

Abstract
This chapter recounts efforts to dissect the cellular and circuit basis of a memory system in the primate cortex with the goal of extending the insights gained from the study of normal brain organization in animal models to an understanding of human cognition and related memory disorders. Primates and humans have developed an extraordinary capacity to process information “on line,” a capacity that is widely considered to underlay comprehension, thinking, and so-called executive functions. Understanding the interactions between the major cellular constituents of cortical circuits—pyramidal and nonpyramidal cells—is considered a necessary step in unraveling the cellular mechanisms subserving working memory mechanisms and, ultimately, cognitive processes. Evidence from a variety of sources is accumulating to indicate that dopamine has a major role in regulating the excitability of the cortical circuitry upon which the working memory function of prefrontal cortex depends. Here, I describe several direct and indirect intercellular mechanisms for modulating working memory function in prefrontal cortex based on the localization of dopamine receptors on the distal dendrites and spines of pyramidal cells and on interneurons in the prefrontal cortex. Interactions between monoamines and a compromised cortical circuitry may hold the key to understanding the variety of memory disorders associated with aging and disease.
Compared with the well-recognized type of memory process referred to variously as associative (1), declarative (2), or episodic (3), “working memory” is a relatively new concept in neuroscience, though it has a longer tradition in human cognitive psychology and antecedence in the more familiar phenomenon of short-term memory (4, 5, 6). Working memory is the ability to hold an item of information transiently in mind in the service of comprehension, thinking, and planning (6, 7). Working memory encompasses both storage and processing functions. In its simplest form, as when holding in mind a phone number long enough to dial it, the memory may decay passively and/or instantaneously. In its most elevated form, working memory serves as a workspace for holding items of information in mind as they are recalled, manipulated, and/or associated to other ideas and incoming information. “Blackboard of the mind” has been a useful metaphor for the limited capacity and processing dynamics of the working memory mechanism (for the origin of this analogy in the speech perception literature, see ref. 8). Mental arithmetic, imaging a sequence of chess moves, constructing a sentence, and creation of music or poetry are examples of the infinite variety of mental gymnastics dependent on the basic operation of working memory. More than any other form of learning or memory, the mechanisms underlying working memory come closest to addressing one of the great issues of neurobiology—the neural basis of mental representation and the workings of the human mind.
Modular Functional Architecture of Prefrontal Cortical Areas
Experimental studies in nonhuman primates indicate that there may be multiple working memory domains, each localized in a different anatomical subdivision of the prefrontal cortex of both human and nonhuman primates, and each having its own specialized processing and content-specific storage mechanisms (9, 10, 11, 12, 13, 14). Visuospatial processes engaged in humans by activities such as chess playing, following maps, recalling one’s location with respect to landmarks, or painting and drawing from memory, and studied in animals by delayed-response tasks rely on the dorsolateral prefrontal convexity both in monkeys (15, 16, 17, 18, 19, 20) and in humans (21, 22). The same cortical areas are consistently activated as human subjects access visuospatial information from long-term storage and/or immediate experience through representation-based action (23, 24, 25, 26, 27, 28, 29, 30). In contrast, working memory for the features of objects or faces engages anatomically different prefrontal regions located in an inferior position in the prefrontal cortex in both species (14, 31, 32, 33, 34). Finally, semantic encoding and retrieval, as well as other verbal processes, engages a more inferior, insular, and/or anterior prefrontal region (35, 36, 37, 38, 39). To date, noninvasive imaging of human subjects performing working memory tasks has failed to identify one common locus of a central “executive” processor (6) or “contention scheduler” (40) that would mediate any and all informational systems. Indeed, based on anatomical, physiological, and lesion evidence in both monkeys and humans, a “central executive” in the sense of an all-purpose polymodal processor may not exist, and to the contrary, a strong case can be made for the view that the substrates of cognition reside in the parallelism of the brain’s modularized information processing systems (9).
Cellular Correlate of Working Memory: Neurons with “Memory Fields”
A major advance in our understanding of prefrontal cortex came in the early 1970s, when electrophysiological studies revealed that neurons in the prefrontal cortex become activated during the delay period of a delayed-response trial when monkeys recalled a visual stimulus that had been presented at the beginning of a trial (18, 19). It quickly became evident that the activity of these prefrontal neurons could be the cellular correlate of a mnemonic event. Indeed, using an oculomotor version of the classical spatial delayed-response paradigm, it has recently been possible to show that prefrontal neurons have memory fields, defined as maximal firing of a neuron to the representation of a visual target in one or a few locations of the visual field—with the same neuron coding the same location trial after trial and different neurons coding different locations (Fig. 1; ref. 17). The neuronal activity displayed in the top part of Fig. 1 is an example; after the brief presentation of a stimulus at the 135° location and the introduction of a 5000-ms delay, the neuron’s activity rises sharply during the delay and remains tonically active in the complete absence of the stimulus or a response until the response is initiated. Importantly, the activation occurs maximally every time the animal has to remember the 135° location but not when the animal is remembering targets presented at other locations (e.g., 45°, 225°, 315°). Neuronal discharge in the absence of stimuli or responses has been recorded for as long as 12–15 s in prefrontal neurons (17, 41). Finally, neuronal activity of a given cell during the delay period is labile and can expand and contract as the delay is lengthened or foreshortened (17, 41), as would be expected if a neuron is engaged in dynamic mnemonic processing. As far as is known, these responses take place within a narrow range of delays (less than 20 s). Information that is retained over longer delays, e.g., tens of seconds, presumably enters intermediate or long-term memory stores and likely depends upon mechanisms beyond working memory, perhaps involving long-term potentiation in the hippocampal formation. The neuronal activation observed in prefrontal neurons is best viewed as a reflection of information that is “on line.”
Figure 1.
