

Abstract
The storage of long-term memory is associated with a cellular program of gene expression, altered protein synthesis, and the growth of new synaptic connections. Recent studies of a variety of memory processes, ranging in complexity from those produced by simple forms of implicit learning in invertebrates to those produced by more complex forms of explicit learning in mammals, suggest that part of the molecular switch required for consolidation of long-term memory is the activation of a cAMP-inducible cascade of genes and the recruitment of cAMP response element binding protein-related transcription factors. This conservation of steps in the mechanisms for learning-related synaptic plasticity suggests the possibility of a molecular biology of cognition.
The remarkable progress in molecular genetics over the last two decades has brought about a new and more unified view of the biological sciences. A major advancement in our understanding of genes, their expression, and the structure of the proteins they encode has led to a better appreciation of the conservation of cellular function at the molecular level that now provides a common conceptual framework for several, previously unrelated, disciplines: cell biology, biochemistry, development, immunology, and cellular neurobiology. A parallel and potentially equally profound unification is occurring between cognitive psychology, the science of the mind, and neural science, the science of the brain. The ability to study the biological basis of mental function is providing a refined impetus for examining cognitive processes, such as perception, language, learning, and memory. To what degree can these two independent and disparate disciplines be brought together? Can molecular biology provide novel insights into the mind? In this brief review we consider the possibility of a molecular biology of cognition, using as examples several elementary forms of learning and memory in both invertebrates and the mammalian brain.
Memory Has at Least Two Major Forms
Modern behavioral and biological studies have shown that learning and memory are not a unitary process—not a single faculty of the mind—but a family of distinct processes, each with its own rules. In the most general sense, learning can be considered the process by which new information about the world is acquired, and memory can be considered the process by which that knowledge is retained. Recent studies have demonstrated that memory can be divided into at least two general categories (1). Explicit or declarative memory is the conscious recall of knowledge about people, places, and things and is particularly well-developed in the vertebrate brain. Implicit or nondeclarative memory is the nonconscious recall of motor skills and other tasks and includes simple associative forms, such as classical conditioning, and nonassociative forms, such as sensitization and habituation. The two types of memory seem to involve different neural circuits in the brain (2). Explicit memory uniquely depends on temporal lobe and diencephalic structures—for example, the hippocampus, subiculum, and entorhinal cortex—whereas implicit memory does not depend on temporal lobe function but rather involves the same sensory, motor, or associational pathways used in the expression of the learning process. Thus, whereas explicit memory is most readily studied in mammals, implicit forms of memory can be effectively studied in both nonmammalian vertebrates and higher invertebrates.
To what degree do these two different forms of memory share common molecular components? One clue to shared mechanisms comes from the study of stages in memory storage. The memory for both implicit and explicit forms of learning is graded and the duration of the memory is related to the number of training trials and is commonly divided into at least two temporally distinct components: short-term memory, lasting minutes to hours, and long-term memory, lasting days, weeks, and, in some cases, even a lifetime. Studies of long-term memory for implicit and explicit learning indicate each employs a cascade of molecular events that occurs during their consolidation period—the initial phase of memory storage—that is labile and highly sensitive to disruption. In both cases the conversion of a transient short-term form that requires only covalent modification of preexisting proteins, to a more stable and self-maintained long-term form that is accompanied by the growth of new synaptic connections, requires a cellular program of gene expression and increased protein synthesis. Here we consider the degree to which these genes and proteins are conserved in the two major forms of memory storage. We first outline some of the molecular insights that have been provided by neurobiological studies of elementary forms of implicit memory in Aplysia and Drosophila. We then briefly consider long-term potentiation (LTP) in the hippocampus, a type of enduring synaptic plasticity thought to be involved in the storage of long-term memory for explicit forms of learning in the mammalian brain.
The Memory for Long-Term Sensitization—an Implicit Form of Memory in Aplysia—Has a Representation in the Monosynaptic Component of the Reflex
Sensitization is an elementary form of nonassociative learning, by which an animal learns about the properties of a single noxious stimulus. The animal learns to strengthen its defensive reflexes and to respond vigorously to a variety of previously neutral or indifferent stimuli after it has been exposed to a potentially threatening or noxious stimulus. In Aplysia, sensitization of the gill- and siphon-withdrawal reflex can be induced by a strong stimulus applied to the tail. This activates facilitatory interneurons, which synapse on the sensory neurons and strengthen the synaptic connection between the sensory neurons and their central target cells (3). As in the case for other defensive withdrawal reflexes, the behavioral memory for sensitization of the gill- and siphon-withdrawal reflex is graded, and retention is proportional to the number of training trials. A single stimulus to the tail gives rise to short-term sensitization lasting minutes to hours. Repetition of the stimulus produces long-term behavioral sensitization that can last days to weeks (refs. 4 and 5; Fig. 1).
Figure 1.
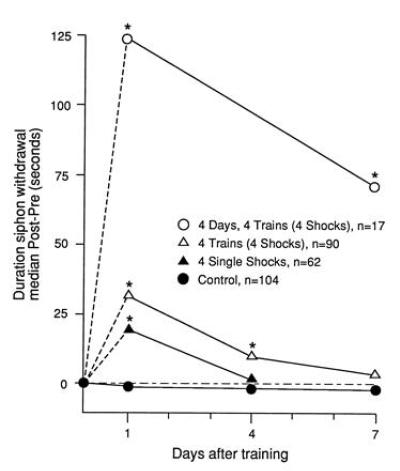
Behavioral long-term sensitization. A summary of the effects of long-term sensitization training on the duration of siphon withdrawal in Aplysia californica. The retention of the memory for sensitization is a graded function proportional to the number of training trials. Experimental animals received either four single shocks for 1 day (▴), four trains of shocks for 1 day (▵), or four trains of shocks a day for 4 days (○). Control animals were not shocked (•). A pretest determined the mean duration of siphon withdrawal for all animals before training. Posttraining testing was carried out 1, 4, or 7 days after the last day of training. The asterisks indicate a significant difference between the duration of siphon withdrawal for the trained and control animals (Mann–Whitney U tests, P < 0.01). N represents the number of animals per group. [Reproduced with permission from Frost et al. (5).]
The memory for both short- and long-term sensitization is represented on a elementary level by the monosynaptic connections between identified mechanoreceptor sensory neurons and their follower cells. Although this component accounts for only a part of the behavioral modification measured in the intact animal, its simplicity has allowed the reduction of the analysis of the short- and long-term memory of sensitization to the cellular and molecular level. For example, this monosynaptic pathway can be reconstituted in dissociated cell culture (6), where serotonin (5-HT), a modulatory neurotransmitter normally released by sensitizing stimuli, can substitute for the shock to the neck or tail used during behavioral training in the intact animal (7). A single application of 5-HT produces short-term changes in synaptic effectiveness, whereas five spaced applications given over a period of 1.5 hr produces long-term changes lasting 1 or more days (8).
