

Abstract
There is extensive evidence that the amygdala is involved in affectively influenced memory. The central hypothesis guiding the research reviewed in this paper is that emotional arousal activates the amygdala and that such activation results in the modulation of memory storage occurring in other brain regions. Several lines of evidence support this view. First, the effects of stress-related hormones (epinephrine and glucocorticoids) are mediated by influences involving the amygdala. In rats, lesions of the amygdala and the stria terminalis block the effects of posttraining administration of epinephrine and glucocorticoids on memory. Furthermore, memory is enhanced by posttraining intra-amygdala infusions of drugs that activate β-adrenergic and glucocorticoid receptors. Additionally, infusion of β-adrenergic blockers into the amygdala blocks the memory-modulating effects of epinephrine and glucocorticoids, as well as those of drugs affecting opiate and GABAergic systems. Second, an intact amygdala is not required for expression of retention. Inactivation of the amygdala prior to retention testing (by posttraining lesions or drug infusions) does not block retention performance. Third, findings of studies using human subjects are consistent with those of animal experiments. β-Blockers and amygdala lesions attenuate the effects of emotional arousal on memory. Additionally, 3-week recall of emotional material is highly correlated with positron-emission tomography activation (cerebral glucose metabolism) of the right amygdala during encoding. These findings provide strong evidence supporting the hypothesis that the amygdala is involved in modulating long-term memory storage.
Keywords: emotional arousal, epinephrine, glucocorticoids, norepinephrine, hippocampus
For decades there has been controversy over the question of whether the amygdala is involved in memory (1, 2, 3, 4). Extensive evidence from many laboratories has now resolved this general controversy. Studies of the effects of lesions of the amygdala in animals and humans leave little doubt that the amygdala is involved in mediating affectively influenced memory (5, 6, 7, 8). However, there remains considerable controversy concerning the specific role (or roles) of the amygdala in affectively influenced memory. The findings of lesion studies suggest that the amygdala may enable the formation of stimulus-reward associations (1, 9, 10, 11) and may be a site of neuroplasticity mediating aversive learning (12, 13, 14, 15). Research from our laboratory using a variety of experimental methods provides extensive additional evidence that the amygdala is involved in memory. However, our findings suggest a somewhat different view of the role of the amygdala in affectively influenced memory. Our findings suggest that the amygdala regulates the storage or consolidation of information in other brain regions (16). The central hypothesis guiding our research is that emotional arousal activates the amygdala and that such activation results in modulation of memory storage processes occurring in brain regions influenced by the amygdala. According to this view, the amygdala is part of a system that serves to regulate the strength of memories in relation to their emotional significance (8, 17). A corollary of this view is that the amygdala is not generally involved in memory but, rather, plays a selective role. Our findings suggest that the amygdala is involved when it is activated by emotional arousal. Furthermore, according to this view, the amygdala is not involved in the retrieval or expression of emotionally influenced memory; the critical role is that of modulating memory consolidation (18, 19). This view of the role of the amygdala in memory is based on evidence from our studies examining the involvement of the amygdala in mediating stress hormone and drug influences on memory and the effects of lesions and temporary inactivation of the amygdala. Although most of the evidence is based on studies using rats, the conclusions are also supported by evidence from several recent studies using human subjects (20, 21, 22).
Stress-Hormone Influences on Memory Storage
There is extensive evidence that many central nervous system stimulants enhance long-term memory when administered to animals shortly after training in inhibitory avoidance tasks as well as other types of learning tasks (18, 23). Such findings support the hypothesis that the drugs influence memory by modulating neurobiological processes underlying memory consolidation. The use of posttraining drug administration obviously excludes effects on acquisition or retrieval because the drug influences occur only after the training is completed, and the drugs used in these experiments are typically active only for a few minutes or hours (24). These findings suggested the possibility that endogenous stress-related hormones released by training experiences may play a role in regulating memory storage (25). There is now extensive evidence supporting this implication (26).
It is well established that, in rats, the adrenal medullary hormone epinephrine is released by the kind of stimulation typically used in learning experiments (27, 28). Furthermore, a large number of studies using a variety of training tasks have demonstrated that posttraining systemic injections of epinephrine enhance memory (28, 29, 30). These findings are consistent with the hypothesis that epinephrine released by training activates brain processes regulating memory storage (31). There is also extensive evidence that retention is modulated by posttraining systemic injections of adrenal glucocorticoids (32). With both types of adrenal stress hormones (i.e., adrenal catecholamines and glucocorticoids) administered after training, the effects on memory are dose-dependent and time-dependent. Enhancement is found with moderate doses and maximal enhancement is obtained with injections administered immediately after training. There is also evidence that these adrenal stress-related hormones interact in modulating memory storage (33). Suppression of the synthesis and subsequent release of glucocorticoids by administering metyrapone, an 11β-hydroxylase inhibitor, prior to training blocks the memory-enhancing effects of epinephrine (34).