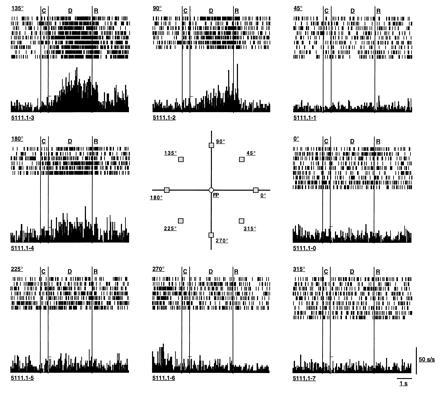
Repeated recordings from one neuron during the many trials over which a monkey performed an oculomotor delayed-response working memory task. Over the course of a testing session, the monkey’s ability to make correct memory-guided responses is tested approximately 10–12 times per target location. The neuron’s response is collated over all the trials for a given target location (e.g., 135°, 45°, etc.) as a histogram of the average response per unit time for that location. The activity is shown in relation to the timed events in the task (C, cue; D, delay; R, response) for each target location. In the example shown, the neuron’s rate of discharge increases maximally during the delay when the target at 135° location is no longer present and the monkey is simply maintaining fixation; the neuronal activation is maintained for more than 5000 ms until the response is made. Delay-period activity is also observed during the delay period for the 90° and 180° targets but is less than that exhibited for the neuron’s “best direction,” indicating that the neuron’s tuning is rather broad. However, this neuron codes the same location trial after trial; different neurons (data not shown) code different locations and have different degrees of tuning in working memory. [Reproduced with permission from ref. 17 (Copyright 1989, The American Physiological Society).]
Microarchitecture: The Neuronal Assembly in Prefrontal Cortex
Subsets of prefrontal neurons in the area of the principal sulcus (i) are activated phasically in the presence of a visual stimulus, (ii) are activated tonically during the delay period over which the stimulus is kept on line, or (iii) show phasic reactivation in relation to the initiation of a memory-guided response (Fig. 2; for review, see ref. 42). Many, if not most, prefrontal neurons respond in more than one phase of the trial, i.e., during the cue, delay, and/or response periods, and their composite profile may be due to inputs from neurons whose activation is simpler and related to only one phase. Thus, prefrontal neuronal activities are differentially time-locked to the running events in a delayed-response trial and temporally phased so as to bridge the time domain, as shown in Fig. 2. The firing profiles of prefrontal neurons are related to the subfunctions of registration, memory, and motor control, respectively. We (42) have hypothesized that the neurons carrying out these component processes occupy distinct positions within the laminar hierarchy of a cortical column (or hypercolumn) which is functionally dedicated to a particular memorandum (in this instance, a particular spatial location), in analogy with the columnar organization of the primary visual cortex. Since the memory-relatedness of prefrontal neurons can be addressed only in the awake, behaving primate, one possible way to address these architectural issues would be to record from multiple units in both vertical and tangential penetrations in prefrontal cortex of trained monkeys. Multiunit recording methods are being developed in a number of laboratories and should be available in the near future to allow more precise mapping of functionally characterized neurons.
Figure 2.
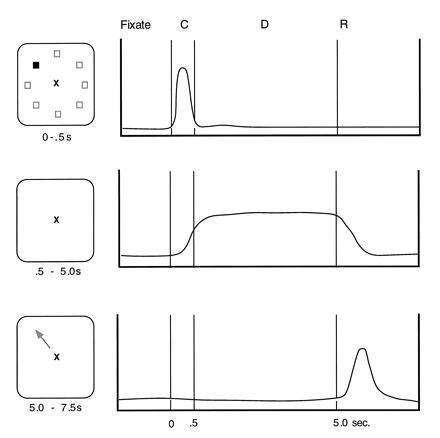
Prefrontal neurons in the region of the principal sulcus exhibit a variety of patterns of activation during the oculomotor tasks. Some respond phasically to the occurrence of a target (Top); some respond in relation to the delay (Middle); and some are activated in relation to the occurrence of a response (Bottom). In all cases, neuronal activity is time-locked to the events of the task and is spatially tuned. The class of neurons with delay period activity is the focus of this essay. (Figure based on refs. 17, 43, and 44.)
Mechanisms for Constructing Memory Fields: Interactions Between Pyramidal and Nonpyramidal Neurons in Prefrontal Cortex
From Cajal on, it has been appreciated that several types of interneurons populate the cerebral cortex and interact with pyramidal cells. We now know that the majority of the interneurons use the inhibitory neurotransmitter, γ-aminobutyric acid (GABA) as their neurotransmitters, whereas pyramidal cells use the excitatory amino acids. Recent evidence indicates that pyramidal–nonpyramidal interactions may be critical to the formation of memory fields in prefrontal cortex just as they are in establishing the orientation specificity of primary visual neurons (for review, see ref. 45). Wilson et al. in this laboratory (46) have succeeded in using wave form analysis to classify functionally characterized neurons as interneurons (thin-spiking neurons) or pyramidal neurons (broader and higher amplitude spikes) in monkeys as they performed the oculomotor delayed-response task. This study showed (i) that interneurons, like pyramidal neurons, express directional preferences (e.g., neuron FS161 in Fig. 3; ref. 46) and (ii) that the patterns of activity expressed by closely adjacent pyramidal and nonpyramidal neurons are often inverse, such that as a nonpyramidal neuron increases its rate of discharge, a nearby pyramidal neuron decreases its rate (compare polar plots of FS161 and RS162 in Fig. 3). These findings provide suggestive evidence that feedforward inhibition may play a role in the construction of a memory field in prefrontal neurons.
Figure 3.
