

Abstract
The imprinting and transcription of the 500 kb genomic region surrounding the mouse Peg3 is predicted to be regulated by the Peg3-differentially methylated region (DMR). In the current study, this prediction was tested using a mutant mouse line lacking this potential imprinting control region (ICR). At the organismal level, paternal and maternal transmission of this knockout (KO) allele caused either reduced or increased growth rates in the mouse, respectively. In terms of the imprinting control, the paternal transmission of the KO allele resulted in bi-allelic expression of the normally maternally expressed Zim2, whereas the maternal transmission switched the transcriptionally dominant allele for Zfp264 (paternal to maternal). However, the allele-specific DNA methylation patterns of the DMRs of Peg3, Zim2 and Zim3 were not affected in the mice that inherited the KO allele either paternally or maternally. In terms of the transcriptional control, the paternal transmission caused a dramatic down-regulation in Peg3 expression, but overall up-regulation in the other nearby imprinted genes. Taken together, deletion of the Peg3-DMR caused global changes in the imprinting and transcription of the Peg3 domain, confirming that the Peg3-DMR is an ICR for this imprinted domain.
INTRODUCTION
Peg3 was the first-imprinted gene identified from the 500 kb domain located on the human chromosome 19q13.4/proximal mouse chromosome 7. Since the discovery of the paternally expressed Peg3, six additional imprinted genes have been identified: three paternally expressed genes (Usp29, Zfp264, APeg3) and three maternally expressed genes (Zim1, Zim2, Zim3) (1,2). Most of these imprinted genes are predicted to be DNA-binding transcription factors based on the presence of zinc finger motifs within their open reading frames. The presence of these imprinted genes was also predicted based on earlier mouse genetic studies involving uniparental disomy. Maternal duplication (paternal deficiency) of proximal mouse chromosome 7 causes neonatal lethality, whereas paternal duplication (maternal deficiency) of the same interval results in reduced growth rates and viability (3). The human PEG3 region is also associated with several types of cancer. In particular, human PEG3 expression is often missing in many cases of glioma, breast and ovarian cancer patients, which are usually caused by complete DNA methylation of the promoter region of PEG3 (4,5,6).
Mouse knockout (KO) experiments have identified small genomic regions as imprinting control regions (ICRs), including the ICRs of H19/Igf2, Kcnq1, Igf2r, Gnas, Gtl2/Dlk1 and Snrpn (7–10). According to these studies, ICRs have three main features. First, ICRs control mono-allelic expression (imprinting) and transcription of their associated imprinted genes in somatic cells. Thus, deletion of these ICRs usually has global effects on the imprinting and transcription of their associated genes. Secondly, imprinted genes are usually associated with CpG-rich regions displaying allele-specific DNA methylation patterns, hence designated differentially methylated regions (DMRs). Only a few DMRs, however, obtain DNA methylation as a parental mark during gametogenesis, and these are thus termed germline DMRs. Interestingly, all the known ICRs are germline DMRs. Thirdly, some of ICRs tend to show tandem repeat sequence structures, and these tandem repeats often contain DNA-binding sites for specific transcription factors, such as CTCF and YY1 (11). A series of subsequent studies have confirmed that these transcription factors play very critical roles in many aspects of imprinting and transcriptional control for the associated imprinted genes (11,12).
The clustering of the seven imprinted genes in the Peg3 domain has been a strong indication that the imprinting of this domain is also regulated through unknown ICRs. DNA methylation analyses have identified three DMRs within this domain, including the DMRs of Peg3, Zim2 and Zim3, but the Peg3-DMR is the only germline DMR obtaining DNA methylation during oogenesis (4,13,14). Also, the Peg3-DMR displays an unusual tandem repeat structure, and the core sequence of these repeats is a binding site for YY1 (15–17). These data suggest that the Peg3-DMR is likely an ICR for this imprinted domain. To test this prediction, we have generated one mutant allele in mice lacking the Peg3-DMR. According to the results from the mutant allele studies, deletion of the Peg3-DMR causes global changes in the imprinting and transcription of the Peg3 domain. The mutant mice also displayed gross-level phenotypes, changes in body weight as well as partial embryonic lethality. These data confirm that the Peg3-DMR is indeed an ICR for this domain.
RESULTS
Generation of a KO allele
To target the Peg3-DMR, we used a KO vector, pTNT (18), which contains positive (neomycin resistance gene) and negative (HSV-thymidine kinase gene) selection markers. The two genomic fragments flanking the 2.5 kb YY1-binding region within the 4 kb Peg3-DMR have been subcloned into the pTNT vector as ‘homologous hooks’. These two fragments include a 3.0 kb genomic fragment surrounding the first exon of Peg3 and another 8.9 kb fragment surrounding the second and third exons of Peg3. The constructed vector (Fig. 1) has been used for ES cell transfection. One targeted ES cell was obtained through screening ∼2000 clones with southern blot and long-distance polymerase chain reaction (PCR) analyses (Fig. 1B). After careful analyses of the identified targeted ES cell, this targeted ES cell was microinjected into the blastocysts of e3.5 embryos from C57BL/6J (B6) mice to produce chimera mice. The obtained male chimeras were bred with female B6 mice to produce F1 mice with germline transmission of the KO allele. The F1 mice with the germline transmission were further bred with transgenic lines expressing germline Cre recombinase (Zp3-cre) to remove the NeoR sequence. Proper deletion of the NeoR sequence was confirmed through long-distance PCR and also through sequencing (data not shown). The mice with confirmed deletion of NeoR have been used for the experiments described in the following section.
Figure 1.

