

Abstract
Aged garlic extract (AGE) is an odorless garlic preparation containing S-allylcysteine (SAC) as its most abundant compound. A large number of studies have demonstrated the antioxidant activity of AGE and SAC in both in vivo—in diverse experimental animal models associated to oxidative stress—and in vitro conditions—using several methods to scavenge reactive oxygen species or to induce oxidative damage. Derived from these experiments, the protective effects of AGE and SAC have been associated with the prevention or amelioration of oxidative stress. In this work, we reviewed different antioxidant mechanisms (scavenging of free radicals and prooxidant species, induction of antioxidant enzymes, activation of Nrf2 factor, inhibition of prooxidant enzymes, and chelating effects) involved in the protective actions of AGE and SAC, thereby emphasizing their potential use as therapeutic agents. In addition, we highlight the ability of SAC to activate Nrf2 factor—a master regulator of the cellular redox state. Here, we include original data showing the ability of SAC to activate Nrf2 factor in cerebral cortex. Therefore, we conclude that the therapeutic properties of these molecules comprise cellular and molecular mechanisms at different levels.
1. Introduction
Garlic (Allium sativum) has been cultivated and used for culinary and medicinal purposes by many cultures for centuries [1, 2]. Even today, garlic cloves are commonly used in Eastern Europe and Asia, whereas garlic supplements are popular in Western Europe and growing in popularity in the US. Garlic is a source particularly rich in organosulfur compounds, which are responsible for its flavor, aroma, and potential health benefits [3]. Recent studies have demonstrated and validated many of the medicinal properties attributed to garlic [4]. Different types of garlic supplements are commercially available, including garlic powders (tablets), garlic oils (capsules), and aged garlic extracts (tablets, capsules, and liquid); each of these sources provide a different profile of organosulfur compounds [5]. One of the better known garlic preparations is aged garlic extract (AGE), which is formed during garlic aging (up to 20 months). During this time, unstable and highly odorous compounds in fresh garlic are converted into more stable and much less odorous compounds [6].
A considerable number of in vivo and in vitro studies have been performed so far in order to test the antioxidant properties of AGE and one of its most abundant organosulfur compounds, S-allylcysteine (SAC). In these studies, different antioxidant mechanisms have been reported, such as their ability to (1) scavenge reactive oxygen (ROS) and nitrogen (RNS) species; (2) increase enzymatic and nonenzymatic antioxidants levels; (3) activate Nrf2 factor; or (4) inhibit some prooxidant enzymes (xanthine oxidase, cyclooxygenase, and NADPH oxidase).
Given that SAC is the most abundant compound in AGE, the present paper brings special attention to the physicochemical characteristics, toxicity, pharmacokinetics, tissue distribution, and metabolism of this compound, as well as on the different antioxidant mechanisms involved in its protective actions in different experimental models of toxicity.
2. Aged Garlic Extract (AGE)
AGE is an odorless product resulting from prolonged extraction of fresh garlic at room temperature; it is highly bioavailable and exerts biological activity in both animals and humans. In AGE, garlic (in 15–20% ethanol) is aged for up to 20 months in stainless steel tanks. The extract is then filtered and concentrated at low temperature [6]. AGE is sold in both dry form and as a liquid containing 10% ethanol [4].
The process of aging gently modifies harsh and irritating compounds from the raw garlic and naturally generates unique and beneficial compounds through both enzymatic and natural chemical reactions (Table 1) [7]. The main changes in AGE during aging process are indicated as follows [4]:
Table 1.
Compositional changes in the AGE.A
| Incubation time (months) | |||||
|---|---|---|---|---|---|
| Compound | 0 | 1 | 3 | 12 | 24 |
| (mg/g dry extract) | |||||
| γ-Glutamyl-S-allylcysteine | 12.7 | 5.8 | 1.1 | 0 | 0 |
| S-Allylcysteine | 0.2 | 5.9 | 7.2 | 7.1 | 7.2 |
| γ-Glutamyl-S-1-propenylcysteine | 15.9 | 3.4 | 0.5 | 0 | 0 |
| S-1-Propenylcysteine | 0.5 | 6.7 | 8.1 | 6.5 | 4.4 |
| S-Allylmercaptocysteine | 0.01 | 0.6 | 1.2 | 1.7 | 1.9 |
ACloves were chopped into pieces (2 × 2 × 1 mm) and placed into 20% ethanol (12 mL/g) in a closed container and stored at room temperature. Synthesized from [4].
complete hydrolysis of the γ-glutamylcysteines to SAC and S-1-propenylcysteine (the content of SAC remains constant after 3 months, but S-1-propenylcysteine steadily decreases);
increase in cystine due to protein hydrolysis and increase in S-allylmercaptocysteine probably due to the reaction of allicin with protein derived cysteine;
initial loss of alliin to thiosulfinates formation (allicin);
complete loss of thiosulfinates after 3 months due to the fact that they are converted into volatile allyl sulfides (diallyl sulfide, diallyl disulfide, diallyl trisulfide), which evaporate almost completely.
Based on typical total SAC content in cloves (41 μmole/g dry wt.), the total SAC content in commercial aged extracts (7.8 μmole/g dry wt. after correcting for 40% excipients) is only 19%, indicating considerable manufacturing losses or the use of cloves with an unusually low content of γ-glutamylcysteines. The commercial aged products are standardized on SAC content, but a specific or even minimum amount has not been declared yet [4].
Recently, new compounds, such as tetrahydro-beta-carbolines (1-methyl-1,2,3,4-tetrahydro-beta-carboline-3-carboxylic acid and 1-methyl-1,2,3,4-tetrahydro-beta-carboline-1,3-dicarboxylic acid) as well as Nα-(1-deoxy-D-fructos-1-yl)-L-arginine, have also been identified in AGE. These compounds increase during the aging process and play an important role as antioxidants [8–10]. Indeed, tetrahydro-beta-carbolines are biologically active alkaloids and are structurally similar to flavonoids. Nα-(1-deoxy-D-fructos-1-yl)-L-arginine is only found in AGE and no other garlic products.
Substantial evidence shows that AGE ameliorates the oxidative damage implicated in aging and a variety of diseases, such as cardiovascular alterations, cancer, stroke, Alzheimer's disease (AD), and other age-related degenerative conditions. In addition, AGE is a commercially available garlic preparation that has been widely studied for its high antioxidant content and its health-protective potential [11–17].
3. S-Allylcysteine (SAC): A Key Compound in Aged Garlic Extract
SAC is formed from γ-glutamyl-S-allylcysteine catabolism (Figure 1) and has been used to standardize commercial AGE [7]. SAC is a white crystalline powder with characteristic odor, it has not hygroscopic ability, and its melting point is 223.3–223.7°C [18]. SAC is a stable compound as it remains unaltered in AGE for up to 2 years [4]. Stored crystal samples show a slight change into a yellowish color, but no transformation or decomposition is observed. Under basic conditions (2 N NaOH, 50°C, 6 days), allylmercaptan and allylsulfide (decomposition products) are observed, suggesting that the C–S bond cleavage occurs. However, under acidic conditions (6 N HCl, 50°C, 5 days), evidence of C–S bond cleavage is not observed. These observations indicate that SAC can be absorbed in gastrointestinal tract after oral administration without any changes [18].
Figure 1.

During the process of aging γ-glutamyl-S-allylcysteine is converted to S-allylcysteine (SAC) by a γ-glutamyltransferase.
3.1. Toxicity
SAC is 30-fold less toxic than other typical garlic compounds such as allicin and diallyldisulfide. The 50% lethal oral dose (LD50) of SAC in female (9.39 g/kg) or male (8.89 g/kg) mice was higher than allicin (female: 0.363 g/kg and male: 0.309 g/kg) and diallyldisulfide (female: 1.3 g/kg and male: 0.145 g/kg) (Reviewed in [6]). Others studies have confirmed the low toxicity of SAC in male mice, since LD50 by oral administration was 8.8 g/kg (Table 2). In male rats, LD50 of SAC by i.p. administration (3.34 g/kg (<20 mM)) is similar to essential L-amino acids such as methionine (29 mM/kg). Gender differences were observed in acute toxicity tests in mice, since LD50 in males was about 1.7 times higher than in females [18].
Table 2.
Toxicity of SAC in mice and rat.A
| LD50 (g/kg body weight)B | |||
|---|---|---|---|
| Oral | i.p. | ||
| Mice | Male | 8.89 | 6.91 |
| Female | 9.39 | 3.65 | |
| Rat | Male | 10.94 | 3.34 |
| Female | 9.50 | 3.34 | |
AAdapted from [18]. B50% lethal dose of a single dose i.p.: intraperitoneal administration.
Negative effects of SAC were observed at high doses (≥500 mg/kg for 1 month, orally) including [18]
decrease of body weight in both genders;
increase of urinary pH in male;
decrease of urinary protein (in female) and urobilinogen (in male) levels. Urobilinogen is a metabolite of bilirubin generated by intestinal bacteria that is reabsorbed. Therefore, these data suggest that SAC may have some effect on the intestinal flora;
increase of serum glucose levels in females, suggesting that SAC induces atrophy of the pancreas and decreases insulin secretion. Atrophy of the pancreatic tissue has been observed only in the females administered with high dosage of SAC;
modifying of renal (decrease blood urea nitrogen and serum creatinine levels) and hepatic (increase serum total cholesterol, protein, lipids, and alkaline phosphatase levels) function in females;
decrease of hematocrit, hemoglobin, mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentration in both genders.
3.2. Pharmacokinetics
In rats, pharmacokinetics of SAC after oral administration shows a three-phase concentration profile: two very fast phases (absorption and distribution) followed by a slow elimination phase. For i.v. administration, SAC pharmacokinetics presents a two-phase concentration profile: a very fast distribution phase and a slow elimination phase [19, 20]. Pharmacokinetic parameters of SAC (100 mg/kg, oral and i.v.) are shown in Table 3.
Table 3.
Pharmacokinetic parameters for oral and i.v. administration of SAC in the serum of rats.A
| Parameter | Oral administration | i.v. administration |
|---|---|---|
| Distribution volume (V d) | 1.2 L | 1.3 L |
| Elimination half-life (T 1/2β) | 2.7 h | 2.6 h |
| Total clearance | 0.3 L/h | 0.3 L/h |
| Peak concentration (C max) | 100.1 mg/L at 30 min | |
| AUC0–t value | 293.5 mg·h/L | 322.6 mg·h/L |
ASynthesized from [19]. AUC0–t : area under the plasma concentration versus time curve from time zero to the last quantifiable concentration.
Oral bioavailability of SAC at 100 mg/kg dose is 91% [19], similar to that reported in other studies where bioavailability was 103.0% in mice, 98.2% in rats, and 87.2% in dogs [20].
