

Abstract
We report the first observation that endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) can decrease when a central nervous system drug acts as an intracellular pharmacological chaperone for its classic receptor. Transient expression of α4β2 nicotinic receptors (nAChRs) in Neuro-2a cells induced the nuclear translocation of activating transcription factor 6 (ATF6), which is part of the UPR. Cells were exposed for 48 h to the full agonist nicotine, the partial agonist cytisine, or the competitive antagonist dihydro-β-erythroidine; we also tested mutant nAChRs that readily exit the ER. Each of these four manipulations increased Sec24D-enhanced green fluorescent protein fluorescence of condensed ER exit sites and attenuated translocation of ATF6-enhanced green fluorescent protein to the nucleus. However, we found no correlation among the manipulations regarding other tested parameters [i.e., changes in nAChR stoichiometry (α42β23 versus α43β22), changes in ER and trans-Golgi structures, or the degree of nAChR up-regulation at the plasma membrane]. The four manipulations activated 0 to 0.4% of nAChRs, which shows that activation of the nAChR channel did not underlie the reduced ER stress. Nicotine also attenuated endogenously expressed ATF6 translocation and phosphorylation of eukaryotic initiation factor 2α in mouse cortical neurons transfected with α4β2 nAChRs. We conclude that, when nicotine accelerates ER export of α4β2 nAChRs, this suppresses ER stress and the UPR. Suppression of a sustained UPR may explain the apparent neuroprotective effect that causes the inverse correlation between a person's history of tobacco use and susceptibility to developing Parkinson's disease. This suggests a novel mechanism for neuroprotection by nicotine.
Introduction
Nicotine seems to cause at least part of the well documented inverse correlation between a person's history of smoking and his or her susceptibility to developing Parkinson's disease (Hernán et al., 2002; Lester et al., 2009; Quik et al., 2011). The use of smoked or smoke-cured tobacco has no medical justification. Therefore, it is essential to understand the mechanistic basis for the apparent neuroprotective action of nicotine.
Nicotine binds to and activates neuronal nicotinic acetylcholine receptors (nAChRs), a family of ligand-gated ion channels comprising homopentameric and heteropentameric combinations of α (α2 to α10) and β (β2 to β4) subunits (Gotti et al., 2007). The substantia nigra pars compacta dopaminergic neurons that degenerate in Parkinson's disease robustly express several nAChR receptor subtypes; some of these subtypes are substantially retained within the endoplasmic reticulum (ER) (Hill et al., 1993; Azam et al., 2002; Commons, 2008), rather than being efficiently trafficked to the plasma membrane (PM). We hypothesized that changes in the accumulation of nAChRs within the ER of dopaminergic neurons could influence a possible unfolded protein response (UPR)/ER stress response (Ron and Walter, 2007; Hetz and Glimcher, 2009). Long-term activation of the UPR can lead to apoptosis (Kim et al., 2006; Li et al., 2006). Post mortem studies have shown that dopaminergic neurons in patients with Parkinson's disease display increases in UPR markers (Hoozemans et al., 2007).
The best-known effect of chronic exposure to nicotine is up-regulation of nAChRs in vivo and in vitro, through cell-delimited, post-translational mechanisms (Schwartz and Kellar, 1983; Peng et al., 1994; Gopalakrishnan et al., 1997; Buisson and Bertrand, 2002; Sallette et al., 2005; Nashmi et al., 2007; Lester et al., 2009; Srinivasan et al., 2011). Emerging data suggest that selective pharmacological chaperoning of acetylcholine receptor number and stoichiometry (SePhaChARNS) by nicotine underlies nicotine-induced nAChR up-regulation (Kuryatov et al., 2005; Sallette et al., 2005; Lester et al., 2009; Miwa et al., 2011; Srinivasan et al., 2011). We hypothesized that SePhaChARNS also could attenuate the UPR and could partly explain the observed neuroprotective effects of nicotine in Parkinson's disease.
To test the ER stress/UPR hypothesis, we used the rich pharmacological features of α4β2 nAChRs. nAChRs may be chaperoned by three distinct classes of α4β2 ligands: full agonists (we chose nicotine itself), partial agonists (we chose cytisine), and antagonists [we chose dihydro-β-erythroidine (DHβE)] (Gopalakrishnan et al., 1997; Whiteaker et al., 1998; Kishi and Steinbach, 2006). We added a fourth, nonpharmacological manipulation: expression of a previously described β2enhanced-ER-export mutant subunit (also called β2-DM), which allows α4β2 nAChRs to exit the ER efficiently (Srinivasan et al., 2011).
We found that exposure to all four manipulations did decrease the UPR, and this correlated well with increased ER exit sites (ERESs). The tested manipulations, however, displayed different effects at all other stages of receptor stabilization and trafficking. Our results point to the possibility that SePhaChARNS can decrease the UPR by increasing cargo export from the ER and altering the physiological features of the ER. The data establish that pharmacological chaperoning of a central nervous system receptor by a drug can decrease ER stress and attenuate the UPR, which is relevant to the apparent neuroprotective effects of nicotine in Parkinson's disease.
Materials and Methods
Reagents.
PfuTurbo Cx Hotstart polymerase was purchased from Agilent Technologies (Santa Clara, CA). Mouse Neuro-2a cells (CCL-131) were obtained from American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle's medium (DMEM) with 4 mM l-glutamine, OptiMEM 1, Leibovitz L-15 imaging medium, Glutamax, Neurobasal medium, Lipofectamine 2000, and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). Nupherin-neuron was purchased from Enzo Life Sciences (Farmingdale, NY). Expressfect was purchased from Denville Scientific (South Plainfield, NJ). Poly-d-lysine–coated and uncoated 35-mm glass-bottomed imaging dishes (coverslip thickness, 0.19 mm) were obtained from MatTek (Ashland, MA). (−)-Nicotine hydrogen tartrate (Pubchem CID 6174), (−)-cytisine (Pubchem CID 22407), and DHβE (Pubchem CID 31762) were purchased from Sigma-Aldrich (St. Louis, MO). Pubchem can be accessed at http://pubchem.ncbi.nlm.nih.gov.
Plasmid Constructs.
Mouse β2-eGFP and β2-mcherry were engineered as described previously (Nashmi et al., 2003). All other plasmids used in this study were described previously (Srinivasan et al., 2011). ATF6-eGFP was obtained from Dr. Ron Prywes (Columbia University, New York, NY) (Shen et al., 2005).
Cell Culture and Transfections.
Mouse Neuro-2a cells were cultured by using standard tissue culture techniques and were maintained in 45% DMEM/45% OptiMEM/10% FBS. Plasmid concentrations used for transfection were as follows: 500 ng of each nAChR subunit, 75 ng of pCS2-mcherry, 250 ng of Sec24D constructs, 100 ng of pDsRed2-ER, 250 ng of GalT-eCFP, and 500 ng of ATF6-eGFP. A total of 90,000 cells were plated in poly-d-lysine–coated 35-mm glass-bottomed imaging dishes (MatTek). The following day, appropriate plasmid DNA concentrations were added to 4 μl of Expressfect transfection reagent in 200 μl of DMEM (final volume), and the mixture was incubated for 20 min at room temperature for formation of cationic lipid-DNA complexes. DMEM and DNA-lipid complexes were added to Neuro-2a cells in 1 ml of DMEM with 10% FBS and were incubated at 37°C for 4 h. Nicotine, cytisine, or DHβE was added at appropriate concentrations during the change of medium after 4 h of incubation with Expressfect-plasmid DNA complexes. Dishes were rinsed twice with DMEM, filled with 3 ml of DMEM with 10% FBS, and incubated at 37°C for 48 h.
Neuronal Transfection and Immunostaining.