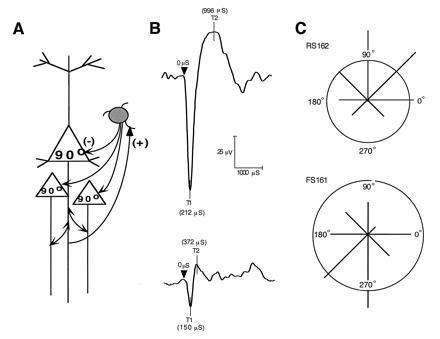
(A) Diagram of a basic pyramidal–interneuronal interaction in cerebral cortex; pyramidal glutamatergic neurons innervate the dendrites of GABAergic interneurons; subsets of GABAergic neurons terminate on various segments of the pyramidal cell. In the diagram, I illustrate a basket cell subtype the terminals of which contact the cell bodies of pyramidal neurons. (B) Action potentials indicative of interneurons (Upper) and pyramidal neurons (Lower) used to define functionally characterized cells in C. (C) Inverse responses of fast-spiking (FS161) and regular-spiking (RS162) pairs of neurons recorded approximately 200 μm apart. Bin width = 40 ms; 10 trials per direction; the vector plot of tuning for the FS161/RS162 pair of neurons for 8 target locations shows that FS161 responded maximally to a stimulus presented 13° above the fixation point (at the 90° location), whereas RS162 responded maximally to stimuli presented at the 270° location below the fixation point. Firing rates were normalized so that the maximum vector length is 100%. The circles represent spontaneous firing rates. [Reproduced with permission from ref. 46 (Copyright 1994, The National Academy of Sciences).]
Recent studies of prefrontal cortex have also begun to elucidate the horizontal connections between groups of pyramidal cells that contribute to local circuits (47, 48). Leavitt et al. (47) made small injections of biocytin into specific layers of the principal sulcus and traced orthogradely transported label. This study revealed narrow (220–400 μm), stripe-like bands of terminal label over 7–8 mm of cortex arising from neurons in layers 2, 3, and 5 at the center of the injection site. Complimentary results have been obtained with the retrograde tracer cholera toxin B subunit. For example, as shown in Fig. 4, injections of this tracer, confined to layer IIIc of prefrontal cortex, labeled clusters of neurons several millimeters distant from the injection site (48), reminiscent of isoorientation columns in the primary visual cortex (49) and anatomical columns formed by long-tract cortico-cortical connections (50). Fig. 5 illustrates a hypothetical modular architecture for spatial working memory in which columns of pyramidal neurons with the same “best directions” (e.g., 90°, 180°, 270°, etc.) are interconnected in a manner analogous to the orientation column system of primary visual cortex (49). The figure also incorporates a basket cell interconnecting two pyramidal cells with opposite best directions—a proposed mechanism of reciprocal feedforward inhibition among columns of pyramidal neurons to accommodate the physiology of spatial working memory (17). According to this scheme, pyramidal cells with opposite best directions communicate through inhibitory interneurons such that a pyramidal neuron with a 90° memory field exhibits enhanced firing during the delay of trials in which the monkey is recalling a 90° target but is inhibited on trials when the memorandum is at the 270° location. A reciprocal pathway allows for a pyramidal neuron with a 270° memory field to inhibit one with a 90° memory field. The proposed arrangement of excitatory–inhibitory units, which could explain the opponent memory fields of neurons in and around the principal sulcus (an example of which is shown in Fig. 1), remains to be tested. However, it is clear from single unit recording that prefrontal neurons have opponent memory fields (17), that pyramidal cells and interneurons interact (46), and (from electronmicroscopic evidence) that pyramidal cells innervate interneurons in the prefrontal cortex (52), and basket cells innervate pyramidal cells (53) in the manner illustrated.
Figure 4.
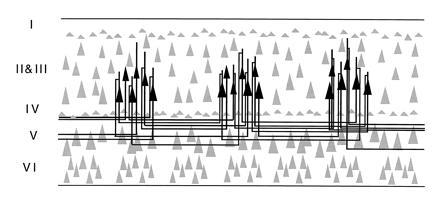
Summary diagram illustrating the patterns of local horizontal intrinsic connections in prefrontal cortex (Walker’s areas 46 and 9) as retrogradely labeled with chholera toxin B subunit. Labeled neurons in layer IIIc in particular form spaced clusters of pyramidal cells with presumed similar best directions. [Reproduced with permission from ref. 48 (Copyright 1995, Wiley–Liss, Inc.).]
Figure 5.
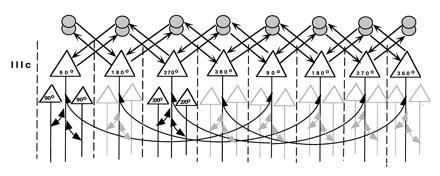
A model of working memory circuitry consisting of modules or clusters of tuned pyramidal neurons (e.g., 90°, 180°, and 270°) arrayed by target location and directly interconnected with each other by their local excitatory axon collaterals (long, thin, curved arrows). Clusters of pyramidal neurons with like best directions are interconnected in a manner similar to isoorientation columns in visual cortex. Two inhibitory interneurons (circles, presumed basket cells in the diagram) provide the reciprocal interconnections (arrows) between pyramidal cells with opposite best directions that could explain the opponent memory fields observed by Funahashi et al. (17). For simplicity, only the 90° to 270° and 270° to 90° ensembles are illustrated. For now, the organization of the pyramidal cells with particular memory fields is hypothetical, as is the reciprocity of the excitatory–inhibitory units. Further analysis of these local circuits is essential for analyzing the neural substrates of working memory. [Reproduced with permission from ref. 51 (Copyright 1995, Cell Press).]