Biophysical studies of this monosynaptic connection suggest that both the similarities and the differences in memory reflect, at least in part, intrinsic cellular mechanisms of the nerve cells participating in memory storage. Thus, studies of the connections between sensory and motor neurons in both the intact animal and in cells in culture indicate that the long-term changes are surprisingly similar to the short-term changes. A component of the increase in synaptic strength observed during both the short- and long-term changes is due, in each case, to enhanced release of transmitter by the sensory neuron, accompanied by an increase in the excitability of the sensory neurons, attributable to the depression of a specific potassium channel (9, 10, 11, 12, 13).
Despite these several similarities, the short-term cellular changes differ from the long-term process in two important ways. First, the short-term change involves only covalent modification of preexisting proteins and an alteration of preexisting connections. Both short-term behavioral sensitization in the animal and short-term facilitation in dissociated cell culture do not require ongoing macromolecular synthesis; the short-term change is not blocked by inhibitors of transcription or translation (14). By contrast, these inhibitors selectively block the induction of the long-term changes both in the semi-intact animal (15) and in primary cell culture (8). Most striking is the finding that the induction of long-term facilitation at this single synapse in Aplysia exhibits a critical time window in its requirement for protein and RNA synthesis characteristic of that necessary for other forms of learning in both vertebrates and invertebrates (16). From a molecular perspective, these studies indicate that the long-term behavioral and cellular changes require the expression of genes and proteins not required for the short term. Second, the long-term process, but not the short-term process, involves a structural change. Bailey and Chen (17, 18, 19) have demonstrated that long-term sensitization training is associated with the growth of new synaptic connections by the sensory neurons onto their follower cells. This synaptic growth can be induced in the intact ganglion by the intracellular injection of adenosine 3′:5′-cyclic phosphate (cAMP), a second messenger activated by 5-HT (20), and can be reconstituted in sensory–motor neuron cocultures by repeated presentations of 5-HT (21, 22). These findings of an elementary, cellular representation of long-term memory now allow us to ask: What are the molecular substrates and regulatory mechanisms that underlie memory storage?
Long-Term Facilitation Requires the Recruitment of cAMP Response Element Binding Protein (CREB)-Related Transcription Factors and the Activation of cAMP-Dependent Gene Expression
Studies by Bernier et al. (23) and Bacskai et al. (24) have shown that 5-HT, acting on the sensory neurons, stimulates the synthesis of cAMP, which then activates the catalytic subunit of protein kinase A (PKA) by releasing its binding to the regulatory subunit. By imaging the free catalytic and regulatory subunits of PKA, Bacskai et al. found that a single pulse of 5-HT increases the concentration of the free catalytic subunit in the cytoplasm of the sensory neuron, especially in the presynaptic terminals. With repeated pulses of 5-HT, the catalytic subunit translocates to the nucleus of the sensory neurons, where it appears to phosphorylate one or more CREB-related transcription factors that activate cAMP-inducible genes.
Dash et al. (25) provided the first experimental evidence that one of the substrates of protein kinase A is a CREB-like protein that binds to the cAMP response element (CRE) by injecting oligonucleotides containing somatostatin CRE into sensory neurons, and blocking long-term facilitation without affecting short-term facilitation. Kaang et al. (26) extended these studies by expressing in sensory neurons a chimeric transactivator consisting of the mammalian CREB activation domain fused to a GAL4 DNA-binding domain, which was able to transactivate a reporter gene in response to repeated 5-HT application. To test whether PKA activity was required for the induction of this 5-HT-dependent response, Kaang et al. compared the activity of the wild-type CREB–GAL4 chimera to a mutant (CREB–GAL4 SA 119) chimera, in which the serine 119 (essential for activation by mammalian CREB), was substituted with an alanine and found that this substitution abolished the ability of 5-HT to induce the transactivation by CREB–GAL4. The kinase essential for this activity is likely to be PKA, since a mutation that inactivates only the PKA phosphorylation site but leaves the Ca2+ calmodulin-dependent kinase consensus site intact (the substitution of arginine 117 with an alanine) blocked the 5-HT-dependent transactivation. These data support the hypothesis that CREB-related transcription factors, activated by PKA-dependent phosphorylation, are required for induction of the long-term process, and suggest that CREB-like proteins are involved in regulation of the new gene expression that accompanies long-term facilitation.
CREB1 Acts Conjointly with CREB2
Does CREB act alone or in concert with other transcription factors? The family of transcription factors to which CREB belongs has the ability to form homo- and heterodimers. These interactions and the DNA binding of resulting dimers are mediated through a bipartite basic leucine zipper domain. Bartsch et al. (27) have used the basic leucine zipper domain from the Aplysia transcription factor ApC/EBP in a two-hybrid screen in yeast and identified two bZIP transcription factors: ApCREB2 and AF-1.
ApCREB2 is expressed in the basal state (without exposure to 5-HT) in Aplysia sensory neurons and is not induced by 5-HT. Although the sequence of this transcription factor does not contain a CREB-like consensus site for phosphorylation by PKA (the KID domain or P box) it does have protein kinase C and several mitogen-activated protein (MAP) kinase sites. In overall primary structure, ApCREB2 is homologous to both human CREB2 and mouse ATF4. This homology is particularly interesting, since human CREB2 has been shown to be a repressor of CREB1-mediated gene expression (28), suggesting that genes regulated by cAMP can function under dual control. On one hand, they can be activated by CREB1 and, on the other, repressed by CREB2. cAMP-induced gene expression may, therefore, involve at least two temporally related steps: (i) activating CREB1 and (ii) relieving the repression of CREB2. If this is so, then the relief of repression might potentiate the activation process. To test directly the learning-related functional and structural corollaries of this hypothesis, we have generated specific antibodies to ApCREB2 and injected them into the nucleus of sensory neurons in sensory–motor neuron cocultures.
ApCREB2 Represses Long-Term Facilitation: Relief of Repression Converts Transient Facilitation into Long-Term Functional and Structural Change
We have found that injection of CREB2 antibodies into sensory neurons allows a single pulse of 5-HT, which normally induces only short-term facilitation lasting minutes, to evoke facilitation lasting >1 day. This facilitation has all of the properties of long-term facilitation: it requires transcription and translation, induces the growth of new synaptic connections, and occludes further facilitation by five pulses of 5-HT.
Fig. 2 illustrates the time course and summary of the effects of injection of ApCREB2 antiserum on short- and long-term facilitation. In both the intact Aplysia and in neuronal cell culture, five pulses of 5-HT induce long-term facilitation in the connections between the sensory and motor neurons lasting 24 hr or more. By contrast, a single pulse of 5-HT produces only short-term facilitation lasting ≈10 min. In the presence of the antiserum, rather than producing short-term facilitation, one pulse of 5-HT now produces facilitation lasting >24 hr. This facilitation is robust and is comparable in magnitude to that seen at 24 hr with five pulses of 5-HT.
