

Abstract
Biological tissues require oxygen to meet their energetic demands. However, the consumption of oxygen also results in the generation of free radicals that may have damaging effects on cells. The brain is particularly vulnerable to the effects of reactive oxygen species due to its high demand for oxygen, and its abundance of highly peroxidisable substrates. Oxidative stress is caused by an imbalance in the redox state of the cell, either by overproduction of reactive oxygen species, or by dysfunction of the antioxidant systems. Oxidative stress has been detected in a range of neurodegenerative disease, and emerging evidence from in vitro and in vivo disease models suggests that oxidative stress may play a role in disease pathogenesis. However, the promise of antioxidants as novel therapies for neurodegenerative diseases has not been borne out in clinical studies. In this review, we critically assess the hypothesis that oxidative stress is a crucial player in common neurodegenerative disease and discuss the source of free radicals in such diseases. Furthermore, we examine the issues surrounding the failure to translate this hypothesis into an effective clinical treatment.
1. Introduction
1.1. Oxidative Stress and Neurodegeneration
It has been long recognised that oxidative stress may be important in the aetiology of a variety of late onset neurodegenerative diseases. Aging has been established as the most important risk factor for the common neurodegenerative diseases, Alzheimer's disease (AD), and Parkinson's disease (PD). Most theories of aging centre are on the idea that cumulative oxidative stress leads to mitochondrial mutations, mitochondrial dysfunction, and oxidative damage [1]. However, as the role of ROS becomes increasingly recognised in aging and age-related diseases, a number of controversies begin to emerge in this field. Is oxidative stress an epiphenomenon of dysfunctional and dying neurons, or does oxidative stress itself cause the dysfunctionality/death of neurons? How does a global event such as oxidative stress result in the selective neuronal vulnerability seen in most neurodegenerative diseases? And finally, if oxidative stress is truly fundamental to pathogenesis then why has the use of antioxidant therapy been thus far largely unsuccessful in such diseases?
In order to address these questions, we first outline the definition of oxidative stress and show how ROS is generated in the human brain (Box 1), as well as the antioxidant defence mechanisms that exist to counteract it (Box 2). We present the evidence that oxidative stress can be found in neurodegenerative disease. Next we address the issue of whether oxidative stress is truly pathogenic in disease models. In order to prove a crucial of ROS, it is necessary to observe oxidative stress as an early event in the disease process, and to further demonstrate that inhibition of ROS production is able to prevent the pathogenic process. We describe the evidence from animal and cellular models of the role of ROS in the major neurodegenerative diseases. We present hypotheses for the interplay between oxidative stress and selective cell death. Finally we study the rationale for the use of antioxidant therapy and the outcome of its use in human disease. Although oxidative stress has been implicated in a range of chronic neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and Amyotrophic Lateral Sclerosis, the two commonest of these diseases, AD and PD, will be discussed in detail in this review, and other neurodegenerative diseases will be referenced where relevant.
2. What Is Oxidative Stress?
Oxygen is essential for the normal function of eukaryotic organisms. Its role in survival is linked to its high redox potential, which makes it an excellent oxidizing agent capable of accepting electrons easily from reduced substrates. Different tissues have different oxygen demands depending on their metabolic needs. Neurons and astrocytes, the two major types of brain cells, are largely responsible for the brain's massive consumption of O2 and glucose; indeed, the brain represents only ~2% of the total body weight and yet accounts for more than 20% of the total consumption of oxygen [2]. Despite the essentiality of oxygen for living organisms, the state of hyperoxia produces toxicity, including neurotoxicity [3, 4]. The toxicity and chemical activity of oxygen depends on its electronic structure. The identical spin states of its two outer orbital electrons render oxygen kinetically stable, except in the presence of appropriate catalysts that scramble electron spin states to produce partially reduced forms of oxygen. Partially reduced forms of oxygen are highly active because the free radical is very unstable and must either accept or be a donor of electrons. There are many different varieties of partially reduced reactive oxygen species (ROS) including superoxide (O2 •−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH•). The modern use of the term “ROS” includes both oxygen radicals and nonradicals that are easily converted into free radicals (O3, H2O2, 1O2) [2]. ROS have different reactive abilities, and one of the most reactive ROS is the hydroxyl radical OH•. Due to the high reactive activity of ROS, they chemically interact with biological molecules leading to changes in cell function and cell death. As a result, oxygen has the potential to be poisonous, and aerobic organisms survive its presence only because they contain antioxidant defences [5]. Brain cells and especially neurons require effective antioxidant protection for several reasons.
They exhibit higher (about 10-fold) oxygen consumption compared to other tissues.
Nondividing cells such as neurons have a long life duration.
Nitric oxide has a prominent role in the brain and can form reactive nitrogen species such as peroxynitrite, in combination with some forms of oxygen such as superoxide. Nitric oxide may take part in nitrosylation of proteins; however, peroxynitrite is a highly reactive nitrogen species that can nitrate tyrosine residues of proteins and alter their function.
Oxidative stress is a condition in which the balance between production of ROS and level of antioxidants is significantly disturbed and results in damage to cells by the excessive ROS. ROS contribute to the development of neurodegeneration by modulating the function of biomolecules. ROS may target several different substrates in the cell, causing protein, DNA, RNA oxidation, or lipid peroxidation (Figure 1). The oxidation products of polyunsaturated fatty acids, especially arachidonic acid and docosahexanoic acid which are abundant in brain, are malondialdehyde and 4-hydroxynonenal. ROS attacks protein, oxidising both the backbone and the side chain, which in turn reacts with amino acid side chains to form carbonyl functions. ROS attacks nucleic acids in a number of ways, causing DNA-protein crosslinks, breaks in the strand, and modifies purine and pyridine bases resulting in DNA mutations.
Figure 1.