Cortical neurons were extracted from day 17 mouse embryos; 150,000 cells were plated in each 35-mm, poly-l-lysine–coated 35-mm glass-bottomed culture dish, in a solution containing Neurobasal medium, B27, and Glutamax supplemented with 3% equine serum. On day 4 of culture, neurons were treated with 1 μM cytosine arabinoside. Neurons were maintained through 50% exchange with feeding medium (Neurobasal medium, B27, and Glutamax) twice per week. Neurons were transfected 5 days after plating, as follows: 1 μg of each nAChR subunit plasmid was incubated with 20 μg of Nupherin-neuron in 400 μl of Neurobasal medium; 10 μl of Lipofectamine 2000 was separately incubated for 15 min in 400 μl of Neurobasal medium. The two solutions were combined, incubated for 45 min at room temperature, and then applied to cells. After 1 h of incubation, the total medium was replaced with 2 ml of fresh medium, with or without 0.1 μM nicotine. For immunostaining, cultures were fixed the following day with 4% paraformaldehyde (10 min), permeabilized in 0.02% Tris-buffered saline (TBS)-Triton X-100 (15 min), and blocked with 10% goat serum (30 min). After two TBS washes, the appropriate primary antibody in 1% goat serum was applied overnight at 4°C. The following day, cultures were rinsed with TBS and incubated with 1% goat serum containing secondary antibody (1:5000, 30 min). Cells were rinsed with TBS and imaged immediately.
Antibodies.
Immunostaining used ATF6 mouse monoclonal antibody (antibody 11909, 1:50 dilution; Abcam) and rabbit polyclonal antibodies to phosphorylated eukaryotic initiation factor 2α (peIF2α) and total eukaryotic initiation factor 2α (eIF2α) (antibodies 9721 and 5324, respectively, both at 1:200 dilution; Cell Signaling Technology, Danvers, MA). We used Alexa Fluor 488-labeled secondary antibodies (donkey anti-mouse and goat anti-rabbit antibodies; Invitrogen).
ATF6-eGFP Translocation Assay.
An Eclipse C1si laser scanning confocal microscope equipped with a VC PlanApo, 63×, 1.4 numerical aperture, oil-immersion objective and a 32-anode photomultiplier tube (Nikon, Melville, NY) was used. All experiments were performed with live cells, 48 h after transfection, at 37°C in a stage-mounted culture dish incubator (Warner Instruments, Hamden, CT). Just before imaging, cell culture medium was replaced with phenol red-free, CO2-independent, Leibovitz L-15 medium. Cells were effectively exposed to L-15 medium for ∼20 to 40 min during imaging. The high amino acid concentrations in L-15 medium are unlikely to affect our observations during this relatively short period of exposure. For quantification of ATF6-eGFP translocation, the cell was focused to a plane where the nucleus was observed most clearly and full emission spectra were acquired after simultaneous excitation of eGFP and mcherry with 488-nm and 561-nm laser lines, respectively. Images were unmixed by using spectra from cells expressing only α4-mcherryβ2-wt nAChRs or only ATF6-eGFP. Regions of interest (ROIs) were manually demarcated for the nucleus and whole cell from the unmixed α4-mcherryβ2-wt image and then applied to the unmixed ATF6-eGFP image of the same cell. Ratios of integrated densities of ATF6-eGFP fluorescence in the nucleus and whole cell were obtained for each cell.
ERES Quantification.
Neuro-2a cells were cotransfected with 500 ng each of α4-mcherry and β2-wt nAChR subunit plasmids and 250 ng of the ERES marker Sec24D-eGFP plasmid. To control for distortions that can occur because of differences in cell size, all imaged cells were between 30 and 40 μm in diameter. Each cell was focused to a plane where the maximal number of ERESs was observed. Sequential images of eGFP and mcherry fluorescence were obtained and linearly unmixed. For quantification, ERES ROIs were demarcated by using intensity-based threshold setting and were counted for each cell by using the particle analysis feature in ImageJ 1.44 (or a later version; http://rsbweb.nih.gov/ij/). The total Sec24D fluorescence in ERES, which constitutes the upper third of total Sec24D-eGFP fluorescence, was quantified for each cell. Total (integrated) Sec24D fluorescence in ERESs was calculated for each cell. Data are presented as the mean of data for 30 to 40 cells imaged under each condition. Error bars for ERES measurements indicate the S.E.M., and p values are based on a two-tailed t test.
Imaging and Quantification of Trans-Golgi Network and ER Fluorescence.
Neuro-2a cells were transfected with α4-eGFP (500 ng), β2-wt (500 ng), and GalT-mcherry (250 ng) or pDSred2-ER (100 ng) plasmids, and cells were incubated for 48 h with appropriate concentrations of nicotine, cytisine, or DHβE. Total internal reflection fluorescence microscopy (TIRFM) was used to observe the PM, trans-Golgi network (TGN), and ER. TIRFM images of 30 to 40 live cells were obtained for each experimental condition. TIRFM imaging was performed by using an inverted microscope (IX71; Olympus America, Center Valley, PA) equipped with an Olympus PlanApo 100×, 1.45 numerical aperture, oil-immersion objective and an actuated stage with a closed-loop controller (Thorlabs, Newton, NJ), to control the position of the fiber optic and TIRFM evanescent field illumination. α4-eGFP was used for observation of α4β2 nAChRs, whereas pDSred2-ER and GalT-mcherry markers (Schaub et al., 2006) were used to label the ER and TGN, respectively. eGFP and mcherry or DSred2 fluorophores were excited with a 488-nm, air-cooled, argon laser (IMA101040ALS; Melles Griot, Carlsbad, CA), and an Optosplit II image splitter (Cairn Research, Faversham, UK) was used to detect red and green fluorescence emissions simultaneously. Images were captured at 14-bit resolution with an iXON DU-897, back-illuminated, electron-multiplying charge-coupled device camera (Andor, South Windsor, CT). Sample exposure rate, percent laser transmission, and gain parameters were adjusted initially and then maintained constant across all samples for each imaging session. The 488-nm argon laser was linearly s-polarized, as revealed by using an achromatic 400- to 800-nm half-wave plate (AQWP05M-600; Thorlabs) (courtesy of Larry Wade).
We used ImageJ 1.44 software to quantify ER area and Metamorph software (Molecular Devices, Sunnyvale, CA) to quantify TGN number, size, and intensity. For quantification of ER area, the background was subtracted for each cell and the DSred2-ER fluorescence signal was subjected to manual threshold setting and demarcation to yield an ER ROI. The total ER area was measured for each cell, and a mean value for ER area was extracted for each experimental group. TGN bodies were subjected to manual threshold-setting and demarcation. With the use of Metamorph software, number, size, and intensity statistics were extracted from demarcated ROIs. The Cairn image splitter plug-in for ImageJ was used for realignment of TIRFM images. TIRFM images were manually realigned by using distinct, obviously colocalized, organelle features in cellular footprints within the red and green emission windows.
Quantification of PM-Localized nAChRs.
Details of the quantification method were described previously (Srinivasan et al., 2011). In brief, PM fluorescence was extracted as follows. Raw TIRFM images were converted to background-subtracted images, and the ER fluorescence was subjected to threshold-setting and selection. ER fluorescence was then subtracted from the original image to generate images with PM fluorescence signals. These procedures yielded a data set of several hundred thousand pixel intensities over the 15 to 50 cells in each experimental group. Fluorescence intensities are simply the sum of pixel values for either the PM, the ER, or the ER plus PM (obtained by using whole TIRFM footprint images, where “footprint” indicates the part of the cell that is in contact with the coverslip). Mean PM fluorescence intensity was derived by dividing the total fluorescence intensity for each experimental group by the number of imaged cells and was normalized to the intensity of untreated cells. To control for effects of cell size on average PM intensity, we imaged cells that had similar footprint areas. Cellular footprints with diameters of >50 μm or <30 μm were not imaged.
Whole-TIRFM footprint (ER plus PM)/ER fluorescence intensity ratios were used to determine the post-Golgi fraction of receptors. For PM integrated-density measurements, we used error bars to depict 99% confidence intervals based on a two-tailed t test, rather than plotting S.E.M. values (which would be indistinguishably small on the plots). Measurements from drug-treated cells were normalized to values for untreated cells imaged on the same day. DHβE TIRFM experiments were performed on a different day than nicotine and cytisine experiments.