Figure 2.
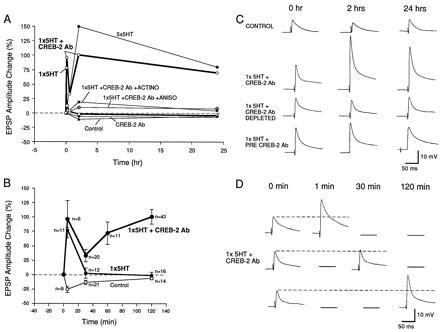
Time course of the effects of injection of ApCREB2 antiserum on short- and long-term facilitation. (A) Time course of excitatory postsynaptic potenial (EPSP) amplitude changes recorded in motor neuron L7 in response to stimulation of the sensory neuron (expressed as percent change in the amplitude of the EPSP) after single and multiple applications of 5-HT to Aplysia sensory–motor neuron cocultures. Changes in EPSP amplitude after application of one 5-min pulse of 5-HT (1× 5-HT, short-term facilitation) and one 5-min pulse of 5-HT paired with injection of anti-ApCREB2 antibodies (1× 5-HT + CREB-2 Ab, both in boldface lines) are compared with changes in EPSP amplitude induced by five pulses of 5-HT (5× 5-HT) at 2 and 24 hr. While the EPSP facilitation decays rapidly after one pulse of 5-HT (with a return to base line after 10 min), pairing one pulse of 5-HT with injection of anti-ApCREB2 antibodies induces a long-term facilitation paralleling that of 5× 5-HT. This long-term facilitation is abolished by the application of the protein synthesis inhibitor anisomycin (1× 5-HT + CREB-2 Ab + ANISO) or the RNA synthesis inhibitor actinomycin D (1× 5-HT + CREB-2 Ab + ACTINO) during the training. The difference in EPSP amplitude at 2 hr between 5× 5-HT and 1× 5-HT + CREB-2 Ab may reflect the transient protein synthesis-dependent, but RNA synthesis-independent, component of long-term facilitation 2 hr after 5-HT stimulation (29). The controls are either untreated (control), or injected with ApCREB2 antiserum without 5-HT administration (CREB-2 Ab). (B) Comparison of the time course of the EPSP amplitude changes in the first 2 hr after application of a single 5-min pulse of 5-HT with or without injection of CREB-2 antibody. The control cells were not exposed to 5-HT. (C) Example of EPSPs recorded in motoneuron L7 after stimulation of the sensory neuron before (0 hr) and 2 and 24 hr after 5-HT treatment. One pulse of 5-HT paired with the injection of an ApCREB2 antiserum induces a significant increase in EPSP amplitude at 2 and 24 hr, but injection the preimmune serum (PRE-CREB-2 Ab) or depleted immune serum does not induce long-term facilitation. (D) Examples of EPSPs recorded at indicated times in cocultures injected with ApCREB2 antiserum paired with one 5-min pulse of 5-HT. [Reproduced with permission from Bartsch et al. (27) (Copyright 1995, Cell Press).]
As mentioned above, long-term facilitation requires new protein and RNA synthesis (8, 22). We therefore examined the effects of protein and RNA synthesis inhibitors on the synaptic modifications produced at 2 and 24 hr after the injection of ApCREB2 antiserum paired with the application of a single pulse of 5-HT. Incubating sensory–motor neuron cocultures with these inhibitors during a single pulse of 5-HT blocks the increase in amplitude of synaptic potential after injection with ApCREB2 antiserum, both at 2 hr after exposure and at 24 hr.
If one pulse of 5-HT in the presence of ApCREB2 antiserum phenocopies long-term facilitation, the injection of antibody should also occlude the effects of five pulses of 5-HT. We found this was the case. In cocultures injected with ApCREB2 antiserum, the facilitation measured 24 hr after five pulses of 5-HT was not significantly greater than the facilitation obtained in cells exposed to five pulses of 5-HT and not injected with antibody or cells treated with five pulses of 5-HT and injected with normal rabbit serum. Thus, the facilitation produced by one pulse of 5-HT in the presence of the antibody has properties similar to that induced by five pulses of 5-HT and occludes the effect of five pulses.
The facilitation produced at 2 hr by five pulses of 5-HT is completely blocked by inhibitors of protein synthesis but is only partially blocked by inhibitors of transcription (29). This suggests that five pulses of 5-HT modulates both transcription and translation. Since ApCREB2 presumably acts only on the transcriptional component of long-term facilitation, one might predict that the pairing of one pulse of 5-HT with injection of ApCREB2 antiserum would produce less facilitation at 2 hr than five pulses of 5-HT. The facilitation at 2 hr produced by one pulse of 5-HT in the presence of ApCREB2 antibody is ≈30% less than that produced by five pulses of 5-HT. Similarly, injection of CRE oligonucleotides, which presumably also affects only the transcriptional component of 2 hr facilitation, was also found to produce a comparable inhibition at 2 hr, thus supporting the notion that the role of ApCREB2 is specific to the 5-HT-induced transcriptional response.
Finally, we examined whether ApCREB2 can also act as a repressor of the morphological changes that accompany long-term facilitation. Here, we injected the ApCREB2 antiserum into sensory neurons and examined, in parallel, the consequences of one pulse of 5-HT on long-term changes in both the strength of the sensory–motor neuron connection and on the number of fluorescence-labeled sensory neuron varicosities contacting the motor neuron (Fig. 3). We found the pairing of a single pulse of 5-HT with the injection of ApCREB2 antiserum 1 hr before training induced significant increases, 24 hr after the injection, in both the strength of the sensory–motor neuron connection and in the number of sensory neuron varicosities. By contrast, control cells receiving just one pulse of 5-HT and no injection of antiserum showed no facilitation and no increase in the number of sensory neuron varicosities 24 hr following training. The magnitude of both the long-term functional and structural changes are comparable to those seen at 24 hr following five pulses of 5-HT (21, 22).
Figure 3.
