

Abstract
Proton coupled electron transfer (PCET) reactions are important in many biological processes. Tyrosine oxidation/reduction can play a critical role in facilitating these reactions. Two examples are photosystem II (PSII) and ribonucleotide reductase (RNR). RNR is essential in DNA synthesis in all organisms. In E. coli RNR, a tyrosyl radical, Y122•, is required as a radical initiator. Photosystem II (PSII) generates molecular oxygen from water. In PSII, an essential tyrosyl radical, YZ•, oxidizes the oxygen evolving center. However, the mechanisms, by which the extraordinary oxidizing power of the tyrosyl radical is controlled, are not well understood. This is due to the difficulty in acquiring high-resolution structural information about the radical state. Spectroscopic approaches, such as EPR and UV resonance Raman (UVRR), can give new information. Here, we discuss EPR studies of PCET and the PSII YZ radical. We also present UVRR results, which support the conclusion that Y122 undergoes an alteration in ring and backbone dihedral angle when it is oxidized. This conformational change results in a loss of hydrogen bonding to the phenolic oxygen. Our analysis suggests that access of water is an important factor in determining tyrosyl radical lifetime and function. TOC graphic
Keywords: UV resonance Raman spectroscopy, electron paramagnetic resonance spectroscopy, photosynthetic water oxidation, DNA synthesis, vibrational spectroscopy
Redox-active tyrosine residues are catalytically important in ribonucleotide reductase (RNR),1 photosystem II (PSII),2,3 prostaglandin H synthase,4 galactose oxidase,5 glyoxyl oxidase,6 and Mycobacterium tuberculosis catalase-peroxidase.7 Redox-active tyrosines mediate long-range electron transfer (ET) and proton coupled electron transfer (PCET) reactions. Participation in PCET is possible because the oxidation of a protonated tyrosine is associated with the deprotonation of the phenolic oxygen.8 A neutral free radical is formed, with spin density distributed in an odd-alternate pattern on the aromatic ring.9,10 Changes in the pKa of the proton-accepting group and in the mechanism of proton/electron transfer can then alter the rate of the reaction11–15 and control catalysis. In particular, differential effects on the pKa of the oxidized, radical state and the reduced, singlet state will have an important impact on rate and energetics, providing exquisite control mechanisms (Figure 1). PCET by the coupled pathway (CPET) in which both proton and electron are transferred in one concerted step, avoids the production of high-energy intermediates (Figure 1). The oxidation and reduction of the tyrosine may be accompanied by significant conformational changes, which can control access to protonatable groups.16 A similar mechanism is involved in vectorial proton transfer in bacteriorhodopsin. 17
Figure 1.
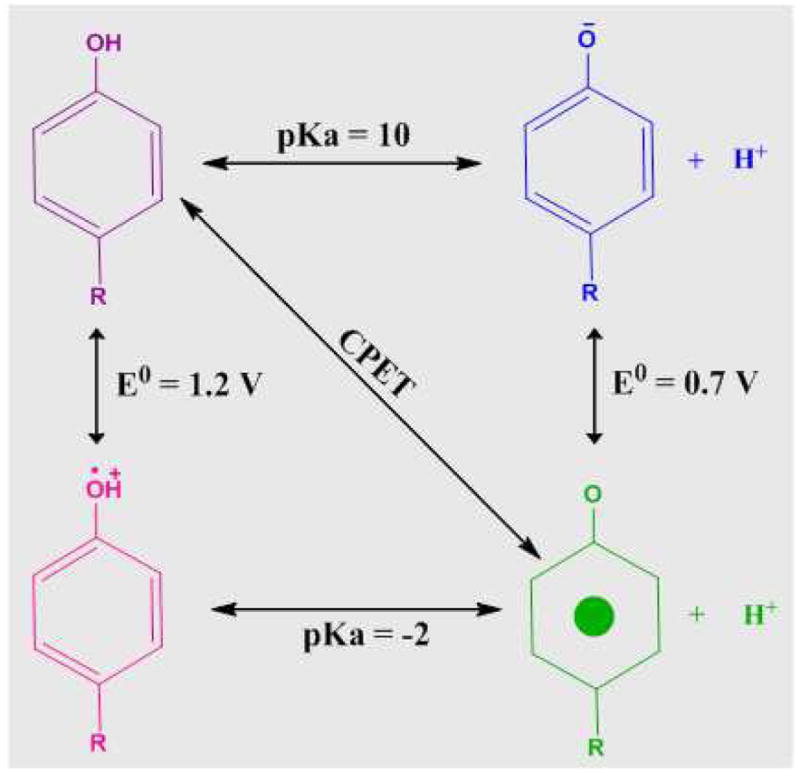
Thermodynamic cycle, illustrating the linkage of pKa and midpoint potential, for tyrosine oxidation. The concerted pathway for proton and electron transfer is labeled as CPET.
In Figure 2, we present the protein environment of four redox active tyrosines in three different proteins, a beta hairpin maquette18,19 (Figure 2A), PSII20 (Figures 2B and C), and the beta subunit of RNR21,22 (Figure 2D). The structures were obtained from NMR or X-ray crystallographic studies of the singlet state. The environments differ markedly in predicted hydrogen bonding and in placement of water and bound metals. The impact of these structural differences on function is discussed below.
Figure 2.

Comparison of the hydrogen bonding environments of four redox active tyrosines. Shown in (A) Y518 in biomimetic beta hairpin (peptide A), in (B) YZ20 in PSII, in (C) YD20 in PSII, and in (D) Y12222,79 in the beta subunit of E. coli RNR. Predicted hydrogen bonds are shown in orange dashed lines, distances are given in black dashed lines. In B, the manganese ions are purple, and the calcium ion is green. In D, the iron ions are blue. Oxygen atoms are shown as red spheres in B–D.
RNR
RNR plays a pivotal role in nucleic acid metabolism and cell division,23 and, therefore, RNR is a target of successful anti-cancer therapies.24 PCET reactions, in which tyrosine side chains are oxidized and reduced, are important in RNR function.12 In class 1a RNR, found in E. coli and mammals, long range PCET links an essential tyrosyl radical, Y122•, in the beta (R2) subunit, and the active site, in the alpha (R1) subunit (Figure 2D).25 Multistep tunneling, involving aromatic amino acid residues, is proposed to occur.26–31 While structures of the alpha and beta subunits are available,21,32,33 there is no X-ray structure of the class 1a RNR holoenzyme (alpha/beta), and there is no structure of a beta subunit containing the tyrosyl radical. Thus, we lack essential structural information about the functional form of RNR. Conformational changes in the substrate-bound quaternary complex must control the reactivity of the tyrosyl radical {see for example, discussions in 34,35}.
PSII
In PSII, there are two redox active tyrosines, YZ (Figure 2B) and YD (Figure 2C), with different protein environments and different roles in electron transfer.2,3 PSII is an enzyme essential for the maintenance of aerobic life on earth. Water oxidation and oxygen production occur at a Mn4Ca cluster, called the oxygen-evolving complex (OEC). PCET reactions involving the redox active tyrosines are light induced, providing advantages in kinetic experiments. Site-directed mutagenesis has shown that YZ is tyrosine 161 of the D1 polypeptide and that tyrosine D is Y160 of the D2 polypeptide.36–40 YZ decays on the microsecond time scale in active PSII preparations, whereas YD forms a stable radical with a decay time on the hours time scale.16 YD is not directly involved in oxygen evolution,2,36 but is in slow redox equilibrium with the oxygen evolving complex (OEC)41 and may be important in assembly of the OEC.42 YZ is essential in photosynthetic oxygen evolution and can be detected transiently as a neutral radical, which mediates electron transfer between the primary chlorophyll donor and the OEC.43,44
A PSII-inspired, beta hairpin maquette
In the beta hairpin model, peptide A (Figure 2A), a single tyrosine residues hydrogen bonds to an arginine and is pi-pi stacked with a histidine.18,19 There is a pi-cation interaction between the tyrosine and a second arginine. This peptide exhibits an interstrand PCET reaction between a pi-stacked histidine and the redox-active tyrosine (Figure 2A) {reviewed in 16}. Mutagenesis of the peptide has been used to systematically explore the effect of these interactions.
