

Abstract
Inflammatory bowel diseases (IBDs) are chronic inflammatory conditions of the gastrointestinal tract that occur in genetically susceptible individuals. Crohn's disease (CD) and ulcerative colitis (UC) are two major types of IBD. In about 20–25% of patients, disease onset is during childhood and pediatric IBD can be considered the best model for studying immunopathogentic mechanisms. The fundamentals of IBD pathogenesis are considered a defective innate immunity and bacterial killing with overaggressive adaptive immune response. A condition of “dysbiosis”, with alterations of the gut microbial composition, is regarded as the basis of IBD pathogenesis. The human gastrointestinal (GI) microbial population is a complex, dynamic ecosystem and consists of up to one thousand different bacterial species. In healthy individuals, intestinal microbiota have a symbiotic relationship with the host organism and carry out important metabolic, “barrier,” and immune functions. Microbial dysbiosis in IBD with lack of beneficial bacteria, together with genetic predisposition, is the most relevant conditions in the pathogenesis of the pediatric IBD.
1. Introduction
IBD are chronic inflammations of the small bowel and/or the colon leading to recurrent diarrhea and abdominal pain. Crohn's disease (CD) and ulcerative colitis (UC) are the two main clinicopathological subtypes of IBD. Despite both being chronic and relapsing inflammatory diseases of the bowel, they can be differentiated by the location of the inflammation in the gastrointestinal tract and by the nature of the histological alterations in the intestinal wall. Epidemiology studies suggest that the prevalence of IBD increases in populations and regions with industrialization [1]. Disease onset appears typically in young adulthood (between the age of 25 and 35 years), but in about 20–25% symptoms begin in pediatric population [2]. Complex interactions between immune system, enteric commensal bacteria/pathogens, and host genotype are thought to underlie the development of IBD [3]. An emerging consensus hypothesis is that intestinal dysbiosis (microbial imbalance) may be a trigger for IBD. In children both mucosal immune system and intestinal flora are still in the developmental stage. Taken together it appears that pediatric IBDs represent a specific group of patients with particular gene defects, phenotypic appearance, drug responsiveness, and intestinal immunopathology [4].
In this paper, we will discuss the meaning of dysbiosis in the pathogenesis of pediatric IBD, the weakening of mucosal defences, and the lack of bacterial clearance by macrophages with the result of a loss of tolerance to commensal flora [5, 6].
2. Microbial Flora and Intestinal Immune System
The human gut is sterile at birth, but colonization with numerous bacterial species starts immediately after birth, thus generating a resident microbiota characterized by unique bacterial profiles and high interindividual and environmental variation [7]. The adult human microbiota consist of around 1014 bacterial cells and up to an estimated 1,000 different bacterial species [8]. Studies have shown that the most abundant bacteria phyla found in healthy human large intestine are Gram-negative Bacteroidetes and Gram-positive low-GC Firmicutes [9]. Microbiota composition varies greatly between individuals, with each individual harbouring a unique collection of bacterial species, which is highly stable over time. The immune regulatory function of the intestinal microbiota consists of priming the mucosal immune system and maintenance of intestinal epithelium homeostasis (Table 1). Studies in germ-free animals have demonstrated that the normal immune function of intestinal mucosa is impaired in the absence of gut microbiota [10]. The “hygiene hypothesis” has been postulated over the years to justify how fundamental lifestyle has changed from one with high to one with low microbial exposure and thus provides an explanation for the higher frequency of IBD [11]. In this condition, the intestinal immune system has smaller Peyer's patches, fewer plasma cells, lower numbers of CD8 intraepithelial lymphocytes with reduced cytotoxicity, and impaired antimicrobial peptide and IgA secretion [12]. The intestinal microbiota are vast and quite diverse at species level. The classification of “normal” microbiota is challenging as each individual possesses a unique collection of microbial species. Firmicutes and Bacteroidetes are the two most predominant bacterial phyla inhabitants in the intestinal tract [13]. The phyla represent the highest taxonomic rank in bacterial classification and are composed of numerous