In addition, a pharmacokinetic study of SAC in humans has been performed by oral administration of garlic preparation containing this compound. The half-life of SAC in humans after oral administration was more than 10 h, and clearance time was more than 30 h [18]. These results are similar to experimental data obtained in dogs, where the half-life of SAC was about 10 h, and clearance time was more than 24 h, albeit these data were different from other experimental results obtained in mice [20]. Total SAC content in the blood of volunteers at T max is about 450 μg (content on T max, 23 ng/mL plasma; body weight: 65 kg; volume of total blood: 1/3 of body weight), suggesting a high bioavailability in humans [18].
3.3. Tissue Distribution
After oral intake, SAC is easily absorbed in the gastrointestinal tract and can be detected in several tissues up to 8 h after dosage [19, 20]. In rats, after a single oral dosage of SAC (50 mg/kg), the area under the plasma concentration versus the time curve from zero to the last quantifiable concentration (AUC0–t )) is 169.2 mg h/kg, whereas, in tissues, the highest peak concentration is observed in the kidney (C max = 65.7 mg/kg at 10 min). The elimination half-life varies among tissues; for instance, liver shows the longest T 1/2 value (2.2 h) the shorted value (1.2 h) was observed in the brain (Table 4). For i.v. administration, the kidney also exhibits the highest SAC exposure (AUC0–8 h) = 171.9 mg h/kg), whereas the heart shows the longest T 1/2 value (2.5 h) [19].
Table 4.
Pharmacokinetic parameters after a single oral dose of SAC.A
| Parameter | C max | AUC0–t | T 1/2 |
|---|---|---|---|
| Tissue | (mg/kg) | mg·h/kg | (h) |
| Kidney | 65.7 | 169.2 | 2.1 |
| Liver | 58.1 | 103.5 | 2.2 |
| Heart | 43.3 | 118.8 | 2.1 |
| Spleen | 43.3 | 100.6 | 1.9 |
| Lung | 35.1 | 94.3 | 1.3 |
| Brain | 26.7 | 70.7 | 1.2 |
ASAC dose of 50 mg/kg. Adapted from [19].
C max: peak concentration at 10 min; AUC0–t : area under the plasma concentration versus time curve from time zero to the last quantifiable concentration; T 1/2: elimination half-life time.
3.4. Metabolism
N-Acetyl-SAC has been identified as a metabolite of SAC in urine of rats, dogs, and humans [20, 21]. Also, small amounts of SAC (less than 1%) have been observed in urine of rats. These data suggest that the absorbed SAC seems to be metabolized to N-acetyl-SAC by N-acetyltransferase, which is mainly found in liver and kidney. However, it has been shown that, when SAC is almost completely eliminated from the liver, it is readily retained at a comparatively high concentration in the kidney. Thus, it can be speculated that SAC may be transformed into N-acetyl-SAC by N-acetyltransferase in the liver, and, then, a portion of N-acetyl-SAC may be deacylated to SAC by acylase in kidney, followed by its reabsorption [20].
4. Antioxidant Mechanisms Associated with the Protective Effect of SAC and/or AGE
Several reports have shown that AGE and SAC inhibit the oxidative damage implicated in aging and a variety of diseases. Derived of these works, different antioxidant mechanisms have been attributed to these compounds and confirmed. Figure 2 shows the antioxidant mechanism associated with the protective effects of SAC.
Figure 2.
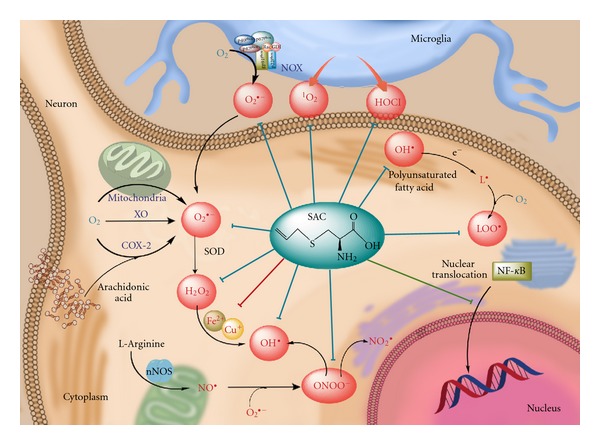
Antioxidant mechanism associated to S-allylcysteine (SAC). SAC can scavenge superoxide anion (O2 •−), hydrogen peroxide (H2O2), hydroxyl radical (OH•), peroxynitrite radical (ONOO−), and peroxyl radical (LOO•) produced in neuronal cells, as well as hypochlorous acid (HOCl) and singlet oxygen (1O2) produced in microglial cells (blue lines). Moreover, SAC also exhibits chelating properties on Fe2+ and Cu2+ ions (red line), hence avoiding Fenton reaction. SAC also inhibits NF-kB translocation into the nucleus (green line), thus preventing apoptotic signaling. COX-2: cyclooxygenase-2, NOX: NADPH oxidase, nNOS: neuronal nitric oxide synthase, SOD: superoxide dismutase, XO: xanthine oxidase.
4.1. AGE and SAC Scavenge Reactive Oxygen Species
SAC contains a thiol group responsible of its antioxidant capacity because this nucleophile can easily donate its proton to an electrophilic species, thereby neutralizing them or making them less reactive.
SAC is the most studied compound of AGE and its antioxidant properties have been reported in several studies. SAC readily prevents lipid [22–26] and protein oxidation [27] and nitration [28], supporting its antioxidant activity. Actually, SAC is known to scavenge superoxide anion (O2 •−) [14, 27, 29], hydrogen peroxide (H2O2) [22, 27, 29–31], hydroxyl radical (•OH) [14, 23, 29, 32], and peroxynitrite anion (ONOO−) [28, 29]. Medina-Campos et al. reported that SAC scavenges hypochlorous acid (HOCl) and singlet oxygen (1O2). Furthermore, these authors compared the scavenging activity of SAC against reference compounds (molecules that scavenge a specific reactive oxygen species) through the IC50 value for each reactive species. SAC scavenged HOCl in a similar manner than lipoic acid, and it was more efficient to scavenge 1O2 than lipoic acid and reduced glutathione [29]. Recently, Maldonado et al. reported that SAC was able to scavenge •OH and peroxyl radical (ROO•) in a concentration-dependent manner, and this effect is reduced when SAC is changed by S-propylcysteine, suggesting that allyl group in SAC is necessary for its scavenging activity [33]. To further support this concept, in our laboratory, we have observed that allyl group is necessary to preserve the scavenging activity of SAC on different reactive oxygen species (unpublished data).
On the other hand, AGE is known to scavenge H2O2 [22, 30] and O2 •− [34, 35]. Consistently, Wei and Lau observed a protective effect of AGE (1–8 mg/mL) in endothelial cells challenged by H2O2 or the O2 •− generator system xantine/xanthine oxidase [36].
Besides SAC, there are other compounds in AGE exhibiting antioxidant properties: (a) S-allylmercaptocysteine scavenges •OH and 1O2 [37]; (b) alliin scavenges •OH [38], O2 •− [32], and H2O2 [22] and inhibits lipid peroxidation [22]; (c) Nα-(1-deoxy-D-fructos-1-yl)-L-arginine scavenges H2O2 [8, 39] and also inhibits the Cu2+-induced low-density lipoprotein (LDL) oxidation and peroxides release during the coincubation of macrophages with oxidized LDL [39]; (d) tetrahydro-beta-carbolines show a strong H2O2 scavenging activity while inhibit 2,2′-azobis(2-amidinopropane) hydrochloride-induced lipid peroxidation and nitrite production induced by lipopolysaccharide in murine macrophages [9, 10].
4.2. AGE and SAC Induce Antioxidant Enzymes and Nrf2 Factor
Hsu et al. evaluated the effect of SAC (1 g/L in drinking water for 4 weeks) on the activity of catalase and glutathione peroxidase in Balb/cA mice. After 4 weeks of treatment, SAC increased glutathione levels in kidney and liver when compared with controls. Moreover, SAC enhanced catalase and glutathione peroxidase activities in kidney and liver [40]. These data suggest that SAC could be acting by recruiting different mechanisms, including radical scavenging and induction of antioxidant enzymes.
Lawal and Ellis reported that AGE prevented death in Cd-treated 1321N1 and HEK293 cells, due to cell AGE reduced Cd-induced lipid peroxidation and LDH leakage, increased GSH levels, expression of the protective enzyme NAD(P)H:quinone oxidoreductase, and the accumulation of the transcription factor Nrf2. The authors suggest that AGE could be beneficial in Cd-induced toxicity, and this protection appears to be mediated via induction of cytoprotective enzymes in a Nrf2-dependent manner. This indicates the AGE potential use as a chemoprevention strategy for Cd toxicity [41].
Kalayarasan et al. reported that SAC (100 mg/kg, i.p. for 3 days) is also able to activate Nrf2 factor in hepatocytes of Wistar rats exposed to chromium [42]. The transcription factor Nrf2 (nuclear factor-E2-related factor 2) is the guardian of redox homeostasis as it regulates basal and inducible expression of antioxidant and cytoprotective genes, providing the level of protection required for normal cellular activities and against various oxidative stress-related pathologies, including ischemic stroke [43–45]. Lee et al. reported the major functional categories of genes responsible for conferring protection against oxidative stress or inflammation [46]. These genes codify for detoxifying enzymes, antioxidant proteins, NADPH-producing proteins, growth factors, defense/immune/inflammation related proteins, and signaling proteins; thus, the activation of Nrf2/ARE (antioxidant response element) pathway could stimulate a synergistic protective effect [47].
Nrf2 is highly expressed in detoxifying organs—such as liver and kidney—and other organs commonly exposed to external environmental conditions—including the skin, lung, and digestive tract—[48], whereas in the brain its levels are low [49].
To date, there is no available information on Nrf2 induction by AGE or SAC in the brain. Here, we show that SAC administration (100 mg/Kg for 5 consecutive days) is able to activate Nrf2 factor in homogenates of cerebral cortex at 24 h (Figure 3). Although the precise mechanism through which SAC is able to evoke this effect in cerebral cortex still remains unclear, a mechanistic proposal is shown in the Figure 4.
Figure 3.
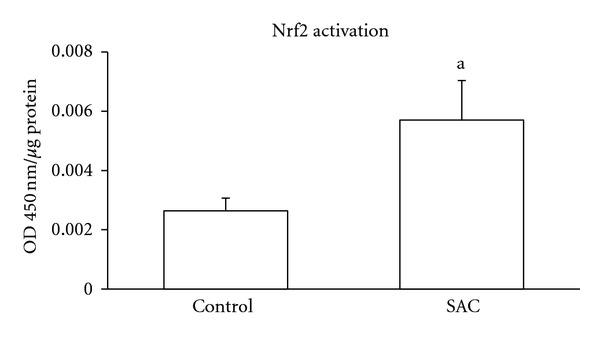
S-Allylcysteine (SAC) induces the activation of the transcription factor nuclear factor-E2-related factor 2 (Nrf2) in cerebral cortex. Animals received SAC 100 mg/kg every day for 5 days. Quantification was made by ELISA at 450 nm in nuclear extracts from frontal cortex of rats at 24 h after the last administration of SAC. Values are expressed as mean ± SEM. n = 4-5. a P < 0.0244 versus control group. Student's t-test. OD: optical density.