Involvement of the Amygdala
There is considerable evidence that epinephrine and glucocorticoid effects on memory are mediated by influences involving the amygdala. These hormones differ, however, in the ways in which they influence the amygdala. It is well established that glucocorticoids readily enter the brain and activate adrenal steroid receptors. However, as epinephrine passes the blood–brain barrier poorly, if at all, epinephrine effects on memory are, as is discussed below, indirectly mediated. We will first review epinephrine effects on memory storage and then review glucocorticoid effects. Additionally, we will discuss these findings in relation to our studies of the interactions of other systems, including opiate, GABAergic and cholinergic systems, in regulating memory storage through influences involving the amygdala.
Epinephrine Effects on Memory Storage.
Our focus on the amygdala as a possible site of influence of epinephrine effects on memory was guided by previous findings that memory storage can be modulated—i.e., either enhanced or impaired by electrical stimulation of the amygdala (35, 36, 37). Furthermore, adrenal demedullation and injections of epinephrine alter the effects of electrical stimulation of the amygdala on memory storage (38). In a series of experiments using inhibitory avoidance training we found that epinephrine effects on memory storage are blocked by lesions of the amygdala (39) as well as by lesions of the stria terminalis (ST) a major amygdala pathway (40). Epinephrine effects on memory storage are also blocked by the β-adrenergic antagonist propranolol infused into the amygdala immediately after training, immediately prior to the peripheral epinephrine injections (41).
The findings that β-adrenergic blockers infused into the amygdala block epinephrine effects suggested that the release of norepinephrine (NE) in the amygdala may play a critical role in mediating epinephrine effects on memory. According to this view, NE infused into the amygdala after training should modulate memory storage. Our findings support this implication. Posttraining infusions of NE produce dose-dependent enhancement (and impairment) of memory storage (42). A further implication of this hypothesis is that stimulation of the kind typically used in training should induce the release of NE in the amygdala. In experiments using in vivo microdialysis and HPLC (see Fig. 1), we found that footshock stimulation similar to that used in inhibitory avoidance training induced a 75% increase in NE release within the amygdala (43).
Figure 1.
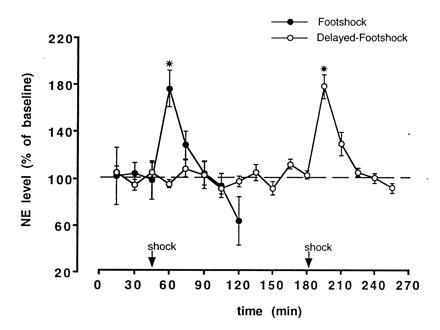
Freely moving rats with a microdialysis probe inserted into a cannula implanted in the amygdala (tip aimed at the border between the Central and Basolateral nuclei) received a single footshock (0.55 mA, 1.0 s) either 45.5 min (n = 5) or 180.5 min (n = 4) after being placed in an apparatus with a grid floor. Dialyzate samples were collected every 15 min and immediately injected into an HPLC with coulometric detection optimized for detection of NE. NE concentration is represented as mean (±SEM) of basal level prior to footshock. NE concentrations in the dialyzate increased to ≈75% above baseline (∗, P < 0.001) following the footshock and returned to baseline within 30 min. [Reproduced with permission from ref. 43 (Copyright 1996, Harcourt Brace.]
Epinephrine effects on memory are also blocked by peripherally administered propranolol (44), as would be expected, because propranolol readily enters the brain. However, the finding that epinephrine effects on memory are blocked by sotalol, a β-adrenergic antagonist that does not readily enter the brain, suggests that epinephrine effects are initiated by activation of peripheral β-adrenergic receptors. The findings of several experiments suggest that epinephrine activates β-adrenergic receptors located on vagal afferents (45) that project to the nucleus of the solitary tract (NTS) and that projections from the NTS release NE within the amygdala (46). Consistent with this hypothesis, inactivation of the NTS with lidocaine blocks the effects of epinephrine on memory (47). Other β-adrenergic agonists that enter the brain, including dipivefrin and clenbuterol enhance memory when administered posttraining (44, 48). Dipivefrin effects are blocked by propranolol, but not by sotalol (44), and clenbuterol effects are blocked by ST lesions (48). Furthermore, as was found with NE, posttraining intra-amygdala infusions of clenbuterol enhance memory storage (48, 49). Considered together, these findings provide strong support for the hypothesis that epinephrine effects on memory storage are mediated by the amygdala and that the effects involve activation of the release of NE within the amygdala.