Direct and Indirect Modes of Regulation of Pyramidal Cell Firing By Dopamine
Although it has been known for several decades that the frontal lobe receives a major dopamine innervation (54, 55, 56), researchers have only recently been able to link dopamine afferents to specific cellular targets and neuronal circuits. Understanding the details of this linkage in prefrontal circuits may be important in resolving the various quandaries concerning the mechanisms of dopamine action or cognitive processes as well as the validity of the dopamine hypothesis of diseases like schizophrenia. An important development in the field that may provide some insight into these issues is the recognition that dopamine’s actions must be viewed within a wider context of interaction with other neurotransmitter systems, notably the excitatory amino acid neurotransmitter glutamate (57, 58), the inhibitory neurotransmitter GABA (59, 60, 61), and the cholinergic neurotransmitters (62). Accordingly, studies in my laboratory have, in recent years, focused on the functional and chemical architecture of the prefrontal cortex with particular emphasis on the dopaminergic modulatory influences impacting pyramidal cell firing in the prefrontal cortex. As the dorsolateral prefrontal cortex in nonhuman primates is among the several areas of the primate brain that both receives a substantial dopamine innervation (54, 56) and is linked at the circuit and cellular levels to working memory functions, it represents an excellent model system for examining these interactions and may provide a clue to the predominance of prefrontal dysfunction in psychopathologies.
Physiological studies have consistently indicated that dopamine has inhibitory actions on prefrontal cortical neurons (63, 64, 65, 66) and have provided powerful evidence of dopamine modulation of pyramidal cell excitability (63, 67, 68, 69, 70). Recent anatomical studies have elucidated at least three distinct substrates that could underlie this interaction. One mode of action appears to be direct through synapses on the spines of cortical pyramidal neurons both in monkey (71) and rodent (72, 73) prefrontal cortex (Fig. 6). Through this mechanism, dopamine axons are placed in direct contact with the major projection neurons of the prefrontal cortex—the cortico-thalamic, cortico-striatal, and cortico-cortical projections—and presumably can modulate their activity directly. There is thus a reasonable degree of “targeting” in the dopaminergic projections to the dorsolateral prefrontal cortex, in addition to the proverbial “sprinkler system” that has characterized dopamine traditionally. However, a second mode of action in prefrontal cortex is undoubtedly nonsynaptic. Nonsynaptic neurotransmission may be a pervasive means of altering pyramidal cell activity as numerous dopamine varicosities are observed in nonsynaptic relationship to cortical elements (74, 75). For example, D1 receptors have been localized to spines of pyramidal cells that appear to lack a dopamine synaptic terminal, but these receptors are invariably apposed to glutamatergic synapses on the same spine (53). Members of the D1 family of dopamine receptors have been found to be particularly prominent in the prefrontal cortex of primates (42, 66, 67) and both D1 and D5 receptor proteins have been localized to the distal dendrites and spines of pyramidal cells (78). As mentioned above, there is evidence that the D1 receptor family may be importantly involved in regulating neurons which subserve specific working memory functions (63).
Figure 6.
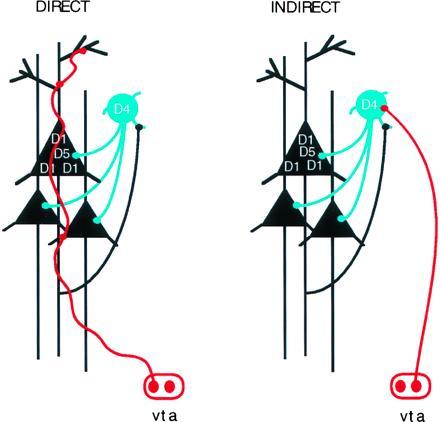
Diagram illustrating two generic synaptic arrangements involving dopamine and major dopamine receptor subtypes CD1, D4, and D5 in the synaptic circuitry of the prefrontal cortex. (Left) Direct mode of pyramidal cell modulation; dopamine (DA) afferents (in red) labeled with a dopamine-specific antibody terminate on the distal dendrites (and spines) of a pyramidal cell (black triangles) in the prefrontal cortex; for further details see Goldman-Rakic et al. (60). vta, Ventral tagmental area (where dopamine cell bodies reside). (Right) Indirect mode of pyramidal cell modulation via dopamine modulation of GABAergic interneurons (blue circle) (81).
A third mode of dopamine action on pyramidal cell firing may be indirect through feedforward inhibition from nonpyramidal neurons (Fig. 6). We have recently shown that pyramidal and nonpyramidal neurons physiologically interact as monkeys hold a particular item of information in working memory (14). The indirect action of dopamine on this circuit derives from two recent discoveries: (i) the identification of dopamine synaptic contacts on nonpyramidal GABAergic neurons in prefrontal cortex, though these contacts appear to be less common than those on pyramidal neurons (71, 75, 79, 80), and (ii) the finding that the D4 member of the D2 family of dopamine receptors is localized postsynaptically on a subset of GABA interneurons (81). D4 receptors are also observed in pyramidal cell spines, though possibly not as densely as are D1 receptors. The localization to interneurons is noteworthy because it suggests that D2 receptor family sites may preferentially inhabit interneurons, whereas the D1 and the D5 receptors appear to be preferentially localized to pyramidal neurons (75, 78). These new results raise the interesting possibility that D4 antagonists may have both direct and indirect effects on pyramidal cell firing. The direct effects could be mediated by D4 receptors directly on cortical pyramidal cells, where, as mentioned, dopamine’s action is primarily inhibitory. The indirect effects could be mediated by the D4 receptor on nonpyramidal cells. Physiological studies have shown that dopamine may inhibit pyramidal cell firing, in part, by activation of certain GABA inhibitory neurons (68, 83) but other actions are also possible. Further, GABA release in the cerebral cortex is known to be modulated by D2-like receptors (83).