Figure 4.
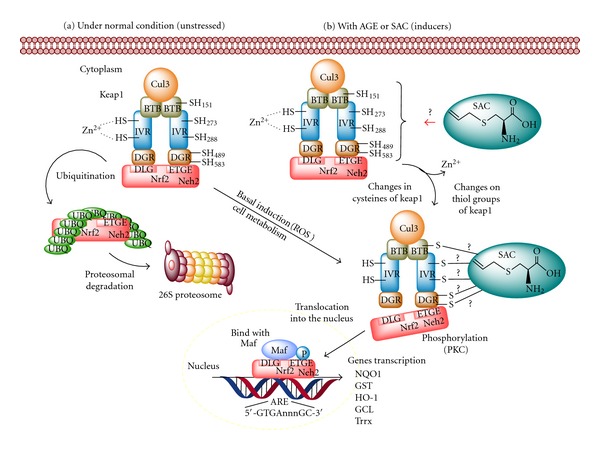
Effect of aged garlic extract (AGE) or S-allylcysteine (SAC) on Nrf2/Keap1 complex. Left panel: Upon unstressed conditions, this complex is dissociated and Nrf2 can either suffer proteosomal degradation or respond to stimuli typical of basal cell metabolism. In the later, Nrf2 is phosphorylated and translocated to the nucleus forming heterodimers with Maf and acting on antioxidant response element (ARE). Right panel: Under stress oxidative conditions, or in the presence of inducers, several cysteine residues suffer changes inducing its Nrf2 dissociation and further translocation of this factor to nucleus, where it will induce phase 2 genes transcription. SAC could modify cysteine residues on Keap1 domain, hence releasing Nrf2 and allowing its transactivation. Nrf2: transcription factor nuclear factor-E2-related factor 2, Keap1: kelch-related erythroid cell-derived protein with CNC homology (ECH) protein 1, UBQ: ubiquitin, ROS: reactive oxygen species, NQO1: NAD(P)H:quinone oxidoreductase 1, GST: glutathione-S-transferase, HO-1: heme oxygenase-1, GCL: glutamate cysteine ligase.
4.3. AGE and SAC Inhibit Prooxidant Enzymes
4.3.1. Nitric Oxide Synthase (NOS)
NOS family is formed by three isoforms: neuronal NOS (nNOS) and endothelial NOS (eNOS) are constitutively expressed and require the formation of a Ca2+-calmodulin complex for their activation. The third isoform, inducible NOS (iNOS), exerts its activity in a Ca2+-independent manner [50]. In the presence of O2, these enzymes catalyze the conversion of L-arginine to L-citrulline plus nitric oxide (NO). Despite increased levels of NO have been involved in degenerative events, NO is also a well-known small molecule that acts as a second messenger in cellular signaling and in a large variety of diverse physiological processes, including neurotransmission, vascular relaxation, blood pressure regulation, defense mechanisms, and immune regulation [51]. Therefore, a dysregulation of NO concentrations compromises the cell survival mechanisms.
SAC is known to inhibit NO production in LPS/cytokine-stimulated macrophages and hepatocytes by suppression of iNOS gene expression [14]. A SAC-induced inhibition of NO production may be related to its ability to decrease NF-κB activation [13, 52], thus leading to cell protection. On the other hand, reports in vivo indicate that AGE increases NO levels through the stimulation of the constitutive NOS isoforms, but not iNOS [15]; however, this mechanism has not been elucidated yet. Altogether, these data suggest that AGE and SAC could be useful in diseases associated with oxidative stress involving dysfunction in NO production.
4.3.2. Xanthine Oxidase (XO)
XO and xanthine dehydrogenase (XDH) are interconvertible forms, and both are members of the molybdenum hydroxylase flavoprotein family; they carry out similar reactions in the purine catabolism in mammals, which are often referred as xanthine oxidoreductase activity. The interconversion from XDH to XO can be either reversible by sulphide reagents (oxidation of important protein thiol group) or irreversible by proteolysis (calcium-dependent proteases) [53]. In particular, XO catalyzes the oxidation of hypoxanthine to xanthine and xanthine to uric acid with the concomitant O2 reduction by a single electron. In turn, XDH catalyzes the same reaction with the reduction of NAD+ by direct two-electron reduction. O2 reduction yields O2 •− and H2O2 as secondary products [54]. O2 •− generated can then react with NO to generate ONOO−, a strong oxidant capable of damage to proteins, lipids, and DNA.
Demirkaya et al. reported that AGE administration (3 mL/kg during 6 weeks, starting 1 week before doxorubicin treatment) decreased XO activity in a model of doxorubicin-induced cardiotoxicity. However, the protective effect observed in this model was not associated with the decrease in XO activity, but to the ability of AGE to increase the levels of some antioxidant enzymes, while decreasing lipid peroxidation [55].
4.3.3. NADPH Oxidase
NADPH oxidase is an enzymatic complex located mainly in the plasmatic membrane of specific granules of phagocytic leukocytes, such as neutrophils and macrophages. It catalyzes the formation of O2 •−, which together with other oxidant species, is produced to kill ingested microorganism (reviewed in [56]). NADPH oxidase overactivation has been related to different pathophysiological events, including renal injury [57], hypertension, atherosclerosis, angiogenesis, and ischemia/reperfusion [56].
Cruz et al. reported a protective effect of SAC (200 mg/kg, i.p.) and AGE (1.2 mL/kg i.p.) administrated every day for 30 days, in 5/6 nephrectomized rats, a model of renal injury and hypertension. In this work, the protective effect exerted by these compounds was associated with their antioxidant properties and their ability to decrease gp91phox and gp22phox levels [58]. However, the mechanism by which SAC and AGE decrease the abundance of gp91phox and gp22phox is unclear. In animals, gp91phox is the catalytic subunit of NADPH oxidase, whereas gp22phox is a membrane protein that forms a heterodimer with gp91phox to produce O2 •−. It is known that NADPH oxidase activation is implicated in the pathogenesis of several renal diseases due to its capacity to produce O2 •−, and its inhibition may be useful to ameliorate and delay the progression of renal injury [57].
4.3.4. Cyclooxygenase (COX)
COX catalyzes the rate-limiting step in the synthesis of prostaglandins and thromboxanes from arachidonic acid [59]. There are two main COX isoforms: COX-1, a constitutive enzyme expressed in many tissues with functions such as gastric mucose protection and regulation of vascular tone [60]. In contrast, the COX-2 isoform is an inducible enzyme primarily associated with inflammatory processes [59, 60]. Several stimuli induce COX-2, including bacterial lipopolysaccharide, interleukin-1 (IL-1), IL-2, and tumor necrosis factor (TNF-α) [60]. COX-2 activity has also been associated with oxidative stress due to its capacity to produce O2 •− from O2.
Colín-González et al. reported the effect of AGE on COX-2 protein levels and activity in a model of cerebral ischemia. They found that AGE administration (1.2 mL/Kg i.p., onset of reperfusion) exerted a neuroprotective effect attributable to its ability to decrease the ischemia-induced increase of 8-hydroxy-2-deoxyguanosine (a marker of oxidative damage to DNA) and TNF-α levels (an inflammation marker). Moreover, AGE decreased COX-2 protein expression and activity, although its mechanism of action remains unclear [61].
Noteworthy, only a few studies have paid attention to the effects of AGE and SAC on the main prooxidant enzymes (XO, NADPH oxidase, or COX), so the mechanisms involved in the possible inhibitory actions of these compounds have been not elucidated yet.
4.4. Chelating Effect of SAC and AGE
Divalent metals (zinc, iron, copper, cobalt, manganese) are involved in a considerable number of physiological processes. However, some of these metals (mostly iron and copper) are involved in the generation of reactive oxygen radicals. A pathological increase in iron or calcium levels has been related with oxidative stress and mitochondrial damage, further leading to neuronal death. Thus, neurodegeneration can be determined by alterations in ionic homoeostasis and/or changes in prooxidative-antioxidant equilibrium, two conditions that are constantly modified in brain cells (reviewed in [62]). Moreover, the observations that iron induces aggregation of α-synuclein and β-amyloid (Aβ) peptides to form toxic aggregates reinforce its critical role in oxidative stress-induced pathogenesis in neurodegeneration (reviewed in [63]). Furthermore, Fenton and Haber-Weiss reactions (Figure 5) are two mechanisms through which free iron or copper produce reactive oxygen species—particularly •OH—to further lead to oxidative degradation of lipids, proteins, and ADN [64–66]. In this regard, it has been reported that the brains of Alzheimer's patients have increased levels of iron, zinc, and copper when compared to control patients [65, 67, 68], whereas the brains of Parkinson's patients show a marked increase in iron and zinc levels [65, 68].
Figure 5.
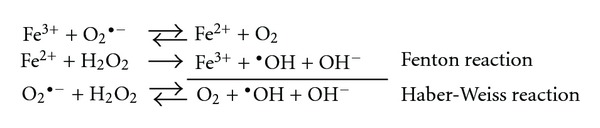
The known ways in which iron is directly involved in the generation of reactive oxygen species. The production of these species is potentially harmful for several cell types and tissues. Adapted from [66].
On the other hand, it has been reported that the interaction between Cu2+ and Aβ produces Aβ-Cu+ and H2O2, and this monovalent form of copper catalyzes the oxidation of Aβ and free radicals production through the Fenton reaction [65, 66, 69] as follows:
| (1) |
Altogether, these data support the concept that a combination of free metal chelation and antioxidant therapies may constitute a valuable approach for neuroprotection.
Dairam et al. found that SAC (30 μg/mL) possesses the property of chelating Fe2+ and Fe3+ in a concentration-dependent manner [70]. Moreover, it has been shown that AGE [34] and SAC [71] are able to inhibit Cu2+-induced LDL oxidation, an effect associated to its ability to chelate Cu2+. In addition, Dillon et al. using a xanthine-XO assay and CuSO4 as inhibitor of XO reported that AGE chelates Cu2+ and restores the activity of XO [72].
5. Neuroprotective Effects of AGE
Technology is increasing exponentially impacting on every aspect of life, improving research, diagnosis, equipment, and procedures in medicine. As a result, life expectancy is increasing, and the population is ageing (in 2007, the overall life expectancy at birth in the US was 77.9 years, representing an increase of 0.2 years from that reported in 2006) [73]. As a consequence, the incidence of neurodegenerative diseases has been increased [74, 75] constituting a global problem that will cause profound economical and social challenges. Nowadays, there are no disease-modifying therapies designed for these disorders, so an intense amount of research is justified.
Oxidative stress is widely implicated in the development of neurodegenerative disorders such as AD, Stroke, Parkinson's disease, and Huntington's disease, among others [76]. Therefore, an increasing number of investigations are expected to appear in a near future studying the effects of AGE and SAC in the central nervous system, probably validating these agents as useful therapeutic strategies.