Glucocorticoid Influences on Memory Storage.
Extensive evidence indicates that activation of adrenal steroid receptors in the hippocampus plays an important role in mediating the effects of glucocorticoids on memory storage (50, 51). However, recent findings from our laboratory strongly indicate that glucorticoids also affect memory storage through influences involving the amygdala. The effects of glucocorticoids on memory for inhibitory avoidance training are remarkably similar to those of studies of epinephrine (32). ST lesions block the memory-enhancing effects of the synthetic glucocorticoid dexamethasone (52). Moreover, as is shown in Fig. 2a, lesions of the amygdala restricted selectively to the basolateral nucleus also block the memory-enhancing effects of posttraining systemic injections of dexamethasone (53). It is important to note that the basolateral lesions alone do not impair inhibitory avoidance retention. As is shown in Fig. 2B, posttraining infusions of a specific glucocorticoid receptor agonist (RU 28362) enhances retention when administered into the basolateral nucleus but are ineffective when administered into the central nucleus (B.R. and J.L.M., unpublished data). The findings of these experiments strongly suggest that the central nucleus is not involved in mediating the memory-modulating effects of glucocorticoids. Additionally, as was found in studies of the effects of epinephrine, the memory-modulating effects of glucocorticoids involve noradrenergic activation within the amygdala. Propranolol infused into the basolateral nucleus blocks the memory-enhancing effects of posttraining systemic injections of dexamethasone (54).
Figure 2.
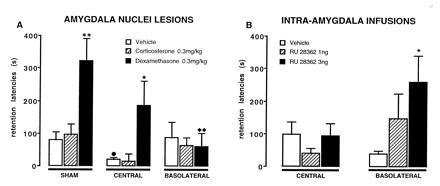
Step-through latencies (mean ± SEM) for a 48-h inhibitory avoidance retention test. (A) Rats with sham, or lesions of either the central or basolateral nucleus had been treated with corticosterone, dexamethasone, or vehicle immediately following training. (B) Rats received posttraining microinfusions of the glucocorticoid agonist RU 28362 into the central or basolateral nucleus. ∗, P < 0.05; ∗∗, P < 0.01 as compared with the corresponding vehicle group; •, P < 0.05 as compared with the corresponding sham lesion-vehicle group; ♦♦, P < 0.01 as compared with the corresponding sham lesion-dexamethasone group. [Reproduced with permission from ref. 53 (Copyright 1996, Harcourt Brace.]
Interactions with Other Neuromodulatory Systems
It is well established that memory storage is also modulated by posttraining systemic injections of drugs affecting opiate, GABAergic, and cholinergic systems (55). These effects, like those of adrenergic and glucocorticoid effects summarized above, also involve the amygdala.
Opioid Influences.
When administered systemically after inhibitory avoidance training, opiate receptor agonists and antagonists impair and enhance, respectively, subsequent retention (56, 57, 58). Furthermore, comparable effects are obtained with posttraining intra-amygdala infusions: naloxone enhances retention and β-endorphin impairs retention (59, 60, 61). The evidence that opioid peptides and opiates inhibit the release of NE (62, 63) suggested that opiate agonists and antagonists may influence memory storage by modulating the release of NE within the amygdala. Our findings provide strong support for this hypothesis. β-Adrenergic antagonists infused into the amygdala block the memory-enhancing effects of naloxone administered either systemically or intra-amygdally (60, 64). Furthermore, in experiments using both inhibitory avoidance and water-maze spatial tasks, intra-amygdala infusions of the β-adrenergic agonist clenbuterol blocked the memory-impairing effects of β-endorphin administered concurrently. Moreover, low, and otherwise ineffective, doses of β-endorphin and propranolol impaired memory when infused together into the amygdala (61).
GABAergic Influences.