It should also be mentioned that the soma and dendrites of nonpyramidal neurons in the prefrontal cortex are a postsynaptic target of serotoninergic axons, although pyramidal neurons also receive some serotoninergic afferents (84, 85). Serotonin 5-HT2 receptors have been reported in cortical interneurons by Morilak et al. (86). The presumed colocalization of D4 and serotonin receptors in nonpyramidal neurons could provide a basis for a synergistic action of these monoamines on cognitive function. Given that the atypical neuroleptic clozapine has a high affinity for the 5-HT2 as well as the D4 receptor (87, 88), the new D4 localization data focuses attention on the nonpyramidal cell as a major target of pharmacological intervention and offers a possible neural explanation for the reported improvements in prefrontal functions (e.g., negative symptoms) by atypical neuroleptics (89). As both dopamine and serotonin have complex effects—modulating pyramidal cell firing directly and indirectly through control of nonpyramidal cell firing—understanding the relative impact of direct and indirect actions of these neurotransmitters on pyramidal cell firing in vivo may hold the key to effective pharmacotherapy of all classes of symptoms in schizophrenia.
Functional–Anatomic Correlations
Dopamine regulation of excitatory neurotransmission in cortical circuits is supported not only by electrophysiological studies in slices of rodent prefrontal cortex (68, 69, 70), but also by studies in human cortical “slices” (67) and also in awake behaving nonhuman primates in which the neurons studied could be functionally characterized while the monkeys performed a working memory task (63). Our study in primates took advantage of the remarkable fact that the process of mental representation can be captured as a sequence of electrophysiological events in prefrontal neurons (17, 18, 19). Analogous to visual receptive fields recorded in primary visual cortex, the memory field of a prefrontal neuron is defined as maximal firing of a neuron during recall or transient storage of a specific item of information, e.g., the location of an object in one or a few locations of the visual field (17, 18, 19), or the memory of a particular face or object (14). In ref. 63, we reported that iontophoretic application of a D1 receptor antagonist enhanced the memory field of the neurons, i.e., increased cell firing only for the cell’s best direction while the cell’s responses were unaltered during behavioral baseline or when nonpreferred targets were recalled. This pattern of selective enhancement of a cell’s mnemonic responses can be accounted for by a juxtaposition in the recorded cell of an excitatory input, e.g., from a parietal afferent, and a dopamine modulatory input such as exists in the triadic synaptic complex described above. The effect we observed was biphasic—both excess stimulation and excess blockade of the D1 receptor inhibited cell firing. Recent in vitro studies by Yang and Seamans (89) are enlarging our understanding of the ionic mechanisms by which D1 stimulation regulates pyramidal cell firing, and such studies together with the findings described above could provide a cellular basis for the commonly observed deficits in working memory consequent to dopamine depletion (91, 92) or, as recently demonstrated, to conditions which result in hyperdopaminergia (93, 94). It is tempting to speculate that fluctuations of dopamine release and dopamine receptor occupancy, and consequent effects on excitatory transmission in information processing pathways, may account for fluctuations in cognitive performance under different conditions of performance or even in relation to symptom expression during the course of schizophrenia both before and during drug treatment. If both too little and too much dopamine D1 stimulation are detrimental to prefrontal function, as the aforementioned studies suggest, then different treatment strategies may be useful at different stages of the disease. Given the evidence that D1 stimulation can modulate excitatory transmission in pyramidal neurons both in vitro and in vivo, the latter on neurons specifically engaged in working memory, it will be important to study the potential functional significance of D1 receptors in the pathophysiology and/or treatment of schizophrenia. The study on the working memory-enhancing potential of low doses of D1 receptor antagonists administered systemically in our laboratory indicate that optimal D1 occupancy for cognitive performance is achieved at low doses (G. V. Williams and P.S.G.-R., unpublished observations). It will also be of value to examine the effects of common drug therapies on working memory and to learn whether cognitive performance can be related to cellular operations altered by these drugs.
Knowledge of cortical architecture should provide new insights into the action of neuroleptics and, by inverse reasoning, the underlying pathophysiology of the disease. As mentioned earlier, the local circuit formed by pyramidal and nonpyramidal neurons constitutes the elements of an information-processing architecture that underlies the capacity to hold a particular item of information in working memory (14). Differential innervation of the two principal components of a functional unit of cortex by dopamine and serotonin, respectively, opens the possibility for an integrated view of cortical dysfunction in schizophrenia—namely that glutamate, GABA, serotonin, or dopamine singly or in combination could disturb the prefrontal circuitry essential for working memory. The same net effect could be produced by alterations in other neurotransmitters, for example, cholinergic and adrenergic neuortransmissions that are as much a part of the cortical circuitry as the neurotransmitters highlighted in this section. Thus, dysfunction in any one of these neurotransmitter systems, their signaling mechanisms and/or biosynthetic pathways could produce the same phenotypic end result. Each is an integral part of an elemental functional circuit whose output we have shown has consequences for information processing. Disturbances in this circuitry could take the form of inadequate maintenance of a representation, or even inadequate cessation of a mental representation in the absence of the triggering stimulus that might qualify as an hallucination. In our view, it is possible to imagine how either dopamine excess or deficiency and/or a pyramidal cell deficiency or both could alter pyramidal cell modulation in a profound way with the net effect being that the pyramidal cell can no longer integrate its myriad informational inputs and no longer maintain information on line. Timing of information will be off kilter, and fragmentation of the thought process could result and/or the brain systems guiding behavior could be thrown into default mode, leading to reliance on automatic, prepotent, and stereotypic responses, and absence of forethought.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: GABA, γ-aminobutyric acid.