5.1. Neurotrophic Activity of AGE
The neurotrophic activity of AGE was revealed in a primary culture of fetal rat hippocampal neurons. In this study, the addition of AGE significantly promoted neuronal survival and the number of branching points per axon [77]. Later on, the neurotrophic molecular mechanisms of AGE were studied through the screening of genes differentially expressed after the addition of AGE (0.67%) to primary cultured rat fetal hippocampal neurons. Quantitative RT-PCR showed that α2-microglobulin-related protein (α2MRP) mRNA levels were remarkably increased at 24 h after incubation with AGE [78]. α2MRP is a member of the acute phase proteins, molecules whose concentrations increase or decrease in response to inflammation, tissue damage, and maintenance of cell homeostasis [79]. This finding indicates that AGE may directly or indirectly activates the expression of important genes for neuronal survival, an effect that could be relevant for the design of pharmacological interventions in neurodegenerative disorders.
5.2. Attenuation of Ischemic Brain Damage by AGE
In the US, cerebral ischemia is the third leading cause of death and the leading cause of disability. Brain injury following ischemia results in the activation of a series of biochemical events eventually leading to neuronal death. Despite recent medical and surgical advances, we are still unable to prevent acute ischemic brain damage, so the development of effective neuroprotective methods is crucial.
The effects of AGE on brain ischemia have been evaluated using different administration schemes in the middle cerebral artery occlusion paradigm: (1) AGE (0.5 mL/Kg i.p.) applied 30 min before the onset of ischemia (1 h ischemia/72 h reperfusion) decreased edema formation [80]; (2) AGE (1.2 mL/Kg i.p.) applied at the beginning of reperfusion (2 h ischemia/2 h reperfusion) decreased infarct area, 3-nytrotirosyne levels, and prevented the decrease in antioxidant enzymes (glutathione peroxidase and superoxide dismutase) [81]; (3) AGE (1.2 mL/Kg i.p.) applied at the beginning of reperfusion (1 h ischemia/24 h reperfusion) diminished infarct area, neuronal deficit, histological alterations, 8-hydroxy-deoxyguanosine, TNF-α, and COX-2 levels [61]. All the above-mentioned findings show that the complex physiopathological mechanisms leading to neuronal injury in cerebral stroke can be modified by AGE treatment at different doses, administration schedules, and ischemia/reperfusion times, thus emphasizing its broad spectrum of therapeutic applications. However, the protective effect exerted by AGE in this model may be primarily associated to the prevention of oxidative stress and inflammatory responses.
5.3. Antiageing Effect of AGE
Oxidative stress and immune dysfunction in biological systems are among the most important causes of ageing [82, 83]. The senescence-accelerated mouse is a genetic murine model for studying ageing and spontaneous senescence, and these animals show a short life span and exhibit several signs of senescence in early age (impairment of learning and memory performance, neurochemical changes, etc.) [84]. Chronic oral administration (2 month) of AGE-enriched food (2% w/w) to senescence-accelerated mouse (1) prevented skin glossiness, coarseness, and hair loss; (2) increased survival ratio; (3) improved the memory acquisition deficit; (4) avoided the decrease of brain weight and the atrophy of the frontal portion of the brain [85–87].
On the other hand, Zhang et al. raised the question on whether AGE could have any effect as immunomodulator in the central nervous system. To answer this question, they used an oral administration of AGE (2% w/w) in senescence-accelerated mouse and found that this compound increased immune responses, suggesting that AGE may also improve age-associated decline in the immune system. Shortly thereafter, they also observed the effect of AGE in thymectomized mice, a model of immunodeficiency and deterioration of learning performance. AGE treatment prevented the reduction of antibody production responses, improved the deterioration of learning behavior, and restored the contents of noradrenaline, 3,4-dihydroxyphenylacetic acid, homovanillic acid, and cholineacetyltransferase activity in the hypothalamus. These data suggest that AGE has the capability of reverting thymectomy-induced alterations to normal levels [88].
Clinical studies have demonstrated that brain lesions in Alzheimer's patients may be associated with abnormal immune reactions [89]. Thus, AGE has also been used in Alzheimer animal models. In 2006, Chauhan investigated the anti-inflammatory effects of dietary AGE (2% w/w) in an Alzheimer's transgenic model harboring Swedish double mutation (KM670/671NL; Tg2576). The Tg2576 brains treated with AGE showed reductions in TNF-α levels accompanied by reductions in IL-1β level, and positive plaque-associated microglia, thereby demonstrating a possible anti-inflammatory effect of this compound [90].
AGE contains multiple ingredients with neurotrophic activity that may directly or indirectly prevent the ischemic damage and the age-related morphological changes. Therefore, further studies, including isolation, purification, and identification of its specific compounds and the mechanisms involved, are needed.
6. Neuroprotective Effects of SAC
SAC has been used in several works in order to find effective, therapeutic, and preventive strategies for intervention in neurodegenerative diseases.
6.1. Neurotrophic Effect of SAC
Moriguchi et al. reported the positive actions of garlic compounds (including SAC) with a thioallyl group in rat hippocampal neurons culture. In this work, SAC increased survival and axonal branching from neurons. Based on these findings, this group suggested that thioallyl group is essential for neurotrophic activity [91]. These data suggest that SAC is a compound that not only acts as an antioxidant agent but also as a neurotrophic molecule.
6.2. SAC in Experimental Models of Alzheimer's Disease (AD)
AD is a devastating neurodegenerative disorder which causes progressive loss of cognitive abilities, the accumulation of Aβ deposits in the basal forebrain, hippocampus, and cortex, together with oxidative stress, have been consistently implicated in the pathogenesis of this disorder [92]. In this regard, the design of a treatment with antioxidant and/or antiamyloidogenic properties represents an approach of considerable therapeutic value for the prevention of the disease progression.
The effect of SAC on PC12 cells exposed to Aβ (25–35) has also been evaluated. SAC suppressed the generation of ROS; attenuated caspase-3 activation, DNA fragmentation, and PARP cleavage eventually protecting against Aβ-induced cell death [93, 94]. Moreover, SAC attenuated cell death induced by Aβ in organotypic hippocampal culture and cultured hippocampal neurons in a concentration-dependent manner, an effect apparently mediated by the caspase-12-dependent pathway [95–97].
On the other hand, some studies have shown that AD patients exhibit an overstimulation of the N-methyl-D-aspartate receptor in some time points of the disease progress [98]. In order to reproduce this state, Aβ and ibotenic acid (a potent N-methyl-D-aspartate agonist) administrations have been used in organotypic hippocampal cultures resulting in a time-dependent neuronal damage, a feature that SAC administration (10 and 100 mM) significantly attenuated in the CA3 area [95].
AD involves misfolding and aggregation of proteins which increased endoplasmic reticulum stress [99]. Tunicamycin, an inhibitor of N-glycosylation in endoplasmic reticulum, reproduces some severe neurological alterations in animals (resembling neurological disorders) through the induction of endoplasmic reticulum stress. The role of SAC (10−8–10−5 M) on tunicamycin-mediated cell death was investigated in PC12 cells and hippocampal neurons, showing a selective neuroprotective effect on the caspase-12-dependent apoptotic pathway [96, 100]. In another work, Aβ plus tunicamycin-induced neurotoxicity was assessed in organotypic hippocampal slice cultures, where SAC (100 μM) improved cell viability in area CA3 and dentate gyrus. Simultaneously, SAC reversed calpain activity as well as the active forms of caspase-12 and caspase-3, without changing the increased levels of endoplasmic reticulum chaperones (GRP94 and −78) or C/EBP homologous protein. The authors suggest that SAC could either directly interact with calpain or alter the environment in the vicinity of the endoplasmic reticulum lumen without affecting unfolded protein responses or the C/EBP homologous protein-mediated signaling pathway induced by Aβ plus tunicamycin [101].
In addition, the protective effect of SAC in Aβ toxicity could not be only related to its antioxidant activity as Gupta and Rao investigated the effect of SAC on Aβ aggregation in vitro. SAC (10–50 M) not only inhibited Aβ fibrillation in a dose-dependent manner and disestablished preformed Aβ-peptide fibrils, but also bound to Aβ fibrils. In a docking protocol, the site suitable for accommodating SAC was identified around the Aβ 40 structure. It was mentioned that SAC could interact with the positively charged Gln15-Lys16 segment. In fact, binding could be induced either by hydrophobic interactions between allyl chain and hydrophobic regions of Aβ (Phe 19 and Val 12), or by the H-bond between the -OH group of the carboxylic group of SAC and donator/acceptor groups of Aβ [102].
Since there is a possibility that AD results from inheritance of an autosomal dominant mutation in the amyloid precursor protein, the introduction of mutant amyloid precursor protein genes into suitable mouse pronuclei is used to build transgenic mouse models of AD. These mutant amyloid precursor protein transgenic models exhibit the progressive Aβ neuritic plaques formation, dystrophic neuritis, and neuroinflammation [103]. Interestingly, the dietary administration of SAC (20 mg/kg for 4 month) in one of these transgenic models decreased Aβ load, IL-1β reactive plaque-associated microglia, Tau2 reactivity, and GSK-3β protein, showing an antiamyloidogenic, anti-inflammatory, and antitangle activity (via GSK-3 β) [90].
Intracerebroventricular injection of streptozotocin to mice impairs brain biochemistry, cerebral glucose, energy metabolism, cholinergic transmission, and increases generation of free radicals, further leading to cognitive deficits; these effects are similar to sporadic dementia in humans. Pretreatment with SAC (30 mg/kg every day for 15 days) in intracerebroventricular streptozotocin-infused mice ameliorated hippocampal neuronal abnormalities, prevented the cognitive and neurobehavioral impairments, restored levels of reduced glutathione and its dependent enzymes (glutathione peroxidase and glutathione reductase), diminished lipid peroxidation, DNA fragmentation, and p53 levels, and increased Bcl2 levels [104].
Finally, SAC (1 g/L for 15 weeks, added to the drinking water) was tested in D-galactose-injected mice. D-Galactose induces AD-like pathological changes in the brain, including increased reactive oxygen species, decreased antioxidant enzyme activity, and enhanced Aβ-peptide expression. SAC decreased the brain levels of Aβ 1–40 and Aβ 1–42, lowered amyloid precursor protein level and BACE1 expressions and activities (both of them being factors responsible for Aβ accumulation and AD progression), retained PKC activity and expression of PKC-α and PKC-γ, lowered Aβ accumulation, reduced the levels of advanced glycation end products (carboxymethyllysine, pentosidine), lowered aldose reductase activity and expression (an enzyme that facilitates the production of sorbitol and fructose, which in turn promote advanced glycation end products formation and glycative stress), and displayed antioxidant protection (evidenced by increased reduced glutathione content and glutathione peroxidase, superoxide dismutase, and catalase activities, accompanied by decreased levels of malondialdehyde, reactive oxygen species, and protein carbonyls) [105].