orders, classes, families, and genera with diverse and broad metabolic, ecological, pathogenic, and symbiotic properties [14]. The description of phylum profiles has only limited biological relevance for understanding host-microbe interactions. Evidence suggests that commensal bacteria play a role in maintaining the integrity of the intestinal epithelium [15]. Intestinal epithelial cells (IECs) provide a physical barrier between luminal microbes and underlying intestinal tissues to control defence and tolerance. IECs express pattern recognition receptors (PRRs) and can recognize microbial pathogen-associated molecular patterns (PAMPs) and respond to intestinal microbes through secretion of cytokines and antimicrobial proteins and up-regulation of surface molecules that mediate intercellular interactions [16]. Peterson et al. have shown that the presence of IgA reduces intestinal proinflammatory signals and drives diversity in gut microbiota [17]. A defective antibacterial, genetically driven barrier allows translocation and regulation of the microbiota. Commensal bacteria can have an anti-inflammatory effect on the developing immune system; for example, in the uterus, T-helper type 2 response is predominant [18]. With gut colonization, a balance between T-helper types 1 and 2 is established to prevent the development of allergic food reactions, and to establish a T-helper type 3 response that provides tolerance to oral protein antigens. Feeding allows for antigenic stimulation and bacterial colonization of the gut. This is required for the development of IgM- and IgA-producing plasma cells in the intestinal lamina propria [19]. Breast milk provides passive protection with antibacterial components such as IgA, lysozyme, and lactoferrin, promotes the development of commensal flora rich in bifidobacteria, and decreases colonization with potential pathogens. Alterations in the normal development of the immune system can lead to chronic disease states. Antibiotic use in the neonatal period and infancy can interfere with the development of a healthy commensal flora and may result in subsequent allergic disease or inflammatory conditions of the intestinal tract (irritable bowel syndrome and IBD) [20].
Table 1.
The effect of commensal bacteria on the development of the immune system.
| Inhibits epithelial NF-kB activation and inflammatory gene expression | |
| Activates CD4 cells in Peyer's patches | |
| Activates CD8 or natural killer cells in intraepithelial leukocyte spaces | |
| Increases numbers of T and B cells, including CD86-positive cells | |
| Organizes the special relationships between T, B, and dendritic cells in the Peyer's patches | |
| Increases the numbers of microfold cells | |
| Increases IgA producing B cells | |
| Hypertrophies Peyer's patches and the development of germinal centers |
In healthy conditions, balanced mechanisms regulate the host's immunological tolerance to the continuous stimulus of resident gut microbiota and their metabolic end products [21]. Hildebrand et al. have shown that pneumonia prior to age 5 years, but not later, and consequent and frequent use of antibiotics were associated with subsequent high risk of CD, and this may represent either susceptibility or causation. The results confirm that early exposures to antibiotics influence immune function through disruption of bowel colonization [22].
3. Microbiota in IBD
The theory is discussed that IBDs represent the consequence of the loss of immunological tolerance against autologous flora. Supporting this theory are a limited number of human studies and a large number of studies in animal models. It is assumed that the presence of bacteria is essential for the development of experimental IBD in most models [23]. Commensal flora appears to exacerbate rather than directly cause disease [24]. In IBD patients, not only is the quantity of commensal bacteria reduced but also the quality of microbiota composition is altered, with reduction of Firmicutes and Bacteroidetes. As a consequence of this dysbiosis, the relative abundance of Enterobacteriacae is increased in IBD patients compared to healthy controls, although their absolute numbers remained unaltered [25]. These findings are present also in several studies, which have observed decreased clostridia concentrations, although not always accompanied by a decrease in Bacteroides [26]. Macfarlane et al. revealed aberrancies in Bifidobacterium populations in rectal biopsies from IBD patients with significant reductions of the counts [27]. Zhang et al. have shown that bacterial diversity of lactobacilli is present in ulcerated tissue compared to nonulcerated tissue in the same UC individuals [28].