Stoichiometry Monitored through Pixel-by-Pixel Normalized Förster Resonance Energy Transfer from Sensitized Acceptor Emission.
General methods for pixel-by-pixel normalized Förster resonance energy transfer (NFRET) from sensitized acceptor emission were described previously (Moss et al., 2009; Son et al., 2009; Srinivasan et al., 2011). For the present NFRET experiments, Neuro-2a cells were transfected with GalT-eCFP, α4-mcherry, and β2-eGFP subunit plasmids. Therefore, all β2 subunits contained eGFP donors and all α4 subunits contained mcherry acceptors. The procedure yielded more-robust signals than those observed in our previous studies that used only fluorescent α subunits or only fluorescent β subunits. GalT-eCFP was used to label the TG/TGN (Schaub et al., 2006).
Cells transfected with α4-mcherryβ2-wt or α4-wtβ2-eGFP nAChRs were included in every imaging session, to control for pixel saturation and spectral bleedthrough (BT). Live cells were imaged by using an Eclipse C1si laser scanning confocal microscope. During image acquisition, cells were focused to a plane where the GalT-eCFP fluorescence was best observed. Images with emission spectra for eCFP, eGFP, and mcherry were acquired in 5-nm bins between 470 and 620 nm; 439-, 488-, and 561-nm laser lines were used to excite eCFP, eGFP, and mcherry, respectively. Images were linearly unmixed by using reference spectra for eCFP, eGFP, and mcherry with emission maxima at 477, 508, and 608 nm, respectively. Reference spectra were acquired from Neuro-2a cells transfected with GalT-eCFP, α4-mcherryβ2-wt, or α4-wtβ2-eGFP, during the same imaging session. Linear unmixing was used to separate all three emission spectra from each image. Linearly unmixed images were compiled into donor spectral BT stacks, acceptor BT stacks, and sample image stacks.
The PixFRET ImageJ plug-in was used to determine the eGFP and mcherry BT values and to calculate the net FRET (eq. 1) and NFRET values at each pixel. With the background and BT corrections set, the net FRET value for each pixel (eq. 1) was calculated and the data were presented as 32-bit images. The net FRET value was divided by the square root of the eGFP and mcherry intensities (eq. 2) to yield the NFRET value at each pixel. FRET normalization was used to control for large differences in fluorophore expression within subcellular regions and between different cells in a dish.
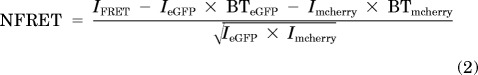 |
where nF is net FRET, IFRET is mcherry-sensitized emission with 488-nm excitation, IeGFP is eGFP emission with 488-nm excitation, BTeGFP is the spectral BT of eGFP emission into mcherry emission spectra at 488-nm excitation, Imcherry is mcherry emission with 561-nm excitation, and BTmcherry is the spectral BT of mcherry emission into eGFP emission spectra at 561-nm excitation. IFRET, IeGFP, and Imcherry were determined from Neuro-2a cells transfected with GalT-eCFP, α4-mcherry, and β2-eGFP. BTeGFP and BTmcherry values were determined from Neuro-2a cells transfected with α4-wt plus β2-eGFP and α4-mcherry plus β2-wt, respectively.
Histograms of NFRET (x-axis) versus the number of pixels (y-axis) for each imaged cell were compiled for either the whole cell sections or the TG/TGN ROIs, which were demarcated by using intensity-based threshold-setting for GalT-eCFP fluorescence. Frequency histograms obtained in this way (omitting values of 0) were merged for all cells in each experimental group and were fitted to two Gaussian components such that a minimal goodness-of-fit (R2) value of 0.995 was achieved. NFRET histograms were fitted to Gaussian components, which yielded the total pixel area for either the A1 (low mean NFRET fit) or A2 (high mean NFRET fit) component. For quantitative measurements of stoichiometry, we compared the fractional area for the high-NFRET component (Whigh) in each case, as given by eq. 3,
 |
where Whigh is the fractional area of the high-NFRET component, A1 is the area under the curve for the low mean NFRET Gaussian component, and A2 is the area under the curve for the high mean NFRET Gaussian component.
Because of variability resulting from cell passage number, we imaged untreated α4-mcherry- plus β2-eGFP-transfected control cells on the same day, to evaluate the effects of drug exposure on stoichiometry. To provide further evidence that the two fitted NFRET components reflect nAChR stoichiometry, we manipulated receptor stoichiometry by transfecting biased ratios of α4-mcherry and β2-eGFP nAChR subunit plasmids into Neuro-2a cells (see Supplemental Fig. 1). We verified that reducing the mole fraction of transfected β2-eGFP subunit cDNA from 0.67 to 0.5 and to 0.33 caused monotonic increases in Whigh and mean NFRET (Supplemental Fig. 1C). The formal Gaussian fits (see Fig. 5 and Supplemental Fig. 1B) could arise either if each component contains a mixture of individual pixels that each contain a pure α43β22 or α42β23 population or if individual pixels contain a mixture of α43β22 and α42β23.
Fig. 5.

The three nicotinic ligands have diverse effects on α4β2 nAChR stoichiometry. A, NFRET measurements were made by using α4-mcherry and β2-eGFP subunits transfected into Neuro-2a cells. Columns indicate the ROIs (whole cell or TG, TGN), and rows indicate drug treatment conditions. For each graph, the blue curve is the overall fit and dashed Gaussian curves are individual fits. Whigh, corresponding to the α43β22 stoichiometry, is shown for each graph. B, representative NFRET images of Neuro-2a cells. Drug treatments are indicated for each cell. Calibration bars are from zero to 20%. Scale bars, 5 μm.
Patch-Clamp Recording.
Neuro-2a cells were plated on 12-mm glass coverslips placed in 35-mm, plastic-bottomed, cell culture dishes. The following day, cells were transfected with 500 ng of α4-eGFP and 500 ng of β2-wt nAChR subunit plasmids. Twenty-four to 48 h later, glass coverslips were transferred to dishes on the microscope stage. Green fluorescence was observed with an upright microscope (BX50WI; Olympus America) with UV illumination with appropriate excitation filters. Agonist-induced currents were tested at a holding potential of −65 mV by using a focal, relatively rapid, drug application system designed to minimize desensitization (Xiao et al., 2009). We applied drugs from the lowest concentration to the highest concentration, at 3-min intervals, to minimize desensitization. Whole-cell patch-clamp recordings were performed with a MultiClamp 700B amplifier, a 1322 analog-to-digital converter, and pCLAMP 9.2 software (all from Molecular Devices). Data were sampled at 10 kHz and were filtered at 2 kHz.
The intrapipette solution contained 135 mM potassium gluconate, 5 mM KCl, 5 mM EGTA, 0.5 mM CaCl2, 10 mM HEPES, 2 mM Mg-ATP, and 0.1 mM GTP; the pH was adjusted to 7.2 with Tris base and the osmolarity was adjusted to 300 mOsM with sucrose. The extracellular solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose; the pH was adjusted to 7.3 with Tris base. The Nernst potential for Cl− in the intrapipette solution was −82.9 mV. The bath was continuously perfused with extracellular solution at room temperature (23 ± 1°C). The patch electrodes had resistances of 5 to 8 MΩ. The junction potential between the patch pipette and the bath solutions was adjusted to null just before a gigaohm seal was formed. Series resistance was monitored and compensated by 70 to 80% throughout recordings. Data were ignored if the series resistance (15–30 MΩ) changed by >20% during the recording session.
Results
Each of the Four Manipulations Inhibits Nuclear Translocation of ATF6.
We hypothesized that nAChR expression and up-regulation could affect the ER stress response/UPR (Srinivasan et al., 2011). During the UPR, ATF6 is translocated from the ER to the Golgi and is cleaved by site 1 and site 2 proteases to release an N-terminal peptide. The N-terminal ATF6 peptide is translocated to the nucleus and initiates transcription of several target UPR genes that enhance ER function (Matsushita et al., 2002; Bommiasamy et al., 2009; Maiuolo et al., 2011).