The effects of GABAergic drugs on memory are highly comparable to those of opiate drugs. Posttraining systemic injections of GABAergic antagonists and agonists enhance and impair, respectively, retention of several types of training (65). Comparable effects are produced by posttraining intra-amygdala infusions of GABAergic drugs (66, 67). Furthermore, lesions of the amygdala (68) and intra-amygdala infusions of propranolol (J. Brioni and J.L.M., unpublished data) block GABAergic influences on memory. We have also examined the involvement of the amygdala in effects of benzodiazepines (BZDs) on memory. BZDs are known to act by binding to the GABAA receptor complex. And, it is well established that BZDs impair memory (69, 70). BZD influences on memory for inhibitory avoidance training appear to be mediated by the amygdala (71). Lesions of the basolateral nucleus block BZD-induced memory impairment but as was noted above, such lesions, alone, do not impair retention (72). In contrast, lesions of the central nucleus do not block BZD-induced memory impairment. Furthermore, we found that infusions of the BZD midazolam into the amygdala induces memory impairment and that intra-amygdala infusions of the GABAergic antagonist bicuculline methiodide block the BZD effect (73, 74). As we found with glucocorticoids, the basolateral nucleus appears to be the critical region of the amygdala for BZD influences on memory. Infusions of midazolam into the basolateral nucleus impair memory, whereas infusions administered into the central nucleus are ineffective (75).
Cholinergic Influences.
As is summarized above, our findings provide extensive evidence suggesting that adrenergic, glucocorticoid, opioid, and GABAergic influences on memory storage are mediated by NE release within the amygdala. Other findings from our laboratory suggest that they involve, at a subsequent step, activation of muscarinic cholinergic receptors within the amygdala. It is well established that, when administered systemically after training, muscarinic cholinergic agonists and antagonists enhance and impair, respectively, retention of a variety of tasks (26, 76). Moreover, highly comparable effects are obtained with posttraining intra-amygdala infusions (49). The hypothesis that NE effects on memory involve subsequent cholinergic activation in the amygdala is supported by evidence that intra-amygdala infusions of propranolol did not block the memory-enhancing effects of posttraining injections of the muscarinic cholinergic agonist oxotremorine administered either systemically or intra-amygdally (49). Furthermore, clenbuterol did not block the memory-impairing effects of the muscarinic cholinergic antagonist atropine when both drugs were infused into the amygdala posttraining (49).
Interactions.
Fig. 3 summarizes the findings of our experiments examining the involvement of the amygdala in mediating the effects of stress-related hormones and drugs on memory storage. The schema is based on the evidence suggesting that several neuromodulatory systems regulate memory storage by influencing the release of NE and the subsequent activation of muscarinic cholinergic receptors within the amygdala. And, as is discussed below, the schema reflects extensive evidence from our laboratory suggesting that these systems influence memory by modulating memory storage in brain regions activated by the amygdala (16, 32, 77).
Figure 3.
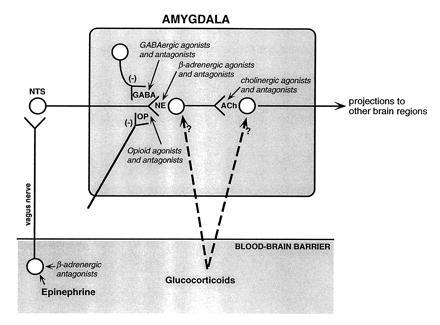
Schematic summarizing the interactions of neuromodulatory systems influencing memory storage suggested by the findings of our experiments. Epinephrine acts at peripheral β-adrenergic receptors located on vagal afferents projecting to the NTS. Activation of the NTS induces NE release in the amygdala. The peripherally acting β-adrenergic antagonist sotalol blocks epinephrine effects. Centrally acting noradrenergic agonists (clenbuterol and dipivefrin) directly activate NE receptors in the amygdala. Opiate and GABAergic agonists inhibit NE release and opiate and GABAergic antagonists induce NE release by blocking the inhibition. Centrally acting β-adrenergic antagonists (e.g., propranolol) block the activation of NE receptors in the amygdala and, thus, block all neuromodulatory influences affecting NE release. A subsequent step involves activation of muscarinic cholinergic receptors within the amygdala. Drugs affecting muscarinic cholinergic activation block the effects of drugs influencing noradrenergic activation. Glucocorticoids act at several sites. The modulatory effects of hormones and drugs on memory storage are mediated by influences on other brain systems.
Locus of Amygdala Influences on Memory Storage
Although findings summarized above strongly suggest that the amygdala is a critical site for integrating the interactions of several neuromodulatory systems influencing memory storage, they do not identify the locus or loci of the influences. The evidence that long-term potentiation (LTP) can be induced in the amygdala (78, 79, 80), as well as the findings indicating that drugs that block LTP also attenuate fear-based learning when administered into the amygdala prior to training suggest the possibility that neural changes mediating fear conditioning may be located within the amygdala (6, 12, 13, 14, 15).