The overview of the Peg3 domain and targeting scheme. (A) Schematic representations of the Peg3 domain (upper panel). Each imprinted gene is indicated with an arrow. The genes with pink are paternally expressed genes, whereas the genes with blue are maternally expressed genes. The three DMRs are indicated with gray boxes. Targeting scheme (lower panel). The 4.0 kb Peg3-DMR contains the first exons of Peg3 and Usp29 and two evolutionarily conserved elements (CSE1 and CSE2 or YY1-binding site) that are located within the first intron of Peg3. The current targeting scheme was designed to delete the 2.5 kb genomic region harboring CSE1 and YY1-binding sites. The transcriptional direction of Peg3 and Usp29 is indicated with arrows, and exons are indicated with thick vertical lines. The region corresponding to the neomycin resistance gene (Neo) along with the two flanking loxP sites within the targeting vector are also indicated by an open box and triangles, respectively. Arrows with numbers underneath ‘Targeted allele’ indicate primers with relative positions that were used for long-distance PCR. (B) Southern blot analyses using the DNA isolated from ES cells that have been transfected with the targeting vector. BamHI-digested DNA was hybridized with the 5′-side probe, showing an additional 5 kb fragment in a targeted clone (marked with *). (C) Western blot analyses using the protein extracts prepared from the neonatal brains of WT and KO pups. This analysis revealed at least 3–4-fold down-regulation of Peg3 in the pup inheriting the KO allele paternally. (D) Picture of 5-day-old WT and KO pups of paternal transmission.
Deletion effects on the survival and birth weight of the mouse
We first performed breeding experiments to analyze the effects of the deletion at the organismal level. We set up three different breeding schemes to test the deletion effects on the mouse: Scheme I, female wild-type littermates × male heterozygotes (+/−) for the paternal transmission of the KO allele; Scheme II, female heterozygotes (+/−) × male wild-type littermates for the maternal transmission of the KO allele; and Scheme III, female and male heterozygotes (+/−) to test the derivation of the homozygotes. The pups derived from these breedings were genotyped, and their body weights were measured and converted into percentile scores as an indicator of their health status (Fig. 2). The results from these breeding experiments are as follows. Paternal transmission of the KO allele has two major effects: partial embryonic lethality and reduced body weight (Fig. 2A). The average litter size of this breeding was 6.3 mice in the 129/B6-mixed background, which is smaller than the normal litter size (∼10) of the same genetic background. Also, the ratio of the heterozygotes to wild-type littermates was 0.52 to 1, significantly deviated from the expected Mendelian ratio of 1 to 1. These results suggest that some of the heterozygotes have died during the gestation period. Also, the majority of the heterozygotes tend to show a much smaller body weight than their littermates (Figs 1D and 2A). This was also much more pronounced in male than in female heterozygotes (Supplementary Material, Fig. S4). This smaller body weight (∼80% of the wild-type littermates; t-test, P= 0.0049) was maintained throughout the developmental stage, even past the weaning to the adult stages.
Figure 2.

Deletion effects on the survival and birth weight of the mouse. Male (A) and female (B) heterozygotes were individually bred with their wild-type littermates for the paternal and maternal transmission of the KO allele. The pups derived from these crosses were analyzed in terms of their litter sizes, mendelian ratios and birth weight percentiles. Birth weight percentiles were calculated by dividing the weight of each pup by the average weight of the litter. The pups inheriting the KO allele paternally showed much lower body weights than their littermates. The values on the X-axis indicate percentile scores, while the values on the Y-axis indicate the number of mice.
To further follow up the observed partial embryonic lethality, we performed a set of timed mating experiments. We obtained three litters of conceptuses, the first litter at 9.5 dpc (nine conceptuses) and the second and third litters at 14.5 dpc (seven and six conceptuses, respectively). Out of the nine conceptuses at the 9.5 dpc set, one embryo was already in the process of resorption and the other one was very small with apparent neurulation defects. Three out of the seven embryos successfully genotyped were heterozygotes. The harvested embryos and placentas from the second and third litters were overall normal in terms of morphology and size, although we did observe two conceptuses in the second litter going through the resorption process. Three and one embryos were heterozygotes among the second and third sets, respectively. Thus, the overall ratio of the heterozygotes to wild-type embryos is 4 to 9, which is significantly deviated from the expected Mendelian ratio of 1 to 1. This suggests that the embryonic lethality may occur around or right after 9.5 dpc.
In contrast, maternal transmission of the KO allele did not cause any significant effect on the survival and body weight of the 1-day-old pups derived from the second breeding scheme (Fig. 2B). The litter size (9.7) was comparable to the average litter size (10) of the 129/B6-mixed background. The ratio of the heterozygotes to wild-type littermates was 0.9 to 1, which is also close to the expected ratio of Mendelian inheritance. However, the majority of the heterozygotes from this breeding were an average of 7% heavier than their wild-type littermates at weaning age (based on the results derived from 5 litters representing 30 mice; t-test, P = 0.0008) (Supplementary Material, Fig. S5). This boosted weight was diminished when the mice reached 2 months of age. This observed boost in growth rate was unexpected since the maternal allele of the Peg3-DMR is normally repressed and non-functional due to DNA methylation. We believe that this unexpected outcome is likely an outcome of some changes in the Peg3 domain that are caused by the deletion of the Peg3-DMR. On the other hand, Breeding Scheme III did not yield any homozygote (wild-types:heterozygotes:homozygotes = 9:16:0) with a slightly smaller litter size (8.3), suggesting potential lethality during the gestation period. These data further suggest that at least one allele, either paternal or maternal, of the Peg3-DMR is required for the survival of the mouse. In sum, the breeding experiments so far indicate that paternal transmission of the KO allele results in partial embryonic lethality and reduced body weight of the mouse, and that the maternal transmission results in slightly increased body weight of the mouse.
Deletion effects on the imprinting status
We analyzed the effects of deleting the 2.5 kb YY1-binding region within the Peg3-DMR on the imprinting status of the Peg3 domain (Fig. 3). For this series of analyses, we generated two sets of F1 hybrids by breeding the 129/B6-mixed KO line with the PWD/PhJ strain. The first F1 set was derived from the crossing of female heterozygotes (+/−) with male PWD for the maternal transmission of the KO allele (129 × PWD), whereas the second set was obtained from the crossing of female PWD with male heterozygotes (+/−) for the paternal transmission (PWD × 129). For the imprinting tests, we have selected neonatal brains as the main tissue to analyze since the majority of the genes in this domain are known to be expressed in this tissue (19–21). The total RNA isolated from two littermates, KO (−M/+ or +/−P) and wild-type (WT) (+/+), were reverse-transcribed and the subsequent cDNAs were amplified with PCR. To differentiate the two alleles for the imprinting tests, we identified several sequence polymorphisms between the 129 and PWD strains for each gene, but used only the informative polymorphisms that can be visualized by restriction enzyme digestion for the actual imprinting tests. The information regarding the polymorphism–restriction enzyme pair used for each gene is available (Supplementary Material, Table S1).