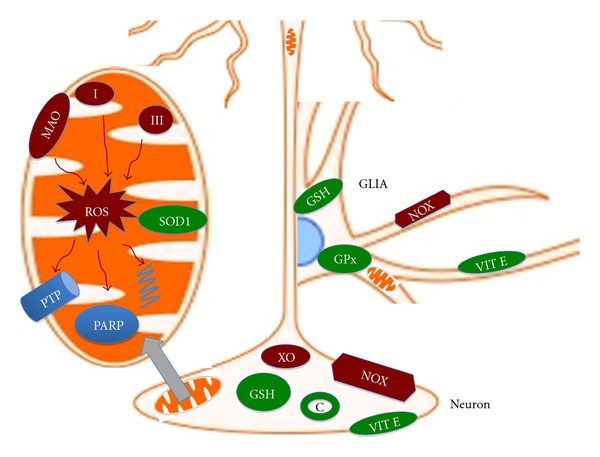
Schematic diagram of main producers of ROS and antioxidant system in neurons and glia. Main ROS producers are shown in red: monoamine oxidase (MAO), complex I and III are major sources within mitochondria. ROS generated in mitochondria target the permeability transition pore (PTP), PARP, and mitochondrial DNA. In the cytosol, NADPH oxidase (NOX) and xanthine oxidase (XO) are the main producers of ROS. The major antioxidant systems are shown in green and include superoxide dismutase (SOD) in the mitochondria, glutathione (GSH), catalase (C), and glutathione peroxidase (GPx).
2.1. Oxidative Stress Occurs in Neurodegenerative Disease
Alzheimer's disease is the most common neurodegenerative disease, affecting approximately 16 million people worldwide. It is characterised by progressive neuronal loss associated with aggregation of protein as extracellular amyloid (βA) plaques, and intracellular tau tangles. AD brains also show evidence of ROS mediated-injury; there is an increase in levels of malondyaldehyde and 4-hydroxynonenal in brain and cerebrospinal fluid of AD patients compared to controls [6]. Protein carbonyl moieties are increased in the frontal and parietal cortices, and hippocampus in AD brain, with sparing of the cerebellum where no AD pathology occurs [7, 8]. There is also an increase in hydroxylated guanine in AD samples compared to age-matched controls [9, 10]. This data from human brain is also supported by data from transgenic animal models of AD in which markers of protein and lipid peroxidation are increased in the cortex and hippocampus prior to the appearance of plaques or tangle pathology [11].
Parkinson's disease is the second most common neurodegenerative disease and is characterised by progressive loss of dopaminergic neurons in the substantia nigra, and aggregation of the protein α-synuclein. In PD brain, the concentration of polyunsaturated free fatty acids in the substantia nigra is reduced, while the levels of lipid peroxidation markers (malondialdehyde and 4-hydroxynonenal) are increased [12]. Protein oxidative damage in the form of protein carbonyls [13] is also evident in PD brain compared to controls, and there is some evidence to suggest a role for nitration and nitrosylation of certain proteins due to reactive nitrogen species in PD brain [14]. In addition to increased levels of 8-hydroxydeoxyguanosine in PD brain [15], it has been reported that there is an increase in the common deletions in mitochondrial DNA in the surviving dopaminergic neurons in PD substantia nigra. Such deletions are believed to be the result of oxidative stress [16].
2.2. Mechanisms of Oxidative Stress: ROS Production by Mitochondrial Dysfunction
Mitochondrial pathology is evident in many neurodegenerative diseases including AD, PD, Huntington's disease, ALS, PSP, Friedreich's ataxia, Neurodegeneration with brain iron accumulation, and optic atrophy. The spectrum of mitochondrial dysfunction is vast and includes respiratory chain dysfunction and oxidative stress, reduced ATP production, calcium dysregulation, mitochondrial permeability transition pore opening, peturbation in mitochondrial dynamics, and deregulated mitochondrial clearance. As many of the functions of the mitochondria are interdependent, many of these pathologies exist together in varying degrees in the different disorders.
The greatest interest in mitochondrial dysfunction and mitochondrial ROS production has been in PD, as demonstrated in human brain, as well as both the toxin and genetic disease models. A reduction in complex I activity has been demonstrated in the substantia nigra, lymphocytes, and platelets of PD patients (reviewed in [17]). The neurotoxin 1-methyl-4-phenyl-l,2,3,6-tetrahydropyridine (MPTP) has been shown to produce parkinsonian symptoms in primates and rodents, and it has therefore been extensively used as an animal model of Parkinson's disease. Studies have indicated that l-methyl-4-phenylpyridinium (MPP+), the active metabolite of MPTP, can block electron transport by binding to the same site as the classic Complex I inhibitor, rotenone, leading to a loss of ATP production. Rotenone or MPP+ also produces superoxide anions in submitochondrial particles, adding further support to the basic premise that MPP+ acts primarily as a mitochondrial toxin [18, 19]. The neurotoxic effects of MPP+ and rotenone are likely to be due to oxidative stress rather than metabolic changes because they can be effectively prevented by antioxidants [20]. Mild uncoupling of mitochondria with UCP2 overexpression reduces ROS production in toxic (MPP+, rotenone) mouse models of Parkinson's disease. UCP2 deficiency also increases the sensitivity of dopamine neurons to MPTP, whereas UCP2 overexpression decreases MPTP-induced nigral dopamine cell loss [21].
The identification of a number of PD-related genes that are strongly associated with mitochondrial function (PINK1, DJ-1, Parkin) further adds weight that mitochondrial dysfunction with resultant oxidative stress is a primary event in PD pathogenesis. Loss of function of DJ-1 results in oxidative stress, and DJ-1 exerts neuroprotection via its antioxidant mechanism in mitochondria [22, 23]. Mutations in PINK1 cause a recessive form of PD. PINK1 is a mitochondrial kinase, and we and other authors have previously demonstrated that PINK1 deficiency results in impaired respiration with inhibition of complex 1, reduced substrate availability, and rotenone-like increased production of ROS in mitochondria [24–26]. PINK1 deficiency also results in an inability to handle cytosolic calcium challenges due to an impairment of mitochondrial calcium efflux, that leads to mitochondrial calcium overload. A combination of ROS production and mitochondrial Ca2+ initiates opening of mitochondrial permeability transition pore (PTP), which allows translocation of proapoptotic molecules from the mitochondria to the cytosol, in order to trigger apoptotic cell death [27, 28]. Early opening of the PTP has been demonstrated in PD models, for example, the toxic (MPP+) model and the genetic PINK1 deficiency model [24, 29]. PINK1 deficiency also results in abnormal mitochondrial morphology reflecting an altered balance of mitochondrial fission/fusion, and in Drosophila models PINK1 appears to genetically interact with the mitochondrial fission/fusion machinery proteins. More recently an emerging function of PINK1 has been its concerted action with parkin in the regulation of clearance of damaged mitochondria via autophagy. Loss of the mitochondrial membrane potential stabilises PINK1 expression, leading to PINK1-dependent recruitment of parkin to the mitochondria. Ubiquitination of a number of substrates by parkin then activates the autophagy of damaged mitochondria. Mutations in either parkin or PINK1 thus result in a failure of mitophagy, and an accumulation of dysfunctional mitochondria with increased ROS production, further increasing the oxidative stress on the neuron [30–32].