However, findings from our laboratory strongly suggest that amygdala influences on memory storage involve amygdala activation of efferents mediated, at least in part, by the ST and, by implication, influences on memory storage in brain regions activated by the amygdala. Although lesions of the ST do not prevent inhibitory avoidance learning, such lesions block the memory-modulating effects of treatments that alter amygdala functioning. For example, ST lesions block the effects, on memory, of electrical stimulation of the amygdala (81) as well as the memory-enhancing and impairing effects of NE infused into the amygdala after inhibitory avoidance training (42). Additionally, as summarized above, lesions of the basolateral nucleus which, alone, do not impair memory, block the effects of BZDs and glucocorticoids on memory storage. Such findings are consistent with the hypothesis that the amygdala is involved in modulating memory storage but is not the locus of neural changes mediating long-term memory of emotionally arousing experiences.
We have used inhibitory avoidance tasks extensively in our research and retention assessed in this task is no doubt based, at least in part, on fear. However, we have found that drugs infused into the amygdala after training also enhance memory that is not based on fear-induced response inhibition, including Y-maze discrimination (82) and behavioral contrast (which assesses memory for increases and decreases in reward) (83, 84) as well as cued and spatial water-maze tasks (85). This latter experiment was based on the findings of “double-dissociation” lesion and drug-infusion studies from our laboratory, as well as other laboratories, indicating that the hippocampus and caudate caudate, respectively, are selectively involved in spatial learning and cue/response (“win–stay”) learning (86, 87, 88, 89, 90, 91). Rats in our study were trained to swim either to a visible cue on a platform located in a different position on each trial or to a submerged platform located in a constant position. Amphetamine or a control solution were microinfused unilaterally into the hippocampus, caudate nucleus or amygdala immediately after the training session and retention was tested the next day. The intrahippocampal and intracaudate infusions produced task-specific enhancement of retention. Hippocampal infusions selectively enhanced retention of spatial training and caudate injections selectively enhanced retention of cue/response training. In contrast, the intra-amygdala infusions enhanced retention of both spatial and cue/response training. Additionally, inactivation of the amygdala with infusions of lidocaine administered prior to the retention test did not block the memory-enhancing effects of infusions of amphetamine administered after the training (85). These findings strongly suggest that the lasting neural changes mediating the enhanced memory for the cued and spatial training were not located within the amygdala.
The findings of several recent studies suggest that the basolateral nucleus influences learning mediated by the hippocampus as well as hippocampal neuroplasticity. We have found that lesions of the basolateral nucleus block the memory-modulating effects (in inhibitory avoidance and spatial learning tasks) of posttraining infusions of glucocorticoid agonists and antagonists infused directly into the dorsal hippocampus (92). Additionally, recent electrophysiological studies indicate that basolateral nucleus lesions attenuate the induction of LTP in the dentate gyrus and that high-frequency stimulation of the basolateral nucleus facilitates the induction of LTP in the dentate gyrus (93, 94, 95).
Other recent findings strongly suggest that the amygdala is also not a critical locus mediating either the acquisition or the retention of inhibitory avoidance. Excitotoxic lesions of the amygdala induced prior to training attenuate, but do not block, inhibitory avoidance retention (96). Experiments in which the amygdala is inactivated prior to a retention test (85) or lesioned after training would seem to provide a direct test of whether the amygdala is a critical site of neural changes underlying memory. In recent experiments, rats received different amounts of training in a footshock escape task in a two-compartment straight alley and subsequently (7 or 30 days later), bilateral sham or excitotoxic (N-methyl-d-aspartate) lesions of the amygdala were induced (97, 98). Retention was first tested by placing the animals in the safe compartment and measuring the latencies to enter the compartment where shock had been delivered on the training trials (i.e., inhibitory avoidance testing). The lesions attenuated, but did not block, retention. In both the sham- and amygdala-lesioned groups, increases in amount of training prior to the lesions resulted in enhanced retention performance. On a subsequent retention test, rats were retained in the alley until they remained in the starting compartment for 100 consecutive seconds. In both the sham-lesioned and amygdala-lesioned groups, animals given increased amounts of original escape training made fewer shock-compartment entries. For rats that had received no prior escape training, the second test assessed acquisition of inhibitory avoidance. The lesions slightly (but significantly) impaired acquisition. Highly comparable results were obtained in an experiment examining the effects of basolateral nucleus lesions induced after escape training (99). Additionally, we found that in animals given footshocks of different intensities on a single inhibitory avoidance training trial, higher intensity footshocks resulted in better retention in sham controls as well as animals in which amygdala lesions were induced 7 days after training (100). Similar results were obtained in experiments examining the effects of lidocaine infusions administered into the amygdala prior to retention tests (101). Considered together, these findings provide strong evidence indicating that an intact amygdala is not required for either the acquisition or retention of footshock-motivated inhibitory avoidance or escape training.