Figure 3.

Deletion effects on the imprinting status. (A and B) Imprinting test in neonatal brains. (C) Imprinting test in 14.5 dpc embryos. Two reciprocal crosses (129 × PWD, PWD × 129) were performed to derive the F1 hybrids with the maternal and paternal transmission of the KO allele, respectively. Each set of imprinting tests used two littermates (KO and WT). The digestion pattern of each gene's RT–PCR product by a given restriction enzyme was compared with that of two parental strains (PWD and 129). In the case of APeg3, total RNA from the littermates (KO and WT) of the PWD × 129 cross was analyzed with a strand-specific RT–PCR scheme (B). Two reactions for each RNA sample, RT(−) and RT(+) indicating without and with reverse transcriptase, respectively, were included to monitor genomic DNA contamination. We placed rectangles on the gene, the imprinting status of which was shown to be affected by the deletion of the Peg3-DMR.
Consistent with previous studies, the majority of the genes in the Peg3 domain except Zim3 are properly imprinted in the neonatal brains of the two F1 hybrids with an intact Peg3-DMR: the paternal expression of Zfp264, Peg3, Usp29 and APeg3, and the maternal expression of Zim1 and Zim2 (WT lanes in Fig. 3A and B). As seen for other ICRs, the deletion of the Peg3-DMR caused changes in the imprinting status of two neighboring genes, Zfp264 and Zim2 (KO lanes). The maternal transmission of the KO allele led to a switch of the dominant allele for Zfp264, from paternal to maternal (KO lane in 129 × PWD). In contrast, the paternal transmission of the KO allele resulted in bi-allelic expression of the normally maternally expressed Zim2 (KO lane in PWD × 129). This series of imprinting tests were repeated using another set of neonatal littermates, which again yielded consistent results as shown in Figure 3A and B. We also repeated a similar series of imprinting tests using 14.5 dpc embryos (Fig. 3C). The expression levels of Zfp264, Zim3 and Zim2 were very marginal, and thus the imprinting status of these genes could not be determined. In contrast, the expression levels of Usp29, Peg3 and Zim1 were readily detectible, and thus the imprinting status of these genes was determine and compared between the two types of embryos: KO (+/−P) and WT (+/+). As seen in neonatal brains, the paternal transmission of the KO allele did not change the imprinting status of these three genes. We also performed a series of imprinting tests using adult testes (data not shown), but we were not able to derive any conclusions relevant to genomic imprinting since adult testes are made of two types of cells, somatic and germ cells. Overall, the deletion of the Peg3-DMR causes changes in the imprinting status of Zfp264 and Zim2 in neonatal brains.
Deletion effects on the DNA methylation
We also analyzed the deletion effects of the 2.5 kb YY1-binding region within the Peg3-DMR on the DNA methylation levels of the Peg3 domain (Fig. 4). For this series of analyses, we isolated DNA from sperm, oocyte and one somatic tissue-type, neonatal tails. The isolated DNA was treated according to the bisulfite conversion protocol, and the modified DNA was used for PCR amplification. The amplified PCR products were further analyzed with COBRA and individual sequencing. For the analyses of the two germ cells, we used three primer sets targeting the promoter region of Peg3 (primer set A), the first intron region harboring two YY1-binding sites (primer set B), and the first intron region with the KO deletion (primer set C). The results from this analysis are as follows. First, the three PCR products of primer sets A, B or C, all are similarly unmethylated in both KO and WT samples of sperm DNA (Fig. 4B), indicating no major effects on the DNA methylation levels of the Peg3-DMR. Secondly, the DNA isolated from mature oocytes also derived a similar result, displaying no major effects caused by the deletion of the Peg3-DMR (Fig. 4C). The PCR product of primer set B representing the first intron region of Peg3 was completely methylated in the WT, and this result was the same for the PCR product of primer set C representing the deleted region in KO. However, the PCR product of primer set A representing the promoter region was not methylated in both WT and KO oocytes. These data suggest that the promoter region of Peg3 may be unmethylated during oogenesis. Thirdly, the DNA isolated from one somatic tissue, tails, showed no major difference between KO and WT (Fig. 4D). We used the DNA isolated from the same set of F1 individual mice that were used for the imprinting tests (Fig. 3), which further allowed us to analyze DNA methylation in an allele-specific manner. In this analysis, we also included another two regions, the Zim3-DMR and Zim2-DMR. As shown in Figure 4D, both sets of DNA show similar levels of DNA methylation between KO and WT, indicating that both the paternal and maternal transmission of the KO allele do not have any major impact on the DNA methylation levels of the three DMRs. In sum, the deletion of the Peg3-DMR does not have any major impact on the DNA methylation levels of the Peg3 domain during gametogenesis or in somatic tissues.
Figure 4.

Deletion effects on the DNA methylation. (A) Relative positions of the PCR primer sets that were used for DNA methylation analyses. (B) DNA methylation analyses using DNA isolated from sperm. The PCR products of primer set A, B, C, were analyzed with COBRA. (C) DNA methylation analyses using DNA isolated from oocytes. Two sets of oocytes were individually isolated from the 2-month-old females with KO (+/−P) and WT (+/+). Each set of eggs was analyzed with primers A and C for KO and primers A and B for WT. Open and closed circles represent unmethylated and methylated status of a given CpG site, respectively. (D) DNA methylation analyses using the DNA derived from the F1 hybrids. Two sets of hybrids, the maternal and paternal transmission of the KO allele (−M/+ and +/−P), were analyzed first with COBRA (left), and later by individual sequencing (right).