The aforementioned evidence serves to suggest that SAC might prevent the progression of AD by multiple mechanisms: antioxidant, antiamyloidogenic, anti-inflammatory, antitangle, and antiglycative activities (Figure 6). However, further studies are essential to determine if SAC is capable of displaying all these proprieties in humans.
Figure 6.
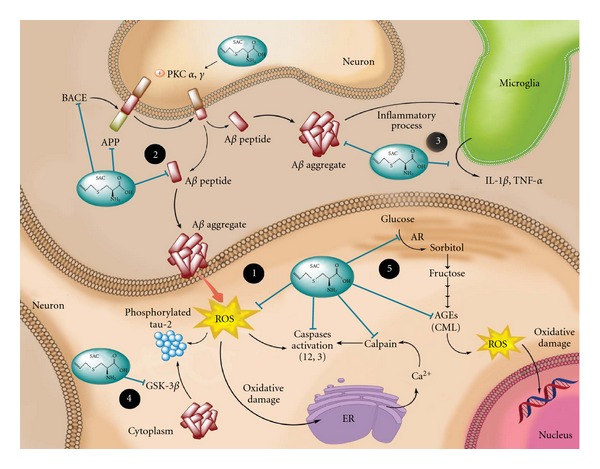
S-allylcysteine (SAC) prevents the progression of Alzheimer's disease (AD) by multiple mechanisms: (1) antioxidant, SAC scavenges free radicals and oxidant specie (direct antioxidant) and restores glutathione peroxidase, glutathione reductase, and superoxide dismutase levels (indirect antioxidant). Consequently, SAC diminishes lipid peroxidation, DNA fragmentation, protein oxidation, and endoplasmic reticulum (ER) stress. The decrease in endoplasmic reticulum stress attenuates Ca2+ release and the subsequent activation of calpain and the caspase-12-dependent pathway, which altogether decrease the cell death; (2) antiamyloidogenic, SAC decreases Aβ formation and/or increases Aβ clearance. SAC lowers amyloid precursor protein (APP) mRNA expression, BACE (β-site APP cleavage enzyme 1) expression and activity and restores PKC activity under AD-like condition, which benefits APP cleavage and decreases the available APP for Aβ. In addition, SAC can bind to Aβ-inhibiting Aβ fibrillation and destabilizing preformed Aβ-peptide fibrils; (3) anti-inflammatory, SAC decreases IL-1β and TNF-α levels and IL-1β-positive plaque-associated microglia; (4) antitangle, SAC reduces tau2 reactivity and its phosphorylation; this reduction in tau appears to involve GSK-3β protein; (5) anti-glycative; SAC declines both activity and mRNA expression of aldose reductase (AR), which subsequently decreases the production of sorbitol and prevents advanced glycation end products (AGEs) formation, such as carboxymethyllysine (CML) and pentosidine, decreasing glycative stress.
6.3. SAC in Ischemic Brain Damage
The effect of SAC in ischemic brain damage was tested at 100, 300, and 600 mg/kg i.p. applied 30 min prior the onset of ischemia in rats. SAC (300 mg/Kg) induced significant reduction of the infarct volume, water content, oxidative stress and improvement in motor performance and memory impairment [23, 80]. Moreover, in a two-vessel occlusion model in gerbils, the oral and i.p. administration of SAC 300 mg/kg increased the number of surviving cells/mm2 of CA1 region [106].
Hypertension is recognized as the most important risk factor for the development of ischemic cerebral infarction in humans [107]. Kim et al. evaluated the effect of SAC in stroke-prone spontaneously hypertensive rats, demonstrating that SAC (5%, 28 day of diet period) reduced mortality and the overall stroke-related behavioral score [108]. In another work, the same authors showed that SAC (300 mg/Kg) reduced the size of infarct area after 2 h occlusion and 22 h reperfusion, prevented neuronal cell death in CA1 region and inhibited the activity of ERK induced by focal ischemia in a middle cerebral artery occlusion model in gerbils, while in vitro exerted scavenging activity on peroxynitrite [28].
Additionally, it has been reported that the positive actions of SAC seen in the cerebral ischemia-induced damage to the hippocampus may be due to the possible modulation of mitochondrial dysfunctions. SAC 300 mg/kg i.p. administered twice (15 min preocclusion and 2 h postocclusion at the time of reperfusion) in the middle cerebral artery occlusion model in rats, produced a significant decrease in mitochondrial lipid peroxidation, protein carbonyl levels, cytochrome c release, and intracellular H2O2 levels. Furthermore, SAC also restored the status of mitochondrial glutathione and glucose 6-phosphate dehydrogenase, ATP content, and the activity of mitochondrial respiratory complexes (I-IV) [109]. In conclusion, the protective effects of SAC on brain ischemic damage may be associated with the decrease of oxidative stress and the modulation of mitochondrial dysfunctions.
6.4. SAC in Experimental Models of Huntington's Disease
Huntington's disease is an autosomal dominant neurodegenerative disorder characterized by the gradual and progressive loss of neurons, predominantly in the striatum. It is caused by a mutation in the huntingtin gen. Its main clinical manifestations are chorea, cognitive impairment, and psychiatric disorders, most of them related to striatal and cortical atrophy. Nowadays, there is no treatment to prevent or reduce the morphological and functional alterations seen in the brains of Huntington's patients. For this reason, it is imperative to look for pharmacological strategies to improve the quality of life of these patients [110].
SAC has been used in two animal models of Huntington's disease: 3-nitropropionic acid and quinolinic acid. SAC (300 mg/kg i.p.) administration to rats infused with 3-nitropropionic acid prevented behavioral alterations, increased manganese and copper/zinc superoxide dismutase activities, and decreased lipid peroxidation and mitochondrial dysfunction [111]. Simultaneously, SAC (0.75 mM) also decreased lipid peroxidation and mitochondrial dysfunction induced by 3-nitropropionic acid in synaptosomal fractions [112].
On the other hand, it has been reported that SAC bonds Fe2+ and Fe3+, preventing the redox cycling of iron, and consequently, the quinolinic acid induced-lipid peroxidation [70]. Alternatively, the effect of SAC was evaluated in a combined model of excitotoxicity/energy deficit produced by the coadministration of quinolinate and 3-nitropropionate acid in brain synaptosomal membranes. SAC abolished the quinolinic acid plus 3-nitropropionate acid-induced lipid peroxidation [113, 114]. The protective effect of SAC in these models has been attributed to its ability to preserve the cell redox status through its antioxidant properties and probably to its iron-binding properties.
6.5. SAC in Experimental Models of Parkinson's Disease
Parkinson's disease is characterized by progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta, with the concomitant formation of intraneuronal fibrillar inclusions (Lewy bodies) and depletion of noradrenaline and serotonin in other brain stem nuclei. In addition, it shows oxidative tissue damage and bioenergetic deficits [115].
1-Methyl-4-phenylpyridinium (MPP+) is the stable metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, and it causes nigrostriatal dopaminergic neurotoxicity, which in turn has been the most widely used model of Parkinson's disease. Rojas et al. reported the neuroprotective effects of SAC against the oxidative stress induced by MPP+ in the mouse striatum. They found that SAC protected dopamine levels, improved hypolocomotion, decreased ROS production and lipid peroxidation, and increased Cu,Zn superoxide dismutase activity in mice injected with MPP+ [116]. Another Parkinsonian model is produced by 6-hydroxydopamine (6-OHDA). In this model, SAC blocked the oxidative damage, prevented dopamine depletion, and returned behavioral performance to basal levels in rats [117]. The protective actions of SAC observed in these models were mainly attributable to its antioxidant activity.
7. AGE in Humans
Altogether, the data presented in this paper clearly shows that AGE and SAC exert antioxidant activity in a variety of experimental models. Of course, these data could be relevant for humans. Indeed, some groups have reported protective effects after chronic administration of AGE to patients with cardiovascular disease and the mechanisms of protection have been associated with the reduction of multiple cardiovascular risk factors, including blood pressure, cholesterol, platelet aggregation and adhesion, and vascular calcification (reviewed in [118, 119]). However, only a few works have been performed in humans bringing attention to the antioxidant effects of AGE, and these are reviewed here.
7.1. Effect of AGE in Atherosclerotic Patients
Cardiovascular mortality is frequently associated with atherosclerosis, a chronic multifactorial disease and a leading cause of death worldwide. Lipid peroxidation and LDLs oxidation, produced by free radicals and end-aldehyde products of lipid hydroperoxide breakdown, play an important role in atherosclerosis. Munday et al. reported that LDLs isolated from subjects receiving 2.4 g of AGE daily for 7 days are more resistant to oxidation than those LDL isolated from subjects receiving no supplementation, suggesting that AGE could be useful in preventing the oxidative stress induced in atherosclerotic disease [120]. Later on, Durak et al. demonstrated that oxidative stress is present in atherosclerotic patients and that the consumption of AGE (1 mL/kg daily; 10 g garlic/day) for 6 months decreased plasma and erythrocyte malondialdehyde levels, with no changes in superoxide dismutase and glutathione peroxidase activities [121].
Currently, there is not enough evidence to determine whether antioxidants can reduce atherogenesis or not; however, it is conceivable that combined antioxidant (AGE) and serum cholesterol-lowering therapies may be useful in reducing the progression of atherosclerosis in humans.
7.2. Effect of AGE in Sickle Cell Anemia Patients
Sickle cell disease is one of the most prevalent hereditary disorders with prominent morbidity and mortality. Clinical manifestations are associated to hemolytic processes leading to severe anemia. It has been reported that oxidative stress plays a significant role in the pathophysiology of sickle cell disease, since it contributes to the sickling process with formation of Heinz bodies that are aggregates of insoluble hemochromes [122].
Takasu et al. reported that AGE (5 mL/day for 4 weeks) diminished the number of Heinz bodies in sickle cell anemia patients and suggested that AGE may be evaluated as a potential therapeutic agent to ameliorate complications of sickle cell anemia [123].
7.3. Effect of AGE in Smoking Individuals
F2-isoprostanes are a family of prostaglandin F2 isomers that are produced in vivo by cyclooxygenase-independent free radical peroxidation of arachidonic acid and are released to plasma in response to cellular activation. Hence, quantification of F2-isoprotanes in plasma and urine is a sensitive and specific indicator of lipid peroxidation in vivo. Smokers are exposed to increased oxidative stress and show increased levels of F2-isoprotanes in plasma and urine. Dillon et al. found that dietary supplementation with AGE (5 mL in a small volume of fruit juice) for 14 days reduced plasma and urinary levels of F2-isoprostane 8-iso-prostaglandin F2α in smokers, suggesting that dietary consumption of AGE may be useful in reducing oxidative stress in humans [17].