Several studies reported that infusions of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropidnic acid (AMPA) receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) into the amygdala prior to retention tests impair inhibitory avoidance performance (102, 103) and fear-potentiated startle (104). As there is evidence that LTP expression involves activation of AMPA receptors (105), such findings are consistent with the view that fear-based learning may be based on LTP in the amygdala. Experiments in our laboratory examined the effects of intra-amygdala infusions of CNQX administered prior to retention tests in experiments using procedures similar to those of our studies of the effects of amygdala lesions induced after escape training (106). The CNQX infusions impaired, but did not block, inhibitory avoidance retention. Further, in both controls and CNQX-treated groups, animals given increased amounts of original training made fewer shock-compartment entries prior to remaining in the safe compartment for 100 consecutive seconds. The CNQX infusions also slightly, but significantly, impaired acquisition in animals that had received no prior escape training. However, we also found that the intra-amygdala CNQX infusions decreased anxiety and footshock sensitivity and increased locomotor activity. Our findings strongly suggest that the impaired retention performance induced by CNQX may have been due to a nonspecific reduction of fear and anxiety and increased locomotor activity. These findings are consistent with evidence that amygdala lesions impair unconditioned responses to stressful stimulation (107, 108). However, our findings clearly indicate that an intact amygdala is not required for the acquisition or expression of retention of footshock-motivated training (16).
Involvement of β-Adrenergic Activation and the Amygdala in Emotionally Influenced Memory in Human Subjects
As we have emphasized above, the findings of our studies using rats provide extensive evidence suggesting that adrenal hormones modulate memory storage and that the effects are mediated through influences involving the amygdala (16). The findings of recent studies of memory in human subjects provide additional evidence for this view. A first experiment (20) examined, in healthy volunteers, the effect of the β-blocker propranolol, or a placebo, on long-term (i.e., 1-week) memory for either an emotionally neutral story or a closely matched, but more emotionally arousing story, each consisting of 12 slides accompanied by narration. The emotionally arousing narration occurred in the middle of the story. The placebo controls showed enhanced memory for the emotional story whereas, in contrast, propranolol selectively impaired memory for the emotionally arousing section but did not impair memory either for the neutral story or for the initial and final portions of the emotionally arousing story. The drug effect could not be attributed to effects on attention, sedation, or emotional reactions of the subjects to the stories. These findings suggest strongly that modulation of memory storage by emotional arousal depends upon activation of β-adrenergic receptors. Nielson and Jensen (109) obtained comparable results in an experiment examining the effects on β-blockers on enhanced memory induced by of physically induced arousal (increased muscle tension). The arousal did not enhance retention in elderly subjects who were taking β-blockers.
The findings of a study using the same general procedures used in the experiment by Cahill et al. (20) indicated that emotional arousal did not enhance long-term memory in a subject with bilateral degenerative lesions of the amygdala (21). In the third study, positron-emission tomography scans assessed cerebral glucose metabolism in healthy volunteers on each of two separate sessions in which the subjects viewed emotionally arousing or emotionally neutral film clips. Memory of the film clips was determined by a surprise free-recall test three weeks later. As is shown in Fig. 4, glucose metabolic rate of the right amygdala induced by viewing the emotional film clips was highly correlated (+0.93) with the number of films recalled (22). These studies clearly suggest that the amygdala in humans is involved with the formation of long-term declarative memory for emotional events.
Figure 4.
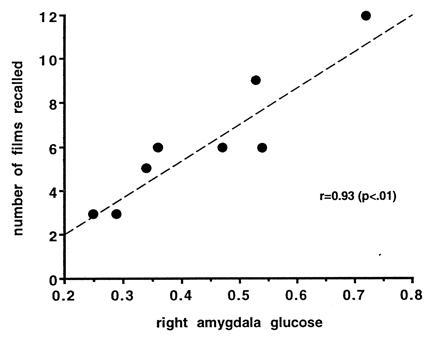
Activity of the amygdala related to emotional memory in healthy human subjects. Scatterplot shows the relationship between relative glucose metabolic rate of the right amygdala in subjects viewing a series of emotionally arousing (aversive) film clips and long-term memory of those clips. Relative glucose metabolic rate in the right amygdala was highly correlated (P < 0.01) with the number of films recalled. [Reproduced with permission from Cahill et al. (22) (Copyright 1996, National Acadeny of Sciences).]