On the other hand, the number of mucosal adherent bacteria, such as invasive E. coli, or Proteobacteria, such as Enterobacteriaceae are increased (Table 2). The possibility that IBDs are a chronic inflammatory response directed against microbial agents has been considered in UC and CD. Several infectious agents, including Mycobacterium avium subspecies paratuberculosis (MAP), adherent invasive E. coli, Yersinia, and Pseudomonas have been implicated as triggering agents of CD [29]. Research has excluded many microorganism including salmonella, campylobacter jejuni, clostridium difficile, adenoviruses, rotaviruses, and mycoplasma as primary etiological agents, although some may be implicated in relapses of CD [30]. One agent that raised a great deal of controversy is Mycobacterium avium subspecies paratuberculosis (MAP), which, for many years, was considered a possible etiologic agent [31]. MAP has been the most enduring infectious candidate to be proposed as a causative agent of CD although its role in etiology of disease has often been questioned. Prevalence studies of MAP in CD patients from many countries worldwide have reported widely divergent results ranging from 0% to 100%. The possible role of MAP in CD has also been supported by the identification of MAP DNA using IS900 polymerase chain reaction (PCR) analysis of media inoculated with peripheral blood mononuclear cells (PBMCs) from patients [32]. Kirkwood et al. described a comprehensive investigation into the presence of MAP in intestinal tissue and PBMC from 142 children presenting with initial symptoms of IBD prior to treatment. The final diagnoses included CD (62 children), UC (26 children), and non-IBD (54 children). There was evidence of MAP infection in biopsy tissue and/or PBMC in a total of 45% of children with CD, 35% of children with UC, and 11% of non-IBD children. The presence of viable MAP in 4/10 CD patients was confirmed by isolation of MAP from biopsy specimens. These results support the hypothesis that MAP infection of intestinal tissue, perhaps associated with bloodborne spread, may be implicated especially in the pathogenesis of pediatric CD [33]. No significant correspondence was found between CD-associated NOD2 polymorphisms, especially in ileal CD, and MAP infection [34]. Recently, another microorganism, Escherichia coli, has been under investigation and associated with ileal CD [35], but there is no evidence that antibiotic treatment against coliforms is efficacious in curing IBD patients. In a number of different mouse models of colitis, it was possible to prevent colitis by raising the mice under germ-free conditions. The hypothesis was developed that physiologic intestinal flora is no longer tolerated in IBD. Since 2001, genom-wide association studies (GWAs) have revealed more than 30 genes that are associated with IBD [36]. Among the identified targets are genes that play an important role for immunological cell-cell interactions and signalling, such as tumor necrosis factor (TNF), TNF-receptor 1 (TNFR1), the interleukin-23 receptor (IL23R) [37], or interleukin-12p40 (IL12B). More importantly, there are genes involved in the immune response to bacteria, such as nucleotide oligomerization domain 2 (NOD2) and the toll-like receptor 4 (TLR4), as well as the autophagy genes autophagy-related like 1 (ATG16L1) and immunity-related GTPase family M (IRGM) [38]. The variants of ATG16L1 and IRGM autophagy genes cause a defective capacity to process cell degradation products as well as bacteria and to eliminate proinflammatory stimuli [39]. Three mutually exclusive theories have been proposed concerning the implication of bacteria in pathogenesis of IBD, such as an involvement of persistent pathogen, an abnormally permeable mucosal barrier leading to excessive bacterial translocation and a breakdown in the balance between putative “protective” as against “harmful” intestinal bacteria which can promote inflammation. Bacteria colonizing the gut mucosa have the ability to strongly adhere to intestinal epithelial cells (IECs), to invade IECs by a mechanism involving actin polymerisation and microtubule recruitment and to induce granuloma formation in vitro [40]. Based on the pathogenic group, this type of E. coli was defined and named AIEC for adherent-invasive E. coli (AIEC). AIEC strains were found to be highly associated with ileal mucosa in CD patients, suggesting that there are specific alterations to the ileal epithelial cells in patients with CD that allow AIEC adhesion. The receptor involved in AIEC colonization, and abnormally expressed on ileal mucosa in 35% of CD patients, was characterized as being the carcinoembryonic antigen-related cell adhesion molecule (CECAM6). In pediatric population, genetics plays an even greater role in disease onset and susceptibility. It does appear, however, that the NOD2 gene is similarly present in 30%–35% of both adult and pediatric CD patients. Although the true pathogenic role of NOD2 in CD remains unknown, it is an important gene involved in innate immunity which lends support to the notion that genetically determined defects in innate, and likely adaptive immunity, alter the way of interaction of mucosal immune system with resident bacterial flora [41]. This dysregulated interaction leads to the adaptive immune response, responsible for the chronic inflammatory lesions, and is more evident in pediatric-age-onset IBD [42].
Table 2.