Figure 2 shows that information concerning the singlet state of redox active tyrosine is readily available. However, acquiring high-resolution structural information about the radical state is still challenging. Because amino acid radicals are such powerful oxidants, they are very reactive, often have a short lifetime, and readily generate oxidized species.45 This leads to instability and subsequent difficulty in determining high-resolution structures. An emerging idea is that the protein environment of the radical responds to the oxidation reaction and that this response may be important in controlling catalysis or triggering a reaction. An example is RNR, where quaternary interactions between the holoenzyme and substrate have been proposed to activate Y122 for rapid electron transfer reactions.28,34,46 Consistent with this proposal, reaction induced FT-IR spectroscopy has detected a secondary structural change when Y122 is reduced,47 and EPR spectroscopy has detected a change in beta-methylene dihedral angle.22
To elucidate these control mechanisms and obtain new fundamental knowledge concerning PCET, the combination of vibrational and EPR spectroscopy is a powerful approach. These approaches define the structural changes associated with the transition from the singlet to the radical state. EPR spectroscopy focuses on radical kinetics by measuring the decay of the electron spin signal.2 Reaction-induced FT-IR spectroscopy is used to identify vibrational bands of the oxidized and reduced tyrosine and accompanying coupled changes in the protein matrix.48 UV resonance Raman spectroscopy (Figure 2) provides information concerning the structure of the radical and singlet states by enhancing their vibrational bands.49–51 The vibrational bands of the phenolic group respond to the oxidation reaction because electron spin migrates to the phenolic oxygen in the radical state. With oxidation, the CO bond length shortens, and the CO stretching vibration shifts up by ~300 cm−1. The bond order was estimated as 0.98 and 1.5 for the singlet and radical states, respectively {see 10 and references therein}. A downshift of the aromatic ring stretching vibration (Y8a) by ~50 cm−1 also occurs (Figure 3). This gives a characteristic vibrational signature of the redox active tyrosine, i.e. a substantial upshift of the CO mode and a substantial downshift of the highest energy, Y8a ring stretching mode.10,52 In tyrosine containing peptides and proteins, shifts in the frequency of adjacent amide vibrations are also observed when the aromatic ring is oxidized. The frequencies reflect the primary sequence and are observed in the reaction induced FT-IR spectrum.47,52,53
Figure 3.

Schematic of the Raman scattering event and of ring stretching (Y8a) vibrational modes, occurring in tyrosine and tyrosyl radical.10
Overview of UV resonance Raman spectroscopy
The Raman spectrum (Figures 3 and 4) reflects inelastic scattering and is induced by a laser probe beam.54 The scattered light is filtered to remove the incident laser energy (Rayleigh line) and dispersed with a grating onto a charge-coupled device (CCD) (Figures 3 and 4). The Raman scattering differs from the incident laser probe by an energy increment, which corresponds to the energy of a molecular vibration. Stokes Raman scattering is observed at decreased energies, compared to the incident laser frequency.
Figure 4.

Schematic of UV resonance Raman spectrometer51 and the vibrational difference method used to study redox active tyrosines. A spectrum acquired at low UV probe power, representing the singlet (light blue), is subtracted from a spectrum acquired at high power (orange), to yield the difference spectrum: radical-minus-singlet (dark blue).
An advantage of Raman spectroscopy is that dilute biological solutions can be studied, because water has a weak Raman signal. Fluorescence from aromatic groups and pigments can be troublesome in Raman spectroscopy, but the use of UV probe wavelengths avoids these background signals by excitation on the high energy side of the electronic transition.54 Through resonance enhancement, UV probe wavelengths select for contributions from aromatic species in proteins.55,56 These species include tyrosine and tryptophan and their corresponding radicals {for recent examples, see 57–61}. To avoid UV damage, a flowing sample is employed. We recently described a micro-flow device, which can be employed with a Raman microprobe to examine 1 ml sample volumes.51 A microscope objective focuses the Raman probe beam on the flowing sample, which forms a jet, and then collects scattered photons in a back-scattering experiment (Figure 4). Difference spectroscopy can be used to study the redox-active tyrosines in model compounds, such as the peptide maquette. In this technique, first described by ref,49 use of high UV probe power generates the radical; use of low UV powers interrogates the structure of the singlet state. The data can then be subtracted, to generate a difference spectrum, presenting the frequency and intensity changes associated with the oxidation reaction. The UV resonance Raman associated with the tyrosine radical in RNR was recently reported.60 This spectrum was obtained by subtraction of data acquired on two different samples, one in which Y122 had been reduced (met-RNR) and one which contained the tyrosyl radical.
Summary of PCET and YD; EPR studies of YD• recombination
We used a proton inventory and EPR spectroscopy to define the PCET pathways for YD (Figure 5).62 Our previous FT-IR spectroscopic studies suggested that a proton is transferred between YD• and His189 in the D2 polypeptide when YD is oxidized and reduced.63 In recent EPR studies, we used 2H2O solvent exchange to demonstrate that YD• PCET mechanism differs at high and low pH,64 consistent with a change from a proton first (PTET) mechanism at low pH to a coupled proton electron transfer (CPET) at high pH. We then utilized the proton inventory technique to study the mechanism at high pH.62 Interestingly, hypercurvature was observed in the proton inventory, demonstrating that multiple protons must be transferred on two different pathways (Figure 5). The pathways were proposed to involve a hydrogen-bonded network including three or more water molecules and a nearby hydrogen-bonded histidine.
Figure 5.

Two possible multi-proton pathways (A and B) for PCET and YD. Pathway B involves a hydrogen bonded chain of water molecules, of which two are shown. This model with two competing pathways, one involving more than three protons, was predicted by a proton inventory experiment at high pH. Reprinted from ref 62.
Summary of PCET and YZ; EPR studies of YZ• recombination
The OEC cycles among five oxidation states, called the Sn states, where n refers to the number of oxidizing equivalents stored.65 Laser flashes can be used to step PSII through the S state transitions. Dark adaptation after a single laser flash prepares PSII in the S1 state. This methodology can be coupled with a freeze quench to prepare PSII in the S2 (one flash), S3 (two flashes), and S0 (three flashes, because S4 converts to S0 in the dark). The transient species, YZ•, is a neutral radical3 and decays on the microsecond to millisecond time scale by reduction from the OEC.43 To slow YZ• reduction and increase the lifetime of the radical, many previous spectroscopic and biochemical studies of YZ• have been performed in PSII preparations, from which the OEC has been removed. Similarly, in many site-directed mutations around the OEC, the OEC is not assembled {reviewed in 16}. The 1.9Å PSII structure predicts a network of hydrogen bonds that connect YZ, His190 in the D1 polypeptide, and water molecules.20 Removal or loss of the OEC will disrupt this network, which plays a potentially critical biological role. Hydrogen bonds are known to facilitate CPET reactions in model compounds.11,66–69 Therefore, to probe the biologically relevant PCET pathway to YZ•, experiments must be conducted in the presence of the OEC.
Recently, we reported that YZ• recombination in the S2 state proceeds by a pH independent CPET mechanism.70 Significantly, this work was conducted on YZ• in the presence of the OEC and in a defined oxidation state of the metal cluster. We used freeze quench techniques to block transfer from the OEC to YZ•, trapping PSII in the S2 state. The YZ• state gives rise to an X-band EPR signal. YZ• was generated by a laser flash, and the amplitude of its signal was monitored to determine the kinetics of YZ• decay. We varied the pH and found that there was minimal pH dependence for this recombination reaction. A significant solvent isotope effect was observed, but this isotope effect was also independent of pH. This is in contrast to our results on recombination of YD, where a significant rate acceleration was observed with decreasing pH, and the solvent isotope effect exhibited pH dependence. To explain the results concerning YZ, we proposed that the network of strong hydrogen bonds around YZ facilitates a CPET reaction, even at low pH. This may be of significance for physiological activity in the chloroplast, avoiding deleterious charge recombination reactions at the low pH of the thylakoid lumen.70 This approach can be used to probe the hydrogen-bonding environment of YZ in other accessible S states, providing a picture of redox interactions between YZ and the OEC.