Deletion effects on the transcriptional levels
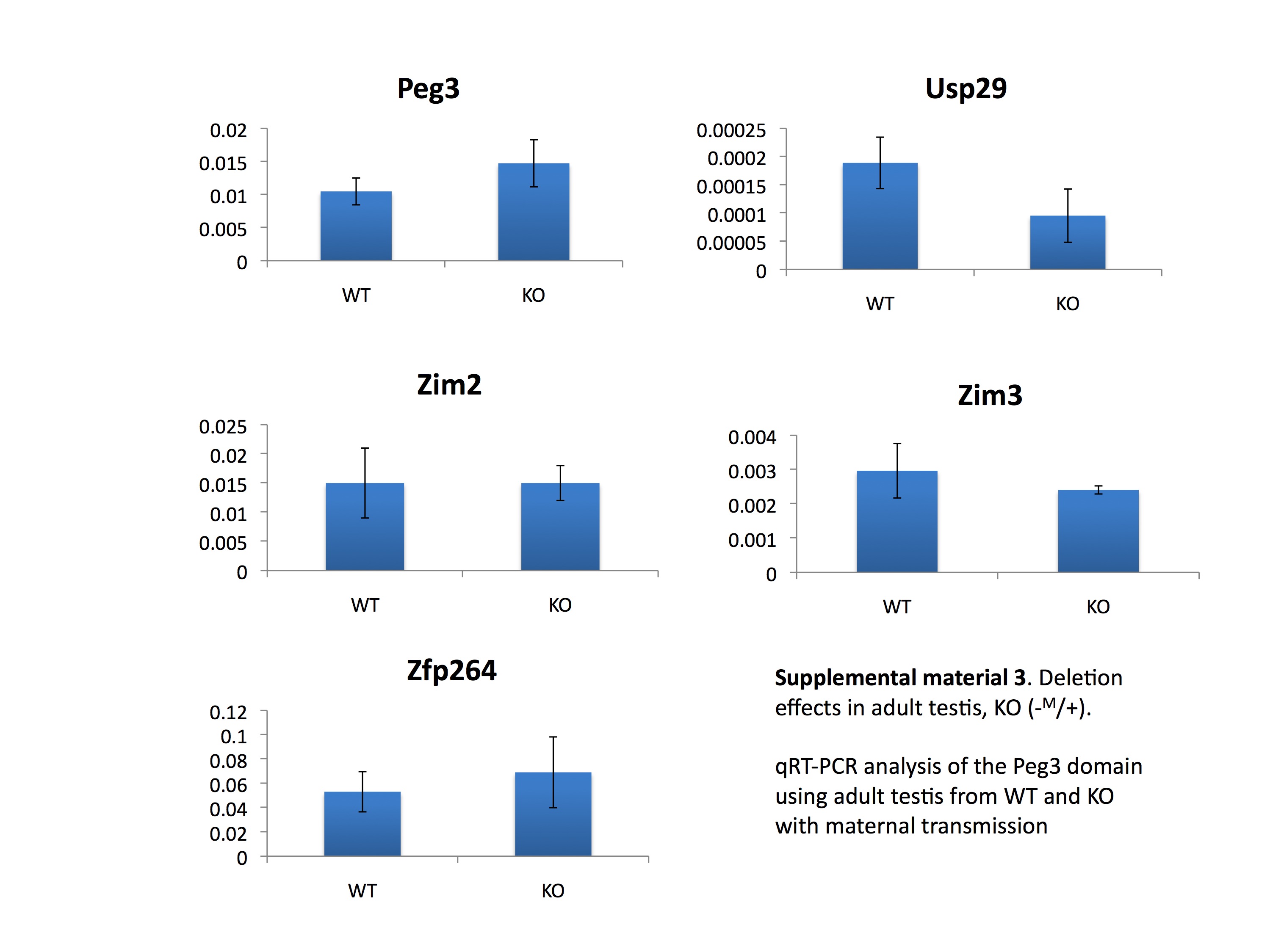
We analyzed the deletion effects of the 2.5 kb YY1-binding region within the Peg3-DMR on the transcriptional levels of the Peg3 domain. For this series of analyses, we isolated total RNA from the neonatal brains and adult testes of the following genotypes: KO (+/−P or −M/+) and WT (+/+) littermates from the paternal or maternal transmission of the KO allele. Total RNA from these mice was first reverse-transcribed and the resulting cDNA was analyzed with qRT–PCR. The results from one representative set out of the three tested sets with the paternal transmission are presented in Figure 5. First, in neonatal brain (Fig. 5A), the deletion of the Peg3-DMR caused a global effect on the transcriptional levels of most of the genes in the Peg3 domain. Zim3 was not included in this analysis since it is expressed only in testis. Among the five genes tested, Peg3 was the only gene showing down-regulation (4-fold decrease) in its transcriptional level. The down-regulation of Peg3 was also confirmed through western blotting (Fig. 1C). The remaining four genes all showed up-regulation with varying levels. In particular, the two maternally expressed genes in the Peg3 domain, Zim1 and Zim2, both displayed up-regulation in KO mice. It is important to point out the observed up-regulation of Zim1 (4-fold increase) since Zim1 usually displays the highest levels of expression among all the genes in the Peg3 domain in embryonic and neonatal stages. We also observed up-regulation of Usp29 and Zfp264, but the up-regulation observed from Zfp264 was not statistically significant. The two other remaining sets also derived similar conclusions as described above. The same series of qRT–PCR was also repeated using the total RNA from the neonatal brain set with the maternal transmission of the KO allele, but we did not observe any significant difference between KO and WT (Supplementary Material, Fig. S2). Secondly, in adult testes (Fig. 5B), the deletion of the Peg3-DMR also caused down-regulation of two genes, Peg3 and Zim2, but the levels of the observed down-regulation were not that dramatic as shown in the neonatal brain in the case of Peg3. The three other genes, Usp29, Zim3 and Zfp264, showed no statistically significant difference between KO and WT. The expression levels of Zim1 are very low in adult testes, and thus Zim1 was not included in this series of analyses. In the case of the adult testis set with the maternal transmission, we did not also observe any major difference between KO and WT (Supplementary Material, Fig. S3). We repeated this series of experiments using another set of adult testes with both paternal and maternal transmission, and the overall conclusions were consistent with those from the first set. In sum, the deletion of the Peg3-DMR caused a global impact on the transcriptional levels of the Peg3 domain when the KO allele was paternally transmitted. In particular, the two adjacent genes, Peg3 and Zim1, are the most significantly affected by the deletion of the Peg3-DMR.
Figure 5.