Composition of commensal microflora in IBD [48].
| Increased | Reduced |
|---|---|
| E. coli | Firmicutes |
| Proteobacteriace | Bacteroidetes |
| Enterobacteriacae | Clostridium ix and iv groups |
| Sulphate-reducing bacteria | Bifidobacteria |
4. Dysbiosis in IBD: Cause or Effect of the Mucosal Inflammation?
In IBD, dysbiosis could be a key factor in the immunopathogenesis of IBD by disrupting the host immune defences against commensal flora microbes at the mucosal border [43]. Increased paracellular intestinal mucosal barrier has long been recognized in IBD with abnormalities in both its structural integrity and mucus barrier functions [44]. Sewell et al. have hypotized that the penetration of gut luminal contents into the altered bowel wall impaired clearance of this material by the innate immune response and propagation of a secondary inflammatory reaction by the adaptive immune system [45]. Bacterial clearance is also altered in IBD, and an interaction between NOD2 and the autophagy system has been elucidated. Frank et al. performed a genotype-phenotype correlation and gene-environment interaction study of IBD patients. The results show that disease phenotype NOD2 composite genotype (Leu1007fs, R702W, G908R alleles) and ATG16L1 genotype (T300A allele) were significantly associated with shifts in microbial compositions with reduced bacterial diversity [46]. Specifically, members of the Lachnospiraceae family (Firmicutes phylum) and Bacteroidales (bacterial order) were depleted in a subset of IBD samples, with a concomitant increase in 16S rRNA sequences of Proteobacteria and Actinobacteria [47]. As a consequence of this dysbiosis, the relative abundance of Enterobacteriaceae was increased in IBD patients compared to healthy controls, although their absolute numbers remained unaltered. More importantly, this study confirmed that Faecalibacterium prausnitzii, a member of the Lachnospiraceae family (clostridial cluster IV and IXa), was reduced in the mucosa of IBD patients [48].
This abnormal microbiota composition shifts complex interactions that occur between microbes and host and its metabolic, trophic, and protective functions, such as immunomodulatory stimulation, strengthening epithelial barrier integrity [49]. In particular, Clostridium and Bacteroides species reduction cause reduction of butyrate and short-chain fatty acid production. F. prausnitzii have anti-inflammatory and anticolitic properties. Overgrowth of a class of microorganisms referred to as sulphate-reducing bacteria (SRB), observed in UC patients, produces substances which are toxic to colonocytes and blocks protective mechanisms in intestinal mucosa [50]. Numerous studies have analysed microbial compositions in individuals with IBD compared to healthy controls analyzing stool samples, but it is well accepted that microbial populations from stool differ from those associated with the mucosa [51–54].
In recent months, however, researchers have been working to characterize the gut microbiota also in pediatric IBD. Richness, evenness, and biodiversity of the gut microbiome were remarkably reduced in 27 children with severe UC compared with healthy controls, and this could include the lack of response to steroid therapy [55].
Darfeuille-Michaud et al. showed that in patients with IBD there was abnormal colonization of the ileal mucosa by AIEC bacteria that induced the release of high amounts of TNFα without leading to host cell apoptosis and with potential ability to induce persistent intestinal inflammation [56]. Some years later, the presence of adhesive invasive bacterial strains was confirmed in a pediatric population with IBD, in inflamed intestinal tissue [57].
It is possible to suggest that microbiota and microbiome are different in different sites of inflamed or non-inflamed gut with loss of tolerance and defective in the production or function of antibacterial peptides, such as defensins by the Paneth cells. There is some evidence that alpha-defensin production is reduced in ileal CD [58] and that, in colonic CD, there is reduced mucosal antimicrobial activity with consistently low antibacterial peptide expression [59]. This quantitative and/or qualitative alteration leads to lower levels of defensins in the numbers and type of intestinal microbiota composition and could promote intestinal inflammation. These alterations cause loss of tolerance to commensal flora and to amplification and maintenance of inflammatory response to intestinal pathogens (Figure 1). In trying to establish a pathogenic role of dysbiosis in IBD, microbial imbalance triggers a range of mechanisms with reduced intraluminal levels of butyrate, with downregulation of epithelial tight junction protein expression and increased epithelial permeability [50]. Killing of bacteria reaching the lamina propria, through the “leaky” epithelium, is also impaired by a genetically predisposed defective phagocytosis by macrophages. Ineffective bacterial clearance leads to excessive TLR stimulation, secretion of proinflammatory cytokines, and activation of innate and T-cell mediated immune responses. In summary, defective killing of phagocytosed organisms, decreased secretion of antimicrobial peptides, increased mucosal permeability, or defective excretion of xenobiotic materials could result in an overwhelming stimulation of adaptive immune responses and loss of immunologic tolerance to commensal bacterial antigens [60]. This disrupted mechanism of tolerance in epithelial cells may recognize dysbiosis as a primum movens. Despite these observations, it is not clear if gut microbial dysbiosis is a cause or a consequence of inflammatory disease [61] as these studies differ in distinct source of microbes and analytical methods.