Comparison of PSII, RNR, and the beta hairpin; energetics of PCET
Because tyrosine is difficult to oxidize, estimates of tyrosine midpoint potential are difficult. Potentiometric titrations of tyrosine are often irreversible, necessitating the use of corrections to give midpoint potentials.71 However, in some cases, these correction factors have been reported to be small.18,72 The midpoint potential of tyrosinate has been reported as 0.7 mV (SHE) at pH 11, with a potential that increases linearly over the pKa. The slope is expected to be 59 mV per pH unit at room temperature because a proton transfer is coupled with the oxidation reaction.71 In agreement, the peak potential has been reported as 1.0 V at pH 5.0 for tyrosine.19 Reversible potentiometric measurements have been acquired for some phenol model compounds, particularly in which the phenol and hydrogen bond partner form an intramolecular bond.11 This work has shown that hydrogen bonding lowers the midpoint potential.11 Pi-cation interactions and hydrogen bonding interactions have also been shown to lower the potential in the beta hairpin maquette, peptide A. For example, a 50 mV potential difference was observed between the beta hairpin peptide and tyrosine solutions at low pH, suggesting that the cross-strand interaction with the protonated histidine lowers the potential. This was corroborated by substitution of the histidine with cyclohexylalanine or valine.18 A 50 mV decrease in potential was also observed when pi-cation interaction and the hydrogen bond were removed.19 Differential pi-cation interactions may contribute to the potential difference observed between YZ and YD.16
For the tyrosine radicals in RNR and PSII, the midpoint potential has been estimated as 1.0 V73 and 1.2 V39 respectively, although a detailed pH dependence has not been reported. The reported 200 mV difference between the YZ and Y122 potential may be due to the proximity of YZ to the OEC.74 When the OEC is removed, the midpoint potential of YZ was reported to decrease by ~120 mV. This may be due to removal of the OEC calcium, which is 5 Å from YZ and linked to YZ by an activated, bound water molecule. This water molecule is predicted to make a strong hydrogen bond (2.9 Å).20 This interaction may destabilize the YZ• state and raise the potential of the YZ•/YZ couple. The midpoint potential of the H2O/O2 couple is ~0.9 mV at pH 5, the likely lumenal pH under physiological conditions.75 Therefore, the driving force for water oxidation by YZ is small. One role of OEC calcium may be to activate YZ and to drive the water oxidation reaction at the low pH of the thylakoid lumen. Other roles for calcium have been suggested.76,77
The midpoint potential of YD has been reported to be substantially lower (0.7 V78) when compared to Y122 and YZ, which again may be attributable to distance (20 Å) from a metal cluster.20,79 There appears to be no correlation between reported midpoint and peak potentials and the lifetimes of the radicals, which vary from the days and hours (YD, PSII and Y122, RNR) to the microsecond time scale (YZ, PSII and Y5, peptide A).16
Comparison of PSII, RNR, and the beta hairpin: kinetics and the access of water
Figure 2 compares the hydrogen bonding interactions of Y5, YZ, YD, and Y122. In the peptide A NMR structure, Y5 is water accessible and hydrogen bonded to the ε-NH of Arg 16. In the X-ray structures of RNR and PSII, the position of assigned water molecules distinguish YZ, YD, and Y122. The X-ray structures are reported at 1.4 Å (beta)79 and 1.9 Å (PSII).20 A water molecule near Y122 is assigned at 3.8 Å; this water is hydrogen bonded to D84, and is bound to the diferric cluster. For comparison, there is a 2.6 Å distance between YZ and the nearest assigned water molecule. In addition, YZ is in a putative strong hydrogen-bonding network including other assigned water molecules and His190. Thus, the environment of Y122 is expected to be more hydrophobic, compared to the YZ environment. Site specific substitution of nitrotyrosine at position 122 provided evidence for this increase in hydrophobicity as a >2 pH unit increase in the pKa of the substituted residue, relative to nitrotyrosine in solution.34 It has been proposed that the iron-bound water molecule may provide a proton to Y122 during the catalytically relevant PCET reaction and may be activated for proton transfer by formation of the quaternary complex. The mechanism of this activation reaction has yet to be determined, but it may result in a decreased distance between the bound water and Y122, which facilitates PCET.34 If substantiated, this hypothesis suggests that changes in hydrogen bonding to water regulates catalysis. A similar idea has been supported by time resolved infrared studies of bacteriorhodopsin, in which transient formation of linear water molecules has been proposed to be critical in light-induced proton transfer.80
Electron density has been assigned to a water molecule near YD; YD is also hydrogen bonded to His189 in the D2 polypeptide.20 The assigned, water molecule has only partial occupancy but is within hydrogen bonding distance (2.7 Å). The electron density may reflect two positions of a single water molecule, one of which is hydrogen bonded to YD (2.73 Å) and one of which is not hydrogen bonded to YD (4.3 Å). The water molecule in the second position may be connected in a hydrogen bonding network, involving Arg 180 of the D2 polypeptide and then a number of water molecules in an exit pathway.20 A proton inventory suggested two pathways for YD• proton transfer, one of which involves multiple protons.62 Based on the structure, the multiple proton pathway most likely involves the hydrogen bonded water chain (Figure 5). Transient access of water molecules may occur, as has been proposed in bacteriorhodopsin.80 Previously, chemical complementation experiments suggested that the YD pocket is relatively accessible.63
To investigate the effect of the proposed hydrogen-bonding network on PCET, we conducted comparative studies of YZ• and YD• recombination. The recombination of YZ• in the S2 state was three orders of magnitude faster than the recombination of YD•.70 Increased access of water and increasing hydrogen bonding to YZ may be correlated with changes in the rate of PCET, when YD and YZ are compared. Comparison to the environment of Y122• in RNR, which is even more stable than YD•, supports a correlation between predicted water access and a decreased lifetime for the radical state.
UV resonance Raman spectra of the radical and singlet states
UV resonance Raman spectroscopy records the vibrational spectrum of the tyrosine in the oxidized and reduced forms. In Figure 6A, we present a UV resonance Raman difference spectrum, associated with the generation of the tyrosyl radical in a flowing tyrosinate solution at pH 11. The radical was photoinduced by the 244 nm probe beam, and the data correspond to an ET reaction. Note that 229 nm excitation was not effective in generating the radical. The difference spectrum in Figure 6A reflects shifts in frequency or amplitude when the tyrosine is oxidized. Positive bands are unique bands of the radical and are observed at 1408, 1516, and 1572 cm−1. These correspond to CH bending/CC stretching, CO stretching (Y7a), and CC ring (Y8a) stretching modes of the radical.10,81 Negative bands are unique bands of the singlet and are observed at 1174, 1207, and 1602 cm−1, assignable to CH bending (Y9a), tyrosine Cring-CH2- (Y7a), and ring stretching (Y8a), respectively.10 See Table 1 for a summary of the observed frequencies and assignments.
Figure 6.

UV resonance Raman spectra of tyrosyl radicals in (A) tyrosinate, (B) a tyrosinate-histidine dipeptide, (C) peptide A, and (D) ribonucleotide reductase (RNR). The data are difference spectra reflecting: radical-minus-singlet, plotted against the Raman shift in cm−1. The spectra in (A–C) correspond to the ET reaction shown. These data were obtained with a 244 nm probe beam, and the radical was photoinduced by increasing the UV power. The difference spectra were acquired by subtracting a low power scan (340μW, 16 scans, 240s/scan) from the high power scan (3.4 mW, 8 scans, 120 s/scan). The amplitude of low intensity scan was multiplied by a factor of 5 before subtraction. The samples were suspended in a 2H2O buffer containing 5 mM sodium borate, p2H 11. In (D), the spectrum corresponds to the PCET reaction shown. The RNR difference spectrum was obtained chemically, by subtracting data obtained from a met-beta (hydroxyurea-treated) sample, which lacks Y122•, from data obtain on a control (untreated) beta sample, which contains Y122•. The data were acquired at pH 7.6 with 229 nm excitation (7 scans, 180s/scan). See ref 60 for more experimental details.
Table 1.
Vibrational frequencies (cm−1) of tyrosyl radical and singlet states
| Radical | Singlet | ||||
|---|---|---|---|---|---|
| Sample | CO stretch Y7a | Ring stretch Y8a | CH bend Y9a | Cring-CH2-Y7a | Ring stretch Y8a |
| Tyr | 1516 | 1572 | 1174 | 1207 | 1602 |
| Tyr-His | 1516 | 1569 | 1174 | 1207 | 1602 |
| Peptide A | 1516 | 1569 | 1174 | 1207 | 1602 |
| RNR | 1499 | 1556 | 1170 | 1199 | 1608 |
Interpretation of singlet spectrum, comparison to model compounds predicts dependence on hydrogen bonding, conformation, and protonation state
For the singlet, model compound data are available, which reveal the factors that influence the vibrational frequencies. For example, the frequencies of the Y7a 1207 (negative) and the Y9a 1174 (negative) cm−1 band are known to be sensitive to hydrogen bonding and conformation, respectively, and the frequency of the Y8a ring stretching (1602 cm−1) mode is known to be sensitive to protonation state {see ref 60 and references therein}. The Y7a Cring-CH2- stretch (1207 cm−1) band acts as a hydrogen bond sensor in proteins, because the normal mode has a ∂COH component. For model tyrosine in solution, this band is observed at ~1210 cm−1 in non-hydrogen bonding or for proton accepting, hydrogen bonded tyrosine and at ~1205 cm−1 in proton donating, hydrogen bonded tyrosine.82 The Y9a band is conformationally sensitive and has been reported to downshift to 1170 cm−1 as the Y–OH hydrogen swings to an out-of-plane configuration.82
Interpretation of the radical spectrum, comparison to model compounds predicts effects of hydrogen bonding
The phenoxyl radical Y7a CO and Y8a ring stretching vibrations have been reported previously at 1502 and 1552 cm−1 in solution.49 In an argon matrix, without hydrogen bonding interactions, the phenoxyl radical bands were reported at 1481 and 1550 cm−1.83 This comparison suggests that a substantial downshift (21 cm−1) of the CO vibration is consistent with a non-interacting environment, while the frequency of the ring stretch is not significantly altered (2 cm−1).