To test the effect of nAChR expression on nuclear ATF6 translocation, we used a previously reported ATF6 construct with an N-terminal eGFP fusion (ATF6-eGFP) (Shen and Prywes, 2005; Shen et al., 2005). Neuro-2a cells were transfected with either ATF6-eGFP alone or ATF6-eGFP and α4-mcherryβ2-wt nAChRs (Fig. 1A). We found that coexpression of α4-mcherryβ2-wt nAChRs increased nuclear ATF6-eGFP translocation (Fig. 1, A and B). For evaluation of whether the observed increase in ATF6-eGFP translocation to the nucleus was an artifact of DNA transfection or protein overexpression, Neuro-2a cells were cotransfected with ATF6-eGFP and α4-mcherry, paired with either β2-wt or a previously described β2enhanced-ER-export mutant subunit (also called β2-DM), which is known to undergo enhanced ER export (Srinivasan et al., 2011). Compared with α4-mcherryβ2-wt nAChRs, expression of α4-mcherryβ2enhanced-ER-export mutants caused a 40% reduction in the ATF6-eGFP nuclear/whole-cell fluorescence ratio in the absence of nicotine (Fig. 1B).
Fig. 1.

Each of the four manipulations decreases ATF6 translocation in Neuro-2a cells. A, representative confocal images of Neuro-2a cells expressing α4-mcherryβ2- wt nAChRs plus ATF6-eGFP or only ATF6-eGFP. Scale bars, 10 μm. B, ATF6-eGFP fluorescence intensity ratios (nucleus/whole cell section). Treatment conditions and transfected subunits are indicated on the x-axis. DM, double-mutant β2enhanced-ER-export subunit. C, ATF6-eGFP fluorescence intensity ratios (nucleus/whole cell section) in the absence of coexpressed nAChRs. Drug treatments are indicated on the x-axis. The p values are based on a 2-tailed t test. **, p < 0.01; ***, p < 0.001. Data were obtained from 30 to 40 cells imaged for each condition. Nic, nicotine; Cyt, cytisine.
In parallel experiments, Neuro-2a cells coexpressing ATF6-eGFP and α4-mcherryβ2-wt nAChRs were treated with nicotine, cytisine (0.1 μM, 48 h), or DHβE (10 or 100 μM, 48 h). Nicotine and cytisine caused ∼25% reductions in the ATF6-eGFP nuclear/whole-cell fluorescence ratio (Fig. 1B), and both concentrations of DHβE reduced the ratio by ∼23% (Fig. 1B). None of the tested ligands affected ATF6-eGFP translocation in the absence of nAChR coexpression (Fig. 1C).
Nicotine Inhibits Endogenous ATF6 Translocation and Prevents eIF2α Phosphorylation in Primary Mouse Cortical Neurons.
We sought to study the effects of nicotine on the UPR in primary neurons in culture. Cells were transfected with α4-mcherryβ2-wt nAChRs and were separately immunostained for endogenous ATF6, total eIF2α, or peIF2α.
A total of 56% of α4-mcherryβ2-wt–expressing neurons showed nuclear ATF6 localization in the absence of nicotine (Fig. 2A), which decreased to 14% after 24-h treatment with 0.1 μM nicotine. Compared with untreated cells, nicotine caused a significant 50% reduction in the ATF6 nuclear/whole-cell fluorescence ratio (Fig. 2B). peIF2α and total eIF2α were localized to the nucleus and cytoplasm of all α4-mcherryβ2-wt–expressing neurons (Fig. 2A). Twenty-four-hour exposure to 0.1 μM nicotine showed a trend toward reductions in whole-cell and cytoplasmic peIF2α fluorescence (p = 0.06), whereas nuclear peIF2α levels were significantly reduced, compared with untreated control cells (Fig. 2C). Nicotine did not affect total eIF2α fluorescence within the cytoplasm or nucleus of neurons (Fig. 2D), which indicates specific nicotine-induced inhibition of eIF2α phosphorylation.
Fig. 2.

Nicotine inhibits ATF6 translocation to the nucleus and eIF2α phosphorylation in mouse cortical neurons. A, representative confocal images of mouse cortical neurons expressing α4-mcherryβ2-wt, with immunostaining for endogenous ATF6, peIF2α, or total eIF2α (scale bars, 10 μm). B, fluorescence intensity ratios for endogenously expressed ATF6, with and without 0.1 μM nicotine (Nic) treatment for 24 h. C, nuclear fluorescence intensity for peIF2α, with and without 0.1 μM nicotine treatment for 24 h. D, fluorescence intensity for total eIF2α. Bars show nicotine treatment conditions. All neurons in B, C, and D expressed transfected α4-mcherry plus β2-wt subunits. The subcellular compartments imaged are indicated on the x-axis. The p values are based on a 2-tailed t test. *, p < 0.05; **, p < 0.01. Data were obtained from 20 to 30 cells imaged for each condition.
Each of the Four Manipulations Increases ERES Formation.
To understand the mechanistic basis for the effects of nicotinic ligands on the UPR, we studied the effects of each ligand on ERES formation. Neuro-2a cells were transfected with α4-mcherryβ2-wt nAChRs, and Sec24D-eGFP was used as a marker to quantify ERES (Fig. 3A). We quantified fluorescence from condensed Sec24D-eGFP ERES structures that contributed to the upper third of total Sec24D-eGFP fluorescence in each cell (Fig. 3B). All three nicotinic ligands caused significant 1.5- to 2-fold increases in the total Sec24D fluorescence in ERES per cell, compared with untreated control cells (Fig. 3C). These data suggest that ligand-induced UPR inhibition arises, at least in part, from increased cargo exit from the ER. Previous data showed that replacement of the β2 subunit by the β2enhanced-ER-export subunit also enhanced the formation of ERES (Srinivasan et al., 2011). We separately measured the area and the number of ERES per cell for all drug treatment conditions, and we observed statistically significant differences (data not shown).
Fig. 3.

Exposure to all three nicotinic ligands increases the formation of ERES. A, representative confocal images of a Neuro-2a cell expressing α4-mcherryβ2-wt nAChRs and Sec24D-eGFP (ERES marker). Scale bars, 10 μm. B, image of the same cell with ERES demarcated for quantification. C, quantification of total Sec24D fluorescence in ERES per cell. Drug treatment conditions are indicated on the x-axis. Error bars indicate S.E.M. The p values are based on a 2-tailed t test. *, p < 0.05; **, p < 0.01. The graphs present total Sec24D fluorescence of ERES per cell, averaged for 30 to 40 cells imaged for each condition. Nic, nicotine; Cyt, cytisine.
The Manipulations Have Diverse Effects on the Morphological Features of the ER and TGN.
TIRFM can be used to determine the subcellular localization of fluorescent protein-labeled receptors at the cellular periphery (Richards et al., 2011). Those experiments showed that a majority of wt α4β2 receptors were localized to the ER (Srinivasan et al., 2011). Here, we used TIRFM to quantify the effects of ligand-induced nAChR up-regulation on the ER and TGN architecture. For quantification, we used fluorescence from specific fluorescent protein-tagged markers of the ER (DSred2-ER) or the TGN (GalT-mcherry) in the presence of coexpressed α4β2 nAChRs. Therefore, the results described here assessed the effects of the ligand-α4β2 interaction on general ER and TGN morphological features.
In a first set of TIRFM experiments, Neuro-2a cells were cotransfected with α4-eGFPβ2-wt nAChRs and DSred2-ER (ER marker). Expressed α4-eGFPβ2-wt nAChRs colocalized almost completely with DSred2-ER fluorescence (Fig. 4A). We used DSred2-ER fluorescence to demarcate and to quantify the average area of peripheral ER. Compared with untreated control cells, nicotine exposure caused a 1.7-fold increase in the average ER area, whereas cytisine caused a significant ∼1.4-fold reduction and DHβE did not significantly affect the ER area (Fig. 4B).