Abnormal aggregation of protein is a characteristic feature of neurodegenerative disease, and it appears that mitochondrial dysfunction and ROS production may influence the aggregation of the protein, α-synuclein, which accumulates in all PD brain. Mutations in α-synuclein gene cause a familial form of autosomal dominant PD. Expression of mutant α-synuclein in mouse models or neurons results in mitochondrial dysfunction and increased ROS production [33, 34]. However, protein oxidation induced by mitochondrial ROS production is also required for α-synuclein oligomerization [35] and toxicity. α-synuclein has recently been shown to bind to mitochondria and induce mitochondrial fragmentation through the inhibition of membrane fusion, a phenotype that interestingly can be rescued by coexpression of the mitochondrial PD proteins PINK1, DJ-1, and parkin [36].
Mitochondrial dysfunction and ROS production may play a role in the pathogenesis of AD. A reduction in complex IV activity has been demonstrated in mitochondria from the hippocampus and platelets of AD patients, as well as in AD animal models and AD cybrid cells in reviewed [37]. Accumulation of βA leads to oxidative stress, mitochondrial dysfunction, and energy failure prior to the development of plaque pathology [38]. Deregulation of calcium homeostasis has been demonstrated in AD, with βA causing increased cytoplasmic calcium levels and mitochondrial calcium overload, resulting in increase in ROS production and opening of the PTP [39, 40]. βA is able to induce opening of PTP in isolated mitochondria [41, 42] and primary astrocytes [43–45]. Furthermore βA may directly interact with cyclophilin D (a PTP component) forming a complex in the mitochondria that has reduced threshold for opening in the presence of mPTP inducers. Prevention of PTP opening by inducing cyclophilin D deficiency (molecular inhibition of PTP opening) is also able to improve mitochondrial function and learning/memory in an aging Alzheimer's disease mouse model [46]. A peturbation in mitochondrial dynamics has also been described in AD human brain and cell models. Fragmented mitochondria are seen in AD hippocampus in association with a downregulation of mitochondrial fusion proteins (MFN-1, MFN-2, OPA-1), with an increase in expression of the mitochondrial fission protein Fis-1 [47].
2.3. Mechanisms of Oxidative Stress: ROS Production via NADPH Oxidase
Although there are several different proposed mechanisms of neurodegeneration in Alzheimer's Disease (AD), there is good evidence for the presence of oxidative stress and the involvement of NADPH oxidase in this disease. Activation of NOX2 has been demonstrated in brains of AD patients [48], with upregulation of NOX1 and NOX3 in early stage postmortem AD brain [49].
The crucial role for NADPH oxidase in AD has also been confirmed in animal models. Thus, NOX2 deficiency improved the outcome in a mouse model of AD. Mice that overexpress the Swedish mutation of APP (Tg2576, which leads to Aβ fragment accumulation) were crossed with NOX2-deficient mice. In this model, the absence of functional NOX2 was protective and prevented the negative effects of βA deposition. Neuronal oxidative stress was abrogated, and behavioural deficits improved in both young and aged Tg2576/NOX2-deficient mice [50, 51].
The role of NADPH oxidase in AD has also been suggested at a cellular level. Amyloid-beta (βA) induced direct activation of NADPH oxidase in rat primary culture of microglial cells and human phagocytes [52]. βA activates microglial NOX through B-class scavenger receptor CD36 [53]. Active NADPH oxidase transfers protons across the membrane and, for normal functioning, requires opening of an ion/anion channel for charge compensation [54]. In βA-activated microglia, inhibition of CLIC1 channel inhibited superoxide production and protected cells by blocking the charge compensatory mechanism of NADPH oxidase [55]. βA-induced stimulation of NADPH oxidase may damage surrounding cells because it occurs in combination with massive NO production and the generation of peroxynitrate [56, 57]. Neuroprotective effect of some endogenous compounds, such a hormone melatonin is induced by its antioxidant properties [58, 59].
βA also activates NADPH oxidase by inducing calcium entry into astrocytes but not neurons [39, 40]. The activation of NADPH oxidase results in the generation of oxidative stress, which depolarises the mitochondrial membrane and, in combination with calcium, induces opening of the mitochondrial permeability transition pore (mPTP; Figure 1) [43, 45] as well as changing membrane structure through activation of phospholipase C [60]. This oxidative stress signal is passed to neighbouring neurons, which are more vulnerable than astrocytes. The mechanism by which this occurs is not certain although neuronal production of glutathione, a key antioxidant, requires glutathione precursors derived from extracellular cleavage of glutathione released from astrocytes. It has therefore been suggested that depletion of GSH in astrocytes due to increased oxidant production by NADPH oxidase could diminish GSH release from astrocytes and consequently deplete GSH in neurons [43, 44]. There is much less information available regarding the direct activation of NADPH oxidase in neurons in AD models although there is some evidence that βA and the presenilins exhibit the ability to activate NADPH oxidase in primary neurons [61–63].
In Parkinson's disease (PD), oxidative stress has been demonstrated in both the rotenone and MPTP-induced toxin models. In these models, activation of NADPH oxidase (NOX2) in microglia occurs. [64, 65]. Furthermore, pharmacological inhibition of NADPH oxidase is able to protect mesencephalic dopaminergic neuronal (N27) cells against MPP+-mediated dopaminergic degeneration [66]. Genetic models of PD also exhibit increased oxidative stress. In one such model, loss of PINK1 function is associated with increased ROS production by NADPH oxidase in midbrain neurons. Interestingly, the NADPH oxidase is activated by high cytosolic calcium concentration, leading to overproduction of superoxide. ROS production from NADPH oxidase inhibits the plasmalemmal glucose transporter resulting in deregulation of mitochondrial metabolism [24, 26].