8. Final Remarks
In summary, the evidence presented in this paper supports the many beneficial health effects attributable to AGE and SAC, as they are able to reduce the risk of stroke and neurodegenerative damage. Moreover, given that SAC is a potent antioxidant agent, a water soluble compound less toxic than other antioxidants, easily absorbed in the gastrointestinal tract, and rapidly detected in several tissues (kidney, liver, lung, brain), it has been suggested that this compound might exhibit prophylactic properties at a clinical level. However, further investigations are still needed to elucidate the precise protective mechanisms exerted by this antioxidant in several toxic paradigms. Finally, further studies in humans are also necessary to support the clinical use of SAC as a drug to prevent the oxidative damage observed in chronic degenerative disorders.
Acknowledgment
This work was supported by CONACYT Grant 103527 (PDM).
References
- 1.Hahn G. History, folk medicine, and legendary uses of garlic. In: Koch HP, Lawson LD, editors. Garlic: The Science and Therapeutic Application of Allium Sativum L and Related Species. Baltimore, Md, USA: Williams & Wilkins; 1996. pp. 1–24. [Google Scholar]
- 2.Block E. The chemistry of garlic and onions. Scientific American. 1985;252(3):114–119. doi: 10.1038/scientificamerican0385-114. [DOI] [PubMed] [Google Scholar]
- 3.Reuter HD, Koch HP, Lawson LD. Therapeutic effects of garlic and its preparations. In: Koch HP, Lawson LD, editors. Garlic: The Science and Therapeutic Application of Allium Sativum L and Related Species. London, UK: Williams & Wilkins; 1996. pp. 13–162. [Google Scholar]
- 4.Lawson LD. Garlic: a review of its medicinal effects and indicated active compounds. In: Lawson LD, Bauer R, editors. Phytomedicines of Europe: Chemistry and Biological Activity. Washington, DC, USA: American Chemical Society; 1998. pp. 179–209. (ACS Symposium Series 691). [Google Scholar]
- 5.Lawson LD, Gardner CD. Composition, stability, and bioavailability of garlic products used in a clinical trial. Journal of Agricultural and Food Chemistry. 2005;53(16):6254–6261. doi: 10.1021/jf050536+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amagase H, Petesch BL, Matsuura H, Kasuga S, Itakura Y. Intake of garlic and its bioactive components. Journal of Nutrition. 2001;131(supplement 3):955S–962S. doi: 10.1093/jn/131.3.955S. [DOI] [PubMed] [Google Scholar]
- 7.Aged Garlic Extract. Research Excerpts from Peer Reviewed Scientific Journals & Scientific Meetings. Mission Viejo, Calif, USA: Wakunaga of America; 2006. [Google Scholar]
- 8.Ryu K, Ide N, Matsuura H, Itakura Y. Nα-(1-deoxy-D-fructos-1-yl)-L-arginine, an antioxidant compound identified in aged garlic extract. Journal of Nutrition. 2001;131(supplement 3):972S–976S. doi: 10.1093/jn/131.3.972S. [DOI] [PubMed] [Google Scholar]
- 9.Ichikawa M, Ryu K, Yoshida J, et al. Antioxidant effects of tetrahydro-β-carboline derivatives identified in aged garlic extract. BioFactors. 2002;16(3-4):57–72. doi: 10.1002/biof.5520160302. [DOI] [PubMed] [Google Scholar]
- 10.Ichikawa M, Yoshida J, Ide N, Sasaoka T, Yamaguchi H, Ono K. Tetrahydro-β-carboline derivatives in aged garlic extract show antioxidant properties. Journal of Nutrition. 2006;136(supplement 3):726S–731S. doi: 10.1093/jn/136.3.726S. [DOI] [PubMed] [Google Scholar]
- 11.Borek C. Antioxidant health effects of aged garlic extract. Journal of Nutrition. 2001;131(3):1010S–1015S. doi: 10.1093/jn/131.3.1010S. [DOI] [PubMed] [Google Scholar]
- 12.Ide N, Lau BHS. Aged garlic extract attenuates intracellular oxidative stress. Phytomedicine. 1999;6(2):125–131. doi: 10.1016/S0944-7113(99)80047-6. [DOI] [PubMed] [Google Scholar]
- 13.Ide N, Lau BHS. Garlic compounds minimize intracellular oxidative stress and inhibit nuclear factor-κ b activation. Journal of Nutrition. 2001;131(supplement 3):1020S–1026S. doi: 10.1093/jn/131.3.1020S. [DOI] [PubMed] [Google Scholar]
- 14.Kim KM, Chun SB, Koo MS, et al. Differential regulation of NO availability from macrophages and endothelial cells by the garlic component S-allyl cysteine. Free Radical Biology and Medicine. 2001;30(7):747–756. doi: 10.1016/s0891-5849(01)00460-9. [DOI] [PubMed] [Google Scholar]
- 15.Morihara N, Sumioka I, Moriguchi T, Uda N, Kyo E. Aged garlic extract enhances production of nitric oxide. Life Sciences. 2002;71(5):509–517. doi: 10.1016/s0024-3205(02)01706-x. [DOI] [PubMed] [Google Scholar]
- 16.Yamasaki T, Lau BH. Garlic compounds protect vascular endothelial cells from oxidant injury. Nihon Yakurigaku Zasshi. 1997;110:138P–141P. doi: 10.1254/fpj.110.supplement_138. [DOI] [PubMed] [Google Scholar]
- 17.Dillon SA, Lowe GM, Billington D, Rahman K. Dietary supplementation with aged garlic extract reduces plasma and urine concentrations of 8-iso-prostaglandin F2α in smoking and nonsmoking men and women. Journal of Nutrition. 2002;132(2):168–171. doi: 10.1093/jn/132.2.168. [DOI] [PubMed] [Google Scholar]
- 18.Kodera Y, Suzuki A, Imada O, et al. Physical, chemical, and biological properties of S-allylcysteine, an amino acid derived from garlic. Journal of Agricultural and Food Chemistry. 2002;50(3):622–632. doi: 10.1021/jf0106648. [DOI] [PubMed] [Google Scholar]
- 19.Yan CK, Zeng FD. Pharmacokinetics and tissue distribution of S-allylcysteine. Asian Journal of Drug Metabolism and Pharmacokinetics. 2005;5(1):61–69. [Google Scholar]
- 20.Nagae S, Ushijima M, Hatono S, et al. Pharmacokinetics of the garlic compound S-allylcysteine. Planta Medica. 1994;60(3):214–217. doi: 10.1055/s-2006-959461. [DOI] [PubMed] [Google Scholar]
- 21.Jandke J, Spiteller G. Unusual conjugates in biological profiles originating from consumption of onions and garlic. Journal of Chromatography. 1987;421(1):1–8. doi: 10.1016/0378-4347(87)80373-0. [DOI] [PubMed] [Google Scholar]
- 22.Ide N, Matsuura H, Itakura Y. Scavenging effect of aged garlic extract and its constituents on active oxygen species. Phytotherapy Research. 1996;10(4):340–341. [Google Scholar]
- 23.Numagami Y, Ohnishi ST. S-allylcysteine inhibits free radical production, lipid peroxidation and neuronal damage in rat brain ischemia. Journal of Nutrition. 2001;131(supplement 3):1100S–1105S. doi: 10.1093/jn/131.3.1100S. [DOI] [PubMed] [Google Scholar]
- 24.Yamasaki T, Li L, Lau BHS. Garlic compounds protect vascular endothelial cells from hydrogen peroxide-induced oxidant injury. Phytotherapy Research. 1994;8(7):408–412. [Google Scholar]
- 25.Ide N, Lau BHS. Garlic compounds protect vascular endothelial cells from oxidized low density lipoprotein-induced injury. Journal of Pharmacy and Pharmacology. 1997;49(9):908–911. doi: 10.1111/j.2042-7158.1997.tb06134.x. [DOI] [PubMed] [Google Scholar]
- 26.Imai J, Ide N, Nagae S, Moriguchi T, Matsuura H, Itakura Y. Antioxidant and radical scavenging effects of aged garlic extract and its constituents. Planta Medica. 1994;60(5):417–420. doi: 10.1055/s-2006-959522. [DOI] [PubMed] [Google Scholar]
- 27.Maldonado PD, Barrera D, Rivero I, et al. Antioxidant S-allylcysteine prevents gentamicin-induced oxidative stress and renal damage. Free Radical Biology and Medicine. 2003;35(3):317–324. doi: 10.1016/s0891-5849(03)00312-5. [DOI] [PubMed] [Google Scholar]
- 28.Kim JM, Lee JC, Chang N, Chun HS, Kim WK. S-Allyl-l-cysteine attenuates cerebral ischemic injury by scavenging peroxynitrite and inhibiting the activity of extracellular signal-regulated kinase. Free Radical Research. 2006;40(8):827–835. doi: 10.1080/10715760600719540. [DOI] [PubMed] [Google Scholar]
- 29.Medina-Campos ON, Barrera D, Segoviano-Murillo S, et al. S-allylcysteine scavenges singlet oxygen and hypochlorous acid and protects LLC-PK1 cells of potassium dichromate-induced toxicity. Food and Chemical Toxicology. 2007;45(10):2030–2039. doi: 10.1016/j.fct.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Ide N, Lau BHS. S-Allylcysteine attenuates oxidative stress in endothelial cells. Drug Development and Industrial Pharmacy. 1999;25(5):619–624. doi: 10.1081/ddc-100102217. [DOI] [PubMed] [Google Scholar]
- 31.Ho SE, Ide N, Lau BHS. S-allyl cysteine reduces oxidant load in cells involved in the atherogenic process. Phytomedicine. 2001;8(1):39–46. doi: 10.1078/0944-7113-00005. [DOI] [PubMed] [Google Scholar]
- 32.Chung LY. The antioxidant properties of garlic compounds: alyl cysteine, alliin, allicin, and allyl disulfide. Journal of Medicinal Food. 2006;9(2):205–213. doi: 10.1089/jmf.2006.9.205. [DOI] [PubMed] [Google Scholar]
- 33.Maldonado PD, Alvarez-Idaboy JR, Aguilar-González A, et al. Role of allyl group in the hydroxyl and peroxyl radical scavenging activity of s-allylcysteine. The Journal of Physical Chemistry B. 2011;115(45):13408–13417. doi: 10.1021/jp208233f. [DOI] [PubMed] [Google Scholar]
- 34.Dillon SA, Burmi RS, Lowe GM, Billington D, Rahman K. Antioxidant properties of aged garlic extract: an in vitro study incorporating human low density lipoprotein. Life Sciences. 2003;72(14):1583–1594. doi: 10.1016/s0024-3205(02)02475-x. [DOI] [PubMed] [Google Scholar]
- 35.Maldonado PD, Barrera D, Medina-Campos ON, Hernández-Pando R, Ibarra-Rubio ME, Pedraza-Chaverrí J. Aged garlic extract attenuates gentamicin induced renal damage and oxidative stress in rats. Life Sciences. 2003;73(20):2543–2556. doi: 10.1016/s0024-3205(03)00609-x. [DOI] [PubMed] [Google Scholar]
- 36.Wei Z, Lau BHS. Garlic inhibits free radical generation and augments antioxidant enzyme activity in vascular endothelial cells. Nutrition Research. 1998;18(1):61–70. [Google Scholar]
- 37.Pedraza-Chaverrí J, Barrera D, Maldonado PD, et al. S-allylmercaptocysteine scavenges hydroxyl radical and singlet oxygen in vitro and attenuates gentamicin-induced oxidative and nitrosative stress and renal damage in vivo . BMC Clinical Pharmacology. 2004;4, article 5 doi: 10.1186/1472-6904-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kourounakis PN, Rekka EA. Effect on active oxygen species of alliin and allium sativum (garlic) powder. Research Communications in Chemical Pathology and Pharmacology. 1991;74(2):249–252. [PubMed] [Google Scholar]
- 39.Ide N, Lau BHS, Ryu K, Matsuura H, Itakura Y. Antioxidant effects of fructosyl arginine, a maillard reaction product in aged garlic extract. Journal of Nutritional Biochemistry. 1999;10(6):372–376. doi: 10.1016/s0955-2863(99)00021-2. [DOI] [PubMed] [Google Scholar]
- 40.Hsu CC, Huang CN, Hung YC, Yin MC. Five cysteine-containing compounds have antioxidative activity in Balb/cA mice. Journal of Nutrition. 2004;134(1):149–152. doi: 10.1093/jn/134.1.149. [DOI] [PubMed] [Google Scholar]
- 41.Lawal AO, Ellis EM. The chemopreventive effects of aged garlic extract against cadmium-induced toxicity. Environmental Toxicology and Pharmacology. 2011;32(2):266–274. doi: 10.1016/j.etap.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 42.Kalayarasan S, Sriram N, Sureshkumar A, Sudhandiran G. Chromium (VI)-induced oxidative stress and apoptosis is reduced by garlic and its derivative S-allylcysteine through the activation of Nrf2 in the hepatocytes of wistar rats. Journal of Applied Toxicology. 2008;28(7):908–919. doi: 10.1002/jat.1355. [DOI] [PubMed] [Google Scholar]
- 43.Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicology and Applied Pharmacology. 2009;244(1):43–56. doi: 10.1016/j.taap.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radical Biology and Medicine. 2004;37(4):433–441. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 45.van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Current Drug Targets. 2005;4(3):267–281. doi: 10.2174/1568007054038238. [DOI] [PubMed] [Google Scholar]
- 46.Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. Journal of Biological Chemistry. 2003;278(14):12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 47.Silva-Islas C, Santana RA, Colín-González AL, Maldonado PD. 2012, NrF-2 activation, an innovative therapeutic alternative in cerebral ischemia. Chapter 20.