Deletion effects on the transcriptional levels. (A) qRT–PCR analyses using the total RNA derived from neonatal brains of the normal littermate (WT) and the heterozygous pup with the paternal transmission of the KO allele (KO). All the results are statistically significant except Zfp264. The two stars (*) indicate two genes, Peg3 and Zim1, the expression levels of which were affected the most by the deletion of the Peg3-DMR. (B) qRT–PCR analyses using the total RNA derived from adult testes of the normal littermate (WT) and the heterozygote with the paternal transmission of the KO allele (KO). The expression level of a given gene was measured in triplicates, and the average with standard deviation was shown on graphs. The numbers on Y-axis indicate the relative values against the expression levels of the internal control, β-actin.
DISCUSSION
In the current study, one mutant allele lacking the 2.5 kb YY1-binding region of the Peg3-DMR has been generated to test whether this region is an ICR for the Peg3 domain. According to the results from the mutant allele, the deletion of the Peg3-DMR caused a global impact on the imprinting and transcription of the Peg3 domain, including changes in the imprinting status of Zim2 and Zfp264 and also in the transcriptional levels of the Peg3 domain. However, the DNA methylation of the Peg3 domain was largely unaffected, suggesting no major effect of the deletion of the Peg3-DMR. The mutant mice also displayed gross-level phenotypes, changes in body weight as well as partial embryonic lethality. Overall, these results confirm that the Peg3-DMR is indeed an ICR responsible for the imprinting and transcription of this 500 kb genomic region.
As predicted, the deletion of the Peg3-DMR resulted in several changes in the imprinting and transcription of the Peg3 domain (Fig. 6). The paternal transmission of the KO allele in neonatal brain caused down-regulation of Peg3, and also bi-allelic expression of the maternally expressed Zim2 (Fig. 6B). First, the observed down-regulation of Peg3 may be an indication that the deleted region, 2.5 kb genomic region (Fig. 1A), may harbor unknown enhancers for the transcription of Peg3. According to the results from previous in vitro studies, this 2.5 kb genomic region contains two evolutionarily conserved sequence elements, CSE1 and CSE2 (15), and CSE2 (the YY1-binding site) has been shown to function as a transcriptional enhancer for Peg3 (22). Thus, the down-regulation of Peg3 may reflect the loss of several YY1-binding sites with transcriptional enhancer activity. Secondly, the bi-allelic expression of the maternally expressed Zim2 is likely linked to the down-regulation of the paternally expressed Peg3. According to previous studies, several genes in this domain, including Peg3, Zim2 and Zfp264, display very similar spatial expression patterns (19), implying that these genes may share unknown enhancers for their expression. If this is the case, Peg3 and Zim2 might compete in cis for these unknown enhancers such that the down-regulation of the transcriptionally dominant Peg3 might allow the expression of Zim2 in the paternal allele, resulting in the bi-allelic expression of Zim2. Similar outcomes have been observed in the mutational studies of other imprinted domains, including H19/Igf2 and Gtl2/Dlk1 (7,8). Taken together, the results described above suggest the presence of transcriptional enhancers for Peg3 within the deleted region, and also a potential linkage between the imprinting regulation of Peg3 and Zim2.
Figure 6.

Schematic summary for the ICR roles of the Peg3-DMR. The deletion effects of the Peg3-DMR are schematically presented: each gene is indicated with an arrow while each DMR is indicated with a box. The open and closed boxes indicate unmethylated and methylated state of a given DMR, whereas an X on the box represents the deleted allele (KO) of the Peg3-DMR. The first illustration summarizes the imprinting status of the Peg3 domain that has been known so far (A). The two illustrations below summarize the deletion effects observed from neonatal brains with the paternal (B) and maternal (C) transmission of the KO allele, respectively.
A pair of two adjacent genes with opposite imprinting is quite common in imprinted domains, and the imprinting and transcription of each pair tend to be linked and co-regulated (7,8). The Peg3/Zim2 pair could be one such example based on the results described above. The paternal transmission of the KO allele in neonatal brains also caused up- and down-regulation of another pair of adjacent genes, Zim1 and Peg3 (Fig. 6B), suggesting a potential regulatory linkage of these two genes. In the case of Zim1, however, the observed up-regulation was still derived from the maternal allele without any contribution from the paternal allele, indicating that the imprinting of Zim1 is not affected by the deletion of the Peg3-DMR. This unexpected outcome is somewhat consistent with the evolutionary history associated with Zim1. Zim1 is found only in a few mammalian species, such as rodents and rabbits, but not in the remaining mammalian species, suggesting the recent formation of Zim1 in these species during evolution (23; Frey et al., unpublished data). Thus, the imprinting mechanisms governing the entire Peg3 domain might have predated the formation of Zim1, and might be independent of the imprinting mechanism of Zim1. This fact might be the reason why the deletion of the Peg3-DMR does not have any impact on the imprinting status of Zim1. If that is the case, then why do the mutant mice still display the coordinated up- and down-regulation of Zim1 and Peg3 although the transcription of these two genes occur from two separate chromosomes, Zim1 on the maternal and Peg3 on the paternal chromosome? One possible scenario would be that Peg3 controls the transcriptional rate of Zim1 as a trans factor. This function is feasible since Peg3 encodes a potential DNA-binding protein with 12 zinc finger motifs (M.M. Thiaville et al., unpublished data). In this case, the down-regulation of Peg3 might produce a reduced amount of the PEG3 protein, which could have a consequence on the transcriptional levels of Zim1 as a potential repressor. If this turns out to be true, the Peg3/Zim1 pair represents a very unusual case in genomic imprinting since Peg3 might interact with its neighbor gene Zim1 through its gene product, a DNA-binding protein, but not through a genetic linkage.
Compared with the paternal transmission, the maternal transmission of the KO allele has a relatively mild impact on the transcription and imprinting of the Peg3 domain (Fig. 6C). According to the results from a series of qRT–PCR, the expression levels of the genes in this domain are mostly unaffected by the deletion of the Peg3-DMR. This observation agrees with the fact that the maternal allele of the Peg3-DMR is usually methylated and thus non-functional. Interestingly, however, the maternal transmission of the KO allele caused a switch in the dominant allele for Zfp264, from the paternal to maternal allele. One possible explanation would be that the maternal allele of the Peg3-DMR might be functional during some unknown stages of development. Thus, the observed change in Zfp264 might reflect an outcome of a chain of events that have been triggered by the deletion of the Peg3-DMR. This idea is further supported by the results derived from our breeding experiments. The homozygotes for the deletion of the Peg3-DMR are not viable, although the homozygotes should be equivalent to the heterozygotes with the paternal transmission of the KO allele if the maternal allele of the Peg3-DMR is truly non-functional. The observed lethality of the homozygotes suggests that the maternal allele of the Peg3-DMR might be functionally required during some stages of development or in certain types of cells. Overall, although we need to first follow up this possibility, these results suggest that the maternal allele of the Peg3-DMR may be required for the imprinting regulation of the Peg3 domain.
