).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- PACAP
- pituitary adenylate cyclase-activating polypeptide
- ADAM
- A disintegrin and metalloprotease
- AG1478
- N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinamine
- AG825
- 3-[3-[(2-benzothiazolylthio)methyl]-4-hydroxy-5-methoxyphenyl]-2-cyano-2-propenamide
- ANOVA
- analysis of variance
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- EC1
- extracellular loop 1
- EGF
- epidermal growth factor
- EGFR
- EGF receptor
- ELISA
- enzyme-linked immunosorbent assay
- ERK
- extracellular signal-regulated kinase
- FBS
- fetal bovine serum
- GM6001
- (R)-N4-hydroxy-N1-[(S)-2-(1H-indol-3-yl)-1-methylcarbamoyl-ethyl]-2-isobutyl-succinamide
- GPCR
- G protein-coupled receptor
- HB
- heparin binding
- H89
- N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide
- HER2
- human epidermal growth factor receptor 2
- IC3
- intracellular loop 3
- IP3
- inositol trisphosphate
- MMP
- matrix metalloprotease
- NAC
- N-acetyl-cysteine
- NSCLC
- non–small-cell lung cancer
- PD168368
- 2S-3-(1H-indol-3-yl)-2-methyl-2-[(4-nitrophenyl) carbamoylamino]-N[(1-pyridin)-2-ylcyclohexyl)methyl] propanamide
- PI
- phosphatidylinositol
- P-EGF
- phosphorylated epidermal growth factor
- PKA
- protein kinase A
- PKC
- protein kinase C
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine
- PY
- phosphotyrosine
- Reg
- regular
- ROS
- reactive oxygen species
- SIT medium
- RPMI 1640 containing 3 × 10−8 M sodium selenite, 5 μg/ml bovine insulin, and 10 μg/ml transferrin
- SV
- splice variant
- TGFα
- transforming growth factor α
- U73122
- 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione
- VIP
- vasoactive intestinal peptide.
Authorship Contributions
Participated in research design: Moody and Jensen.
Conducted experiments: Moody, Osefo, and Ridnour.
Performed data analysis: Moody.
Wrote or contributed to the writing of the manuscript: Moody, Nuche-Berenguer, Wink, and Jensen.
References
- Arimura A. (1992) Pituitary adenylate cyclase activating polypeptide (PACAP): discovery and current status of research. Regul Pept 37:287–303 [PubMed] [Google Scholar]
- Bhola NE, Grandis JR. (2008) Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci 13:1857–1865 [DOI] [PubMed] [Google Scholar]
- Buscail L, Cambillau C, Seva C, Scemama JL, De Neef P, Robberecht P, Christophe J, Susini C, Vaysse N. (1992) Stimulation of rat pancreatic tumoral AR4–2J cell proliferation by pituitary adenylate cyclase-activating peptide. Gastroenterology 103:1002–1008 [DOI] [PubMed] [Google Scholar]
- Cattaneo F, Iaccio A, Guerra G, Montagnani S, Ammendola R. (2011) NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic Biol Med 51:1126–1136 [DOI] [PubMed] [Google Scholar]
- Draoui M, Hida T, Jakowlew S, Birrer M, Zia F, Moody TW. (1996) PACAP stimulates c-fos mRNAs in small cell lung cancer cells. Life Sci 59:307–313 [DOI] [PubMed] [Google Scholar]
- Germano PM, Lieu SN, Xue J, Cooke HJ, Christofi FL, Lu Y, Pisegna JR. (2009) PACAP induces signaling and stimulation of 5-hydroxytryptamine release and growth in neuroendocrine tumor cells. J Mol Neurosci 39:391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, Coldren C, Barón A, Zeng C, Franklin WA, et al. (2006) Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res 12:7117–7125 [DOI] [PubMed] [Google Scholar]
- Landi L, Cappuzzo F. (2011) Targeted therapies: Front-line therapy in lung cancer with mutations in EGFR. Nat Rev Clin Oncol 8:571–573 [DOI] [PubMed] [Google Scholar]
- Leyton J, Coelho T, Coy DH, Jakowlew S, Birrer MJ, Moody TW. (1998) PACAP(6–38) inhibits the growth of prostate cancer cells. Cancer Lett 125:131–139 [DOI] [PubMed] [Google Scholar]
- Lui VW, Thomas SM, Zhang Q, Wentzel AL, Siegfried JM, Li JY, Grandis JR. (2003) Mitogenic effects of gastrin-releasing peptide in head and neck squamous cancer cells are mediated by activation of the epidermal growth factor receptor. Oncogene 22:6183–6193 [DOI] [PubMed] [Google Scholar]
- Lutz EM, Ronaldson E, Shaw P, Johnson MS, Holland PJ, Mitchell R. (2006) Characterization of novel splice variants of the PAC1 receptor in human neuroblastoma cells: consequences for signaling by VIP and PACAP. Mol Cell Neurosci 31:193–209 [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350:2129–2139 [DOI] [PubMed] [Google Scholar]
- May V, Lutz E, MacKenzie C, Schutz KC, Dozark K, Braas KM. (2010) Pituitary adenylate cyclase-activating polypeptide (PACAP)/PAC1HOP1 receptor activation coordinates multiple neurotrophic signaling pathways: Akt activation through phosphatidylinositol 3-kinase γ and vesicle endocytosis for neuronal survival. J Biol Chem 285:9749–9761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata A, Arimura A, Dahl RR, Minamino N, Uehara A, Jiang L, Culler MD, Coy DH. (1989) Isolation of a novel 38-residue hypothalamic polypeptide which stimulated adenylate cyclase in pituitary cells. Biochem Biophys Res Commun 164:567–574 [DOI] [PubMed] [Google Scholar]
- Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, Wink DA, Chan D, Giaccone G, Jensen RT. (2010) Neuromedin B receptors regulate EGF receptor tyrosine phosphorylation in lung cancer cells. Eur J Pharmacol 637:38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody TW, Chan D, Fahrenkrug J, Jensen RT. (2003) Neuropeptides as autocrine growth factors in cancer cells. Curr Pharm Des 9:495–509 [DOI] [PubMed] [Google Scholar]
- Moody TW, Jensen RT. (2006) VIP and PACAP as autocrine growth factors in breast and lung cancer, in Handbook of Biologically Active Peptides (Kastin A. ed) pp 493–498, Elsevier Academic Press, Amsterdam [Google Scholar]
- Moody TW, Leyton J, Casibang M, Pisegna J, Jensen RT. (2002) PACAP-27 tyrosine phosphorylates mitogen activated protein kinase and increases VEGF mRNAs in human lung cancer cells. Regul Pept 109:135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody TW, Leyton J, Jensen RT. (2012) Pituitary adenylate cyclase-activating polypeptide causes increased tyrosine phosphorylation of focal adhesion kinase and paxillin. J Mol Neurosci 46:68–74 [DOI] [PubMed] [Google Scholar]
- Moody TW, Zia F, Bitar K, Coy DH.(1994) PACAP(6–38) is a PACAP type I receptor antagonist on small cell lung cancer cells, in VIP, PACAP and Related Regulatory Peptides: From Molecular Biology to Clinical Applications (Rosselin G. ed) pp527–534, World Scientific Publishing Co., Singapore [Google Scholar]
- Moody TW, Zia F, Makheja A. (1993) Pituitary adenylate cyclase activating polypeptide receptors are present on small cell lung cancer cells. Peptides 14:241–246 [DOI] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304:1497–1500 [DOI] [PubMed] [Google Scholar]
- Pisegna JR, Leyton J, Coelho T, Hida T, Jakowlew S, Birrer M, Fridkin M, Gozes I, Moody TW. (1997) PACAP hybrid: a new PACAP receptor antagonist. Life Sci 61:631–639 [DOI] [PubMed] [Google Scholar]
- Pisegna JR, Wank SA. (1993) Molecular cloning and functional expression of the pituitary adenylate cyclase-activating polypeptide type I receptor. Proc Natl Acad Sci U S A 90:6345–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisegna JR, Wank SA. (1996) Cloning and characterization of the signal transduction of four splice variants of the human pituitary adenylate cyclase activating polypeptide receptor. Evidence for dual coupling to adenylate cyclase and phospholipase C. J Biol Chem 271:17267–17274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubi JC, Läderach U, Waser B, Gebbers JO, Robberecht P, Laissue JA. (2000) Vasoactive intestinal peptide/pituitary adenylate cyclase-activating polypeptide receptor subtypes in human tumors and their tissues of origin. Cancer Res 60:3105–3112 [PubMed] [Google Scholar]
- Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, et al. (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361:958–967 [DOI] [PubMed] [Google Scholar]
- Sekido Y, Fong KM, Minna JD. (2005) Molecular biology of lung cancer, in Cancer: Principles and Practice of Oncology (DeVita VT, Helman S, Rosenberg SA. eds) pp 745–752, Lippincott Williams and Wilkins, Philadelphia [Google Scholar]
- Sherwood NM, Krueckl SL, McRory JE. (2000) The origin and function of the pituitary adenylate cyclase-activating polypeptide (PACAP)/glucagon superfamily. Endocr Rev 21:619–670 [DOI] [PubMed] [Google Scholar]
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, Journot L. (1993) Differential signal transduction by five splice variants of the PACAP receptor. Nature 365:170–175 [DOI] [PubMed] [Google Scholar]
- Ushiyama M, Ikeda R, Sugawara H, Yoshida M, Mori K, Kangawa K, Inoue K, Yamada K, Miyata A. (2007) Differential intracellular signaling through PAC1 isoforms as a result of alternative splicing in the first extracellular domain and the third intracellular loop. Mol Pharmacol 72:103–111 [DOI] [PubMed] [Google Scholar]
- Ushiyama M, Ikeda R, Yoshida M, Mori K, Kangawa K, Sugawara H, Inoue K, Yamada K, Miyata A. (2010) Alternative splicing of the pituitary adenylate cyclase-activating polypeptide (PACAP) receptor contributes to function of PACAP-27. J Mol Neurosci 42:341–348 [DOI] [PubMed] [Google Scholar]
- Valdehita A, Bajo AM, Schally AV, Varga JL, Carmena MJ, Prieto JC. (2009) Vasoactive intestinal peptide (VIP) induces transactivation of EGFR and HER2 in human breast cancer cells. Mol Cell Endocrinol 302:41–48 [DOI] [PubMed] [Google Scholar]
- Valdehita A, Carmena MJ, Bajo AM, Prieto JC. (2012) RNA interference-directed silencing of VPAC1 receptor inhibits VIP effects on both EGFR and HER2 transactivation and VEGF secretion in human breast cancer cells. Mol Cell Endocrinol 348:241–246 [DOI] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Bourgault S, Basille M, Burel D, Wurtz O, Fournier A, Chow BK, Hashimoto H, Galas L, et al. (2009) Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharm Rev 61:283–357 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, Kamens J, Gooding WE, Grandis JR. (2004) SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res 64:6166–6173 [DOI] [PubMed] [Google Scholar]
- Zia F, Fagarasan M, Bitar K, Coy DH, Pisegna JR, Wank SA, Moody TW. (1995) PACAP receptors regulate the growth of non-small cell lung cancer cells. Cancer Res 55:4886–4891 [PMC free article] [PubMed] [Google Scholar]