References
- 1.Gormezano I. In: Experimental Methods and Instrumentation in Psychology. Sidowski J B, editor. New York: McGraw–Hill; 1966. pp. 385–428. [Google Scholar]
- 2.Squire L, Cohen N J. In: Neurobiology of Memory and Learning. Lynch G, McGaugh J L, Weinberger N M, editors. New York: Guilford; 1984. pp. 3–64. [Google Scholar]
- 3.Tulving E. Episodic and Semantic Memory. New York: Academic; 1972. pp. 381–403. [Google Scholar]
- 4.Atkinson R C, Shiffrin R M. In: The Psychology of Learning and Motivation: Advances in Research and Theory. Spence K W, editor. Vol. 2. New York: Academic; 1968. pp. 89–195. [Google Scholar]
- 5.Norman D A. Models of Human Memory. New York: Academic; 1970. [Google Scholar]
- 6.Baddeley A. Working Memory. London: Oxford Univ. Press; 1986. [Google Scholar]
- 7.Carpenter P A, Just M A. In: Complex Information Processing: The Impact of Herbert A. Simon. Klahr D, Kotovsky K, editors. Hillsdale, NJ: Lawrence Erlbaum Associates; 1988. [Google Scholar]
- 8.Reddy D R. In: Perception and Production of Fluent Speech. Cole R A, editor. Hillsdale, NJ: Lawrence Erlbaum Associates; 1980. pp. 215–242. [Google Scholar]
- 9.Goldman-Rakic P S. In: Handbook of Physiology. Plum F, Mountcastle V, editors. Vol. 5. Bethesda: Am. Physiol. Soc.; 1987. pp. 373–417. [Google Scholar]
- 10.Goldman-Rakic P S. Annu Rev Neurosci. 1988;11:137–156. doi: 10.1146/annurev.ne.11.030188.001033. [DOI] [PubMed] [Google Scholar]
- 11.Selemon L D, Goldman-Rakic P S. J Neurosci. 1988;8:4049–4068. doi: 10.1523/JNEUROSCI.08-11-04049.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavada C, Goldman-Rakic P S. J Comp Neurol. 1989;287:393–421. doi: 10.1002/cne.902870402. [DOI] [PubMed] [Google Scholar]
- 13.Cavada C, Goldman-Rakic P S. J Comp Neurol. 1989;287:422–445. doi: 10.1002/cne.902870403. [DOI] [PubMed] [Google Scholar]
- 14.Wilson F A W, O’Scalaidhe S P, Goldman-Rakic P S. Science. 1993;260:1955–1958. doi: 10.1126/science.8316836. [DOI] [PubMed] [Google Scholar]
- 15.Friedman H R, Goldman-Rakic P S. J Neurosci. 1988;8:4693–4706. doi: 10.1523/JNEUROSCI.08-12-04693.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman H R, Goldman-Rakic P S. J Neurosci. 1994;14:2775–2788. doi: 10.1523/JNEUROSCI.14-05-02775.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funahashi S, Bruce C J, Goldman-Rakic P S. J Neurophysiol. 1989;61:331–349. doi: 10.1152/jn.1989.61.2.331. [DOI] [PubMed] [Google Scholar]
- 18.Fuster J M, Alexander G E. Science. 1971;173:652–654. doi: 10.1126/science.173.3997.652. [DOI] [PubMed] [Google Scholar]
- 19.Kubota K, Niki H. J Neurophysiol. 1971;34:337–347. doi: 10.1152/jn.1971.34.3.337. [DOI] [PubMed] [Google Scholar]
- 20.MacAvoy M G, Gottlieb J P, Bruce C J. Cereb Cortex. 1991;1:95–102. doi: 10.1093/cercor/1.1.95. [DOI] [PubMed] [Google Scholar]
- 21.Freedman M, Oscar-Berman M. Behav Neurosci. 1986;100:337–342. doi: 10.1037//0735-7044.100.3.337. [DOI] [PubMed] [Google Scholar]
- 22.Verin M, Partiot A, Pillon B, Agid Y, Dubois B. Neuropsychologia. 1993;31:1279–1296. doi: 10.1016/0028-3932(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 23.Baker S C, Frith C D, Frackowiak R S J, Dolan R J. Cereb Cortex. 1996;6:612–619. doi: 10.1093/cercor/6.4.612. [DOI] [PubMed] [Google Scholar]
- 24.Gold J M, Berman K F, Randolph C, Goldberg T E, Weinberger D R. Neuropsychology. 1966;10:3–10. [Google Scholar]
- 25.Goldberg T E, Randolph C, Berman K F, Gold J M, Weinberger D R. NeuroImage. 1996;3:69–78. doi: 10.1006/nimg.1996.0008. [DOI] [PubMed] [Google Scholar]
- 26.Nichelli P, Grafman J, Pietrini P, Alway D, Carton J C, Miletich R. Nature (London) 1994;369:191. doi: 10.1038/369191a0. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy G, Blamire A M, Puce A, Nobre A C, Bloch G, Hyder F, Goldman-Rakic P S, Shulman R G. Proc Natl Acad Sci USA. 1994;91:8690–8694. doi: 10.1073/pnas.91.18.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owen A M, Evans A C, Petrides M. Cereb Cortex. 1996;6:31–38. doi: 10.1093/cercor/6.1.31. [DOI] [PubMed] [Google Scholar]
- 29.Smith E E, Jonides J, Koeppe R A. Cereb Cortex. 1996;6:11–20. doi: 10.1093/cercor/6.1.11. [DOI] [PubMed] [Google Scholar]
- 30.Sweeney J A, Mintun M A, Kwee B S, Wiseman M B, Brown D L, Rosenberg D R, Carl J R. J Neurophysiol. 1996;75:454–468. doi: 10.1152/jn.1996.75.1.454. [DOI] [PubMed] [Google Scholar]
- 31.Adcock R A, Constable R T, Gore J C, Goldman-Rakic P S. NeuroImage. 1996;3:S526. [Google Scholar]