Interpretation of the singlet and radical spectrum, normal mode calculations predict conformation dependence
The conformation dependence has also been described in previous computational studies.10,81 In ref10, vibrational frequencies were calculated for several conformations of the singlet and the radical using B3LYP analysis. The singlet was modeled as an anion (negatively charged carboxylate), with a protonated phenolic oxygen. Deprotonated phenolic forms (the dianion) gave positive eigenvalues for some of the occupied orbitals. The radical was also modeled as an anion. The relevant conformations were determined by performing constrained geometry optimizations at 10° increments over the backbone (C-Cα-Cβ-C1′) and ring (Cα-Cβ-C1′-C2′) dihedral angles. Several unconstrained geometry optimizations were then run near each minimum. Unique minima were then reoptimized, and vibrational analysis was performed. The results predicted that there are two stable conformers of the singlet (A and B), with ring dihedral angles of −102° and −110° (Figure 7). The C conformer, with a ring dihedral of 147°, was predicted to be 1.3–2.0 kcal/mol higher in energy. Subsequent ESEEM studies indicated that the B singlet state was the predominate state frozen in at pH 11 in powders.84 The A and B states of the singlet are similar in energy (0.1–0.6) (Figure 7), but the B conformer has a larger, predicted dipole moment (7.2D for B, compared to 6.3 D for A). Solvent stabilization would therefore favor the B singlet state.
Figure 7.

Ring {r} and backbone {bb} dihedral angles, plus Y9a (singlet), Y7a (radical), and Y8a (radical) vibrational frequencies, for the lowest energy conformers of tyrosinate and tyrosyl radical.10
For the radical, the lowest energy conformation was predicted to be A, with a ring dihedral angle of 85°C (Figure 7). The B, C, and D conformers were substantially higher in energy (2.5 kcal/mole for B, 2.8–2.9 kcal/mole for C and D).10 The A radical conformer would be expected to be the predominant species in solution. Therefore, with reduction in solution, a change in conformation is expected, with a transition from the B state (singlet) to the A state (radical) (Figure 7).
Vibrational frequencies were then predicted for the different singlet conformers. The UVRR active Y9a (1174 cm−1) band exhibited conformation dependence, with the frequency changing 16 cm−1 when the A and C conformers were compared (Figure 7). The Y8a ring stretching mode, which overlapped with a NH2 bending mode, exhibited less conformation sensitivity (2 cm−1).10 This result agrees with the expectations derived from model compound studies (discussed above).
For the radical, this approach was used to predict the four lowest energy conformers and the vibrational frequencies of the CO (Y7a) and ring stretching (Y8a) modes (Figure 7). The results predicted the expected substantial decrease in frequency of both modes, when compared to the singlet state. In addition, conformational dependence was described. DFT calculations predicted downshifts of 9 (CO, Y7a) and 4 cm−1 (ring stretching, Y8a), respectively, when the A and B conformers were compared (Figure 7). The transition from the A to the C conformer was accompanied by an 11 cm−1 upshift of Y7a and a 7 cm−1 upshift of Y8a. The transition from the A to D conformer was accompanied by a 6 cm−1 downshift of Y7a and a 3 cm−1 downshift of Y8a.
UV resonance Raman spectra of a tyrosine-histidine dipeptide and peptide A
In Figure 6B and C, the difference spectra of the tyrosyl radical was generated in a tyrosine histidine dipeptide and in the beta hairpin maquette. The data correspond to an ET reaction. A 244 nm probe was required to generate the radical in peptide A. The spectra are similar to those obtained from tyrosinate (Figure 6A). From this, we conclude that conformation and hydrogen bonding of the radical and singlet state are similar, when tyrosinate, the dipeptide, and peptide A are compared.
UV resonance Raman spectrum of Y122 in RNR
Figure 6D presents the UV resonance Raman spectrum associated with reduction of Y122• in the beta subunit of RNR. The data correspond to a PCET reaction. To enhance Raman scattering, a 229 nm probe beam was employed, instead of the 244 nm band used in Figure 6A–C. Visible probe wavelengths are associated with a much higher fluorescence background from the protein. The assignments are summarized in Table 1. Significant differences are observed when these frequencies are compared to the models in Figure 6A–C, showing that the UV spectrum is a sensitive probe of the protein environment. The RNR spectrum exhibits negative bands at 1608, 1199, and 1170 cm−1 from the reduced tyrosine, Y122. Two positive bands are observed at 1499 and 1556 cm−1. The 1499 cm−1 band shifts −18 cm−1 with 2H4 ring isotopic labeling.47 This isotope shift agrees with predictions from DFT calculations on the same isotopomer, confirming assignment to the CO vibration.10 A 1499 cm−1 band was also observed in a visible Raman study of E. coli RNR.85
Considering first the negative bands from the RNR singlet state, when compared to the tyrosine singlet, Y7a and Y9a are downshifted. The band at 1199 cm−1 is consistent with Y122 forming a hydrogen bond, as predicted in the X-ray structure.22 The band at 1170 cm−1 is consistent with the out-of-plane configuration for the Y–OH hydrogen, also as predicted in the X-ray structure. 22 The negative 1608 cm−1 band is assigned to the Y8a mode of the singlet. Y122 is protonated in the reduced state,34 and weakly hydrogen bonded to D84.22 However, the observed 1608 cm−1 frequency (Table 1) is between the frequency expected for a protonated (1614 cm−1) and a deprotonated (1606 cm−1) tyrosine.82
Considering next the positive bands from the RNR radical state, the CO and ring stretching vibrational bands are observed. While there is no X-ray structure of the Y122• containing form of RNR, it has been reported that the radical is not hydrogen bonded.86 When compared to tyrosine models, the Y7a CO and Y8a ring stretching modes of Y122• are downshifted, by 17 and 9 cm−1 respectively. In the experiments of ref 83, a 21 cm−1 downshift of the phenoxyl radical Y7a was observed in an argon matrix, but the phenoxyl radical Y8a ring stretching vibration was downshifted by only 2 cm−1. Therefore, an additional interaction is necessary to explain the downshifted Y8a modes of Y122•.
Conformational dependence is predicted to give up to ~10 cm−1 shifts of Y8a and ~20 cm−1 shifts of Y7a in the tyrosyl radical10 (Figure 6). The UV Raman spectra of tyrosine, the dipeptides, and peptide A (Figure 6A–C) were obtained in solution at room temperature. These spectra are expected to represent the lowest energy A conformer of the radical (Figure 6A–C). In RNR, Y122• is constrained by non-covalent contacts and may be generated in a higher energy conformation. Examining the predicted vibrational frequencies in ref10, we find that conformational selection to give the next higher energy B conformer explains the downshifted CO and ring stretching modes observed in Figure 6D. The B conformer is predicted to be 2.5 kcal/mole higher in energy, corresponding to an energy storage equivalent to one hydrogen bond. This energy could be used to drive conformational rearrangement during PCET reactions.
We can describe the effects of this change in conformation in RNR, starting with the structure of singlet Y122.22 The X-ray coordinates predict a ring dihedral angle of −47° and a backbone dihedral of −178° for the singlet. On the calculated potential energy surface, this conformation is closest to the A conformer.10 Modeling the conformational rotation to the B form gives dihedral angles of 80° and −70° and shifts the radical out of hydrogen bonding distance from D84 (Figure 8). Therefore, this conformational change could be important in control of PCET.
Figure 8.

Structure22 of singlet Y122 (blue) and predicted structure of Y122• radical (green) based on the observed CO and ring stretching frequency, which are consistent with the B conformer.10 The conformational change from the B conformer (radical, green) to A conformer (singlet, blue) is predicted to rotate Y122 (singlet) into hydrogen bonding distance with Asp 84 (D84). The blue spheres represent the iron cluster. Distances between D84 and Y122 are in dotted lines and in Angstroms. Ring and backbone (Bckbn) angles for the radical (green) and singlet (blue) are shown.