Figure 1.
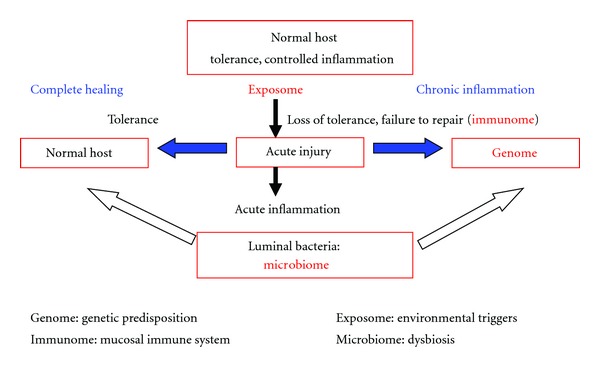
Pathogenesis of IBD: host response and loss of tolerance in intestine.
5. Conclusions and Perspectives
There have been new findings in identifying the pathogenesis of IBD over the last year, but environment, genetic makeup, commensal flora, and immune response can be considered the key factors. Dysbiosis can be considered an important pathogenetic factor with advancement growth of invasive pathogenic bacteria. It can also facilitate bacterial translocation through the intestinal mucosa barrier to the mesenteric lymph nodes. Analysis of the microbiota of CD and UC has so far resulted in diverging views of the importance of the particular bacteria in pathogenesis of IBD. Not only is the quantity of commensal bacteria in the IBD intestine reduced but also the quality and diversity of the commensal composition are altered. On the other hand, the number of mucosal adherent bacteria, such as invasive E. coli, or Proteobacteria, such as Enterobacteriaceae, is increased, resulting in the so-called state of “dysbiosis.” This condition may have a pathogenetic role which is more important in pediatric IBD, where interaction with genetic predisposition is more significant.
Further studies are needed to better define the true composition of the microbiota in patients with IBD and to understand if dysbiosis is a predisposing condition or a consequence of chronic intestinal inflammation.
References
- 1.Sauer CG, Kugathasan S. Pediatric inflammatory bowel disease: highlighting pediatric differences in IBD. Gastroenterology Clinics of North America. 2009;38(4):611–628. doi: 10.1016/j.gtc.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Kugathasan S, Judd RH, Hoffmann RG, et al. Epidemiologic and clinical characteristics of children with newly diagnosed inflammatory bowel disease in Wisconsin: a statewide population-based study. Journal of Pediatrics. 2003;143(4):525–531. doi: 10.1067/s0022-3476(03)00444-x. [DOI] [PubMed] [Google Scholar]
- 3.Sun L, Nava GM, Stappenbeck TS. Host genetic susceptibility, dysbiosis, and viral triggers in inflammatory bowel disease. Current Opinion in Gastroenterology. 2011;27(4):321–327. doi: 10.1097/MOG.0b013e32834661b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker AW, Sanderson JD, Churcher C, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiology. 2011;11, article 7 doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 6.Kaser A, Zeissig S, Blumberg RS. Inflammatorybowel disease. Annual Review of Immunology. 2009;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Domínguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(26):11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Domínguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140(6):1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Seminars in Immunology. 2007;19(2):59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. New England Journal of Medicine. 2002;347(12):911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 12.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nature Reviews Immunology. 2009;9(5):313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahowalda MA, Reya FE, Seedorfa H, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knights D, Costello EK, Knight R. Supervised classification of human microbiota. FEMS Microbiology Reviews. 2011;35(2):343–359. doi: 10.1111/j.1574-6976.2010.00251.x. [DOI] [PubMed] [Google Scholar]
- 15.Chung H, Kasper DL. Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Current Opinion in Immunology. 2010;22(4):455–460. doi: 10.1016/j.coi.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Abraham C, Medzhitov R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology. 2011;140(6):1729–1737. doi: 10.1053/j.gastro.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host and Microbe. 2007;2(5):328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan JL, Shi HN, Walker WA. The role of microbes in developmental immunologic programming. Pediatric Research. 2011;69(6):465–472. doi: 10.1203/PDR.0b013e318217638a. [DOI] [PubMed] [Google Scholar]