Much less information is available about the role of Xanthine Oxidase (XO) in Alzheimer's or Parkinson's diseases. Oxypurinol was able to reduce the production of ROS and protect neurons in the genetic presenilin 2 mouse model of AD [67]. In addition, βA is able to activate production of H2O2 in cytosol of neocortical neurons [68]. Inhibitor of XO allopurinol significantly suppressed *OH generation in rat striatum of toxic models of Parkinson's disease induced by paranonylphenol and MPP(+) [69], suggesting a potential role for XO in the oxidative stress associated with PD.
2.4. Oxidative Stress Results in Selective Neuronal Degeneration
One of the major features of neurodegenerative disease is the selective vulnerability of different neuronal populations that are affected in a progressive and often stereotyped manner. However, the susceptible neuronal population varies between diseases, despite oxidative stress being implicated as the major pathogenic process in all of them. Thus, the most vulnerable regions of the brain affected in AD such as the entorhinal cortex and the hippocampus CA1 region differ from the most vulnerable regions affected in PD which includes the dopaminergic neurons in the substantia nigra. Even within a region, the subset of neurons affected will be adjacent to a subset of neurons that are spared; for example, in PD, the SNpc dopaminergic neurons are affected while the VTA neurons are spared. Therefore, in addition to a global oxidative stress that affects all neurons, there must be additional factors that determine the selective cell death in each disease.
Certain neuronal groups have high intrinsic levels of oxidative stress and are therefore more vulnerable to additional disease-related oxidative stress. Neurons that have long axons and multiple synapses have high bioenergetic requirements for axonal transport or long-term plasticity. A high ATP demand combined with relative mitochondrial dysfunction will render these groups of neurons far more sensitive to degeneration than other neuronal groups. Different neuronal groups exhibit different degrees of oxidative stress. For example, in the hippocampus CA1 neurons generate higher levels of superoxide anion than CA3 neurons and exhibit higher levels of expression of both antioxidant and ROS-producing genes [70]. Neurons that are exposed to higher levels of cytosolic dopamine; that is, dopaminergic neurons are also exposed to additional oxidative stress produced by the metabolism of dopamine by MAO (which generates hydrogen peroxide) as well as the autooxidation of dopamine (which generates superoxide). Thus endogenous dopamine, as well as exogenous treatment with levodopa (used in PD) may be a further source of oxidative stress that may worsen pathogenesis [71, 72]. Higher levels of intrinsic oxidative stress may result in mitochondrial dysfunction, which further results in the production of more free radicals and an exacerbation of the cycle of oxidative stress. However, it should be noted that the MAO-induced metabolism of dopamine and production of hydrogen peroxide have an important role in physiological calcium signaling in astrocytes and is not solely a pathological process [73]. Therefore, it is possible that antioxidant treatment may also have an effect on normal signal transmission in the brain.
One interesting hypothesis for the vulnerability of specific neuronal groups in Parkinson's disease has emerged from the discovery that adult substantia nigra pars compacta dopaminergic neurons have an autonomous pacemaker mechanism that utilizes L-type calcium channels resulting in intracellular calcium oscillations. This creates a metabolic stress for such neurons as the repeated and persistent entry of calcium into cells needs to be counterbalanced by ATP demanding pumps to restore the calcium concentration. In fact it has been demonstrated that the opening of these L-type ion channels results in higher levels of oxidative stress in the mitochondria of such neurons [74]. Moreover, other nondopaminergic groups of neurons that are also selectively affected in PD such as the dorsal motor nucleus of the vagus, the serotonergic neurons of the raphe nuclei, and neurons of the locus coeruleus all engage the same L-type calcium channel pacing mechanism. Therefore, this represents an example of a cell-specific factor that renders certain neuronal groups highly vulnerable to the disease process.
2.5. Use of Antioxidant Therapy in Neurodegenerative Disease
Based on the hypothesis that oxidative stress is pathogenic in neurodegenerative disease, the rationale for the use of antioxidants as therapies is clear. And indeed the initial demonstration of the benefits of antioxidants in animal and cell models of disease was promising. Perhaps the most widely studied of these antioxidant therapies have been vitamin E (the major scavenger of lipid peroxidation in brain), vitamin C (intracellular reducing molecule), and coenzyme Q10 (transfers electrons from complexes I and II to complex III in respiratory chain).
Vitamin E supplementation in an AD mouse model resulted in improved cognition and reduced βA deposition [75]. The reduction of amyloid deposition was particularly noted in young AD mice [76]. Daily injections of vitamin C in the APP/presenilin 1 mouse model significantly reduced memory deficits.
Conversely, vitamin E has not been shown to have a protective effect in the commonly used toxin-based model of MPTP-induced PD. However, the molecule coenzyme Q10 has been shown to have multiple protective effects within the mitochondria and therefore has been widely tested as a potential therapy. Administration of CoQ10 protects MPTP-treated mice from dopaminergic neuronal loss and also attenuated α-synuclein aggregation. Neuroprotection by CoQ10 in an MPTP-primate model has also been reported [77].
Despite the promise of these animal studies, there has been no proven benefit for the use of vitamin E and/or vitamin C in either AD or PD from large randomised controlled trials [78]. These trials have been reviewed in detail in [79, 80]. Furthermore, a large meta-analysis of vitamin E clinical trials, CoQ10 trials, and a glutathione trial in PD concluded that there were only minor treatment benefits in the CoQ10 trials that may have been due to improvement in the respiratory chain deficit rather than a direct antioxidant action [81]. None of the trials have shown significant benefit to warrant recommendation for use in the clinical setting.
The disappointing translation of the oxidative stress hypothesis into useful therapy in human disease raises several issues regarding extrapolation of results from animal studies to the clinical setting. All animal models are limited in recreating the human disease as they do not recapitulate the long-time frame and gradual accumulation of age-related changes that characterise late onset sporadic neurodegenerative diseases in humans. From much of the animal model data, it appears that antioxidants must be administered at an early stage in the disease where the process influences pathogenesis most, and therefore the use of antioxidants in established late disease in humans may be ineffective. There are several pharmacodynamic and pharmacokinetic considerations such as the bioavailability of reducing molecules in the human brain in the doses used in animal models and furthermore the effective targeting of such molecules to the mitochondria in human brain. Many therapies have used antioxidants that act as scavengers, rather than blocking the source of the ROS production, and this may be less effective. Finally it is possible that there are several different producers of oxidative stress in each disease, and that these may need to be targeted separately but simultaneously by multiple therapies in order to effectively reduce oxidative stress and slow disease progression.