Comparison of mouse and E. coli tyrosyl radicals
The proposal that Y122• is in a conformationally strained state is supported by comparison of the Y• radical in the mouse and E. coli subunits. The essential tyrosyl radical in the mouse β subunit is Y177.87 This radical exhibits a significantly shorter lifetime in the presence of reductants, when compared to the E. coli radical {reviewed in 34,87}. The radical has been reported to be hydrogen bonded in mouse RNR.88 A previous visible Raman spectrum has been obtained.89 This experiment revealed that the Y177• CO vibrational frequency is at 1516 cm−1, with a 6 cm−1 downshift in 2H2O, consistent with hydrogen bonding to the radical. This is in contrast to Y122•, which has been reported to be non-hydrogen bonded86 and has a 1499 cm−1 CO vibration (Figure 6D). The mouse CO frequency (1516 cm−1) is the same as tyrosinate and our model peptides (Figure 6A–C). From this, we conclude that the Y177• radical in the mouse beta subunit is hydrogen bonded and in the lowest energy A conformer. Previously, EPR studies have detected a small change in β-methylene proton dihedral angle when mouse and E. coli RNR are compared.88
Future directions
The central role of RNR in cell proliferation, DNA synthesis, and repair makes it an important target in anticancer and antiviral therapies.90 One drug is a radical scavenger, hydroxyurea (HU), used in the treatment of melanoma, leukemia, and cervical cancer.91 HU has also shown promise in the treatment of HIV.92 HU reduces Y122•93 in a clinically significant reaction94 but the pathway(s) of this reaction is not known. To be effective, HU must be used in high concentration and then has significant side effects. A design of more selective radical scavenging inhibitors would be aided by a comparison of E. coli RNR with the human RNR proteins. Like E. coli and mouse RNR, human RNR is a class 1a enzyme and contains a tyrosyl radical, (Y176) and an iron cluster. However, there are differences between human and E. coli RNR in oligomeric state, sequence, and structure.95–98 Two forms of the beta subunit are produced in human cells.99 One, human beta, is expressed in a S phase dependent fashion, while the second, p53 beta, is activated by p53, a tumor suppressor protein {reviewed in 33}. p53 beta is produced in response to DNA damage,100,101 is responsible for DNA synthesis in proliferating cells,102 and may be important in mitochondrial DNA replication.103,104 The two forms of the human beta subunit share ~80% sequence similarity, and their structures have been determined.33 X-ray structures, EPR studies, and site-directed mutagenesis suggest that there are differences in hydrogen bonding and water accessibility around the tyrosyl radical,105 which could be important in reactions with radical scavengers.106 Both subunits produce an EPR detectable tyrosyl radical.105,106 Figure 9 illustrates some of the structural differences between the E. coli (Figure 9A), human (Figure 9B), and human p53-inducible (Figure 9C) beta subunits.
Figure 9.

Comparison of the environments, near the redox active tyrosines, in (A) E. coli, (B) human, and (C) human p53-induced RNR (beta subunit). The structures of the singlet state are shown. The blue and red spheres represent iron and water/oxygen. Distances are in black dotted lines and in Angstroms. The resolutions of the solved X-ray structures are (A) 1.4 Å79 (B) 2.8 Å33 and (C) 2.8 Å.33
The lifetimes of the tyrosyl radicals are different when human and E. coli RNR are compared. E. coli Y122• persists for days,34 but the tyrosyl radicals from mammals exhibit much shorter lifetimes.90 As shown in Figure 9, solvent accessibility, which is governed by the hydrophobic environment, may be responsible for the observed radical stabilities. Another structural difference is that the conserved aspartate residue (D84 in E. coli) that forms hydrogen bonds with E. coli Y122 in the reduced state. In p53 beta, this residue is substituted with a glutamate (Figure 9C). The metal ligation of the diiron centers is also predicted to be significantly different (Figure 9A–C). In addition, there is metal center heterogeneity in the X-ray structures of the human beta subunits.33
UV resonance Raman spectroscopy can be applied to these different forms of RNR to test the hypothesis that access of water and conformation are critical in controlling radical lifetime. A comparative study of the E. coli, human, and human p53-inducible beta subunits will be of great value, because the structures show differences in the placement of water molecules (Figure 9) in the vicinity of the stable tyrosyl radical.33 Differences in structure and sequence may lead to alterations in radical scavenger pathways.90 The human p53-inducible beta subunit provides an opportunity for targeted cancer therapies, because it is differentially expressed, when compared to human beta.107
Acknowledgments
The research results from the Barry group were supported by NIH GM43273 (B.A.B.). The authors thank Brandon Polander, Ashley Zuniga, and Tina Dreaden for helpful discussions.
Biographies
Bridgette Barry is a professor in the School of Chemistry/Biochemistry at Georgia Tech. She received her A.B. from Oberlin College, her Ph.D. from UC Berkeley, and she was a postdoctoral fellow at Michigan State University. Her research focuses on mechanisms of long distance electron transfer and oxidative damage/repair in proteins.
Jun Chen received his Ph.D. from Dalian Institute of Chemical Physics. He was a postdoctoral fellow at The University of Tokyo and at Georgia Tech. Currently, he is an assistant professor at Dalian Institute of Chemical Physics in China, and his research focuses on water splitting by hybrid photosystems.
James Keough graduated from Georgia Tech in 2006 with a B.S. in chemistry and is currently conducting graduate studies in the Barry group. His research interests include electron paramagnetic resonance spectroscopy, proton-coupled electron transfer mechanisms, photosystem II, and incorporation of non-natural amino acids into cyanobacteria.
David Jenson studied chemistry at Virginia Commonwealth University and obtained his Ph.D. from Georgia Tech in the Barry group. Currently, he is laboratory instructor for undergraduate analytical and physical chemistry in the School of Chemistry and Biochemistry at Georgia Tech. His research interest is in the mechanism of biological electron transfer.
Adam Offenbacher graduated with a B.S. from Ohio Northern University in 2005 and a Ph.D. from Georgia Tech, where he conducted research on ribonucleotide reductase in the Barry group. He is currently a postdoctoral fellow at Georgia Tech and is studying PCET mechanisms in photosystem II.
Cynthia Pagba obtained a degree in Chemistry from the University of the Philippines (UP). She received her Ph. D. from Rutgers University and did postdoctoral work at the UC-Davis Center for Biophotonics. She is currently studying proton-coupled electron transfer and biomimetic peptides in the Barry group.
References
- 1.Stubbe J. Ribonucleotide Reductases: Amazing and Confusing. J Biol Chem. 1990;265:5329–5332. [PubMed] [Google Scholar]
- 2.Barry BA, Babcock GT. Tyrosine Radicals are Involved in the Photosynthetic Oxygen-Evolving system. Proc Nat Acad Sci. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boerner RJ, Barry BA. Isotopic Labeling and EPR Spectroscopy Show that a Tyrosine Residue is the Terminal Electron Donor, Z, in Manganese-depleted Photosystem II Preparations. J Biol Chem. 1993;268:17151–17154. [PubMed] [Google Scholar]
- 4.Kulmacz RJ, Ren Y, Tsai AL, Palmer G. Prostaglandin H synthase: Spectroscopic Studies of the Interaction with Hydroperoxides and with Indomethacin. Biochemistry. 1990;29:8760–8771. doi: 10.1021/bi00489a037. [DOI] [PubMed] [Google Scholar]