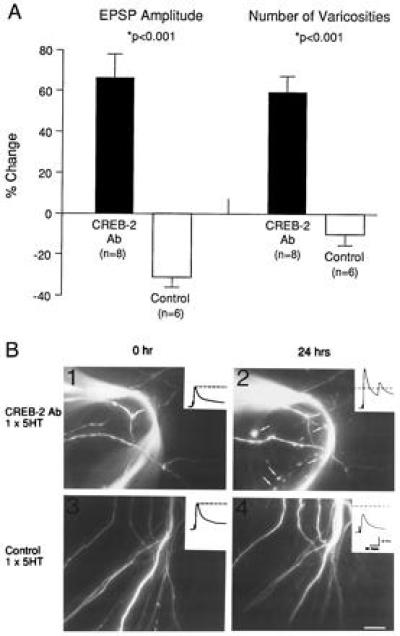
Long-term functional and structural changes evoked by one pulse of 5-HT paired with injection of ApCREB2 antiserum. (A) Summary of the structural and functional changes induced by one pulse of 5-HT. Injection of the ApCREB2 antiserum paired with one pulse of 5-HT 24 hr later results in a significant enhancement of the EPSP amplitude and a concomitant significant increase in the number of varicosities. (B) Examples of structural changes evident 24 hr after one pulse of 5-HT paired with injection of ApCREB2 antiserum. The fluorescent micrographs taken from the same regions of sensory neurites contacting the axon hillock of L7 before (1 and 3) and 24 hr after treatment (2 and 4). Arrows in 2 illustrate examples of some of the new varicosities present 1 day after one pulse of 5-HT paired with the injection of the ApCREB2 antiserum. The EPSPs, evoked before (0 hr) and after (24 hr) one pulse of 5-HT in the pictured neurons are indicated in the insets. (Bar = 20 μm.) [Reproduced with permission from Bartsch et al. (27) (Copyright 1995, Cell Press).]
Initiation of Long-Term Facilitation Requires the Coordinated Regulation of Both CREB1 And CREB2
Our data provide evidence that ApCREB2 is a functional repressor of long-term facilitation. These data and the parallel work in Drosophila provide the first molecular evidence for a possible role of activators and repressors in memory storage. Overexpression of an inhibitory form of the Drosophila CREB1 homologue, dCREB2b, blocks the formation of long-term memory in transgenic flies (30). Recently, Yin et al. (31) demonstrated that overexpressing an activating form of the Drosophila CREB1 homologue, dCREB2a greatly reduces the number of training trials needed to establish long-term memory. This gain of function, where a single massed training trial is sufficient to achieve long-term memory, which normally requires spaced training trials, greatly strengthens the earlier evidence from Drosophila (30, 31), Aplysia (25, 26), and mice (32) that CREB1 is of central importance in initiating the long-term process.
The results in Aplysia point to a parallel importance for ApCREB2 in this process. Injection of ApCREB2 antibodies paired with a single training trial, which normally produces only short-term facilitation, results in induction of long-term facilitation. This gain of function resembles overexpression of the dCREB2a activator in Drosophila and suggests the interesting possibility that removal of ApCREB2-mediated repression may be limiting in regulating the long-term increase in synaptic strength.
How might the repression of ApCREB1 by ApCREB2 be relieved? Since we do not detect a significant degradation of the ApCREB2 protein after exposure to 5-HT, the repressive action of ApCREB2 is most likely relieved by a covalent modification induced by the repeated pulses of 5-HT. Indeed, we have detected changes in phosphorylation of ApCREB2 following repeated exposure to 5-HT. According to this view, the physiological role of ApCREB2 may be two-fold: first, it may prevent the long-term process from being turned on adventitiously without repeated exposures to 5-HT; and, second, it may regulate the amplitude of synaptic change by integrating the activation of ApCREB1 by PKA with signals from additional second messenger pathways.
The molecular mechanisms of ApCREB2 derepression under physiological conditions are not known. However, it is interesting that ApCREB2 shares MAP kinase phosphorylation sites with its homologues human CREB2 and mouse ATF4. Furthermore, MAP kinase is activated by both 5-HT and forskolin in Aplysia neurons and, like PKA, translocates to the nucleus with prolonged activity (K. Martin, personal communication). The necessity to translocate both PKA and MAP kinase to the nucleus may provide some insight into why long-term facilitation requires repeated pulses of 5-HT. These may be needed to allow persistent activation of PKA and MAP kinase and to translocate PKA and perhaps MAP kinase to the nucleus, so as to activate the activators and relieve the repressors. In addition, the pathways regulating stimulation of the activator and relief of the repressor may have distinct kinetics. Such differences in kinetics could define the optimal time window separating training trials and account for the well-established difference between massed and spaced training. Perhaps the reason that spaced training is more effective than massed training is that only spaced training allows the coordinated activation of ApCREB1 and the derepression of ApCREB2.
Aplysia CCAAT Enhancer-Binding Protein (ApC/EBP) Is an Immediate-Early Gene Induced During the Consolidation Phase of Long-Term Facilitation
Which genes are downstream from CREB1? To address this question, we next focused on cAMP-regulated transcription factors. Some of the transcription factors known to be activated by cAMP belong to a family known as CCAAT enhancer-binding proteins (C/EBPs). A member of this family, C/EBPβ, is expressed in the rat pheochromocytoma PC12 cell line, where it has been shown to be activated by cAMP and to regulate the expression of the c-fos gene by binding to the enhancer response element (ERE) in the c-fos promoter (33). Since Aplysia neurons contain specific binding activity for ERE, Alberini et al. (34) used the ERE binding sequence and isolated a clone that interacted specifically with C/EBP DNA binding elements. The Aplysia C/EBP mRNA is expressed at low levels in the basal state, but it is rapidly and transiently induced by 5-HT and cAMP, even in the presence of protein synthesis inhibitors, suggesting that ApC/EBP is an immediate-early gene.
Is activation of ApC/EBP critical for the conversion of short- to long-term facilitation? To address this question, Alberini et al. injected ERE oligonucleotides into sensory neurons in sensory–motor neuron cocultures. This selectively blocked the 5-HT-induced long-term facilitation without affecting short-term facilitation (Fig. 4). Similar results were obtained by microinjection of either ApC/EBP antisense RNA or antibody to ApC/EBP. These results suggest that Aplysia C/EBP, an immediate-early gene activated during the consolidation phase of long-term facilitation, serves as part of a molecular switch for converting short-term to long-term memory.
Figure 4.
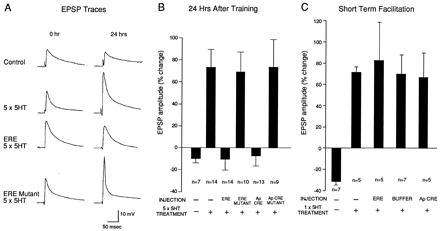
Injection Of ERE oligonucleotides blocks 5-HT-induced long- but not short-term facilitation in sensory motor synapses. (A) Examples of EPSPs recorded in motoneuron L7 after stimulation of the sensory neuron before (0 hr) and 24 hr after 5-HT treatment. Injection of the ERE oligonucleotide but not of the corresponding mutant (ERE Mutant) blocks the 5-HT-induced increase in EPSP amplitude at 24 hr. The control culture did not receive 5-HT applications or oligonucleotide injections. (B) Bar graph representing the effects of oligonucleotide injections in long-term facilitation. The height of each bar corresponds to the mean percentage change ± SEM in EPSP amplitude tested 24 hr after 5-HT treatment. Five pulses of 5-HT significantly increase the EPSP amplitude in noninjected cells, as well as in ERE mutant or ApCRE mutant injected cells, relative to the control (not 5-HT-treated and noninjected cells). On the contrary, the EPSP amplitude change in ERE or ApCRE injected cells was not significantly different from that of control cells that were neither injected nor treated. (C) Bar graph representing the mean EPSP amplitude percentage change ± SEM of short-term facilitated cells injected with ERE oligonucleotides, with ApCRE, or with buffer. A single pulse of 5-HT had no significant effect on EPSP amplitude in noninjected cells or cells injected with ERE, or ApCRE. [Reproduced with permission from Alberini et al. (34) (Copyright 1995, Cell Press).]