Concluding Comments
The findings of our studies using human subjects are consistent with those of our other studies using animal subjects in indicating that memory storage is influenced by activation of β-adrenergic systems and the amygdala (19, 55, 110). Considered together, these findings provide strong evidence supporting the hypothesis that the amygdala, especially the basolateral nucleus, plays a central role in modulating the consolidation of long-term memory of emotionally arousing experiences.
Acknowledgments
This research supported by research Grant MH12526 from National Institute of Mental Health, by a grant from the National Institute on Drug Abuse (J.L.M.), and by the R.W. and L. Gerard Fellowship (B.R.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ST, stria terminalis; NE, norepinephrine; NTS, nucleus of the solitary tract; BZD, benzodiazepine; LTP, long-term potentiation; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione.
References
- 1.Weiskrantz L. J Comp Physiol Psychol. 1956;49:381–391. doi: 10.1037/h0088009. [DOI] [PubMed] [Google Scholar]
- 2.Aggleton J P, editor. The Amygdala. New York: Wiley–Liss; 1992. [Google Scholar]
- 3.Scoville W B, Milner B. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zola-Morgan S, Squire L R, Amaral D G. J Neurosci. 1989;9:1922–1936. doi: 10.1523/JNEUROSCI.09-06-01922.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis M. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- 6.LeDoux J E. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- 7.Maren S, Fanselow M S. Neuron. 1996;16:237–240. doi: 10.1016/s0896-6273(00)80041-0. [DOI] [PubMed] [Google Scholar]
- 8.McGaugh J L. In: Memory Distortion: How Minds, Brains, and Societies Reconstruct the Past. Schacter D L, Coyle J T, Mesulam M-M, Sullivan L E, editors. Cambridge, MA: Harvard Univ. Press; 1995. pp. 255–273. [Google Scholar]
- 9.Everitt B J, Robbins T W. In: The Amygdala. Aggleton J P, editor. New York: Wiley–Liss; 1992. pp. 401–430. [Google Scholar]
- 10.Kesner R P, Williams J M. Neurobiol Learn Mem. 1995;64:237–244. doi: 10.1006/nlme.1995.0006. [DOI] [PubMed] [Google Scholar]
- 11.White N M, McDonald R J. Behav Brain Res. 1993;55:269–281. doi: 10.1016/0166-4328(93)90122-7. [DOI] [PubMed] [Google Scholar]
- 12.Falls W A, Miserendino M J D, Davis M. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fanselow M S, Kim J J. Behav Neurosci. 1994;108:210–212. doi: 10.1037//0735-7044.108.1.210. [DOI] [PubMed] [Google Scholar]
- 14.Kim M, McGaugh J L. Brain Res. 1992;585:35–48. doi: 10.1016/0006-8993(92)91188-k. [DOI] [PubMed] [Google Scholar]
- 15.Miserendino M J D, Sananes C B, Melia K R, Davis M. Nature (London) 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 16.McGaugh J L, Cahill L, Parent M B, Mesches M H, Coleman-Mesches K, Salinas J A. In: Plasticity in the Central Nervous System: Learning and Memory. McGaugh J L, Bermudez-Rattoni F, Prado-Alcala R A, editors. Hillsdale, NJ: Lawrence Erlbaum; 1995. pp. 17–40. [Google Scholar]
- 17.McGaugh J L. Psychol Sci. 1990;1:15–25. [Google Scholar]
- 18.McGaugh J L. Brain Res Bull. 1989;23:339–345. doi: 10.1016/0361-9230(89)90220-7. [DOI] [PubMed] [Google Scholar]
- 19.Cahill L, McGaugh J L. Curr Opin Neurobiol. 1996;6:237–242. doi: 10.1016/s0959-4388(96)80078-x. [DOI] [PubMed] [Google Scholar]
- 20.Cahill L, Prins B, Weber M, McGaugh J L. Nature (London) 1994;371:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- 21.Cahill L, Babinsky R, Markowitsch H J, McGaugh J L. Nature (London) 1995;377:296. doi: 10.1038/377295a0. [DOI] [PubMed] [Google Scholar]