- 48.Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294(1-2):1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 49.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nature Reviews Neuroscience. 2007;8(10):766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 51.Chirino YI, Orozco-Ibarra M, Pedraza-Chaverrí J. Role of peroxynitrite anion in different diseases. Revista de Investigacion Clinica. 2006;58(4):350–358. [PubMed] [Google Scholar]
- 52.Geng Z, Rong Y, Lau BHS. S-allyl cysteine inhibits activation of nuclear factor κ B in human T cells. Free Radical Biology and Medicine. 1997;23(2):345–350. doi: 10.1016/s0891-5849(97)00006-3. [DOI] [PubMed] [Google Scholar]
- 53.Pritsos CA. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chemico-Biological Interactions. 2000;129(1-2):195–208. doi: 10.1016/s0009-2797(00)00203-9. [DOI] [PubMed] [Google Scholar]
- 54.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radical Biology and Medicine. 2002;33(6):774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 55.Demirkaya E, Avci A, Kesik V, et al. Cardioprotective roles of aged garlic extract, grape seed proanthocyanidin, and hazelnut on doxorubicin-induced cardiotoxicity. Canadian Journal of Physiology and Pharmacology. 2009;87(8):633–640. doi: 10.1139/y09-051. [DOI] [PubMed] [Google Scholar]
- 56.Ago T, Kuroda J, Kamouchi M, Sadoshima J, Kitazono T. Pathophysiological roles of NADPH oxidase/nox family proteins in the vascular system review and perspective. Circulation Journal. 2011;75(8):1791–1800. doi: 10.1253/circj.cj-11-0388. [DOI] [PubMed] [Google Scholar]
- 57.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxidants and Redox Signaling. 2006;8(9-10):1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 58.Cruz C, Correa-Rotter R, Sánchez-González DJ, et al. Renoprotective and antihypertensive effects of S-allylcysteine in 5/6 nephrectomized rats. American Journal of Physiology. 2007;293(5):F1691–F1698. doi: 10.1152/ajprenal.00235.2007. [DOI] [PubMed] [Google Scholar]
- 59.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annual Review of Pharmacology and Toxicology. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 60.Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. The FASEB Journal. 1998;12(12):1063–1073. [PubMed] [Google Scholar]
- 61.Colín-González AL, Ortiz-Plata A, Villeda-Hernández J, et al. Aged garlic extract attenuates cerebral damage and cyclooxygenase-2 induction after ischemia and reperfusion in rats. Plant Foods for Human Nutrition. 2011;66(4):348–354. doi: 10.1007/s11130-011-0251-3. [DOI] [PubMed] [Google Scholar]
- 62.Pelizzoni I, Macco R, Zacchetti D, Grohovaz F, Codazzi F. Iron and calcium in the central nervous system: a close relationship in health and sickness. Biochemical Society Transactions. 2008;36(6):1309–1312. doi: 10.1042/BST0361309. [DOI] [PubMed] [Google Scholar]
- 63.Mandel S, Amit T, Reznichenko L, Weinreb O, Youdim MBH. Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Molecular Nutrition and Food Research. 2006;50(2):229–234. doi: 10.1002/mnfr.200500156. [DOI] [PubMed] [Google Scholar]
- 64.Sayre LM, Perry G, Smith MA. Redox metals and neurodegenerative disease. Current Opinion in Chemical Biology. 1999;3(2):220–225. doi: 10.1016/S1367-5931(99)80035-0. [DOI] [PubMed] [Google Scholar]
- 65.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Current Medicinal Chemistry. 2005;12(10):1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 66.Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Molecular and Cellular Biochemistry. 2010;345(1-2):91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- 67.Drew SC, Barnham KJ. The heterogeneous nature of Cu2+ interactions with Alzheimer’s amyloid-β peptide. Accounts of Chemical Research. 2011;44(11):1146–1155. doi: 10.1021/ar200014u. [DOI] [PubMed] [Google Scholar]
- 68.Barnham KJ, Bush AI. Metals in Alzheimer’s and Parkinson’s Diseases. Current Opinion in Chemical Biology. 2008;12(2):222–228. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 69.Huang X, Cuajungco MP, Hartshorn MA, et al. Cu(II) potentiation of Alzheimer aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. Journal of Biological Chemistry. 1999;274(52):37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 70.Dairam A, Fogel R, Daya S, Limson JL. Antioxidant and iron-binding properties of curcumin, capsaicin, and S-allylcysteine reduce oxidative stress in rat brain homogenate. Journal of Agricultural and Food Chemistry. 2008;56(9):3350–3356. doi: 10.1021/jf0734931. [DOI] [PubMed] [Google Scholar]
- 71.Huang CN, Horng JS, Yin MC. Antioxidative and antiglycative effects of six organosulfur compounds in low-density lipoprotein and plasma. Journal of Agricultural and Food Chemistry. 2004;52(11):3674–3678. doi: 10.1021/jf0307292. [DOI] [PubMed] [Google Scholar]
- 72.Dillon SA, Burmi RS, Lowe GM, Billington D, Rahman K. Antioxidant properties of aged garlic extract: an in vitro study incorporating human low density lipoprotein. Life Sciences. 2003;72(14):1583–1594. doi: 10.1016/s0024-3205(02)02475-x. [DOI] [PubMed] [Google Scholar]
- 73.Arias E. United States life tables, 2007. National Vital Statistics Reports. 2011;59(9):1–60. [PubMed] [Google Scholar]
- 74.Access Economics. Projections of dementia prevalence and incidence in NSW: 2009–2005. 2009, http://www.health.nsw.gov.au/pubs/2009/adhc_dementia.html.