Fig. 4.

The three nicotinic ligands have diverse effects on ER and TGN architecture. A, representative TIRFM images of a cell coexpressing α4-eGFPβ2-wt nAChRs and pDSred2-ER marker. The merged image shows nearly complete colocalization of fluorescence from nAChRs and DSred2-ER. Scale bars, 10 μm. B, quantification of average ER area determined by using DSred2-ER fluorescence. Drug treatments are indicated on the x-axis. C, representative TIRFM images of a cell expressing α4-eGFPβ2-wt nAChRs and GalT-mcherry. The merged image shows colocalization of GalT-mcherry with nAChRs. Scale bars, 10 μm. D, quantification of the number of TGN bodies observed by using GalT-mcherry under each treatment condition, as indicated on the x-axis. E, quantification of the average TGN body fluorescence intensity observed by using GalT-mcherry under each treatment condition, as indicated on the x-axis. Error bars indicate S.E.M. The p values are based on a 2-tailed t test. *, p < 0.05; **, p < 0.01. Data were obtained from 30 to 40 cells imaged for each condition. Nic, nicotine; Cyt, cytisine; NS, not significant.
In a second set of TIRFM experiments, Neuro-2a cells were cotransfected with α4-eGFPβ2-wt and GalT-mcherry to demarcate the TGN (Fig. 4C). In this case, we quantified the average number and intensity of GalT-mcherry–labeled TGN bodies. Nicotine caused a significant ∼2-fold increase in the average number of TGN bodies per cell, compared with untreated cells, whereas cytisine and DHβE failed to affect the number of TGN bodies significantly (Fig. 4D). In addition, nicotine exposure did not affect the TGN intensity, whereas cytisine and DHβE significantly reduced the average intensity of TGN bodies (Fig. 4E). These data show that receptor chaperoning by nicotine, cytisine, and DHβE alters the morphological features of the ER and the TGN in a ligand-dependent manner.
The Four Manipulations Have Diverse Effects on nAChR Stoichiometry.
These experiments implemented an improved NFRET method for monitoring nAChR stoichiometry (see Materials and Methods and Supplemental Fig. 1). We tested the effects of the three ligands on the stoichiometry (α42β23 versus α43β22) of assembled pentameric nAChRs, primarily in organelles. The fractional area of the high-NFRET component (Whigh) was used to quantify nAChRs with α43β22 stoichiometry. Neuro-2a cells were transfected with α4-mcherry, β2-eGFP, and a trans-Golgi (TG), TGN marker, GalT-eCFP. Whole-cell and TG, TGN Whigh values for untreated cells were 0.46 and 0.47, respectively, whereas whole-cell and TG, TGN Whigh values decreased to 0.26 and 0.20, respectively, in nicotine-treated cells (Fig. 5A). In contrast to nicotine-treated cells, cytisine-treated cells showed net increases in Whigh values for the whole cell (Whigh = 0.70) and the TG, TGN (Whigh = 0.58), compared with untreated control cells (Fig. 5A). DHβE did not affect nAChR stoichiometry at concentrations ranging from 0.1 to 1 μM, but 10 μM DHβE increased the whole-cell Whigh to 0.55 and increased the TG, TGN Whigh to 0.63 (Fig. 5A). It should be noted that, compared with untreated, nicotine-treated, or cytisine-treated cells, treatment with either 10 or 100 μM DHβE produced a 3- to 5-fold reduction in the number of NFRET-positive pixels for the whole cell and TG, TGN (Fig. 5B).
These results demonstrate that nicotine and cytisine stabilize the assembly of α42β23 and α43β22 receptors, respectively, whereas DHβE weakly favors the assembly of α43β22 receptors. Previous data showed that replacement of the β2 subunit with the β2enhanced-ER-export subunit also stabilized the α42β23 stoichiometry (Srinivasan et al., 2011). All four manipulations produced changes in stoichiometry before export of receptors from the ER to the Golgi.
The Tested Agonist Concentrations Minimally Activate α4β2 nAChRs.
Because the observed effects of the agonists nicotine and cytisine on the cellular secretory pathway occurred at 0.1 μM, we sought to determine whether, and to what extent, this agonist concentration activates α4β2 nAChRs. Whole-cell electrophysiological responses to focal nicotine and cytisine puffs were measured in Neuro-2a cells expressing α4-eGFPβ2-wt nAChRs. Figure 6A shows representative traces with 0.1 μM nicotine or cytisine. Measured current amplitudes were normalized to the maximal current amplitude obtained through puffing of 500 μM nicotine (Fig. 6A). With 0.1 μM nicotine, we observed nicotine-evoked currents that were 0.44 ± 0.07% (n = 4) of maximal currents induced by 500 μM nicotine. In the case of cytisine, 0.1 μM activated <0.1% (n = 7) of maximal currents induced by 500 μM nicotine. These data indicate that the effects of nicotine and cytisine on nAChR stoichiometry are likely to be independent of nAChR activation and ion flux.
Fig. 6.

The three nicotinic ligands have diverse effects on PM-localized α4β2 nAChRs. A, representative traces showing whole-cell currents induced by puffs of 0.1 and 500 μM nicotine or 0.1 μM cytisine and 500 μM nicotine. B, representative TIRFM images of cells expressing α4-eGFPβ2-wt nAChRs with treatment with the indicated drugs for 48 h. Scale bars, 10 μm. C, bar graph showing normalized PM fluorescence intensity from TIRFM images, normalized as described in Materials and Methods. Drug treatment conditions are shown on the x-axis. Error bars indicate 99% confidence intervals, which show that all differences are highly significant. D, footprint/ER ratios from TIRFM images. Drug treatment conditions are shown on the x-axis. Error bars indicate relative S.E. Data were obtained from 30 to 40 cells imaged for each condition. Nic, nicotine; Cyt, cytisine.
The Four Manipulations Have Diverse Effects on Up-regulation of PM nAChRs.
We used TIRFM to quantify the effects of nicotine, cytisine, and DHβE on nAChRs at the PM of Neuro-2a cells. Pixel-by-pixel TIRFM quantification is a direct measure of PM-localized receptors (Srinivasan et al., 2011). In addition, whole-footprint/ER-localized nAChR fluorescence ratios (footprint/ER ratios) provide quantitative measures of nAChRs in the PM versus the peripheral ER (Srinivasan et al., 2011). For these experiments, cells were transfected with α4-eGFPβ2-wt, and α4-eGFP fluorescence was used to quantify nAChRs. Figure 6B shows representative TIRFM images of nAChR fluorescence after 48-h exposure to each drug.
Compared with untreated control cells, nicotine and cytisine caused 2- and 3-fold increases, respectively, whereas DHβE caused a small but significant increase in PM-localized nAChRs (Fig. 6C). To address the possibility of distortions arising from differences in cell footprint size, we also analyzed the TIRFM data by summing pixel fluorescence values for all cells within an experimental group and dividing the result by the number of pixels imaged, to obtain the average fluorescence intensity per pixel. The trend among the three ligands resembled that depicted in Fig. 6C. Compared with untreated cells, nicotine, cytisine, and DHβE increased average PM pixel fluorescence values by 1.4-, 1.85-, and 1.1-fold, respectively.
The footprint/ER ratio decreased by ∼2-fold after nicotine treatment, whereas cytisine- and DHβE-treated cells showed ∼1.5-fold increases in the footprint/ER ratio, compared with untreated cells (Fig. 6D). Previous data showed that replacement of the β2 subunit with the β2enhanced-ER-export subunit also markedly increased the footprint/ER ratio (Srinivasan et al., 2011).