- 5.Whittaker MM, Whittaker JW. Tyrosine-derived Free Radical in Apogalactose Oxidase. J Biol Chem. 1990;265:9610–9613. [PubMed] [Google Scholar]
- 6.Whittaker MM, Kersten PJ, Nakamura N, Sanders-Loehr J, Schweizer ES, Whittaker JW. Glyoxal Oxidase from Phanerochaete chrysosporium is a New Radical-copper Oxidase. J Biol Chem. 1996;271:681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- 7.Chouchane S, Girotto S, Yu S, Magliozzo RS. Identification and Characterization of Tyrosyl Radical Formation in Mycobacterium tuberculosis Catalase-peroxidase (KatG) J Biol Chem. 2002;277:42633–42638. doi: 10.1074/jbc.M207916200. [DOI] [PubMed] [Google Scholar]
- 8.Dixon WT, Murphy D. Determination of the Acidity Constants of Some Phenol Radical Cations by Means of Electron Spin Resonance. J Chem Soc London, Faraday Trans II. 1976;72:1221–1230. [Google Scholar]
- 9.Barry BA, El-Deeb MK, Sandusky PO, Babcock GT. Tyrosine Radicals in Photosystem II and Related Model Compounds. J Biol Chem. 1990;265:20139–20143. [PubMed] [Google Scholar]
- 10.Range K, Ayala I, York D, Barry BA. Normal Modes of Redox-active Tyrosine: Conformation Dependence and Comparison to Experiment. J Phys Chem. 2006;110:10970–10981. doi: 10.1021/jp061503f. [DOI] [PubMed] [Google Scholar]
- 11.Rhile IJ, Markle TF, Nagao H, DiPasquale AG, Lam OP, Lockwood MA, Rotter K, Mayer JM. Concerted Proton-electron Transfer in the Oxidation of Hydrogen Bonded Phenols. J Am Chem Soc. 2006;128:6075–6088. doi: 10.1021/ja054167+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Proton-coupled Electron Transfer: The Mechanistic Underpinning for Radical Transport and Catalysis in Biology. Phil Trans Roy Soc B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moser C, Page C, Farid R, Dutton P. Biological Electron Transfer. J Bioenergetics Biomem. 1995;27:263–274. doi: 10.1007/BF02110096. [DOI] [PubMed] [Google Scholar]
- 14.Gray HB, Winkler JR. Long-range Electron Transfer. Proc Nat Acad Sci. 2005;102:3534–3539. doi: 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dempsey JL, Winkler JR, Gray HB. Proton-coupled Electron Flow in Protein Redox Machines. Chem Rev. 2010;110:7024–7039. doi: 10.1021/cr100182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barry BA. Proton Coupled Electron Transfer and Redox Active Tyrosines in Photosystem II. J Photochem Photobiol B. 2011;104:60–71. doi: 10.1016/j.jphotobiol.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramaniam S, Henderson R. Molecular Mechanism of Vectorial Proton Translocation by Bacteriorhodopsin. Science. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 18.Sibert RS, Josowicz M, Porcelli F, Veglia G, Range K, Barry BA. Proton-coupled Electron Transfer in Biomimetic Peptide as a Model of Enzyme Regulatory Mechanism. J Am Chem Soc. 2007;129:4393–4400. doi: 10.1021/ja068805f. [DOI] [PubMed] [Google Scholar]
- 19.Sibert RS, Josowicz M, Barry BA. Control of Proton and Electron Transfer in de novo Designed, Biomimetic β Hairpins. ACS Chem Biol. 2010;5:1157–1168. doi: 10.1021/cb100138m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal Structure of Oxygen-evolving Photosystem II at a Resolution of 1. 9 Angstrom. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 21.Nordlund P, Sjöberg BM, Eklund H. Three-dimensional Structure of the Free Radical Protein of Ribonucleotide Reductase. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 22.Högbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzlan F. Displacement of the Tyrosyl Radical Cofactor in Ribonucleotide Reductase Obtained by Single-crystal High-field EPR and 1.4-A X-ray Data. Proc Nat Acad Sci. 2003;100:3209–3214. doi: 10.1073/pnas.0536684100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan A, Reichard P. Ribonucleotide Reductases. Annu Rev Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. [DOI] [PubMed] [Google Scholar]
- 24.Cotruvo JA, Jr, Stubbe J. Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair In Vitro and In Vivo. Annu Rev Biochem. 2010 doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stubbe J, Nocera DG, Yee CS, Chang MCY. Radical Initiation in the Class I Ribonucleotide Reductase: Long-range Proton-coupled Electron Transfer? Chem Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 26.Rova U, Goodtzova K, Ingemarson R, Behravan G, Graslund A, Thelander L. Evidence by Site-directed Mutagenesis Supports Long-range Electron Transfer in Mouse Ribonucleotide Reductase. Biochemistry. 1995;34:4267–4275. doi: 10.1021/bi00013a016. [DOI] [PubMed] [Google Scholar]
- 27.Rova U, Adrait A, Pötsch S, Gräslund A, Thelander L. Evidence by Mutagenesis that Tyr370 of the Mouse Ribonucleotide Reductase R2 protein is the Connecting Link in the Intersubunit Radical Transfer Pathway. J Biol Chem. 1999;274:23746– 23751. doi: 10.1074/jbc.274.34.23746. [DOI] [PubMed] [Google Scholar]
- 28.Seyedsayamdost MR, Xie J, Chan CTY, Schultz PG, Stubbe J. Site-specific Insertion of 3-aminotyrosine into Subunit Alpha 2 of E-coli Ribonucleotide Reductase: Direct Evidence for Involvement of Y-730 and Y-731 in Radical Propagation. J Am Chem Soc. 2007;129:15060–15071. doi: 10.1021/ja076043y. [DOI] [PubMed] [Google Scholar]
- 29.Yokoyama K, Uhlin U, Stubbe J. A Hot Oxidant, 3-NO2Y122 radical, Unmasks Conformational Gating in Ribonucleotide Reductase. J Am Chem Soc. 2010;132:15368–15379. doi: 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minnihan EC, Seyedsayamdost MR, Uhlin U, Stubbe J. Kinetics of Radical Intermediate Formation and Deoxynucleotide Production in 3-aminotyrosine-substituted Escherichia coli Ribonucleotide Reductases. J Am Chem Soc. 2011;133:9430–9440. doi: 10.1021/ja201640n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seyedsayamdost MR, Yee CS, Stubbe J. Use of 2,3,5-F(3)Y-beta2 and 3-NH(2)Y-alpha2 to Study Proton-coupled Electron Transfer in Escherichia coli Ribonucleotide Reductase. Biochemistry. 2011;50:1403–1411. doi: 10.1021/bi101319v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlin U, Eklund H. The Ten-stranded beta/alpha barrel in Ribonucleotide Reductase Protein R1. J Mol Biol. 1996;262:358–369. doi: 10.1006/jmbi.1996.0519. [DOI] [PubMed] [Google Scholar]
- 33.Smith P, Zhou B, Ho N, Yuan YC, Su L, Tsai SC, Yen Y. 2.6 A X-ray Crystal Structure of Human p53R2, a p53-inducible Ribonucleotide Reductase. Biochemistry. 2009;48:11134–11141. doi: 10.1021/bi9001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokoyama K, Uhlin U, Stubbe J. Site-specific Incorporation of 3-nitrotyrosine as a Probe of pKa Perturbation of Redox-active Tyrosines in Ribonucleotide Reductase. J Am Chem Soc. 2010;132:8385–8397. doi: 10.1021/ja101097p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang W, Xie J, Varano PT, Krebs C, Bollinger JM., Jr Two Distinct Mechanisms of Inactivation of the Class Ic Ribonucleotide Reductase from Chlamydia trachomatis by Hydroxyurea: Implications for the Protein Gating of Intersubunit Electron Transfer. Biochemistry. 2010;49:5340–5349. doi: 10.1021/bi100037b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Debus RJ, Barry BA, Babcock GT, McIntosh L. Site-specific Mutagenesis Identifies a Tyrosine Radical Involved in the Photosynthetic Oxygen-evolving Complex. Proc Nat Acad Sci. 1988;85:427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vermaas WFJ, Rutherford AW, Hansson Ö. Site-directed Mutagenesis in Photosystem II of the Cyanobacterium Synechocystis sp. PCC 6803: Donor D is a Tyrosine Residue in the D2 Protein. Proc Nat Acad Sci. 1988;85:8477–8481. doi: 10.1073/pnas.85.22.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debus RJ, Barry BA, Sithole I, Babcock GT, McIntosh L. Directed Mutagenesis Indicates that the Donor to P680+ in Photosystem II is Tyr-161 of the D1 polypeptide. Biochemistry. 1988;27:9071–9074. doi: 10.1021/bi00426a001. [DOI] [PubMed] [Google Scholar]