- 32.Cohen J D, Forman S D, Braver T S, Casey B J, Servan-Schreiber D, Noll D C. Hum Brain Mapp. 1994;1:293–304. doi: 10.1002/hbm.460010407. [DOI] [PubMed] [Google Scholar]
- 33.Courtney S M, Ungerleider L G, Keil K, Haxby J V. Cereb Cortex. 1996;6:39–49. doi: 10.1093/cercor/6.1.39. [DOI] [PubMed] [Google Scholar]
- 34.McCarthy G, Puce A, Constable R T, Krystal J H, Gore J C, Goldman-Rakic P. Cereb Cortex. 1996;6:600–611. doi: 10.1093/cercor/6.4.600. [DOI] [PubMed] [Google Scholar]
- 35.Raichle M E, Fiez J A, Videen T O, MacLeod A K, Pardo J V, Fox P T, Petersen S E. Cereb Cortex. 1994;4:8–26. doi: 10.1093/cercor/4.1.8. [DOI] [PubMed] [Google Scholar]
- 36.Demb J B, Desmond J E, Wagner A D, Vaidya C J, Glover G H, Gabrieli J D E. J Neurosci. 1995;15:5870–5878. doi: 10.1523/JNEUROSCI.15-09-05870.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiez J A, Raife E A, Balota D A, Schwarz J P, Raichle M E, Petersen S E. J Neurosci. 1996;16:808–822. doi: 10.1523/JNEUROSCI.16-02-00808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulesu E, Frith C D, Frackowiak R S J. Nature (London) 1993;362:342–345. doi: 10.1038/362342a0. [DOI] [PubMed] [Google Scholar]
- 39.Price C J, Wise R J S, Frackowiak R S J. Cereb Cortex. 1996;6:62–69. doi: 10.1093/cercor/6.1.62. [DOI] [PubMed] [Google Scholar]
- 40.Shallice T. Philos Trans R Soc London B. 1982;298:199–209. doi: 10.1098/rstb.1982.0082. [DOI] [PubMed] [Google Scholar]
- 41.Kojima S, Goldman-Rakic P S. Brain Res. 1982;248:43–49. doi: 10.1016/0006-8993(82)91145-3. [DOI] [PubMed] [Google Scholar]
- 42.Goldman-Rakic P S, Lidow M S, Gallager D W. J Neurosci. 1990;10:2125–2138. doi: 10.1523/JNEUROSCI.10-07-02125.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funahashi S, Bruce C J, Goldman-Rakic P S. J Neurophysiol. 1990;63:814–831. doi: 10.1152/jn.1990.63.4.814. [DOI] [PubMed] [Google Scholar]
- 44.Funahashi S, Bruce C J, Goldman-Rakic P S. J Neurophysiol. 1991;65:1464–1483. doi: 10.1152/jn.1991.65.6.1464. [DOI] [PubMed] [Google Scholar]
- 45.Sillito A M, Murphy P C. In: Neurotransmitters and Cortical Function. Avioli M, Reader T A, Dykes R W, Gloor P, editors. New York: Plenum; 1986. pp. 167–186. [Google Scholar]
- 46.Wilson F A W, O’Scalaidhe S P, Goldman-Rakic P S. Proc Natl Acad Sci USA. 1994;91:4009–4013. doi: 10.1073/pnas.91.9.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leavitt J B, Lewis D A, Yoshioka T, Lund J S. J Comp Neurol. 1993;338:360–376. doi: 10.1002/cne.903380304. [DOI] [PubMed] [Google Scholar]
- 48.Kritzer M K, Goldman-Rakic P S. J Comp Neurol. 1995;359:131–143. doi: 10.1002/cne.903590109. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert C D. Cereb Cortex. 1993;3:373–386. doi: 10.1093/cercor/3.5.373. [DOI] [PubMed] [Google Scholar]
- 50.Goldman P S, Nauta W J H. Brain Res. 1977;l22:393–413. doi: 10.1016/0006-8993(77)90453-x. [DOI] [PubMed] [Google Scholar]
- 51.Goldman-Rakic P S. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 52.Williams S M, Goldman-Rakic P S, Leranth C. J Comp Neurol. 1992;320:353–369. doi: 10.1002/cne.903200307. [DOI] [PubMed] [Google Scholar]
- 53.Somogyi P, Kisvardy Z F, Martin K A C, Whitteridge D. Neuroscience. 1983;10:261–294. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- 54.Brown R M, Crane A, Goldman P S. Brain Res. 1979;168:133–150. doi: 10.1016/0006-8993(79)90132-x. [DOI] [PubMed] [Google Scholar]
- 55.Lewis D A, Foote S L, Goldstein M, Morrison J H. Brain Res. 1988;449:225–243. doi: 10.1016/0006-8993(88)91040-2. [DOI] [PubMed] [Google Scholar]
- 56.Williams S M, Goldman-Rakic P S. Cereb Cortex. 1993;3:199–222. doi: 10.1093/cercor/3.3.199. [DOI] [PubMed] [Google Scholar]
- 57.Carlsson M, Carlsson A. Schizophrenia Bull. 1990;16:425–432. doi: 10.1093/schbul/16.3.425. [DOI] [PubMed] [Google Scholar]
- 58.Olney J W, Farber N B. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 59.Benes F M, McSparren J, Bird E D, San Giovanni J P, Vincent S L. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- 60.Bourdelais A J, Deutch A Y. Cereb Cortex. 1994;4:69–77. doi: 10.1093/cercor/4.1.69. [DOI] [PubMed] [Google Scholar]
- 61.Vincent S L, Adamec E, Sorensen I, Benes F M. Synapse. 1994;17:26–35. doi: 10.1002/syn.890170104. [DOI] [PubMed] [Google Scholar]
- 62.Day J, Fibiger H C. Neuroscience. 1993;54:643–648. doi: 10.1016/0306-4522(93)90235-8. [DOI] [PubMed] [Google Scholar]
- 63.Williams G V, Goldman-Rakic P S. Nature (London) 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 64.Bunney B S, Aghajanian G K. Life Sci. 1976;19:1783–1792. doi: 10.1016/0024-3205(76)90087-4. [DOI] [PubMed] [Google Scholar]