According to the results from DNA methylation analyses (Fig. 4), the deletion has almost no impact on the Peg3 domain. This is somewhat surprising given the fact that similar KO experiments tend to disrupt the allele-specific DNA methylation pattern of other ICRs (7,8). Although speculative, this might be related to the fact that the current KO scheme deletes only part of the entire DMR, the 2.5 kb first intron region of Peg3, which contains two conserved elements, CSE1 and CSE2 (YY1-binding sites). In fact, the entire Peg3-DMR spans a 4 kb region harboring the first exons of Peg3 and Usp29 as well as the two CSEs in the first intron of Peg3 (Fig. 1A). In retrospect, an ideal KO scheme should have been deleting the entire 4 kb region. At the beginning, however, this scheme was regarded as a risky approach since this scheme would not only delete potential regulatory elements for imprinting but also inactivate the transcription of both Peg3 and Usp29. Thus, a next logical step for further characterizing this ICR should be generating conditional KO alleles that can delete the entire Peg3-DMR, which we plan to perform in the near future.
Besides the changes described above, the paternal transmission of the KO allele caused gross-level phenotypes: partial embryonic lethality and reduced body weight of the mouse. The major contributors to these observed phenotypes are Peg3 and Zim1 since these two genes are changed the most in the mutant mice (Fig. 5). According to the results from another KO allele targeting Peg3 (24–26), the mutant mice also exhibit smaller body size than their wild-type littermates, but this mutant model does not display partial embryonic lethality. It is relevant to note that the expression levels of Zim1 are very high during embryonic stages and also in the placenta (27). Thus, it is possible that the partial embryonic lethality might be contributed by the up-regulation of Zim1. However, we have also observed slightly increased body weight of the mouse in the maternal transmission of the KO allele. It is difficult to explain this phenotype at the moment since none of the imprinted genes appears to be significantly affected by the maternal transmission of the KO allele at their transcriptional levels. However, one possible scenario might be de-repression of the Peg3-DMR in a small subset of cells either in the brain or during early embryogenesis, which is triggered by the deletion. If this turns out to be the case, then this supports an idea that the deleted region may be required for the maintenance of the repressed state of the Peg3-DMR. It will be of great interest to investigate this possibility in the near future.
MATERIALS AND METHODS
Generation of a KO allele
The Peg3-DMR has been targeted using a KO vector, pTNT (18), which contains positive (neomycin resistance gene) and negative (HSV-thymidine kinase gene) selection markers. The two genomic fragments flanking the 2.5 kb YY1-binding region of the Peg3-DMR have been subcloned into the pTNT vector as ‘homologous hooks’. A 3.0 kb genomic fragment surrounding the first exon of Peg3 has been subcloned into the NotI site of this vector; another 8.9 kb fragment surrounding the second and third exons of Peg3 was subcloned into the HindIII site of the same vector. These two DNA fragments were amplified from one mouse bacterial artificial chromosome clone (RPCI23-588F20) of the 129/SvJ origin using the following two primer sets: Peg3-KO-51 (5′-ttgcggccgcACGGTGGTCACATTCTTCCATGAG-3′) and Peg3-KO-52 (5′- ttgcggccgcACAGTGCCGCGCTTAGCGGTAGATG-3′) for the 3.0 kb genomic fragment, and Peg3-KO-53 (5′-ttaagcttGGTGTCTGAATTTTTCACTGTGGA-3′) and Peg3-KO-54 (5′-ttaagCTTGAAACTCTCCAACGGAGTGGTGA-3′) for the 8.9 kb genomic fragment. The constructed vector was first linearized with SalI digestion, and subsequently transfected into the AB2.2 ES cell line of the 129/SvJ origin (http://www.bcm.edu/dtmc/, Darwin Transgenic Mouse Core facility of Baylor College of Medicine). ES cells were first screened with a long-distance PCR scheme using the following primer set: Peg3-KO-59 (5′-GCATCTGCTGTTACGGGGATTGTTGA-3′) and Peg3-KO-61 (5′-CTCCAGTCACCTATGTGTGGATGT-3′). Later, potential targeted clones were further analyzed with Southern blot using the two genomic fragments as probes. These two genomic fragments were amplified using the following two primer sets: Peg3-KO-57 (5′-CAGGAAGACACCCTGATACAG-3′) and Peg3-KO-58 (5′-GATATCCTACTCACTATATCC-3′) for the 5′-side probe, and Peg3-KO-63 (5′-ACCTTCCACTAGATTTCACCTCCT-3′) and Peg3-KO-64 (5′-CACTGCCAAAAGCATGAGATGGTC-3′) for the 3′-side probe. One targeted ES cell was microinjected into the blastocysts of e3.5-embryos of the C57BL/6J (B6) mouse. Twenty chimeras with varying degrees of coat color contribution were obtained, and 5 of these chimeras were bred with 10 B6 females, finally deriving 17 F1 mice with the germline transmission of the KO allele. Some of these F1 mice were further bred with one Zp3-cre line to remove the NeoR sequence (Jackson Lab; Stock No. 003651; Strain name, C57BL/6-Tg(Zp3-cre)93Knw/J).