- 39.Metz JG, Nixon PJ, Rögner M, Brudvig GW, Diner BA. Directed Alteration of the D1 Polypeptide of Photosystem II: Evidence that Tyrosine-161 is the Redox Component, Z, Connecting the Oxygen-evolving Complex to the Primary Electron Donor, P680. Biochemistry. 1989;28:6960–6969. doi: 10.1021/bi00443a028. [DOI] [PubMed] [Google Scholar]
- 40.Noren GH, Barry BA. The YF161D1 Mutant of Synechocystis 6803 Exhibits an EPR Signal from a Light-induced Photosystem II Radical. Biochemistry. 1992;31:3335–3342. doi: 10.1021/bi00128a005. [DOI] [PubMed] [Google Scholar]
- 41.Styring S, Rutherford AW. In the Oxygen-evolving Complex of Photosystem II the S0 State is Oxidized to the S1 State by D+ (Signal IIslow) Biochemistry. 1987;26:2401–2405. [Google Scholar]
- 42.Ananyev GM, Sakiyan I, Diner BA, Dismukes GC. A Functional Role for Tyrosine-D in Assembly of the Inorganic Core of the Water Oxidase Complex of Photosystem II and the Kinetics of Water Oxidation. Biochemistry. 2002;41:974–980. doi: 10.1021/bi011528z. [DOI] [PubMed] [Google Scholar]
- 43.Babcock GT, Blankenship RE, Sauer K. Reaction Kinetics for Positive Charge Accumulation on the Water Side of Chloroplast Photosystem II. FEBS Lett. 1976;61:286–289. doi: 10.1016/0014-5793(76)81058-7. [DOI] [PubMed] [Google Scholar]
- 44.Gerken S, Brettel K, Schlodder E, Witt HT. Optical Characterization of the Immediate Donor to Chlorophyll aII+ in O2-evolving Photosystem II Complexes. FEBS Lett. 1988;237:69–75. [Google Scholar]
- 45.MacDonald GM, Steenhuis JJ, Barry BA. A Difference Infrared Spectroscopic Study of Chlorophyll Oxidation in Hydroxylamine Treated Photosystem II. J Biol Chem. 1995;270:8420–8428. doi: 10.1074/jbc.270.15.8420. [DOI] [PubMed] [Google Scholar]
- 46.Ge J, Yu G, Ator MA, Stubbe J. Pre-steady-state and Steady-state Kinetic Analysis of E. coli class I Ribonucleotide Reductase. Biochemistry. 2003;42:10071–10083. doi: 10.1021/bi034374r. [DOI] [PubMed] [Google Scholar]
- 47.Offenbacher AR, Vassiliev IR, Seyedsayamdost MR, Stubbe J, Barry BA. Redox-linked Structural Changes in Ribonucleotide Reductase. J Am Chem Soc. 2009;131:7496–7497. doi: 10.1021/ja901908j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacDonald GM, Bixby KA, Barry BA. A difference FT-IR study of Two Redox-Active Tyrosine Residues in Photosystem II. Proc Nat Acad Sci. 1993;90:11024–11028. doi: 10.1073/pnas.90.23.11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson CR, Ludwig M, Asher SA. Ultraviolet Resonance Raman Characterization of Photochemical Transients of Phenol, Tyrosine, and Tryptophan. J Am Chem Soc. 1986;108:905–912. [Google Scholar]
- 50.Mukherjee A, McGlashen ML, Spiro TG. Ultraviolet Resonance Raman Spectroscopy and General Valence Force Field Analysis of Phenolate and Phenoxyl rRdical. J Phys Chem. 1995;99:4912–4917. [Google Scholar]
- 51.Chen J, Barry B. Ultraviolet Resonance Raman Microprobe Spectroscopy of Photosystem II. Photochem Photobiol. 2008;84:815–818. doi: 10.1111/j.1751-1097.2008.00298.x. [DOI] [PubMed] [Google Scholar]
- 52.Ayala I, Range K, York D, Barry BA. Spectroscopic Properties of Tyrosyl Radicals in Dipeptides. J Am Chem Soc. 2002;124:5496–5505. doi: 10.1021/ja0164327. [DOI] [PubMed] [Google Scholar]
- 53.Vassiliev IR, Offenbacher AR, Barry BA. Redox-active Tyrosine Residues in Pentapeptides. J Phys Chem B. 2005;109:23077–23085. doi: 10.1021/jp054159f. [DOI] [PubMed] [Google Scholar]
- 54.Oladepo SA, Xiong K, Hong Z, Asher SA. Elucidating Peptide and Protein Structure and Dynamics: UV resonance Raman spectroscopy. J Phys Chem Lett. 2011;2:334–344. doi: 10.1021/jz101619f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rava RP, Spiro TG. Selective Enhancement of Tyrosine and Tryptophan Resonance Raman Spectra via Ultraviolet Laser Excitation. J Am Chem Soc. 1984;106:4062–4064. [Google Scholar]
- 56.Rava RP, Spiro TG. Resonance Enhancement in the Ultraviolet Raman Spectra of Aromatic Amino Acids. J Phys Chem. 1985;89:1856–1861. [Google Scholar]
- 57.Chen J, Bender SL, Keough JM, Barry BA. Tryptophan as a Probe of Photosystem I Electron Transfer Reactions: a UV resonance Raman Study. J Phys Chem B Lett. 2009;113:11367–11370. doi: 10.1021/jp906491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shafaat HS, Leigh BS, Tauber MJ, Kim JE. Resonance Raman Characterization of a Stable Tryptophan Radical in an Azurin Mutant. J Phys Chem B. 2009;113:382–388. doi: 10.1021/jp809329a. [DOI] [PubMed] [Google Scholar]
- 59.Schlamadinger DE, Gable JE, Kim JE. Hydrogen Bonding and Solvent Polarity Markers in the UV Resonance Raman Spectrum of Tryptophan: Application to Membrane Proteins. J Phys Chem B. 2009;113:14769–14778. doi: 10.1021/jp905473y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Offenbacher AR, Chen J, Barry BA. Perturbations of Aromatic amino acids are Associated with Iron Cluster Assembly in Ribonucleotide Reductase. J Am Chem Soc. 2011;133:6978–6988. doi: 10.1021/ja107918g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halsey CM, Oshokoya OO, Jiji RD, Cooley JW. Deep-UV Resonance Raman Analysis of the Rhodobacter capsulatus Cytochrome bc1 Complex Reveals a Potential Marker for the Transmembrane Peptide Backbone. Biochemistry. 2011;50:6531–6538. doi: 10.1021/bi200596w. [DOI] [PubMed] [Google Scholar]
- 62.Jenson D, Barry BA. Proton-coupled Electron Transfer in Photosystem II: Proton Inventory of a Redox Active Tyrosine. J Am Chem Soc. 2009;131:10567–10573. doi: 10.1021/ja902896e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S, Liang J, Barry BA. Chemical Complementation Identifies a Proton Acceptor for Redox-active Tyrosine D in Photosystem II. Proc Nat Acad Sci. 1997;94:14406–14411. doi: 10.1073/pnas.94.26.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jenson D, Evans A, Barry BA. Proton-coupled Electron Transfer and Tyrosine D of Photosystem II. J Phys Chem B. 2007;111:12599–12604. doi: 10.1021/jp075726x. [DOI] [PubMed] [Google Scholar]
- 65.Joliot P, Kok B. Oxygen Evolution in Photosynthesis. In: Govindjee, editor. Bioenergetics of Photosynthesis. Academic Press; New York: 1975. pp. 388–412. [Google Scholar]
- 66.Irebo T, Reece SY, Sjödin M, Nocera DG, Hammarström L. Proton-coupled Electron Transfer of Tyrosine Oxidation: Buffer Dependence and Parallel Mechanisms. J Am Chem Soc. 2007;129:15462–15464. doi: 10.1021/ja073012u. [DOI] [PubMed] [Google Scholar]
- 67.Sjödin M, Irebo T, Utas JE, Lind J, Merényi G, Akermark B, Hammarström L. Kinetic Effects of Hydrogen Bonds on Proton-coupled Electron Transfer from Phenols. J Am Chem Soc. 2006;128:13076–13083. doi: 10.1021/ja063264f. [DOI] [PubMed] [Google Scholar]
- 68.Hammes-Schiffer S, Soudackov AV. Proton-coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J Phys Chem B. 2008;112:14108–14123. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonin J, Costentin C, Louault C, Robert M, Routier M, Savéant JM. Intrinsic Reactivity and Driving Force Dependence in Concerted Proton-electron Transfers to Water Illustrated by Phenol Oxidation. Proc Nat Acad Sci. 2010;107:3367–3372. doi: 10.1073/pnas.0914693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keough J, Jenson DL, Zuniga A, Barry BA. Proton Coupled Electron Transfer and Redox-active Tyrosine Z in the Photosynthetic Oxygen Evolving Complex. J Am Chem Soc. 2011;133:11084. doi: 10.1021/ja2041139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Harriman A. Further Comments on the Redox Potentials of Tryptophan and Tyrosine. J Phys Chem. 1987;91:6102–6104. [Google Scholar]
- 72.Tommos C, Skalicky J, Pilloud DL, Wand AJ, Dutton LP. De Novo Proteins as Models of Radical Enzymes. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 73.Silva KE, Elgren TE, Que L, Jr, Stankovich MT. Electron Transfer Properties of the R2 protein of Ribonucleotide Reductase from Escherichia coli. Biochemistry. 1995;34:14093–14103. doi: 10.1021/bi00043a014. [DOI] [PubMed] [Google Scholar]
- 74.Yerkes CT, Babcock GT, Crofts AR. A Tris-induced Change in the Midpoint Potential of Z, the Donor to Photosystem II, as Determined by the Kinetics of the Back Reaction. FEBS Lett. 1983;158:359–363. [Google Scholar]