- 65.Fitzgerald L W, Deutch A Y, Gasic G, Heinemann S F, Nestler E J. J Neurosci. 1995;15:2453–2461. doi: 10.1523/JNEUROSCI.15-03-02453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferron A, Thierry A-M, Le Dourarin C, Glowinski J. Brain Res. 1984;302:257–265. doi: 10.1016/0006-8993(84)90238-5. [DOI] [PubMed] [Google Scholar]
- 67.Cepeda C, Radisavljevic Z, Peacock W, Levine M S, Buchwald N A. Synapse. 1992;11:330–341. doi: 10.1002/syn.890110408. [DOI] [PubMed] [Google Scholar]
- 68.Gellman R L, Aghajanian G K. Brain Res. 1993;600:63–73. doi: 10.1016/0006-8993(93)90402-9. [DOI] [PubMed] [Google Scholar]
- 69.Law-Tho D, Hirsch J C, Crepel F. Neurosci Res. 1994;21:151–160. doi: 10.1016/0168-0102(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 70.Yang C R, Seamans J K. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldman-Rakic P S, Leranth C, Williams S M, Mons N, Geffard M. Proc Natl Acad Sci USA. 1989;86:9015–9019. doi: 10.1073/pnas.86.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seguela P, Watkins K C, Descarries L. J Comp Neurol. 1989;289:129–142. doi: 10.1002/cne.902890111. [DOI] [PubMed] [Google Scholar]
- 73.Van Eden C G, Hoorneman E M D, Buijs R M, Matthijssen M A H, Geffard M, Uylings H B M. Neuroscience. 1987;22:849–862. doi: 10.1016/0306-4522(87)92964-2. [DOI] [PubMed] [Google Scholar]
- 74.Smiley J F, Levey A I, Ciliax B J, Goldman-Rakic P S. Proc Natl Acad Sci USA. 1994;91:5720–5724. doi: 10.1073/pnas.91.12.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smiley J F, Goldman-Rakic P S. Cereb Cortex. 1993;3:223–238. doi: 10.1093/cercor/3.3.223. [DOI] [PubMed] [Google Scholar]
- 76.Lidow M S, Goldman-Rakic P S, Gallager D W, Rakic P. Neuroscience. 1991;40:657–671. doi: 10.1016/0306-4522(91)90003-7. [DOI] [PubMed] [Google Scholar]
- 77.Lidow M S, Goldman-Rakic P S, Rakic P, Innis R B. Proc Natl Acad Sci USA. 1989;86:6412–6416. doi: 10.1073/pnas.86.16.6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bergson C, Mrzljak L, Smiley J F, Pappy M, Levenson R, Goldman-Rakic P S. J Neurosci. 1995;15:7821–7837. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smiley J F, Williams S M, Szigeti K, Goldman-Rakic P S. J Comp Neurol. 1992;321:325–335. doi: 10.1002/cne.903210302. [DOI] [PubMed] [Google Scholar]
- 80.Sesack S R, Snyder C L, Lewis D A. J Comp Neurol. 1995;363:264–280. doi: 10.1002/cne.903630208. [DOI] [PubMed] [Google Scholar]
- 81.Mrzljak L, Bergson C, Pappy M, Levenson R, Huff R, Goldman-Rakic P S. Nature (London) 1996;381:245–248. doi: 10.1038/381245a0. [DOI] [PubMed] [Google Scholar]
- 82.Pirot S, Godbout R, Mantz J, Tassin J-P, Glowinski J, Thierry A-M. Neuroscience. 1992;49:857–865. doi: 10.1016/0306-4522(92)90362-6. [DOI] [PubMed] [Google Scholar]
- 83.Retaux S, Besson M J, Penit-Soria J. Neuroscience. 1991;42:61–71. doi: 10.1016/0306-4522(91)90150-m. [DOI] [PubMed] [Google Scholar]
- 84.Smiley J F, Goldman-Rakic P S. J Comp Neurol. 1996;367:431–443. doi: 10.1002/(SICI)1096-9861(19960408)367:3<431::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 85.Jakab R L, Goldman-Rakic P S. Neurosci Abstr. 1996;22:905. [Google Scholar]
- 86.Morilak D A, Garlow S K, Ciaranello R D. Neuroscience. 1993;54:701–717. doi: 10.1016/0306-4522(93)90241-7. [DOI] [PubMed] [Google Scholar]
- 87.Meltzer H Y, Matsubara S, Lee J-C. J Pharmacol Exp Ther. 1989;251:238–246. [PubMed] [Google Scholar]
- 88.Leysen J E, Schotte A, Janssen F, Gommeren W, Van Gompel P, Lesage A S, De Backer M D, Luytten W H M L, Amlaiky N, Megens A A H P. In: Schizophrenia: An Integrated View. Fog R, Gerlach J, Hemmingsen R, editors. Copenhagen: Munksgaard; 1995. pp. 344–356. [Google Scholar]
- 89.Lee M A, Thompson P A, Meltzer H Y. J Clin Psychiatry. 1994;55:82–87. [PubMed] [Google Scholar]
- 90.Yang C R, Seamans J K. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brozoski T, Brown R M, Rosvold H E, Goldman P S. Science. 1979;205:929–932. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- 92.Schneider J S, Kovelowski C J, II. Brain Res. 1990;519:122–128. doi: 10.1016/0006-8993(90)90069-n. [DOI] [PubMed] [Google Scholar]
- 93.Murphy B L, Arnsten A F T, Goldman-Rakic P S, Roth R H. Proc Natl Acad Sci USA. 1996;93:1325–1329. doi: 10.1073/pnas.93.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Verma A, Moghaddam B. J Neurosci. 1996;16:373–379. doi: 10.1523/JNEUROSCI.16-01-00373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]