There are many processes that have been implicated in the pathogenesis of neurodegeneration including protein misfolding and aggregation, abnormal kinase-signalling pathways, neuronal calcium dysregulation, and impaired synaptic transmission. Many of these interact with, and are exacerbated by, oxidative stress. With the mounting robust evidence of the role of oxidative stress in pathogenesis, it remains likely that the hypothesis that oxidative damage is critical in disease is indeed true, and moreover that careful targeting of this process should serve to ameliorate neurodegenerative disease.
3. BOX 1: ROS Producers in Mammalian Brain
3.1. NADPH Oxidase
NADPH oxidase is a multisubunit enzyme complex that was first described in phagocytes [82]. NADPH oxidase is a member of the NOX gene family, also called NOX2 and phagocytic oxidase (PHOX). Seven NOX genes have been identified: NOX1 to 5 and DUOX1 and 2. Very little is known about the role of NOX5 and DUOX1 and 2 in the CNS. The majority of the NOX enzyme expressed in the brain is NOX2 although some evidence exists for the CNS localization of NOX1, NOX4, and possibly also NOX3. The NOX2 enzyme complex consists of the membrane-bound cytochrome b558 (p22PHOX and the enzymatic subunit, gp91PHOX), several cytosolic proteins (p47PHOX, p67PHOX, and p40PHOX), and the Rac G-protein. NADPH oxidase is activated when the cytosolic subunits are phosphorylated and Rac is activated in the cytosol, resulting in their translocation to the membrane and formation of the active NADPH oxidase complex with cytochrome b558 [83]. The enzyme transfers the proton across the membrane, and the end product of the enzyme is superoxide. The NOX family of proteins is expressed on diverse cell types, and NADPH oxidase is present in microglia [84], neurons, and astrocytes [85, 86]. The physiological role of NOX enzymes in brain cells is still unclear and described mostly as an effect of superoxide produced by NADPH oxidase [87].
3.2. Xanthine Oxidase
Xanthine oxidase or xanthine dehydrogenase are two convertible forms of Xanthine oxidoreductase. It is a complex molybdoflavoenzyme that is readily available from milk and widely distributed in mammalian tissues and is generally accepted to be a key enzyme of purine catabolism [88]. XO catalyses the oxidation of a wide range of substrates and can pass electrons to molecular oxygen to produce uric acid, superoxide, and hydrogen peroxide. Under normal conditions the enzyme exists in the form of xanthine dihydrogenase. Ca2+-stimulated proteases cause the irreversible partial cleavage of xanthine dehydrogenase to xanthine oxidase, which in turn catalyzes the oxidation of hypoxanthine to xanthine.
The role of XO in ischaemic cell injury was demonstrated in the 1980's. Deprivation of oxygen leads to the metabolism of ATP and accumulation of hypoxanthine. Hypoxia induces a rise in the levels of intracellular calcium, which activates a protease that converts XDH, predominantly in vivo, into XO. Concomitantly, purines are catabolised, and hypoxanthine accumulates. On reperfusion, oxygen is again available and, in the presence of XO and hypoxanthine, is reduced to hydrogen peroxide and superoxide [88, 89].
3.3. Mitochondria
It has become dogma that mitochondria are a major source of ROS. In pathological conditions, this organelle actually produces less-free radicals than cytosolic enzymes such as NADPH oxidase. However, mitochondria (electron transport chain-ETC), in contrast to other cellular producers of ROS, generate free radicals all the time. Mitochondria, which harbor the bulk of oxidative pathways, are packed with various redox carriers and centers that can potentially leak single electrons to oxygen and convert it into superoxide anion, a progenitor ROS. The initial observations of ROS production in mitochondrial fragments came as early as 1966 but passed almost unnoticed until 1971 when Loschen, Flohe, and Chance demonstrated for the first time that succinate supported H2O2 production by intact pigeon heart mitochondria [90–92]. Depending on the metabolic conditions, isolated mitochondria produced superoxide in respiratory complex I (in the direction of the matrix), complex III (in direction of matrix and to outside of matrix) [90]. ROS in mitochondria can also be generated in several enzymes including aconitase and α-ketoglutarate dehydrogenase complex [90, 93]. The production of superoxide by the ETC in mitochondria is dependent on the value of mitochondrial membrane potential.
Because of the constant production of free radicals, mitochondria possess a very efficient antioxidant system in the matrix. Overproduction of ROS in mitochondria or in other sources and changes in the antioxidant system lead to imbalance and induce oxidative stress and neurodegeneration.
Inhibition of neuronal respiration by oxygen deprivation or chemical ischemia leads to a significant increase in the generation of ROS in mitochondria [94]. This effect can be reduced by the application of mitochondrial uncouplers, and a number of reports have demonstrated significant neuroprotection by mild uncoupling with UCP2 in cerebral stroke [95]. In addition, mutations in mitochondrial complexes I–IV induce inhibition of respiration and activation of ROS production [96, 97] and selective neuronal cell death [98].
3.4. Monoamine Oxidase
The mitochondrially located (outer membrane) flavoenzymes monoamine oxidase A (MAO A) and monoamine oxidase B (MAOB) represent one of the most extensively studied enzyme group. This long-term interest stems from their role in the oxidative catabolism of important amine neurotransmitters, including serotonin, dopamine, and epinephrine [99]. MAO-A and MAO-B are encoded by separate genes that correspond to different amino acid sequences that are ~70% identical. In the CNS they are expressed in neurons (MAO-A) and glial cells (MAO A and B). MAO breaks down monoamines using FAD and results in the production of aldehydes. The FAD-FADH2 cycle generates hydrogen peroxide.
4. BOX 2: The Antioxidant System
4.1. Antioxidant System
Cellular levels of ROS are controlled by antioxidant enzymes and small-molecule antioxidants.
4.1.1. Superoxide Dismutase
As major antioxidant enzymes, superoxide dismutases (SODs), play a crucial role in scavenging O2 •−. The superoxide dismutase family is specialized in eliminating superoxide anion radicals derived from extracellular stimulants, including ionizing radiation and oxidative insults, together with those primarily produced within the mitochondrial matrix as byproducts of oxygen metabolism through the electron transport chain [100]. Three distinct isoforms of SOD have been identified and characterized in mammals: copper-zinc superoxide dismutase (Cu/ZnSOD; encoded by the sod1 gene), manganese superoxide dismutase (MnSOD; encoded by the sod2 gene), and extracellular superoxide dismutase (ECSOD; encoded by the sod3 gene). These forms of SOD exhibit similar functions, but characteristics of their protein structure, chromosome localization, metal cofactor requirements, gene distribution, and cellular compartmentalization are distinctly different from one another [100].