Discussion
The data establish, for the first time, that pharmacological chaperoning of a central nervous system receptor by a drug can decrease ER stress and attenuate the UPR. Parkinson's disease is associated with increased ER stress/UPR in dopaminergic neurons. Upon persistent activation, the UPR can cause apoptosis (Silva et al., 2005; Hoozemans et al., 2007). Because nicotine is potentially neuroprotective in Parkinson's disease (Hernán et al., 2002; Lester et al., 2009; Quik et al., 2011), we assessed the subcellular effects of pharmacological chaperoning by nicotinic ligands. Table 1 summarizes data obtained for the four manipulations [three exemplar nicotinic ligands and nAChRs containing β2enhanced-ER-export mutant subunits]. All four manipulations suppressed ATF6 translocation, which serves as a key sensor for ER stress and the UPR (Ron and Walter, 2007; Hetz and Glimcher, 2009; Maiuolo et al., 2011), but the only other consistent phenomenon was the increased total Sec24D fluorescence in ERES. Therefore, we suggest that increased ER exit of nAChRs underlies the suppression of ATF6 translocation.
TABLE 1.
The four manipulations all reduce ATF6 translocation and increase condensed ERES but have diverse effects on other aspects of α4β2 stoichiometry, organelle structure, and PM up-regulation
Numbers indicate fold change compared with untreated cells. Arrows indicate the fold increase or decrease from observed values in untreated cells. Some of the data for β2-DM are from Srinivasan et al. (2011).
| Tested Parameter | Nicotine | Cytisine | DHβE | β2-DM |
|---|---|---|---|---|
| ATF6 (nucleus/whole cell) | ↓1.4 | ↓1.4 | ↓1.4 | ↓1.6 |
| Total Sec24D fluorescence in ERES | ↑1.9 | ↑2 | ↑2.1 | ↑2 |
| Peripheral ER area | ↑1.8 | ↓1.5 | ↓1.2 | N.D. |
| nAChR footprint/ER ratio | ↓2 | ↑2 | ↑1.5 | ↑2.3 |
| No. of TGN bodies | ↑2 | N.E. | N.E. | N.D. |
| TGN intensity | N.E. | ↓3 | ↓3 | N.D. |
| Major stoichiometry | α42β23 | α43β22 | α43β22 | α42β23 |
| PM nAChRs | ↑2 | ↑3 | ↑1.2 | ↑2.5 |
N.D., not done; N.E., no effect; ↑, increase; ↓, decrease.
Dopaminergic neurons show robust expression of several nAChR subtypes (Champtiaux et al., 2003) that are localized to the ER under physiological conditions (Hill et al., 1993; Commons, 2008). Therefore, nAChRs are in a key position to affect the ER stress response by influencing the exit of cargo from the ER. In the present experiments, ER stress was caused by expression of the α4β2 nAChRs themselves (Fig. 1, A and B), but it is plausible to suggest that increasing the ER exit of nAChRs could reorganize the COPII vesicle population and relieve ER stress caused by other environmental or genetic factors. Dopaminergic neurons contain potentially toxic metabolites of dopamine (Ahmadi et al., 2008; Miyazaki and Asanuma, 2009) and relatively high intracellular Ca2+ levels (Surmeier et al., 2011). These factors probably lead to increased levels of ER stress under physiological conditions (Egawa et al., 2011). Additional ER stress in catecholaminergic cell lines and in cultured dopaminergic neurons occurs by means of dopaminergic toxins or the overexpression of α-synuclein, which is linked to Parkinson's disease (Ryu et al., 2002; Holtz and O'Malley, 2003; Thayanidhi et al., 2010).
Several points suggested that the enhanced ATF6 translocation was not related to mere nonspecific overexpression. First, transient expression of ATF6-eGFP alone did not cause ATF6 translocation to the nucleus (Fig. 1B), and nicotinic ligands did not influence ATF6 localization (Fig. 1C). Second, nAChRs containing mutant β2 subunits with enhanced ER export prevented ATF6 translocation, even in the absence of nicotine (Fig. 1B). Third, our experiments used a well characterized expression system in Neuro-2a cells, which express membrane proteins at rather lower levels, compared with more-commonly studied human embryonic kidney 293 cells (Moss et al., 2009). Likewise, neurons typically express transfected genes at only modest levels. It should be noted that, in most of our experiments, two or three α4 subunits per receptor contained a fluorescent protein label, which enabled us to identify nAChR-expressing Neuro-2a cells or neurons. Previous experiments with both cultured Neuro-2a cells and knock-in mice indicated that nAChRs containing such labeled α4 subunits were normal in every way studied (Nashmi et al., 2003, 2007; Fonck et al., 2009; Son et al., 2009). We cannot rule out the possibility that a large intracellular fluorophore in the α4 subunit can affect nAChR assembly, thereby enhancing an ER stress response.
Nicotine also inhibited ATF6 translocation in primary mouse cortical neurons transfected with α4β2 nAChRs (Fig. 2, A and B). Therefore, the observed effects of nicotinic ligands on ATF6 localization occur in at least two cell types (neurons and a neuroblastoma cell line). These effects do not require ATF6 overexpression or the use of a non-native promoter to drive ATF6 gene expression. During the UPR, protein kinase RNA-like endoplasmic reticulum kinase (PERK) induces eIF2α phosphorylation, which results in global inhibition of protein translation (Harding et al., 2000). A sustained increase in phosphorylated eIF2α can cause C/EBP homologous protein (CHOP) mediated apoptosis (Harding et al., 2000; Scheuner et al., 2001; Galehdar et al., 2010). Previous reports described eIF2α in the nucleus, which may function to regulate transcriptional and translational processes (Goldstein et al., 1999; DuRose et al., 2009; Tejada et al., 2009). We found that nicotine exposure significantly inhibited nuclear eIF2α phosphorylation in α4β2-overexpressing neurons, without affecting total eIF2α expression (Fig. 2, C and D). Nicotine also caused a nearly significant reduction of peIF2α in the cytoplasm (p = 0.06). Together, these data indicate that nicotinic ligands can inhibit the UPR via their interaction with nAChRs.
ERES formation is generally linked to some aspects of ER function. Chronic exposure to nicotine, cytisine, and DHβE increased the accumulation of Sec24D in ERES by ∼2-fold in the presence of coexpressed nAChRs (Fig. 3C). In the context of Parkinson's disease neuroprotection, dopaminergic neurons exposed chronically to nicotine would increase the formation of ERES in a nAChR-dependent manner. We suggest that these events could cause a generalized exit of cargo from the ER, which would result in reduced signals for sensors of ER stress.
The specification of nAChR stoichiometry is a basic aspect of nAChR assembly that occurs before ER exit. Of the four manipulations tested, two (nicotine and inclusion of the β2export-ER-export subunit) preferentially stabilized receptors with α42β23 stoichiometry, whereas cytisine and DHβE preferentially stabilized the assembly of α43β22 receptors (Fig. 5). Cytisine and DHβE bind strongly at the α4-α4 interface that exists only in α43β22 nAChRs, which provides the possible structural basis for the preferential stabilization of this stoichiometry (Mazzaferro et al., 2011). It is noteworthy that both types of stabilization increased ERES and suppressed ATF6 translocation. Apparently the nature of the subunit in the fifth or “auxiliary” position is not crucial for interactions with the COPII complex or for ER exit. Nicotine increased whereas cytisine reduced the area of peripheral ER, although both drugs decreased the UPR (Fig. 4B). Therefore, the attenuation of ATF6 translocation is apparently not sufficient to suppress the ER proliferation triggered by nAChRs. However, the attenuation of ATF6 translocation is sufficient to suppress ER proliferation when expression of a tail-anchored protein is sensed within the lipid bilayer (Maiuolo et al., 2011).