Breeding experiments
Three different breeding schemes were set up to test the deletion effects on the mouse: Scheme I, female wild-type littermates × male heterozygotes (+/−) for the paternal transmission of the KO allele; Scheme II, female heterozygotes (+/−) × male wild-type littermates for the maternal transmission of the KO allele; and Scheme III, female × male heterozygotes (+/−) to test the derivation of the homozygotes. One-day-old pups derived from these breeding were genotyped using the following primer set: Peg3-6R (5′-CTTAGGGTCTTAGGGCTTTAGGT-3′) and Peg3-KO-61 (5′-CTCCAGTCACCTATGTGTGGATGT-3′). The genders of these pups were determined using the following primer set: mSry-F (5-GTCCCGTGGTGAGAGGCACAAG-3) and mSry-R (5-GCAGCTCTACTCCAGTCTTGCC-3). The body weights of the pubs were also measured as an indicator for their health status. For genotyping and gender determination, genomic DNA was isolated from either clipped tails or ears by incubating the tissues overnight at 55°C in the lysis buffer (0.1 m Tris-Cl, pH 8.8, 5 mm ethylenediaminetetraacetic acid pH 8.0, 0.2% sodium dodecyl sulfate 0.2 m NaCl, 20 μg/ml Proteinase K).
Imprinting tests
For imprinting tests, male and female heterozygotes (+/−) were bred individually with female and male mice of the PWD/PhJ strain (Jackson Lab, Stock No. 004660). One-day-old F1 pups from these crosses were sacrificed and used for isolating total RNA. The isolated RNA was reverse-transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen), and the resulting cDNA was used as a template for the amplification of each imprinted gene using the Maxime PCR premix kit (Intron Biotech). Hybrid embryos at 14.5 dpc harvested from timed mating were also used for imprinting tests. A separate table is available for all the detailed information regarding the primer sets and conditions for the amplification of each imprinted gene as well as the restriction enzyme that has been used to visualize a RFLP for each imprinted gene (Supplementary Material, Table S1).
DNA methylation analysis
DNA methylation levels of each DMR were analyzed using genomic DNA isolated from the tissues of the F1 mice as well as two germ cells, sperm and oocyte. Detailed protocols for isolating sperm and oocyte were described previously (28). Briefly, the sperm was isolated from the epididymus of 2-moth-old mice using the ‘swim-up’ method (29). Mature oocytes were isolated from 2-month-old females after superovulation with PMS and hCG treatment (30,31). The isolated DNA was treated with the bisulfite conversion reaction according to the manufacturer's protocol (EZ DNA methylation kit, Zymo Research). The converted DNA was used as a template for the PCR reaction using specific primers that were designed for amplifying each DMR. Each PCR product was further analyzed using the following two approaches: (i) the restriction enzyme digestion-based COBRA (32) and (ii) subcloning and sequencing. For the COBRA analysis, each PCR product was digested with a series of restriction enzymes. The PCR product was also individually subcloned into the pGEM T-Easy vector (Promega), and 10–20 clones were subsequently sequenced to survey its DNA methylation levels at each locus. The detailed information regarding oligonucleotide sequences, sequence polymorphisms and COBRA is also available (Supplementary Material, Table S1).
Quantitative RT–PCR analyses
Total RNA was isolated from the tissues of neonates and adults using a commercial kit (Trizol, Invitrogen). As described earlier, the isolated RNA was first reverse-transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen), and the subsequent cDNAs were used as a template for quantitative real-time PCR. This analysis was performed with the iQ SYBR green supermix (Bio-Rad) using the iCycler iQTM multicolor real-time detection system (Bio-Rad). All qRT–PCR reactions were carried out for 40 cycles under standard PCR conditions. The analyses of the results derived from qRT–PCR were described previously (33). The information regarding individual primer sequences and PCR conditions is also available (Supplementary Material, Table S1).
Western blot analysis
For western blotting, the tissues were first homogenized using lysis buffer (0.25 m Tris–HCl, pH 7.8, plus 0.1% NP-40). Cellular debris was removed by centrifugation for 10 min. Protein concentrations were determined by the Bradford assay kit (Pierce). Twenty micrograms of lysates were separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and transferred to PVDF membranes (Hybond-P, Amersham) using a Mini Trans-Blot transfer cell (Bio-Rad). Membranes were blocked for 1 h in the Tris-buffered saline containing 5% skim milk and 0.05% Tween 100, and incubated at 4°C overnight with anti-PEG3 or anti-USP29 antibodies (34). These blots were incubated for an additional 1 h with the secondary antibody linked to horseradish peroxidase (Sigma). The blots were developed using a western blot detection system according to the manufacturer's protocol (Intron Biotech).
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by National Institutes of Health (J.K. R01-GM066225 and R15-ES019118).
Supplementary Material
Acknowledgements
We would like to thank Dr Jeong Do Kim and Isabel Lorenzo at Baylor College of Medicine for their technical help for generating a mouse KO allele. We also thank Michelle M. Thiaville for careful reading and discussion of the manuscript.
Conflict of Interest statement. None declared.