4.1.2. Glutathione Peroxidases
Glutathione peroxidase is the general name for a family of multiple isozymes that catalyze the reduction of H2O2 or organic hydroperoxides to water or corresponding alcohols using reduced glutathione (GSH) as an electron donor (H2O2 + 2GSH → GS-SG + 2H2O). In mammalian tissues, there are four major selenium-dependent glutathione peroxidases (GPX) and phospholipid hydroperoxide glutathione peroxidase, which incorporates cysteine instead of selenocysteine in the conserved catalytic motif [101]. GPX1 is known to localize primarily in glial cells, in which GP activity is tenfold higher than in neurons [101].
4.1.3. Catalase
Catalase is a ferriheme-containing enzyme that is responsible for the conversion of hydrogen peroxide (but not other peroxides) to water [5]. It is localised in peroxisomes and may also be found in cytoplasm and mitochondria. It has a minor role at low levels of hydrogen peroxide generation but becomes more important at higher levels of hydrogen peroxide production.
4.2. Nonenzymatic Antioxidants
4.2.1. GSH
The main antioxidant in CNS, glutathione (GSH), is the most abundant small molecule, nonprotein thiol in cells (present in millimolar concentration in the brain) [102]. It consists of a tripeptide of glutamate, cysteine and glycine characterized by a reactive thiol group and γ-glutamyl bond. Reduced GSH can nonenzymatically act directly with free radicals, notably superoxide radicals, hydroxyl radicals, nitric oxide, and carbon radicals for their removal. GSH peroxidase and GSH reductase can act enzymatically to remove H2O2 and maintain GSH in a reduced state [102].
4.2.2. Vitamin E
The role of vitamin E in the central nervous system is not fully understood although it is a lipid soluble molecule with antioxidant function. It appears to neutralize the effect of peroxide and prevent lipid peroxidation in membranes.
Acknowledgment
This work was supported by the Wellcome/MRC Parkinson's Disease Consortium grant to UCL/IoN; AYA is Parkinson's UK Senior Research Fellow. Funding to pay the open access publication charges for this article was provided by the Wellcome/MRC Parkinson's Disease Consortium grant to UCL/IoN.
References
- 1.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry. 2006;97(6):1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 3.Ahdab-Barmada M, Moossy J, Nemoto EM, Lin MR. Hyperoxia produces neuronal necrosis in the rat. Journal of Neuropathology and Experimental Neurology. 1986;45(3):233–246. [PubMed] [Google Scholar]
- 4.Davydov BI, Drobyshev VI, Ushakov IB, Fyodorov VP. Morphological analysis of animal brain reactions to short-term hyperoxia. Kosmicheskaya Biologiya i Aviakosmicheskaya Meditsina. 1988;22(2):56–62. [PubMed] [Google Scholar]
- 5.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 6.Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology. 1995;45(8):1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 7.Hensley K, Butterfield DA, Hall N, et al. Reactive oxygen species as causal agents in the neurotoxicity of the Alzheimer’s disease-associated amyloid beta peptide. Annals of the New York Academy of Sciences. 1996;786:120–134. doi: 10.1111/j.1749-6632.1996.tb39057.x. [DOI] [PubMed] [Google Scholar]
- 8.Hensley K, Hall N, Subramaniam R, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. Journal of Neurochemistry. 1995;65(5):2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 9.Mecocci P, Cherubini A, Polidori MC, Cecchetti R, Chionne F, Senin U. Oxidative stress and dementia: new perspectives in AD pathogenesis. Aging. 1997;9(4):51–52. doi: 10.1007/BF03339704. [DOI] [PubMed] [Google Scholar]
- 10.Mecocci P, Polidori MC, Ingegni T, et al. Oxidative damage to DNA in lymphocytes from AD patients. Neurology. 1998;51(4):1014–1017. doi: 10.1212/wnl.51.4.1014. [DOI] [PubMed] [Google Scholar]
- 11.Praticò D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Annals of the New York Academy of Sciences. 2008;1147:70–78. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 12.Dalfo EP, Portero-Otin MMP, Ayala VP, Martinez A, Pamplona RM, Ferrer IM. Evidence of oxidative stress in the neocortex in incidental lewy body disease. Journal of Neuropathology & Experimental Neurology. 2005;64:816–830. doi: 10.1097/01.jnen.0000179050.54522.5a. [DOI] [PubMed] [Google Scholar]
- 13.Beal MF. Oxidatively modified proteins in aging and disease. Free Radical Biology and Medicine. 2002;32(9):797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 14.Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochimica et Biophysica Acta. 2004;1658(1-2):44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Seet RCS, Lee CYJ, Lim ECH, et al. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radical Biology and Medicine. 2010;48(4):560–566. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 16.Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genetics. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 17.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. The Lancet Neurology. 2008;7(1):97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 18.Lotharius J, O’Malley KL. The Parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation: a novel mechanism of toxicity. Journal of Biological Chemistry. 2000;275(49):38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- 19.Smith TS, Bennett JP. Mitochondrial toxins in models of neurodegenerative diseases. I: in vivo brain hydroxyl radical production during sytemic MPTP treatment or following microdialysis infusion of methylpyridinium or azide ions. Brain Research. 1997;765(2):183–188. doi: 10.1016/s0006-8993(97)00429-0. [DOI] [PubMed] [Google Scholar]
- 20.González-Polo RA, Soler G, Rodríguezmartín A, Morán JM, Fuentes JM. Protection against MPP+ neurotoxicity in cerebellar granule cells by antioxidants. Cell Biology International. 2004;28(5):373–380. doi: 10.1016/j.cellbi.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Andrews ZB, Horvath B, Barnstable CJ, et al. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson’s disease. Journal of Neuroscience. 2005;25(1):184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lev N, Ickowicz D, Melamed E, Offen D. Oxidative insults induce DJ-1 upregulation and redistribution: implications for neuroprotection. NeuroToxicology. 2008;29(3):397–405. doi: 10.1016/j.neuro.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Li HM, Niki T, Taira T, Iguchi-Ariga SMM, Ariga H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radical Research. 2005;39(10):1091–1099. doi: 10.1080/10715760500260348. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell. 2009;33(5):627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piccoli C, Sardanelli A, Scrima R, et al. Mitochondrial respiratory dysfunction in familiar Parkinsonism associated with PINK1 mutation. Neurochemical Research. 2008;33(12):2565–2574. doi: 10.1007/s11064-008-9729-2. [DOI] [PubMed] [Google Scholar]