- 19.Feng T, Elson CO, Cong Y. Treg cell-IgA axis in maintenance of host immune homeostasis with microbiota. International Immunopharmacology. 2011;11(5):589–592. doi: 10.1016/j.intimp.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ekbom A, Adami HO, Helmick CG, Jonzon A, Zack MM. Perinatal risk factors for inflammatory bowel disease: a case-control study. American Journal of Epidemiology. 1990;132(6):1111–1119. doi: 10.1093/oxfordjournals.aje.a115754. [DOI] [PubMed] [Google Scholar]
- 21.Matricon J, Barnich N, Ardid D. Immunopathogenesis of inflammatory bowel disease. Self/Nonself. 2010;1(4):299–309. doi: 10.4161/self.1.4.13560. Landes Bioscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hildebrand H, Malmborg P, Askling J, Ekbom A, Montgomery SM. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scandinavian Journal of Gastroenterology. 2008;43(8):961–966. doi: 10.1080/00365520801971736. [DOI] [PubMed] [Google Scholar]
- 23.Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunological Reviews. 2005;206:260–276. doi: 10.1111/j.0105-2896.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 24.Scaldaferri F, Fiocchi C. Inflammatory bowel disease: progress and current concepts of etiopathogenesis. Journal of digestive diseases. 2007;8(4):171–178. doi: 10.1111/j.1751-2980.2007.00310.x. [DOI] [PubMed] [Google Scholar]
- 25.Sokol H, Lay C, Seksik P, Tannock GW. Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflammatory Bowel Diseases. 2008;14(6):858–867. doi: 10.1002/ibd.20392. [DOI] [PubMed] [Google Scholar]
- 26.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host and Microbe. 2008;3(6):417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macfarlane S, Steed H, Macfarlane GT. Intestinal bacteria and inflammatory bowel disease. Critical Reviews in Clinical Laboratory Sciences. 2009;46(1):25–54. doi: 10.1080/10408360802485792. [DOI] [PubMed] [Google Scholar]
- 28.Zhang M, Liu B, Zhang Y, Wei H, Lei Y, Zhao L. Structural shifts of mucosa-associated lactobacilli and Clostridium leptum subgroup in patients with ulcerative colitis. Journal of Clinical Microbiology. 2007;45(2):496–500. doi: 10.1128/JCM.01720-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflammatory Bowel Disease. 2012;18(5):968–984. doi: 10.1002/ibd.21866. [DOI] [PubMed] [Google Scholar]
- 30.de Hertogh G, Geboes K. Crohn’s disease and infections: a complex relationship. MedGenMed Medscape General Medicine. 2004;6(3, article 14) [PMC free article] [PubMed] [Google Scholar]
- 31.Chiodini RJ, van Kruiningen HJ, Thayer R, et al. Possible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn’s disease. Digestive Diseases and Sciences. 1984;29(12):1073–1079. doi: 10.1007/BF01317078. [DOI] [PubMed] [Google Scholar]
- 32.Bull TJ, McMinn EJ, Sidi-Boumedine K, et al. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s disease. Journal of Clinical Microbiology. 2003;41(7):2915–2923. doi: 10.1128/JCM.41.7.2915-2923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkwood CD, Wagner J, Boniface K, et al. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn’s disease. Inflammatory Bowel Diseases. 2009;15(11):1643–1655. doi: 10.1002/ibd.20967. [DOI] [PubMed] [Google Scholar]
- 34.Bernstein CN, Wang MH, Sargent M, Brant SR, Collins MT. Testing the interaction between NOD-2 status and serological response to Mycobacterium paratuberculosis in cases of inflammatory bowel disease. Journal of Clinical Microbiology. 2007;45(3):968–971. doi: 10.1128/JCM.02062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins MD, Lawson PA, Willems A, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. International Journal of Systematic Bacteriology. 1994;44(4):812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- 36.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nature Genetics. 2007;39(2):207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 37.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314(5804):1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scharl M, Rogler G. Microbial sensing by the intestinal epithelium in the pathogenesis of inflammatory bowel disease. International Journal of Inflammation. 2010;2010:12 pages. doi: 10.4061/2010/671258. Article ID 671258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deretic V. Links between autophagy, innate immunity, inflammation and Crohn’s disease. Digestive Diseases. 2009;27(3):246–251. doi: 10.1159/000228557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meconi S, Vercellone A, Levillain F, et al. Adherent-invasive Escherichia coli isolated from Crohn’s disease patients induce granulomas in vitro. Cellular Microbiology. 2007;9(5):1252–1261. doi: 10.1111/j.1462-5822.2006.00868.x. [DOI] [PubMed] [Google Scholar]