References
- 1.Choo J.H., Kim J.D., Kim J. Imprinting of an evolutionarily conserved antisense transcript gene APeg3. Gene. 2008;409:28–33. doi: 10.1016/j.gene.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim J., Stubbs L. Rapidly Evolving Imprinted Loci, Encyclopedia of Genetics, Genomics, Proteomics and Bioinformatics. West Sussex, UK: John Wiley Publishers; 2005. [Google Scholar]
- 3.Cattanach B.M., Beechey C.V., Peters J. Interactions between imprinting effects in the mouse. Genetics. 2004;168:397–413. doi: 10.1534/genetics.104.030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J.M., Kim J. DNA methylation analysis of mammalian Peg3 imprinted domain. Gene. 2009;442:18–25. doi: 10.1016/j.gene.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maegawa S., Yoshioka H., Itaba N., Kubota N., Nishihara S., Shirayoshi Y., Nanba E., Oshimura M. Epigenetic silencing of PEG3 gene expression in human glioma cell lines. Mol. Carcinogen. 2001;31:1–9. doi: 10.1002/mc.1034. [DOI] [PubMed] [Google Scholar]
- 6.Dowdy S.C., Gostout B.S., Shridhar V., Wu X., Smith D.I., Podratz K.C., Jiang S.W. Biallelic methylation and silencing of paternally expressed gene 3 (PEG3) in gynecologic cancer cell lines. Gynecol. Oncol. 2005;99:126–34. doi: 10.1016/j.ygyno.2005.05.036. [DOI] [PubMed] [Google Scholar]
- 7.Edwards C.A., Ferguson-Smith A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007;19:281–289. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Ideraabdullah F.Y., Vigneau S., Bartolomei M.S. Genomic imprinting mechanisms in mammals. Mutat. Res. 2008;647:77–85. doi: 10.1016/j.mrfmmm.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters J., Robson J.E. Imprinted noncoding RNAs. Mamm. Genome. 2008;19:493–502. doi: 10.1007/s00335-008-9139-4. [DOI] [PubMed] [Google Scholar]
- 10.Spahn L., Barlow D.P. An ICE pattern crystallizes. Nat. Genet. 2003;35:11–12. doi: 10.1038/ng0903-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J. Multiple YY1 and CTCF binding sites in imprinting control regions. Epigenetics. 2008;3:115–118. doi: 10.4161/epi.3.3.6176. [DOI] [PubMed] [Google Scholar]
- 12.Bartolomei M.S. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124–2133. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucifero D., Mertineit C., Clarke H.J., Bestor T.H., Trasler J.M. Methylation dynamics of imprinted genes in mouse germ cells. Genomics. 2002;79:530–538. doi: 10.1006/geno.2002.6732. [DOI] [PubMed] [Google Scholar]
- 14.Lucifero D., Mann M.R., Bartolomei M.S., Trasler J.M. Gene-specific timing and epigenetic memory in oocyte imprinting. Hum. Mol. Genet. 2004;13:839–849. doi: 10.1093/hmg/ddh104. [DOI] [PubMed] [Google Scholar]
- 15.Kim J., Kollhoff A., Bergmann A., Stubbs L. Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum. Mol. Genet. 2003;12:233–245. doi: 10.1093/hmg/ddg028. [DOI] [PubMed] [Google Scholar]
- 16.Kim J.D., Hinz A.K., Bergmann A., Huang J.M., Ovcharenko I., Stubbs L., Kim J. Identification of clustered YY1 binding sites in imprinting control regions. Genome Res. 2006;16:901–911. doi: 10.1101/gr.5091406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J.D., Kim J. YY1′s longer DNA-binding motif. Genomics. 2009;93:152–158. doi: 10.1016/j.ygeno.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinz J.M., Nham P.B., Urbin S.S., Jones I.M., Thompson L.H. Disparate contributions of the Fanconi anemia pathway and homologous recombination in preventing spontaneous mutagenesis. Nucleic Acids Res. 2007;35:3733–3740. doi: 10.1093/nar/gkm315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J., Noskov V., Lu X., Bergmann A., Ren X., Warth T., Richardson P., Kouprina N., Stubbs L. Discovery of a novel, paternally expressed ubiquitin-specific processing protease gene through comparative analysis of an imprinted region of mouse chromosome 7 and human chromosome 19q13.4. Genome Res. 2000;10:1138–1147. doi: 10.1101/gr.10.8.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J., Bergmann A., Wehri E., Lu X., Stubbs L. Imprinting and evolution of two Kruppel-type zinc-finger genes, Zim3 and ZNF264, located in the PEG3/USP29-imprinted domain. Genomics. 2001;77:91–98. doi: 10.1006/geno.2001.6621. [DOI] [PubMed] [Google Scholar]
- 21.Kim J., Bergmann A., Lucas S., Stone R., Stubbs L. Lineage-specific imprinting and evolution of the zinc-finger gene ZIM2. Genomics. 2004;84:47–58. doi: 10.1016/j.ygeno.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Kim J.D., Yu S., Choo J.H., Kim J. Two evolutionarily conserved sequence elements for Peg3/Usp29 transcription. BMC Mol. Biol. 2008;9:108. doi: 10.1186/1471-2199-9-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J., Bergmann A., Choo J.H., Stubbs L. Genomic organization and imprinting of the Peg3 domain in bovine. Genomics. 2007;90:85–92. doi: 10.1016/j.ygeno.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Curley J.P., Barton S., Surani M.A., Keverne E.B. Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc. Biol. Sci. 2004;271:1303–1309. doi: 10.1098/rspb.2004.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curley J.P., Pinnock S.B., Dickson S.L., Thresher R., Miyoshi N., Surani M.A., Keverne E.B. Increased body fat in mice with a targeted mutation of the paternally expressed imprinted gene Peg3. FASEB J. 2005;19:1302–1304. doi: 10.1096/fj.04-3216fje. [DOI] [PubMed] [Google Scholar]
- 26.Li L.L., Szeto I.Y., Cattanach B.M., Ishino F., Surani M.A. Organization and parent-of-origin-specific methylation of imprinted Peg3 gene on mouse proximal chromosome 7. Genomics. 2000;63:333–340. doi: 10.1006/geno.1999.6103. [DOI] [PubMed] [Google Scholar]
- 27.Kim J., Lu X., Stubbs L. Zim1, a maternally expressed mouse Kruppel-type zinc-finger gene located in proximal chromosome 7. Hum. Mol. Genet. 1999;8:847–854. doi: 10.1093/hmg/8.5.847. [DOI] [PubMed] [Google Scholar]
- 28.Kim J.D., Kang K., Kim J. YY1′s role in DNA methylation of Peg3 and Xist. Nucleic Acids Res. 2009;37:5656–5664. doi: 10.1093/nar/gkp613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunch P.O., Saling P.M. Generation of a mouse membrane fraction with zona receptor activity. Biol. Reprod. 1991;44:672–680. doi: 10.1095/biolreprod44.4.672. [DOI] [PubMed] [Google Scholar]
- 30.Eppig J.J., Telfer E. Isolation and culture of oocytes. Meth. Enzymol. 1993;225:77–84. doi: 10.1016/0076-6879(93)25008-p. [DOI] [PubMed] [Google Scholar]
- 31.Hogan B., Beddington R., Constantini F., Lacy E. Manipulating the Mouse Embryo. 2nd edn. 1994. pp. 296–298. Cold Spring Harbor, NY. [Google Scholar]
- 32.Xiong Z., Laird P.W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J.D., Hinz A.K., Choo J.H., Stubbs L., Kim J. YY1 as a controlling factor for the Peg3 and Gnas imprinted domains. Genomics. 2007;89:262–269. doi: 10.1016/j.ygeno.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J., Kim J.D. In vivo YY1-knockdown effects on genomic imprinting. Hum. Mol. Genet. 2008;17:391–401. doi: 10.1093/hmg/ddm316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}