Events downstream from Golgi exit are not thought to influence the UPR markedly, and these events varied among the manipulations. Nicotine increased the number of TGN bodies per cell, whereas cytisine and DHβE reduced the average intensity of the TGN bodies (Fig. 4, D and E). Nicotine caused a ∼2-fold increase in PM-localized nAChRs and a reduction in the footprint/ER integrated density ratio (Fig. 6, C and D). In contrast, cytisine treatment resulted in ∼3.5-fold up-regulation of PM receptors and caused a ∼2-fold increase in the footprint/ER ratio, whereas DHβE exposure led to no marked effects on these parameters (Fig. 6, C and D). These observations may indicate that the two nAChR stoichiometries stabilized by nicotinic ligands use distinct mechanisms for trafficking from Golgi to PM, through different interactions between the subunits' M3-M4 loops and cytoskeletal or trafficking proteins (Xu et al., 2006; Kabbani et al., 2007).
Our data (Fig. 6B) agree with earlier reports that DHβE produces comparatively minor up-regulation of PM α4β2 nAChRs (Yang and Buccafusco, 1994; Gopalakrishnan et al., 1997) and that up-regulation requires DHβE concentrations of 10 to 100 μM (Gopalakrishnan et al., 1997). Our data also suggest that DHβE shifts the nAChR stoichiometry toward the α43β22 form, which is usually thought to have comparatively low acetylcholine sensitivity (reviewed by Miwa et al., 2011). A discrepant study by Buisson and Bertrand (2001) was performed with the same cell line as studied by Gopalakrishnan et al. (1997). However, the electrophysiological results reported by Buisson and Bertrand (2001) are unique in several ways; 10 nM DHβE produced up-regulation, and this was the most up-regulation measured among the ligands assessed. The concentration of 10 nM is a small fraction of the IC50 for DHβE, and this treatment increased the fraction of high-sensitivity nAChRs. We cannot explain the differences between the DHβE effects reported by Buisson and Bertrand (2001) and the consistent DHβE effects reported by Yang and Buccafusco (1994) and Gopalakrishnan et al. (1997) and in the present study.
Nicotine may also regulate nAChR interactions with the ubiquitin-proteasome system (Rezvani et al., 2007, 2010). The present study shows reduction of ER stress at nicotine concentrations 500-fold lower than those required to suppress UPR pathways in tunicamycin-treated PC12 cells, for which direct actions of nicotine on the ubiquitin-proteasome system were suggested (Sasaya et al., 2008). PC12 cells lack α4β2 nAChRs but express lower-affinity α3β4 nAChRs, which may be subject to chaperoning at relatively high nicotine concentrations.
None of the four tested manipulations produce appreciable nAChR activation, which rules out mechanisms in which ion flux associated with receptor activation affects intracellular signaling cascades, receptor assembly, stoichiometry, and consequent effects on the UPR. Our electrophysiological experiments confirmed, in the transfected Neuro-2a cells under study, that 0.1 μM nicotine or cytisine activated only 0.4% or <0.1% of the total receptor population, respectively (Fig. 6A). Moreover, the nicotine exposure used in our experiments probably desensitizes α4β2 nAChRs. DHβE activated no receptors, and the mutant α4β2enhanced-ER-export receptors displayed no constitutive activity. These data agree with previous studies that showed that up-regulation is independent of surface nAChR activation (Sallette et al., 2005; Corringer et al., 2006). Although these ligand concentrations activate no nAChRs when applied for a few seconds, these concentrations exceed, by factors of 10 to 100, the equilibrium binding dissociation constants measured with incubations of minutes to hours (Gopalakrishnan et al., 1997; Warpman et al., 1998; Whiteaker et al., 1998). Evidently, these ligand concentrations stabilize the formation of fully assembled ligand-receptor complexes, which can interact with ER-associated export machinery more readily than with ER-associated degradation.
Parkinson's disease becomes clinically apparent after a degenerative process has operated for a decade or longer. The apparent neuroprotective effects of smoking also begin decades before the clinical diagnosis (Ritz et al., 2007). Therefore, nicotine may exert a cumulative protective effect, which counteracts the cumulative degenerative mechanism. The data establish that pharmacological chaperoning of a central nervous system receptor by a drug can decrease ER stress and attenuate the UPR, and it is plausible to suggest that this mechanism exerts the required cumulative protective effect. Reductions in ER stress can occur without activation of PM nAChRs, which suggests a therapeutic strategy for neuroprotection without the potential for abuse. We do not yet know whether similar reductions in ER stress and UPR occur when nicotine interacts intracellularly with nAChRs at endogenous levels in the neurons of intact brains.
Supplementary Material
Acknowledgments
We thank Johannes Schwarz for discussion, Elisha D. W. Mackey for assistance with cloning, and Sheri McKinney for assistance with primary neuronal cultures.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant NS11756]; the National Institutes of Health National Institute on Aging [Grant AG033954]; National Institutes of Health National Institute on Drug Abuse Kirschstein National Research Service Award [Grant DA030877] (to C.I.R.); Targacept; Louis and Janet Fletcher; the Michael J. Fox Foundation; a California Tobacco-Related Disease Research Program postdoctoral fellowship [Grant 18FT-0066] (to R.S.); a Beckman Institute fellowship (to C.I.R.); and a California Tobacco-Related Disease Research Program New Investigator Award [Grant 19KT-0032] (to J.M.M.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- ATF6
- activating transcription factor 6
- BT
- bleedthrough
- CHOP
- C/EBP homologous protein
- DHβE
- dihydro-β-erythroidine
- eCFP
- enhanced cyan fluorescent protein
- eGFP
- enhanced green fluorescent protein
- eIF2α
- eukaryotic initiation factor 2α
- peIF2α
- phosphorylated eukaryotic initiation factor 2α
- ER
- endoplasmic reticulum
- ERES
- endoplasmic reticulum exit site(s)
- Whigh
- fractional area of high-normalized Förster resonance energy transfer component
- FRET
- Förster resonance energy transfer
- nAChR
- nicotinic acetylcholine receptor
- NFRET
- normalized Förster resonance energy transfer
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- PM
- plasma membrane
- ROI
- region of interest
- SePhaChARNS
- selective pharmacological chaperoning of acetylcholine receptor number and stoichiometry
- TG
- trans-Golgi
- TGN
- trans-Golgi network
- TIRFM
- total internal reflection fluorescence microscopy
- UPR
- unfolded protein response
- wt
- wild-type
- GalT
- galactosyltransferase
- FBS
- fetal bovine serum
- DMEM
- Dulbecco's modified Eagle's medium
- TBS
- Tris-buffered saline.
Authorship Contributions
Participated in research design: Srinivasan, Richards, Pantoja, and Lester.
Conducted experiments: Srinivasan, Richards, Xiao, Rhee, and Pantoja.
Contributed new reagents or analytic tools: Srinivasan, Richards, and Miwa.
Performed data analysis: Srinivasan, Richards, Xiao, and Lester.
Wrote or contributed to the writing of the manuscript: Srinivasan, Richards, Dougherty, and Lester.