How long does this transcription factor need to be active? Is the binding of ApC/EBP to its target sequences required throughout the entire maintenance period or does the facilitation become self-perpetuating as a result of subsequent expression of later, more stable effector genes? To distinguish between these hypotheses, Alberini et al. injected ERE oligonucleotides into sensory cells at various times after 5-HT treatment. They found the blocking effect was progessively reduced when the injection was performed at longer intervals after the training, with facilitation no longer affected by the injection at 12 hr after the training. Therefore, the induction of ApC/EBP during the 5-HT treatment leads to the activation of a cascade of self-perpetuating events essential for the late phase of long-term facilitation.
Structural Changes Stabilize the Late Phase of Long-Term Facilitation
The products of this network of genes, only a few of which have been identified so far, lead to the growth of additional synapses between the sensory neurons and their follower cells, which stabilize the self-maintained long-term memory process (17, 18, 19). Indeed, the stability of long-term facilitation seems to result from the persistence of these structural changes in the synapses between the sensory and motor neurons, the decay of which parallels the decay of the behavioral memory (35).
What are the molecular mechanisms underlying the learning-related formation of new synaptic connections? The 5-HT-induced synaptic growth in sensory–motor neuron cocultures is associated with a down-regulation of nerve cell adhesion molecule (NCAM)-related Aplysia cell adhesion molecules (apCAMs) on the surface membrane of the sensory neuron (36). Down-regulation is particularly prominent at sites where the processes of the sensory neurons contact one another and is achieved there by the protein synthesis-dependent activation of a coordinated program of clathrin-mediated endocytosis, leading to the internalization and apparent degradation of apCAM (37). Aplysia expresses two isoforms of apCAM, a membrane form and a phosphoinositol-linked form. Which of the two apCAM isoforms is internalized? To address this question, Bailey et al. (38) selectively expressed epitope-tagged constructs of the two isoforms in cultured sensory neurons. By combining thin-section electron microscopy with gold-conjugated antibodies, they found that 5-HT elicits a 68% decrease in the density of gold-labeled complexes bound to the transmembrane form of apCAM at the surface membrane and a 24-fold increase in their internalization. By contrast, 5-HT has no effect on either the surface distribution or internalization of the phosphatidylinositol-linked isoform of apCAM. The selective internalization of the transmembrane form highlights the potential regulatory significance of its intracellular domain, which contains a sequence rich in proline, glutamic acid, serine, and threonine residues thought to mediate protein degradation (PEST) and has two consensus sites for MAP kinase phosphorylation. Deletions of, or mutations in, the cytoplasmic tail should allow determination of which part of this molecule triggers internalization and which part targets degradation.
The ability of 5-HT to modify the structure of the surface and internal membrane systems of sensory neurons in Aplysia, by initiating a rapid and protein synthesis-dependent sequence of steps, bears a striking similarity to the ruffling of the cell surface and membrane remodeling induced in nonneuronal systems by epidermal growth factor and other well-characterized growth factors (39) or by nerve growth factor in PC12 cells (40). These similarities suggest that modulatory transmitters important for learning, such as 5-HT, may serve a double function. In addition to producing a transient regulation of the excitability of neurons, repeated or prolonged exposure of a modulatory transmitter can also produce an action comparable to that of a growth factor, resulting in a more persistent alteration in the architecture of the neuron.
Based on these findings, Bailey et al. (37) have suggested that the 5-HT-induced internalization of apCAM and consequent endocytically activated membrane remodeling may represent the first morphological steps in the structural program underlying long-term facilitation. According to this view, learning-related synapse formation is preceded by and perhaps requires endocytic activation, which can then serve a double function. First, the removal of cell adhesion molecules from the neuronal surface at sites of apposition may destabilize adhesive contacts and facilitate defasciculation, a process that may be important in disassembly. Second, the massive endocytic activation might lead to a redistribution of membrane components that favors synapse formation.
A Three-Step Molecular Model of the Transition of Short- to Long-Term Memory in Aplysia: Initiation, Consolidation, and Stabilization
Fig. 5 illustrates a schematic summary of the three steps that we have delineated in the conversion of short- to long-term presynaptic facilitation. In this model, 5-HT, a modulatory transmitter released by facilitating interneurons that end presynaptically on the sensory neuron, acts to initiate separate memory processes with different durations. Short-term facilitation, which has a time course of minutes, begins with the binding of 5-HT to its surface receptors. This activates adenylyl cyclase, which catalyzes the synthesis of cAMP. cAMP binds to the regulatory subunit of PKA, leading to the release and activation of its catalytic subunit. PKA acts on at least two classes of substrates to enhance transmitter release (41). First, it phosphorylates K+ channels or associated proteins, which leads to a reduction of the outward K+ current and results in a broadening of the action potential and increased Ca2+ influx into the presynaptic neuron (9, 10, 11, 12, 13). Second, PKA also seems to act directly on the machinery involved in the exocytotic release of transmitter. These modifications take place in the presynaptic terminals and are independent of new macromolecular synthesis, and their duration determines the time course of short-term facilitation.
Figure 5.
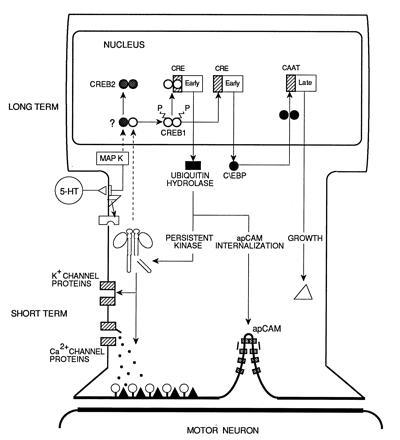
Molecular pathways underlying short- and long-term presynaptic facilitation in Aplysia. See text for details.