- 41.Biswas A, Petnicki-Ocwieja T, Kobayashi KS. Nod2: a key regulator linking microbiota to intestinal mucosal immunity. Journal of Molecular Medicine. 2012;90(1):15–24. doi: 10.1007/s00109-011-0802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nature Genetics. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckburg PB, Bik EM, Bernstein CN, et al. Microbiology: diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marks DJB. Defective innate immunity in inflammatory bowel disease: a Crohn’s disease exclusivity? Current Opinion in Gastroenterology. 2011;27(4):328–334. doi: 10.1097/MOG.0b013e3283463b45. [DOI] [PubMed] [Google Scholar]
- 45.Sewell GW, Marks DJ, Segal AW. The immunopathogenesis of Crohn’s disease: a three-stage model. Current Opinion in Immunology. 2009;21(5):506–513. doi: 10.1016/j.coi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank DN, Robertson CE, Hamm CM, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflammatory Bowel Diseases. 2011;17(1):179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, Acero D, Garcia-Gil LJ. Abnormal microbiota composition in the ileocolonic mucosa of Crohn’s disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflammatory Bowel Diseases. 2006;12(12):1136–1145. doi: 10.1097/01.mib.0000235828.09305.0c. [DOI] [PubMed] [Google Scholar]
- 49.Gilbert JA, Meyer F, Jansson J, et al. The Earth microbiome project: meeting report of the “1st EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6th 2010. Standards in Genomic Sciences. 2010;3(3):249–253. doi: 10.4056/aigs.1443528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fava F, Danese S. Intestinal microbiota in inflammatory bowel disease: friend of foe? World Journal of Gastroenterology. 2011;17(5):557–566. doi: 10.3748/wjg.v17.i5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joossens M, Huys G, Cnockaert M, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 52.Seksik P, Rigottier-Gois L, Gramet G, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52(2):237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scanlan PD, Shanahan F, O’Mahony C, Marchesi JR. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. Journal of Clinical Microbiology. 2006;44(11):3980–3988. doi: 10.1128/JCM.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang S, Denman SE, Morrison M, et al. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflammatory Bowel Diseases. 2010;16(12):2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 55.Michail S, Durbin M, Turner D, et al. Alterations in the gut microbiome of children with severeulcerative colitis. doi: 10.1002/ibd.22860. Inflammatory Bowel Diseases. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 57.Negroni A, Costanzo M, Vitali R, et al. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflammatory Bowel Diseases. 2012;18(5):913–924. doi: 10.1002/ibd.21899. [DOI] [PubMed] [Google Scholar]
- 58.Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell α-defensins in ileal Crohn’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(50):18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nuding S, Fellermann K, Wehkamp J, Stange EF. Reduced mucosal antimicrobial activity in Crohn’s disease of the colon. Gut. 2007;56(9):1240–1247. doi: 10.1136/gut.2006.118646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sartor RB, Muehlbauer M. Microbial host interactions in IBD: implications for pathogenesis and therapy. Current Gastroenterology Reports. 2007;9(6):497–507. doi: 10.1007/s11894-007-0066-4. [DOI] [PubMed] [Google Scholar]
- 61.Gareau MG, Sherman PM, Walker WA. Probiotics and the gut microbiota in intestinal health and disease. Nature Reviews. Gastroenterology & Hepatology. 2010;7(9):503–514. doi: 10.1038/nrgastro.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]