References
- Ahmadi FA, Grammatopoulos TN, Poczobutt AM, Jones SM, Snell LD, Das M, Zawada WM. (2008) Dopamine selectively sensitizes dopaminergic neurons to rotenone-induced apoptosis. Neurochem Res 33:886–901 [DOI] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan UH, Chen Y, Leslie FM. (2002) Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444:260–274 [DOI] [PubMed] [Google Scholar]
- Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ, et al. (2009) ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 122:1626–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. (2001) Chronic exposure to nicotine upregulates the human α4β2 nicotinic acetylcholine receptor function. J Neurosci 21:1819–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. (2002) Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci 23:130–136 [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Léna C, Clementi F, Moretti M, Rossi FM, Le Novère N, et al. (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820–7829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons KG. (2008) α4 containing nicotinic receptors are positioned to mediate postsynaptic effects on 5-HT neurons in the rat dorsal raphe nucleus. Neuroscience 153:851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer PJ, Sallette J, Changeux JP. (2006) Nicotine enhances intracellular nicotinic receptor maturation: a novel mechanism of neural plasticity? J Physiol Paris 99:162–171 [DOI] [PubMed] [Google Scholar]
- DuRose JB, Scheuner D, Kaufman RJ, Rothblum LI, Niwa M. (2009) Phosphorylation of eukaryotic translation initiation factor 2α coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol Cell Biol 29:4295–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. (2011) The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem 286:7947–7957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonck C, Nashmi R, Salas R, Zhou C, Huang Q, De Biasi M, Lester RA, Lester HA. (2009) Demonstration of functional α4-containing nicotinic receptors in the medial habenula. Neuropharmacology 56:247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. (2010) Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci 30:16938–16948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein EN, Owen CR, White BC, Rafols JA. (1999) Ultrastructural localization of phosphorylated eIF2α [eIF2α(P)] in rat dorsal hippocampus during reperfusion. Acta Neuropathol 98:493–505 [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Molinari EJ, Sullivan JP. (1997) Regulation of human α4β2 neuronal nicotinic acetylcholine receptors by cholinergic channel ligands and second messenger pathways. Mol Pharmacol 52:524–534 [PubMed] [Google Scholar]
- Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. (2007) Heterogeneity and complexity of native brain nicotinic receptors. Biochem Pharmacol 74:1102–1111 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108 [DOI] [PubMed] [Google Scholar]
- Hernán MA, Takkouche B, Caamaño-Isorna F, Gestal-Otero JJ. (2002) A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 52:276–284 [DOI] [PubMed] [Google Scholar]
- Hetz C, Glimcher LH. (2009) Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol Cell 35:551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Jr, Zoli M, Bourgeois JP, Changeux JP. (1993) Immunocytochemical localization of a neuronal nicotinic receptor: the β2-subunit. J Neurosci 13:1551–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. (2003) Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 278:19367–19377 [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. (2007) Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun 354:707–711 [DOI] [PubMed] [Google Scholar]
- Kabbani N, Woll MP, Levenson R, Lindstrom JM, Changeux JP. (2007) Intracellular complexes of the β2 subunit of the nicotinic acetylcholine receptor in brain identified by proteomics. Proc Natl Acad Sci USA 104:20570–20575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, Emi M, Tanabe K, Murakami S. (2006) Role of the unfolded protein response in cell death. Apoptosis 11:5–13 [DOI] [PubMed] [Google Scholar]
- Kishi M, Steinbach JH. (2006) Role of the agonist binding site in up-regulation of neuronal nicotinic α4β2 receptors. Mol Pharmacol 70:2037–2044 [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. (2005) Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol Pharmacol 68:1839–1851 [DOI] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC. (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J 11:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS. (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281:7260–7270 [DOI] [PubMed] [Google Scholar]
- Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N. (2011) Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci USA 108:7832–7837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita N, Okada H, Yasoshima Y, Takahashi K, Kiuchi K, Kobayashi K. (2002) Dynamics of tyrosine hydroxylase promoter activity during midbrain dopaminergic neuron development. J Neurochem 82:295–304 [DOI] [PubMed] [Google Scholar]
- Mazzaferro S, Benallegue N, Carbone A, Gasparri F, Vijayan R, Biggin PC, Moroni M, Bermudez I. (2011) Additional acetylcholine (ACh) binding site at α4/α4 interface of α43β22α4 nicotinic receptor influences agonist sensitivity. J Biol Chem 286:31043–31054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa JM, Freedman R, Lester HA. (2011) Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron 70:20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M. (2009) Approaches to prevent dopamine quinone-induced neurotoxicity. Neurochem Res 34:698–706 [DOI] [PubMed] [Google Scholar]
- Moss FJ, Imoukhuede PI, Scott K, Hu J, Jankowsky JL, Quick MW, Lester HA. (2009) GABA transporter function, oligomerization state, and anchoring: correlates with subcellularly resolved FRET. J Gen Physiol 134:489–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Dickinson ME, McKinney S, Jareb M, Labarca C, Fraser SE, Lester HA. (2003) Assembly of α4β2 nicotinic acetylcholine receptors assessed with functional fluorescently labeled subunits: effects of localization, trafficking, and nicotine-induced upregulation in clonal mammalian cells and in cultured midbrain neurons. J Neurosci 23:11554–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, et al. (2007) Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 27:8202–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. (1994) Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol 46:523–530 [PubMed] [Google Scholar]
- Quik M, Bordia T, Huang L, Perez X. (2011) Targeting nicotinic receptors for Parkinson's disease therapy. CNS Neurol Disord Drug Targets 10:651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, De Biasi M. (2010) The ubiquitin-proteasome system regulates the stability of neuronal nicotinic acetylcholine receptors. J Mol Neurosci 40:177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Teng Y, Shim D, De Biasi M. (2007) Nicotine regulates multiple synaptic proteins by inhibiting proteasomal activity. J Neurosci 27:10508–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CI, Srinivasan R, Xiao C, Mackey ED, Miwa JM, Lester HA. (2011) Trafficking of α4* nicotinic receptors revealed by superecliptic phluorin: effects of a β4 ALS-associated mutation and chronic exposure to nicotine. J Biol Chem 286:31241–31249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA, Ross GW, Strickland D, Van Den Eeden SK, Gorell J. (2007) Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol 64:990–997 [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529 [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 22:10690–10698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. (2005) Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron 46:595–607 [DOI] [PubMed] [Google Scholar]
- Sasaya H, Utsumi T, Shimoke K, Nakayama H, Matsumura Y, Fukunaga K, Ikeuchi T. (2008) Nicotine suppresses tunicamycin-induced, but not thapsigargin-induced, expression of GRP78 during ER stress-mediated apoptosis in PC12 cells. J Biochem 144:251–257 [DOI] [PubMed] [Google Scholar]
- Schaub BE, Berger B, Berger EG, Rohrer J. (2006) Transition of galactosyltransferase 1 from trans-Golgi cisterna to the trans-Golgi network is signal mediated. Mol Biol Cell 17:5153–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7:1165–1176 [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. (1983) Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220:214–216 [DOI] [PubMed] [Google Scholar]
- Shen J, Prywes R. (2005) ER stress signaling by regulated proteolysis of ATF6. Methods 35:382–389 [DOI] [PubMed] [Google Scholar]
- Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. (2005) Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol 25:921–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, et al. (2005) CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 95:974–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son CD, Moss FJ, Cohen BN, Lester HA. (2009) Nicotine normalizes intracellular subunit stoichiometry of nicotinic receptors carrying mutations linked to autosomal dominant nocturnal frontal lobe epilepsy. Mol Pharmacol 75:1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Pantoja R, Moss FJ, Mackey ED, Son CD, Miwa J, Lester HA. (2011) Nicotine up-regulates α4β2 nicotinic receptors and ER exit sites via stoichiometry-dependent chaperoning. J Gen Physiol 137:59–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. (2011) The origins of oxidant stress in Parkinson's disease and therapeutic strategies. Antioxid Redox Signal 14:1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejada S, Lobo MV, García-Villanueva M, Sacristán S, Pérez-Morgado MI, Salinas M, Martín ME. (2009) Eukaryotic initiation factors (eIF) 2α and 4E expression, localization, and phosphorylation in brain tumors. J Histochem Cytochem 57:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. (2010) α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell 21:1850–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warpman U, Friberg L, Gillespie A, Hellström-Lindahl E, Zhang X, Nordberg A. (1998) Regulation of nicotinic receptor subtypes following chronic nicotinic agonist exposure in M10 and SH-SY5Y neuroblastoma cells. J Neurochem 70:2028–2037 [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Sharples CG, Wonnacott S. (1998) Agonist-induced up-regulation of α4β2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol 53:950–962 [PubMed] [Google Scholar]
- Xiao C, Nashmi R, McKinney S, Cai H, McIntosh JM, Lester HA. (2009) Chronic nicotine selectively enhances α4β2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J Neurosci 29:12428–12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Heinemann SF. (2006) Identification of sequence motifs that target neuronal nicotinic receptors to dendrites and axons. J Neurosci 26:9780–9793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Buccafusco JJ. (1994) Effect of chronic central treatment with the acetylcholine analog methylcarbamylcholine on cortical nicotinic receptors: correlation between receptor changes and behavioral function. J Pharmacol Exp Ther 271:651–659 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.