By contrast to the short-term effects, repeated activation of the serotonergic interneurons triggers long-term facilitation that requires the induction of new proteins and that lasts for >1 day. With repeated or prolonged application of 5-HT, the PKA catalytic subunit translocates to the nucleus, where it acts on nuclear substrates, which include transcription factors of the CREB/activating transcription factor family. Thus, the activity of CRE-binding proteins is necessary for the initiation of long-term facilitation. Specifically, this initiation component of the switch to long-term facilitation requires the coordinated regulation of at least two transcription factors: the activation of CREB1 and the relief of repression of CREB2. This initiation component leads to the rapid induction of the immediate-early genes ubiquitin hydrolase and ApC/EBP and suggests that the consolidation component of long-term facilitation requires the activation of a network of genes with constitutively active proteins regulating the expression of immediate-response genes. Some of these are early regulatory genes that, in turn, appear to be lead to the expression of late (presumably structural) genes responsible for the stabilization phase. The switching on of a self-maintaining mechanism by immediate-early genes explains why the characteristic protein synthesis-dependent phase is brief: the induction of regulatory factors is the limiting step that allows the expression of late phase events. In addition to regulatory factors, early effectors also are synthesized during the consolidation phase. Among these effectors is the C-terminal ubiquitin hydrolase, which seems to participate in the proteolytic cleavage of the PKA regulatory subunit, maintaining the enzymatic activity of the catalytic subunit in the absence of a cAMP increase (42, 43) and light chains of clathrin, which may be involved in the removal of apCAM from the cell surface through activation of the endocytic pathway and thus, may contribute to the initial stages of synaptic growth (37, 44).
Although PKA enzymatic activity is necessary for the first 10 hr following repeated 5-HT application, it is not maintained (45). Rather, the late phase is characterized by structural changes that appear within 1 hr after 5-HT or tail shock training (46) and persist for days or weeks (35). Indeed, the induced regulatory genes may, in turn, initiate further rounds of transcriptional activation generating a cascade of sequential gene expression affecting genes encoding proteins such as BiP (47) and calreticulin (48), which may reflect a general response designed to meet the posttranslational demands of increased protein synthesis, as well as structural proteins necessary for the construction of the new synaptic arbors associated with long-term sensitization. Memory lasting hours is retained by the half-life of the effector proteins or by functional modifications, such as phosphorylation, of these proteins. Certain of the early effector proteins may also serve to reinforce and maintain the initial response, for example, by proteolytic activation of a protein kinase. Memory lasting days, weeks, or months (longer than the half-life of effector proteins) is initiated by the early regulatory genes, whose protein products trigger the maintained expression of late effector genes, which may contribute to and stabilize the growth of new synaptic connections coincident with the maintenance phase of long-term memory.
The cAMP Cascade Is Also Used for Implicit Forms of Memory in Drosophila
How general is the cAMP-triggered, CREB-mediated cascade of gene activation for memory storage? Another simple form of implicit memory, that for classical conditioning, has been examined in Drosophila using olfactory cues paired with electric shock. Several single gene mutants have been isolated that cannot learn the task although their behavior is otherwise normal. Three mutations have been analyzed in particular detail and each involves a step in the cAMP cascade. dunce involves a defect in the cAMP phosphodiesterase (49), rutabaga is defective in the Ca2+/calmodulin-dependent adenylyl cyclase (50), and amnesiac lacks a pituitary adenylyl cyclase activating peptide (PACAP)-like peptide transmitter that stimulates the adenylyl cyclase (51). Expression of an inhibitor of PKA using a heat shock promoter also blocks the learning (52).
Recently, Tully et al. (53) have shown that spaced training gives rise to a long-term memory that lasts at least 7 days and is blocked by inhibitors of protein synthesis. This long-term memory is selectively blocked by the heat shock-induced expression of a dominant negative inhibitor of CREB, a cAMP response element modulator (CREM)-like transcription repressor (30). By contrast, overexpression of CREB activator leads to immediate long-term memory. Thus, several forms of long-term memory for different forms of learning in Drosophila require CREB- and cAMP-induced gene expression.
Shared Molecular Mechanisms for Long-Term Memory in Implicit and Explicit Learning
The studies in Aplysia and Drosophila suggest that part of the molecular switch required for consolidation of long-term memory during elementary, implicit forms of learning involves the cAMP-mediated induction of immediate early genes. Is there a similar set of molecular steps for memory consolidation of more complex, explicit forms of learning in the mammalian brain?
Memory storage for explicit forms of learning, both in humans and experimental animals, is critically dependent upon structures within the temporal lobe, such as the hippocampus (2). What are the cellular mechanisms used within the hippocampus for explicit memory storage? This question was first addressed in 1973, when Timothy Bliss and Terry Lømo demonstrated that hippocampal neurons display enduring, plastic capabilities of the kind that would be required for long-term memory storage (54). Brief, high-frequency stimulation in any one of the three best characterized neural pathways within the hippocampus results in an increase in synaptic efficacy that can endure for hours or days. This strengthening is called LTP.
LTP in the mammalian hippocampus shares some of the same mechanisms used for synaptic facilitation in Aplysia. For example, mossy fiber LTP, which occurs at synapses between the dentate gyrus granule cells and CA3 pyramidal cells, involves a cAMP-dependent enhancement of transmitter release from the presynaptic terminals. By contrast, Schaffer collateral LTP in CA1 is much more complex. Here the inductive phase is a postsynaptic event and involves calcium influx through the N-methyl-d-aspartate receptor channel and the recruitment of several second-messenger pathways involving tyrosine kinases, protein kinase C, and calcium/calmodulin kinase II. In addition to these early steps in the postsynaptic cell, Schaffer collateral LTP also involves an enhancement of transmitter release from the presynaptic neuron (55, 56, 57), which may be mediated by retrograde messenger signals (perhaps nitric oxide or carbon monoxide) that diffuse from the postsynaptic cell (58, 59, 60).
Similar to the presynaptic facilitation in Aplysia, both mossy fiber and Schaffer collateral LTP have distinct temporal phases, each with a cellular representation. The early phase is produced by a single tetanic stimulation, lasts 1–3 hr, and requires only covalent modification of preexisting proteins. By contrast, the late phase is induced by repeated tetanic stimulation, persists for several hours, and is dependent on new protein and RNA synthesis (61, 62, 63, 64). As is the case with long-term memory in Aplysia, on the cellular level there is a consolidation switch, and the requirement for transcription in LTP has a critical time window (64). In addition, the late transcription-dependent phase of LTP is blocked by inhibitors of PKA (61, 62). Recent studies by Nguyen and Kandel (65) now indicate that these features of LTP also apply to a third major hippocampal pathway, the medial perforant pathway, which originates in the entorhinal cortex and ends onto granule cells in the dentate gyrus. LTP in this pathway exhibits both an early, transient phase, which does not require protein or RNA synthesis and is independent of PKA activation, and a late phase, which requires the synthesis of proteins and RNA and can be selectively blocked by inhibitors of PKA. Thus, as in Aplysia presynaptic facilitation, cAMP-mediated transcription appears to be a common mechanism for the late form of LTP in all three pathways within the hippocampus (Fig. 6). Consistent with these observations are the findings of Bourtchuladze et al. (32), who have studied a mouse line with selective ablation of the CREB Δ isoform. Although these mice show normal acquisition and short-term retention (30 min) of contextual learning, they are selectively defective for long-term memory tested at 1 hr or 24 hr.
Figure 6.