- 75.Kramer DM, Sacksteder CA, Cruz JA. How Acidic is the Lumen? Photosyn Res. 1999;60:151–163. [Google Scholar]
- 76.McEvoy JP, Brudvig GW. Water-splitting Chemistry of Photosystem II. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 77.Miqyass M, van Gorkom HJ, Yocum CF. The PSII Calcium Site Revisited. Photosynth Res. 2007;92:275–287. doi: 10.1007/s11120-006-9124-2. [DOI] [PubMed] [Google Scholar]
- 78.Boussac A, Etienne AL. Midpoint Potential of Signal II (Slow) in Tris-washed Photosystem-II particles. Biochim Biophys Acta. 1984;766:576–581. [Google Scholar]
- 79.Nordlund P, Eklund H. Structure and Function of the Escherichia coli Ribonuclease Reductase Protein R2. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 80.Freier E, Wolf S, Gerwert K. Proton Transfer via a Transient Linear Water-molecule Chain in a Membrane Protein. Proc Natl Acad Sci. 2011;108:11435–11439. doi: 10.1073/pnas.1104735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qin Y, Wheeler RA. Density-functional Methods Give Accurate Vibrational Frequencies and Spin Densities for Phenoxyl Radical. J Chem Phys. 1995;102:1689–1698. [Google Scholar]
- 82.Takeuchi H, Watanabe N, Satoh Y, Harada I. Effects of Hydrogen Bonding on the Tyrosine Raman Bands in the 1300–1150 cm−1 Region. J Raman Spec. 1989;20:233–237. [Google Scholar]
- 83.Spanget-Larsen J, Gil M, Gorski A, Blake DM, Waluk J, Radziszewski JG. Vibrations of the Phenoxyl Radical. J Am Chem Soc. 2001;123:11253–11261. doi: 10.1021/ja0113596. [DOI] [PubMed] [Google Scholar]
- 84.McCracken J, Vassiliev IR, Yang EC, Range K, Barry BA. ESEEM studies of Peptide Nitrogen Hyperfine Coupling in Tyrosyl Radicals and Model Peptides. J Phys Chem B. 2007;111:6586–6592. doi: 10.1021/jp071402x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Backes G, Sahlin M, Sjoberg BM, Loehr TM, Sanders-Loehr J. Resonance Raman Spectroscopy of Ribonucleotide Reductase. Evidence for a Deprotonated Tyrosyl Radical and Photochemistry of the Binuclear Iron Center. Biochemistry. 1989;28:1923–1929. doi: 10.1021/bi00430a074. [DOI] [PubMed] [Google Scholar]
- 86.Bender CJ, Sahlin M, Babcock GT, Barry BA, Chandrashekar TK, Salowe SP, Stubbe JA, Lindstrom B, Petersson L, Ehrenberg A, Sjoberg BM. An ENDOR Study of the Tyrosyl Free Radical in Ribonucleotide Reductase fromEscherichia coli. J Am Chem Soc. 1989;111:8076–8083. [Google Scholar]
- 87.Kauppi B, Nielsen BB, Ramaswamy S, Larsen IK, Thelander M, Thelander L, Eklund H. The Three-dimensional Structure of Mammalian Ribonucleotide Reductase Protein R2 Reveals a More-accessible Iron-radical Site than Escherichia coli R2. J Mol Biol. 1996;262:706–720. doi: 10.1006/jmbi.1996.0546. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt PP, Andersson KK, Barra AL, Thelander L, Graslund A. High field EPR Studies of Mouse Ribonucleotide Reductase Indicate Hydrogen Bonding of the Tyrosyl Radical. J Biol Chem. 1996;271:23615–23618. doi: 10.1074/jbc.271.39.23615. [DOI] [PubMed] [Google Scholar]
- 89.Hanson MA, Schmidt PP, Strand KR, Graslund A, Solomon EI, Andersson KK. Resonance Raman Evidence for a Hydrogen-bonded Oxo bBridge in the R2 Protein of Ribonucleotide Reductase from Mouse. J Am Chem Soc. 1999;121:6755–6756. [Google Scholar]
- 90.Shao J, Zhou B, Chu B, Yen Y. Ribonucleotide Reductase Inhibitors and Future Drug Design. Curr Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 91.Saban N, Bujak M. Hydroxyurea and Hydroxamic acid Derivatives as Antitumor Drugs. Cancer Chemother Pharmacol. 2009;64:213–221. doi: 10.1007/s00280-009-0991-z. [DOI] [PubMed] [Google Scholar]
- 92.Zachary KC, Davis B. Hydroxurea for HIV Infection. AIDS Clin Care. 1998;10:26–26. [PubMed] [Google Scholar]
- 93.Karlsson M, Sahlin M, Sjoberg BM. Escherichia coli Ribonucleotide Reductase - Radical Susceptibility to Hydroxyurea is Dependent on the Regulatory State of the Enzyme. J Biol Chem. 1992;267:12622–12626. [PubMed] [Google Scholar]
- 94.Sneeden JL, Loeb LA. Mutations in the R2 Subunit of Ribonucleotide Reductase that Confer Resistance to Hydroxyurea. J Biol Chem. 2004;279:40723–40728. doi: 10.1074/jbc.M402699200. [DOI] [PubMed] [Google Scholar]
- 95.Kashlan OB, Scott CP, Lear JD, Cooperman BS. A Comprehensive Model for the Allosteric Regulation of Mammalian Ribonucleotide Reductase. Functional Consequences of ATP- and dATP-induced Oligomerization of the Large Subunit. Biochemistry. 2002;41:462–474. doi: 10.1021/bi011653a. [DOI] [PubMed] [Google Scholar]
- 96.Qiu W, Zhou B, Darwish D, Shao J, Yen Y. Characterization of Enzymatic Properties of Human Ribonucleotide Reductase Holoenzyme Reconstituted in vitro from hRRM1, hRRM2, and p53R2 Subunits. Biochem Biophys Res Commun. 2006;340:428–434. doi: 10.1016/j.bbrc.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 97.Wang J, Lohman GJ, Stubbe J. Mechanism of Inactivation of Human Ribonucleotide Reductase with p53R2 by Gemcitabine 5′-diphosphate. Biochemistry. 2009;48:11612–11621. doi: 10.1021/bi901588z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, Dealwis CG. Structural Basis for Allosteric Regulation of Human Ribonucleotide Reductase by Nucleotide-induced Oligomerization. Nat Struct Mol Biol. 2011;18:316–322. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. A Ribonucleotide Reductase Gene Involved in a p53-dependent Cell-cycle Checkpoint for DNA Damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 100.Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, Arakawa H. Impaired Function of p53R2 in Rrm2b-null Mice Causes Severe Renal Failure through Attenuation of dNTP Pools. Nat Genet. 2003;34:440–445. doi: 10.1038/ng1212. [DOI] [PubMed] [Google Scholar]
- 101.Chang L, Zhou B, Hu S, Guo R, Liu X, Jones SN, Yen Y. ATM-mediated Serine 72 Phosphorylation Stabilizes Ribonucleotide Reductase Small Subunit p53R2 Protein against MDM2 to DNA Damage. Proc Nat Acad Sci. 2008;105:18519–18524. doi: 10.1073/pnas.0803313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guittet O, Hakansson P, Voevodskaya N, Fridd S, Graslund A, Arakawa H, Nakamura Y, Thelander L. Mammalian p53R2 Protein Forms an Active Ribonucleotide Reductase In Vitro with the R1 Protein, which is Expressed Both in Resting Cells in Response to DNA Damage and in Proliferating Cells. J Biol Chem. 2001;276:40647–40651. doi: 10.1074/jbc.M106088200. [DOI] [PubMed] [Google Scholar]
- 103.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A. Mutation of RRM2B, Encoding p53-controlled Ribonucleotide Reductase (p53R2), Causes Severe Mitochondrial DNA Depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 104.Pontarin G, Ferraro P, Rampazzo C, Kollberg G, Holme E, Reichard P, Bianchi V. Deoxyribonucleotide Metabolism in Cycling and Resting Human Fibroblasts with a Missense Mutation in p53R2, a Subunit of Ribonucleotide Reductase. J Biol Chem. 2011;286:11132–11140. doi: 10.1074/jbc.M110.202283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou B, Su L, Yuan YC, Un F, Wang N, Patel M, Xi B, Hu S, Yen Y. Structural Basis on the Dityrosyl-diiron Radical Cluster and the Functional Sifferences of Human Ribonucleotide Reductase Small Subunits hp53R2 and hRRM2. Mol Cancer Ther. 2010;9:1669–1679. doi: 10.1158/1535-7163.MCT-10-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shao J, Zhou B, Zhu L, Qiu W, Yuan YC, Xi B, Yen Y. In vitro Characterization of Enzymatic Properties and Inhibition of the p53R2 Subunit of Human Ribonucleotide Reductase. Cancer Res. 2004;64:1–6. doi: 10.1158/0008-5472.can-03-3048. [DOI] [PubMed] [Google Scholar]
- 107.Zhang K, Wu J, Wu X, Wang X, Wang Y, Zhou N, Kuo M-l, Liu X, Zhou B, Chang L, Ann D, Yen Y. p53R2 Inhibits the Proliferation of Human Cancer Cells in Association with Cell-cycle Arrest. Mol Cancer Therapuetics. 2011;10:269–278. doi: 10.1158/1535-7163.MCT-10-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]