- 75.Melo A, Monteiro L, Lima RM, de Oliveira DM, de Cerqueira MD, El-Bachá RS. Oxidative stress in neurodegenerative diseases: mechanisms and therapeutic perspectives. Oxidative Medicine and Cellular Longevity. 2011;2011:14 pages. doi: 10.1155/2011/467180. Article ID 467180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40(8):959–975. doi: 10.1016/s0028-3908(01)00019-3. [DOI] [PubMed] [Google Scholar]
- 77.Moriguchi T, Nishiyama N, Saito H, Katsuki H. Trophic effects of aged garlic extract (AGE) and its fractions on primary cultured hippocampal neurons from fetal rat brain. Phytotherapy Research. 1996;10(6):468–472. [Google Scholar]
- 78.Sumi S, Tsuneyoshi T, Matsuo H, Yoshimatsu T. Isolation and characterization of the genes up-regulated in isolated neurons by aged garlic extract (AGE) Journal of Nutrition. 2001;131(supplement 3):1096S–1099S. doi: 10.1093/jn/131.3.1096S. [DOI] [PubMed] [Google Scholar]
- 79.Cray C. Acute phase proteins in animals. Progress in Molecular Biology and Translational Science. 2012;105:113–150. doi: 10.1016/B978-0-12-394596-9.00005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Numagami Y, Sato S, Ohnishi ST. Attenuation of rat ischemic brain damage by aged garlic extracts: a possible protecting mechanism as antioxidants. Neurochemistry International. 1996;29(2):135–143. doi: 10.1016/0197-0186(95)00117-4. [DOI] [PubMed] [Google Scholar]
- 81.Aguilera P, Chánez-Cárdenas ME, Ortiz-Plata A, et al. Aged garlic extract delays the appearance of infarct area in a cerebral ischemia model, an effect likely conditioned by the cellular antioxidant systems. Phytomedicine. 2010;17(3-4):241–247. doi: 10.1016/j.phymed.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 82.Zimniak P. Relationship of electrophilic stress to aging. Free Radical Biology and Medicine. 2011;51(6):1087–1105. doi: 10.1016/j.freeradbiomed.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corona AW, Fenn AM, Godbout JP. Cognitive and behavioral consequences of impaired immunoregulation in aging. Journal of NeuroImmune Pharmacology. 2012;7(1):7–23. doi: 10.1007/s11481-011-9313-4. [DOI] [PubMed] [Google Scholar]
- 84.Takeda T, Hosokawa M, Takeshita S, et al. A new murine model of accelerated senescence. Mechanisms of Ageing and Development. 1981;17(2):183–194. doi: 10.1016/0047-6374(81)90084-1. [DOI] [PubMed] [Google Scholar]
- 85.Moriguchi T, Takashina K, Chu PJ, Saito H, Nishiyama N. Prolongation of life span and improved learning in the senescence accelerated mouse produced by aged garlic extract. Biological and Pharmaceutical Bulletin. 1994;17(12):1589–1594. doi: 10.1248/bpb.17.1589. [DOI] [PubMed] [Google Scholar]
- 86.Moriguchi T, Saito H, Nishiyama N. Anti-ageing effect of aged garlic extract in the inbred brain atrophy mouse model. Clinical and Experimental Pharmacology and Physiology. 1997;24(3-4):235–242. doi: 10.1111/j.1440-1681.1997.tb01813.x. [DOI] [PubMed] [Google Scholar]
- 87.Nishiyama N, Moriguchi T, Saito H. Beneficial effects of aged garlic extract on learning and memory impairment in the senescence-accelerated mouse. Experimental Gerontology. 1997;32(1-2):149–160. doi: 10.1016/s0531-5565(96)00062-9. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Y, Moriguchi T, Saito H, Nishiyama N. Functional relationship between age-related immunodeficiency and learning deterioration. European Journal of Neuroscience. 1998;10(12):3869–3875. doi: 10.1046/j.1460-9568.1998.00393.x. [DOI] [PubMed] [Google Scholar]
- 89.di Bona D, Scapagnini G, Candore G, et al. Immune-inflammatory responses and oxidative stress in Alzheimer’s disease: therapeutic implications. Current Pharmaceutical Design. 2010;16(6):684–691. doi: 10.2174/138161210790883769. [DOI] [PubMed] [Google Scholar]
- 90.Chauhan NB. Effect of aged garlic extract on APP processing and tau phosphorylation in Alzheimer’s transgenic model Tg2576. Journal of Ethnopharmacology. 2006;108(3):385–394. doi: 10.1016/j.jep.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 91.Moriguchi T, Matsuura H, Kodera Y, et al. Neurotrophic activity of organosulfur compounds having a thioallyl group on cultured rat hippocampal neurons. Neurochemical Research. 1997;22(12):1449–1452. doi: 10.1023/a:1021946210399. [DOI] [PubMed] [Google Scholar]
- 92.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radical Biology and Medicine. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 93.Peng Q, Buz’Zard AR, Lau BH. Neuroprotective effect of garlic compounds in amyloid-β peptide-induced apoptosis in vitro . Medical Science Monitor. 2002;8(8):BR328–BR337. [PubMed] [Google Scholar]
- 94.Ito Y, Kosuge Y, Sakikubo T, et al. Protective effect of S-allyl-L-cysteine, a garlic compound, on amyloid β-protein-induced cell death in nerve growth factor-differentiated PC12 cells. Neuroscience Research. 2003;46(1):119–125. doi: 10.1016/s0168-0102(03)00037-3. [DOI] [PubMed] [Google Scholar]
- 95.Ito Y, Ito M, Takagi N, Saito H, Ishige K. Neurotoxicity induced by amyloid β-peptide and ibotenic acid in organotypic hippocampal cultures: protection by S-allyl-L-cysteine, a garlic compound. Brain Research. 2003;985(1):98–107. doi: 10.1016/s0006-8993(03)03173-1. [DOI] [PubMed] [Google Scholar]
- 96.Kosuge Y, Koen Y, Ishige K, et al. S-allyl-L-cysteine selectively protects cultured rat hippocampal neurons from amyloid β-protein- and tunicamycin-induced neuronal death. Neuroscience. 2003;122(4):885–895. doi: 10.1016/j.neuroscience.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 97.Ishige K, Takagi N, Imai T, et al. Role of caspase-12 in amyloid β-peptide-induced toxicity in organotypic hippocampal slices cultured for long periods. Journal of Pharmacological Sciences. 2007;104(1):46–55. doi: 10.1254/jphs.fp0061533. [DOI] [PubMed] [Google Scholar]
- 98.Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochemistry International. 2004;45(5):583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 99.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. Journal of Neuroinflammation. 2009;6, article 41 doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kosuge Y, Sakikubo T, Ishige K, Ito Y. Comparative study of endoplasmic reticulum stress-induced neuronal death in rat cultured hippocampal and cerebellar granule neurons. Neurochemistry International. 2006;49(3):285–293. doi: 10.1016/j.neuint.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 101.Imai T, Kosuge Y, Ishige K, Ito Y. Amyloid β-protein potentiates tunicamycin-induced neuronal death in organotypic hippocampal slice cultures. Neuroscience. 2003;147(3):639–651. doi: 10.1016/j.neuroscience.2007.04.057. [DOI] [PubMed] [Google Scholar]
- 102.Gupta VB, Rao KS. Anti-amyloidogenic activity of S-allyl-l-cysteine and its activity to destabilize Alzheimer’s β-amyloid fibrils in vitro . Neuroscience Letters. 2007;429(2-3):75–80. doi: 10.1016/j.neulet.2007.09.042. [DOI] [PubMed] [Google Scholar]
- 103.King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’ disease through 19 months. Physiology & Behavior. 2002;75(5):627–642. doi: 10.1016/s0031-9384(02)00639-x. [DOI] [PubMed] [Google Scholar]
- 104.Javed H, Khan MM, Khan A, et al. S-allyl cysteine attenuates oxidative stress associated cognitive impairment and neurodegeneration in mouse model of streptozotocin-induced experimental dementia of Alzheimer’s type. Brain Research. 2011;1389:133–142. doi: 10.1016/j.brainres.2011.02.072. [DOI] [PubMed] [Google Scholar]
- 105.Tsai SJ, Chiu CP, Yang HT, Yin MC. S-allyl cysteine, S-ethyl cysteine, and S-propyl cysteine alleviate β-amyloid, glycative, and oxidative injury in brain of mice treated by D-galactose. Journal of Agricultural and Food Chemistry. 2011;59(11):6319–6326. doi: 10.1021/jf201160a. [DOI] [PubMed] [Google Scholar]
- 106.Chun HS, Kim JM, Choi EH, Chang N. Neuroprotective effects of several Korean medicinal plants traditionally used for stroke remedy. Journal of Medicinal Food. 2008;11(2):246–251. doi: 10.1089/jmf.2007.542. [DOI] [PubMed] [Google Scholar]
- 107.Hayakawa T, Waltz AG, Jacobson RL. Hypertension and acute focal cerebral ischemia. Infarction and edema after occlusion of a middle cerebral artery in cats. Stroke. 1979;10(3):263–267. doi: 10.1161/01.str.10.3.263. [DOI] [PubMed] [Google Scholar]
- 108.Kim JM, Chang N, Kim WK, Chun HS. Dietary S-allyl-L-cysteine reduces mortality with decreased incidence of stroke and behavioral changes in stroke-prone spontaneously hypertensive rats. Bioscience, Biotechnology, and Biochemistry. 2006;70(8):1969–1971. doi: 10.1271/bbb.50697. [DOI] [PubMed] [Google Scholar]
- 109.Atif F, Yousuf S, Agrawal SK. S-Allyl L-cysteine diminishes cerebral ischemia-induced mitochondrial dysfunctions in hippocampus. Brain Research C. 2009;1265:128–137. doi: 10.1016/j.brainres.2008.12.077. [DOI] [PubMed] [Google Scholar]
- 110.Bano D, Zanetti F, Mende Y, Nicotera P. Neurodegenerative processes in Huntington’s disease. Cell Death and Disease. 2011;2, article e228 doi: 10.1038/cddis.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Herrera-Mundo MN, Silva-Adaya D, Maldonado PD, et al. S-Allylcysteine prevents the rat from 3-nitropropionic acid-induced hyperactivity, early markers of oxidative stress and mitochondrial dysfunction. Neuroscience Research. 2006;56(1):39–44. doi: 10.1016/j.neures.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 112.La Cruz VPD, González-Cortés C, Pedraza-Chaverrí J, Maldonado PD, Andrés-Martínez L, Santamaría A. Protective effect of S-allylcysteine on 3-nitropropionic acid-induced lipid peroxidation and mitochondrial dysfunction in rat brain synaptosomes. Brain Research Bulletin. 2006;68(5):379–383. doi: 10.1016/j.brainresbull.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 113.Elinos-Calderón D, Robledo-Arratia Y, La Cruz VPD, et al. Antioxidant strategy to rescue synaptosomes from oxidative damage and energy failure in neurotoxic models in rats: protective role of S-allylcysteine. Journal of Neural Transmission. 2010;117(1):35–44. doi: 10.1007/s00702-009-0299-5. [DOI] [PubMed] [Google Scholar]
- 114.La Cruz VPD, Konigsberg M, Pedraza-Chaverri J, et al. Cytoplasmic calcium mediates oxidative damage in an excitotoxic/energetic deficit synergic model in rats. European Journal of Neuroscience. 2008;27(5):1075–1085. doi: 10.1111/j.1460-9568.2008.06088.x. [DOI] [PubMed] [Google Scholar]
- 115.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience. 2011;198:221–231. doi: 10.1016/j.neuroscience.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rojas P, Serrano-García N, Medina-Campos ON, Pedraza-Chaverri J, Maldonado PD, Ruiz-Sánchez E. S-Allylcysteine, a garlic compound, protects against oxidative stress in 1-methyl-4-phenylpyridinium-induced parkinsonism in mice. The Journal of Nutritional Biochemistry. 2011;22(10):937–944. doi: 10.1016/j.jnutbio.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 117.Garcia E, Limon D, La Cruz VPD, et al. Lipid peroxidation, mitochondrial dysfunction and neurochemical and behavioural deficits in different neurotoxic models: protective role of S-allylcysteine. Free Radical Research. 2008;42(10):892–902. doi: 10.1080/10715760802506356. [DOI] [PubMed] [Google Scholar]
- 118.Rahman K. Historical perspective on garlic and cardiovascular disease. Journal of Nutrition. 2001;131(3):977S–979S. doi: 10.1093/jn/131.3.977S. [DOI] [PubMed] [Google Scholar]
- 119.Budoff MJ, Takasu J, Flores FR, et al. Inhibiting progression of coronary calcification using aged garlic extract in patients receiving statin therapy: a preliminary study. Preventive Medicine. 2004;39(5):985–991. doi: 10.1016/j.ypmed.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 120.Munday JS, James KA, Fray LM, Kirkwood SW, Thompson KG. Daily supplementation with aged garlic extract, but not raw garlic, protects low density lipoprotein against in vitro oxidation. Atherosclerosis. 1999;143(2):399–404. doi: 10.1016/s0021-9150(98)00293-7. [DOI] [PubMed] [Google Scholar]
- 121.Durak I, Aytaç B, Atmaca Y, et al. Effects of garlic extract consumption on plasma and erythrocyte antioxidant parameters in atherosclerotic patients. Life Sciences. 2004;75(16):1959–1966. doi: 10.1016/j.lfs.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 122.Rice-Evans C, Omorphos SC, Baysal E. Sickle cell membranes and oxidative damage. Biochemical Journal. 1986;237(1):265–269. doi: 10.1042/bj2370265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takasu J, Uykimpang R, Sunga M, Amagase H, Niihara Y. Aged garlic extract therapy for sickle cell anemia patients. BMC Blood Disorders. 2002;2, article 3 doi: 10.1186/1471-2326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]