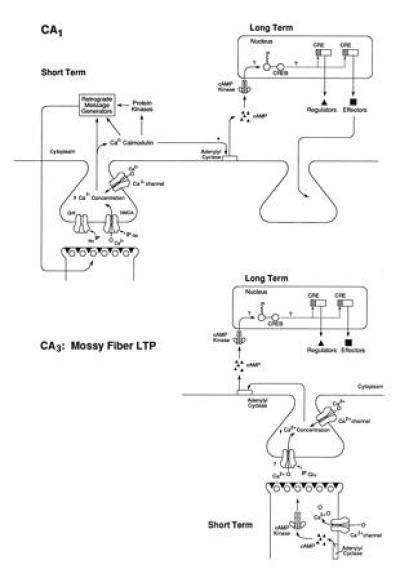
A working model of the molecular mechanisms underlying the early and late phases of LTP in hippocampal regions CA1 And CA3. See text for details.
An Overall View
One of the unifying principles emerging from these molecular studies of implicit and explicit memory storage is that, despite the different ways by which each form is induced, the subsequent genetic switch required for conversion of their short-term memory to one of longer duration may be similar. Thus, molecular studies of cognition are revealing, on a mechanistic level, previously unanticipated relationships between different classes of learning, and suggest the attractive possibility that the storage of long-term memory may utilize common genes and proteins.
In Aplysia, these mechanisms include a sequence of three steps. First, the initiation step involves the activation of CREB1 and the derepression of CREB2. Second, the consolidation step involves the induction by CREB1 of a set of immediate-early genes, such as the C-terminal ubiquitin hydrolase and the transcription factor C/EBP. Third, the stabilization step involves the down-regulation of apCAMs and the recruitment of a growth process. Since a number of studies in the vertebrate brain have shown that immediate-early genes are induced in the hippocampus and certain regions of the neocortex by treatments that lead to LTP (66, 67, 68), it will be of particular interest to investigate whether cAMP-dependent transcription factors, perhaps of the C/EBP family, are also required for long-term synaptic modifications in mammals.
The apparent similarity in some of the molecular steps that underlie learning-related synaptic plasticity may reflect the fact that long-term memory for both implicit and explicit storage is associated with structural changes (69). This stable and self-sustaining late phase is switched on by the cAMP-mediated induction of a cascade of immediate-early genes.
That the late phase of mossy fiber, Schaffer collateral, and medial perforant pathway LTP also involves cAMP raises the interesting possibility that, in the hippocampus as well, cAMP and protein kinase A are recruited, because they may be able to access the molecular machinery for long-term structural changes. By delineating the genes and proteins recruited by the N-methyl-d-aspartate-dependent and -independent forms of LTP, this possibility can now be tested. Thus, whereas animals and humans are capable of a wide variety of learning processes that utilize a number of different second messenger cascades, they may recruit a much more restricted set of molecular mechanisms for long-term memory storage.
Acknowledgments
Portions of the work cited in this review were supported by the Howard Hughes Medical Institute to E.R.K. and National Institutes of Health Grants MH37134 and GM32099 to C.H.B.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: LTP, long-term potentiation; 5-HT, serotonin; CRE, cAMP response element; CREB, CRE binding protein; PKA, protein kinase A; MAP, mitogen-activated protein; C/EBP, CCAAT enhancer-binding protein; ApC/EBP, Aplysia C/EBP; ERE, enhancer response element; EPSP, excitatory postsynaptic potential.
References
- 1.Polster M R, Nadel L, Schachter D L. J Cognit Neurosci. 1991;3:95–116. doi: 10.1162/jocn.1991.3.2.95. [DOI] [PubMed] [Google Scholar]
- 2.Squire L R. Psychol Rev. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- 3.Hawkins R D, Castellucci V F, Kandel E R. J Neurophysiol. 1981;45:315–326. doi: 10.1152/jn.1981.45.2.315. [DOI] [PubMed] [Google Scholar]
- 4.Pinsker H M, Hening W A, Carew T J, Kandel E R. Science. 1973;182:1039–1042. doi: 10.1126/science.182.4116.1039. [DOI] [PubMed] [Google Scholar]
- 5.Frost W N, Castellucci V F, Hawkins R D, Kandel E R. Proc Natl Acad Sci USA. 1985;82:8266–8269. doi: 10.1073/pnas.82.23.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rayport S G, Schacher S. J Neurosci. 1986;6:759–763. doi: 10.1523/JNEUROSCI.06-03-00759.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glanzman D L, Mackey S L, Hawkins R D, Dyke A M, Lloyd P O, Kandel E R. J Neurosci. 1989;9:4200–4213. doi: 10.1523/JNEUROSCI.09-12-04200.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montarolo P G, Goelet P, Castellucci V F, Morgan J, Kandel E R, Schacher S. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- 9.Klein M, Kandel E R. Proc Natl Acad Sci USA. 1980;77:6912–6916. doi: 10.1073/pnas.77.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochner B, Schacher S, Kandel E R. Proc Natl Acad Sci USA. 1986;83:8410–8414. doi: 10.1073/pnas.83.21.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dale N, Kandel E R, Schacher S. J Neurosci. 1987;7:2232–2238. doi: 10.1523/JNEUROSCI.07-07-02232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scholz K P, Byrne J H. Science. 1987;235:685–687. doi: 10.1126/science.2433766. [DOI] [PubMed] [Google Scholar]
- 13.Dale N, Schacher S, Kandel E R. Science. 1988;239:282–285. doi: 10.1126/science.2892269. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz J H, Castellucci V F, Kandel E R. J Neurophysiol. 1971;34:939–953. doi: 10.1152/jn.1971.34.6.939. [DOI] [PubMed] [Google Scholar]
- 15.Castellucci V F, Blumenfeld H, Goelet P. J Neurobiol. 1989;20:1–9. doi: 10.1002/neu.480200102. [DOI] [PubMed] [Google Scholar]
- 16.Davis H P, Squire L R. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- 17.Bailey C H, Chen M. Science. 1983;220:91–93. doi: 10.1126/science.6828885. [DOI] [PubMed] [Google Scholar]
- 18.Bailey C H, Chen M. Proc Natl Acad Sci USA. 1988;85:2373–2377. doi: 10.1073/pnas.85.7.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey C H, Chen M. Proc Natl Acad Sci USA. 1988;85:9356–9359. doi: 10.1073/pnas.85.23.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nazif F A, Byrne J H, Cleary L J. Brain Res. 1991;539:324–327. doi: 10.1016/0006-8993(91)91638-h. [DOI] [PubMed] [Google Scholar]
- 21.Glanzman D L, Kandel E R, Schacher S. Science. 1990;249:799–802. doi: 10.1126/science.2389145. [DOI] [PubMed] [Google Scholar]