- 26.Wood-Kaczmar A, Gandhi S, Yao Z, et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE. 2008;3(6) doi: 10.1371/journal.pone.0002455. Article ID e2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84(2-3):143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 28.Giorgio V, Soriano ME, Basso E, et al. Cyclophilin D in mitochondrial pathophysiology. Biochimica et Biophysica Acta. 2010;1797(6-7):1113–1118. doi: 10.1016/j.bbabio.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleren C, Starkov AA, Calingasan NY, Lorenzo BJ, Chen J, Beal MF. Promethazine protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Neurobiology of Disease. 2005;20(3):701–708. doi: 10.1016/j.nbd.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 31.Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biology. 2010;8(1) doi: 10.1371/journal.pbio.1000298. Article ID e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(1):378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. Journal of Biological Chemistry. 2008;283(14):9089–9100. doi: 10.1074/jbc.M710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin LJ, Pan Y, Price AC, et al. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. Journal of Neuroscience. 2006;26(1):41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esteves AR, Arduíno DM, Swerdlow RH, Oliveira CR, Cardoso SM. Oxidative stress involvement in α-synuclein oligomerization in Parkinson’s disease cybrids. Antioxidants and Redox Signaling. 2009;11(3):439–448. doi: 10.1089/ars.2008.2247. [DOI] [PubMed] [Google Scholar]
- 36.Kamp F, Exner N, Lutz AK, et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO Journal. 2010;29(20):3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(43):18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caspersen C, Wang N, Yao J, et al. Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. The FASEB Journal. 2005;19(14):2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 39.Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. Journal of Neuroscience. 2003;23(12):5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochimica et Biophysica Acta. 2004;1742(1-3):81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Parks JK, Smith TS, Trimmer PA, Bennett JP, Davis Parker W. Neurotoxic Aβ peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. Journal of Neurochemistry. 2001;76(4):1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- 42.Shevtzova EF, Kireeva EG, Bachurin SO. Effect of β-amyloid peptide fragment 25-35 on nonselective permeability of mitochondria. Bulletin of Experimental Biology and Medicine. 2001;132(6):1173–1176. doi: 10.1023/a:1014559331402. [DOI] [PubMed] [Google Scholar]
- 43.Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. Journal of Neuroscience. 2004;24(2):565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abramov AY, Duchen MR. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philosophical Transactions of the Royal Society B. 2005;360(1464):2309–2314. doi: 10.1098/rstb.2005.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abramov AY, Fraley C, Diao CT, et al. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(46):18091–18096. doi: 10.1073/pnas.0708959104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiology of Aging. 2011;32(3):398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Su B, Siedlak SL, et al. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(49):19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimohama S, Tanino H, Kawakami N, et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochemical and Biophysical Research Communications. 2000;273(1):5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 49.De La Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2006;9(2):167–181. doi: 10.3233/jad-2006-9209. [DOI] [PubMed] [Google Scholar]
- 50.Park L, Anrather J, Zhou P, et al. NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. Journal of Neuroscience. 2005;25(7):1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park L, Zhou P, Pitstick R, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(4):1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Della Bianca V, Dusi S, Bianchini E, Dal Prà I, Rossi F. β-amyloid activates the O2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. Journal of Biological Chemistry. 1999;274(22):15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 53.Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CYD, Landreth G. Fibrillar β-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. Journal of Biological Chemistry. 2006;281(30):20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- 54.DeCoursey TE, Morgan D, Cherny VV. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422(6931):531–534. doi: 10.1038/nature01523. [DOI] [PubMed] [Google Scholar]
- 55.Milton RH, Abeti R, Averaimo S, et al. CLIC1 function is required for β-amyloid-induced generation of reactive oxygen species by microglia. Journal of Neuroscience. 2008;28(45):11488–11499. doi: 10.1523/JNEUROSCI.2431-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abramov YA, Kasymov VA, Zinchenko VP. Beta-amyloid activates synthesis of nitric oxide in hyppocampal astrocytes and causes death of neurons. Biologicheskie Membrany. 2008;25(1):11–17. [Google Scholar]
- 57.Brown GC. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochemical Society Transactions. 2007;35(5):1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- 58.Ionov M, Burchell V, Klajnert B, Bryszewska M, Abramov AY. Mechanism of neuroprotection of melatonin against beta-amyloid neurotoxicity. Neuroscience. 2011;180:229–237. doi: 10.1016/j.neuroscience.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 59.Reiter R, Tang L, Garcia JJ, Muñoz-Hoyos A. Pharmacological actions of melatonin in oxygen radical pathophysiology. Life Sciences. 1997;60(25):2255–2271. doi: 10.1016/s0024-3205(97)00030-1. [DOI] [PubMed] [Google Scholar]
- 60.Hicks JB, Lai Y, Sheng W, et al. Amyloid-β peptide induces temporal membrane biphasic changes in astrocytes through cytosolic phospholipase A2. Biochimica et Biophysica Acta. 2008;1778(11):2512–2519. doi: 10.1016/j.bbamem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hashimoto Y, Niikura T, Yuko ITO, Kita Y, Terashita K, Nishimoto I. Neurotoxic mechanisms by Alzheimer’s disease-linked N141l mutant presenilin 2. Journal of Pharmacology and Experimental Therapeutics. 2002;300(3):736–745. doi: 10.1124/jpet.300.3.736. [DOI] [PubMed] [Google Scholar]
- 62.Hashimoto Y, Tsukamoto E, Niikura T, et al. Amino- and carboxyl-terminal mutants of presenilin 1 cause neuronal cell death through distinct toxic mechanisms: study of 27 different presenilin 1 mutants. Journal of Neuroscience Research. 2004;75(3):417–428. doi: 10.1002/jnr.10861. [DOI] [PubMed] [Google Scholar]
- 63.Shelat PB, Chalimoniuk M, Wang JH, et al. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. Journal of Neurochemistry. 2008;106(1):45–55. doi: 10.1111/j.1471-4159.2008.05347.x. [DOI] [PubMed] [Google Scholar]