- 22.Bailey C H, Montarolo P G, Chen M, Kandel E R, Schacher S. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- 23.Bernier L, Castellucci V F, Kandel E R, Schwartz J H. J Neurosci. 1982;2:1682–1691. doi: 10.1523/JNEUROSCI.02-12-01682.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacskai B J, Hochner B, Mahaut-Smith M, Adamas S R, Kaang B-K, Kandel E R, Tsien R Y. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- 25.Dash P K, Hochner B, Kandel E R. Nature (London) 1990;345:718–721. doi: 10.1038/345718a0. [DOI] [PubMed] [Google Scholar]
- 26.Kaang B K, Kandel E R, Grant S G N. Neuron. 1993;10:427–435. doi: 10.1016/0896-6273(93)90331-k. [DOI] [PubMed] [Google Scholar]
- 27.Bartsch D, Ghirardi M, Skehel P A, Karl K A, Herder S P, Chen M, Bailey C H, Kandel E R. Cell. 1995;83:979–992. doi: 10.1016/0092-8674(95)90213-9. [DOI] [PubMed] [Google Scholar]
- 28.Karpinski B A, Morle G D, Huggenvik J, Uhler M D, Lenden J M. Proc Natl Acad Sci USA. 1992;89:4820–4824. doi: 10.1073/pnas.89.11.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghirardi M, Montarolo P G, Kandel E R. Neuron. 1995;14:413–420. doi: 10.1016/0896-6273(95)90297-x. [DOI] [PubMed] [Google Scholar]
- 30.Yin J C P, Wallach J S, Del Vecchio M, Wilder E L, Zhou H, Quinn W G, Tully T. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 31.Yin J C P, Del Vecchio M, Zhou H, Tully T. Cell. 1995;81:107–115. doi: 10.1016/0092-8674(95)90375-5. [DOI] [PubMed] [Google Scholar]
- 32.Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva A J. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 33.Metz R, Ziff E. Genes Dev. 1991;5:1754–1766. doi: 10.1101/gad.5.10.1754. [DOI] [PubMed] [Google Scholar]
- 34.Alberini C M, Ghirardi M, Metz R, Kandel E R. Cell. 1994;76:1099–1114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- 35.Bailey C H, Chen M. J Neurosci. 1989;9:1774–1780. doi: 10.1523/JNEUROSCI.09-05-01774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayford M, Barzilai A, Keller F, Schacher S, Kandel E R. Science. 1992;256:638–644. doi: 10.1126/science.1585176. [DOI] [PubMed] [Google Scholar]
- 37.Bailey C H, Chen M, Keller F, Kandel E R. Science. 1992;256:645–649. doi: 10.1126/science.1585177. [DOI] [PubMed] [Google Scholar]
- 38.Bailey C H, Kaang B-K, Chen M, Kandel E R. Soc Neurosci Abstr. 1994;20:1072. [Google Scholar]
- 39.Bretscher A. J Cell Biol. 1989;108:921–930. doi: 10.1083/jcb.108.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connolly J L, Green S A, Greene L A. J Cell Biol. 1984;98:457–465. doi: 10.1083/jcb.98.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byrne J H, Kandel E R. J Neurosci. 1995;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergold P J, Sweatt J D, Winicov I, Weiss K R, Kandel E R, Schwartz J H. Proc Natl Acad Sci USA. 1990;87:3788–3791. doi: 10.1073/pnas.87.10.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hegde A N, Goldberg A L, Schwartz J H. Proc Natl Acad Sci USA. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Y, Barzilai A, Chen M, Bailey C H, Kandel E R. Neuron. 1993;10:921–929. doi: 10.1016/0896-6273(93)90207-8. [DOI] [PubMed] [Google Scholar]
- 45.Montarolo P G, Ghirardi M, Kandel E R. Soc Neurosci Abstr. 1992;18:712. [Google Scholar]
- 46.Bailey C H, Chen M, Kandel E R, Schacher S. Soc Neurosci Abstr. 1993;19:16. [Google Scholar]
- 47.Kuhl D, Kennedy T E, Barzilai A, Kandel E R. J Cell Biol. 1992;119:1069–1076. doi: 10.1083/jcb.119.5.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kennedy T E, Gawinowicz M A, Barzilai A, Kandel E R, Sweatt J D. Proc Natl Acad Sci USA. 1988;85:7008–7012. doi: 10.1073/pnas.85.18.7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Byers D, Davis R L, Kiger J A. Nature (London) 1981;289:79–81. doi: 10.1038/289079a0. [DOI] [PubMed] [Google Scholar]
- 50.Levin L R, Han P L, Hwang P M, Feinstein P G, Dins R L, Reed R R. Cell. 1992;68:479–489. doi: 10.1016/0092-8674(92)90185-f. [DOI] [PubMed] [Google Scholar]
- 51.Feany M B, Quinn W G. Science. 1995;268:869–873. doi: 10.1126/science.7754370. [DOI] [PubMed] [Google Scholar]
- 52.Drain P, Folkers E, Quinn W G. Neuron. 1991;6:71–82. doi: 10.1016/0896-6273(91)90123-h. [DOI] [PubMed] [Google Scholar]
- 53.Tully T, Preat T, Boynton S C, Del Vecchio M. Cell. 1994;79:35–47. doi: 10.1016/0092-8674(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 54.Bliss T V P, Lomo T. J Physiol (London) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bekkers J M, Stevens C F. Nature (London) 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 56.Malinow R, Tsien T W. Nature (London) 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- 57.Malgaroli A, Tsien R W. Nature (London) 1992;357:134–139. doi: 10.1038/357134a0. [DOI] [PubMed] [Google Scholar]
- 58.O’Dell T J, Hawkins R D, Kandel E R, Arancio O. Proc Natl Acad Sci USA. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuman E M, Madison D V. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 60.O’Dell T J, Huang P L, Dawson T M, Dinerman J L, Snyder S H, Kandel E R, Fishman M C. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- 61.Frey U, Huang Y-Y, Kandel E R. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 62.Huang Y-Y, Li X-C, Kandel E R. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 63.Huang Y-Y, Kandel E R. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- 64.Nguyen P V, Abel T, Kandel E R. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- 65.Nguyen, P. V. & Kandel, E. R. (1996) J. Neurosci. 16, in press. [DOI] [PMC free article] [PubMed]
- 66.Cole A J, Saffen D W, Baraban J M, Worley P F. Nature (London) 1989;340:474–476. doi: 10.1038/340474a0. [DOI] [PubMed] [Google Scholar]
- 67.Dragunow M, Currie R W, Faull R L M, Robertson H A, Jansen K. Neurosci Behav Rev. 1989;13:301–313. doi: 10.1016/s0149-7634(89)80066-1. [DOI] [PubMed] [Google Scholar]
- 68.Sonnenberg J L, Rauscher F J, III, Morgan J I, Curran T. Science. 1989;246:1622–1625. doi: 10.1126/science.2512642. [DOI] [PubMed] [Google Scholar]
- 69.Bailey C H, Kandel E R. Annu Rev Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]