- 64.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. Journal of Neuroscience. 2003;23(15):6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu DC, Teismann P, Tieu K, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG. Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. NeuroToxicology. 2007;28(5):988–997. doi: 10.1016/j.neuro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abe Y, Hashimoto Y, Tomita Y, et al. Cytotoxic mechanisms by M239V presenilin 2, a little-analyzed Alzheimer’s disease-causative mutant. Journal of Neuroscience Research. 2004;77(4):583–595. doi: 10.1002/jnr.20163. [DOI] [PubMed] [Google Scholar]
- 68.Kaminsky YG, Kosenko EA. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radical Research. 2008;42(6):564–573. doi: 10.1080/10715760802159057. [DOI] [PubMed] [Google Scholar]
- 69.Obata T, Kubota S, Yamanaka Y. Allopurinol suppresses para-nonylphenol and 1-methyl-4-phenylpyridinium ion (MPP+)-induced hydroxyl radical generation in rat striatum. Neuroscience Letters. 2001;306(1-2):9–12. doi: 10.1016/s0304-3940(01)01828-6. [DOI] [PubMed] [Google Scholar]
- 70.Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in Aging Neuroscience. 2010;2:p. 12. doi: 10.3389/fnagi.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Müller T. Motor complications, levodopa metabolism and progression of Parkinson’s disease. Expert Opinion on Drug Metabolism and Toxicology. 2011;7(7):847–855. doi: 10.1517/17425255.2011.575779. [DOI] [PubMed] [Google Scholar]
- 72.Müller T, Muhlack S. Cysteinyl-glycine reduction as marker for levodopa-induced oxidative stress in Parkinson’s disease patients. Movement Disorders. 2011;26(3):543–546. doi: 10.1002/mds.23384. [DOI] [PubMed] [Google Scholar]
- 73.Vaarmann A, Gandhi S, Abramov AY. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. Journal of Biological Chemistry. 2010;285(32):25018–25023. doi: 10.1074/jbc.M110.111450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Goldberg JA. The origins of oxidant stress in parkinson’s disease and therapeutic strategies. Antioxidants and Redox Signaling. 2011;14(7):1289–1301. doi: 10.1089/ars.2010.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conte V, Uryu K, Fujimoto S, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. Journal of Neurochemistry. 2004;90(3):758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- 76.Sung S, Yao Y, Uryu K, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. The FASEB Journal. 2004;18(2):323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 77.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochimica et Biophysica Acta. 2010;1802(1):198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dumont M, Lin MT, Beal MF. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer’s disease. Journal of Alzheimer’s Disease. 2010;20(2):S633–S643. doi: 10.3233/JAD-2010-100507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H. Targeting mitochondrial dysfunction in neurodegenerative disease: part II. Expert Opinion on Therapeutic Targets. 2010;14(5):497–511. doi: 10.1517/14728221003730434. [DOI] [PubMed] [Google Scholar]
- 80.Müller T. New small molecules for the treatment of Parkinson’s disease. Expert Opinion on Investigational Drugs. 2010;19(9):1077–1086. doi: 10.1517/13543784.2010.504711. [DOI] [PubMed] [Google Scholar]
- 81.Weber CA, Ernst ME. Antioxidants, supplements, and Parkinson’s disease. Annals of Pharmacotherapy. 2006;40(5):935–938. doi: 10.1345/aph.1G551. [DOI] [PubMed] [Google Scholar]
- 82.Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. Journal of Clinical Investigation. 1973;52(3):741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Babior BM. NADPH oxidase. Current Opinion in Immunology. 2004;16(1):42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Letters. 1987;223(2):284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 85.Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. Journal of Neuroscience. 2005;25(40):9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. The Journal of Neuroscience. 2000;20(23):p. RC111. doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radical Biology and Medicine. 2009;47(9):1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radical Biology and Medicine. 2002;33(6):774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 89.Granger DN, Hollwarth ME, Parks DA. Ischemia-reperfusion injury: role of oxygen-derived free radicals. Acta Physiologica Scandinavica. 1986;126(548):47–63. [PubMed] [Google Scholar]
- 90.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 91.Jensen PK. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles I. pH dependency and hydrogen peroxide formation. Biochimica et Biophysica Acta. 1966;122(2):157–166. doi: 10.1016/0926-6593(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 92.Loschen G, Flohé L, Chance B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Letters. 1971;18(2):261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 93.Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. Journal of Neuroscience. 2004;24(36):7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. Journal of Neuroscience. 2007;27(5):1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mehta SL, Li PA. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. Journal of Cerebral Blood Flow and Metabolism. 2009;29(6):1069–1078. doi: 10.1038/jcbfm.2009.4. [DOI] [PubMed] [Google Scholar]
- 96.Huang G, Chen Y, Lu H, Cao X. Coupling mitochondrial respiratory chain to cell death: an essential role of mitochondrial complex I in the interferon-β and retinoic acid-induced cancer cell death. Cell Death and Differentiation. 2007;14(2):327–337. doi: 10.1038/sj.cdd.4402004. [DOI] [PubMed] [Google Scholar]
- 97.Iuso A, Scacco S, Piccoli C, et al. Dysfunctions of cellular oxidative metabolism in patients with mutations in the NDUFS1 and NDUFS4 genes of complex I. Journal of Biological Chemistry. 2006;281(15):10374–10380. doi: 10.1074/jbc.M513387200. [DOI] [PubMed] [Google Scholar]
- 98.Abramov AY, Smulders-Srinivasan TK, Kirby DM, et al. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain. 2010;133(3):797–807. doi: 10.1093/brain/awq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Edmondson DE, Binda C, Wang J, Upadhyay AK, Mattevi A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry. 2009;48(20):4220–4230. doi: 10.1021/bi900413g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Miao L, Clair DKS. Regulation of superoxide dismutase genes: implications in disease. Free Radical Biology and Medicine. 2009;47(4):344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M. Glutathione peroxidase family—an evolutionary overview. FEBS Journal. 2008;275(15):3959–3970. doi: 10.1111/j.1742-4658.2008.06542.x. [DOI] [PubMed] [Google Scholar]
- 102.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biological Chemistry. 2003;384(4):505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]