- 22.Cahill L, Haier R J, Fallon J, Alkire M, Tang C, Keator D, Wu J, McGaugh J L. Proc Natl Acad Sci, USA. 1996;93:8016–8021. doi: 10.1073/pnas.93.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGaugh J L. Annu Rev Pharmacol. 1973;13:229–241. doi: 10.1146/annurev.pa.13.040173.001305. [DOI] [PubMed] [Google Scholar]
- 24.McGaugh J L, Herz M J. Memory Consolidation. San Francisco: Albion; 1972. [Google Scholar]
- 25.Gold P E, McGaugh J L. In: Short-Term Memory. Deutsch D, Deutsch J A, editors. New York: Academic; 1975. pp. 355–378. [Google Scholar]
- 26.McGaugh J L. Annu Rev Neurosci. 1989;12:255–287. doi: 10.1146/annurev.ne.12.030189.001351. [DOI] [PubMed] [Google Scholar]
- 27.McCarty R, Gold P E. Horm Behav. 1981;15:168–182. doi: 10.1016/0018-506x(81)90026-x. [DOI] [PubMed] [Google Scholar]
- 28.McGaugh J L, Gold P E. In: Psychoendocrinology. Brush R B, Levine S, editors. New York: Academic; 1989. pp. 305–339. [Google Scholar]
- 29.Gold P E, van Buskirk R. Behav Biol. 1975;13:247–260. doi: 10.1016/s0091-6773(75)91784-8. [DOI] [PubMed] [Google Scholar]
- 30.McGaugh J L. Annu Rev Psychol. 1983;34:297–323. doi: 10.1146/annurev.ps.34.020183.001501. [DOI] [PubMed] [Google Scholar]
- 31.McGaugh J L. In: Handbook of Emotion and Memory: Current Research and Theory. Christianson S-A, editor. Hillsdale, NJ: Lawrence Erlbaum; 1992. pp. 245–268. [Google Scholar]
- 32.Roozendaal B, Cahill L, McGaugh J L. In: Brain Processes and Memory. Ishikawa K, McGaugh J L, Sakata H, editors. Amsterdam: Elsevier; 1996. pp. 39–54. [Google Scholar]
- 33.Borrell J, de Kloet E R, Versteeg D H G, Bohus B. Behav Neural Biol. 1983;39:241–258. doi: 10.1016/s0163-1047(83)90910-x. [DOI] [PubMed] [Google Scholar]
- 34.Roozendaal B, Carmi O, McGaugh J L. Proc Natl Acad Sci USA. 1996;93:1429–1433. doi: 10.1073/pnas.93.4.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goddard G V. J Comp Physiol Psychol. 1964;58:23–30. doi: 10.1037/h0049256. [DOI] [PubMed] [Google Scholar]
- 36.Kesner R P, Wilburn M. Behav Biol. 1974;10:259–293. doi: 10.1016/s0091-6773(74)91894-x. [DOI] [PubMed] [Google Scholar]
- 37.McGaugh J L, Gold P E. In: Neural Mechanisms of Learning and Memory. Rosenzweig M R, Bennett E L, editors. Cambridge, MA: MIT Press; 1976. pp. 549–560. [Google Scholar]
- 38.Liang K C, Bennett C, McGaugh J L. Behav Brain Res. 1985;15:93–100. doi: 10.1016/0166-4328(85)90056-7. [DOI] [PubMed] [Google Scholar]
- 39.Cahill L, McGaugh J L. Psychobiology. 1991;19:206–210. [Google Scholar]
- 40.Liang K C, McGaugh J L. Behav Brain Res. 1983;9:49–58. doi: 10.1016/0166-4328(83)90013-x. [DOI] [PubMed] [Google Scholar]
- 41.Liang K C, Juler R G, McGaugh J L. Brain Res. 1986;368:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- 42.Liang K C, McGaugh J L, Yao H-Y. Brain Res. 1990;508:225–233. doi: 10.1016/0006-8993(90)90400-6. [DOI] [PubMed] [Google Scholar]
- 43.Galvez, R., Mesches, M. & McGaugh, J. L. (1996) Neurobiol. Learn. Mem., in press. [DOI] [PubMed]
- 44.Introini-Collison I, Saghafi D, Novack G, McGaugh J L. Brain Res. 1992;572:81–86. doi: 10.1016/0006-8993(92)90454-h. [DOI] [PubMed] [Google Scholar]
- 45.Schreurs J, Seelig T, Schulman H. J Neurochem. 1986;46:294–296. doi: 10.1111/j.1471-4159.1986.tb12961.x. [DOI] [PubMed] [Google Scholar]
- 46.Ricardo J A, Koh E T. Brain Res. 1978;153:1–26. doi: 10.1016/0006-8993(78)91125-3. [DOI] [PubMed] [Google Scholar]
- 47.Williams C L, McGaugh J L. Behav Neurosci. 1993;107:1–8. [PubMed] [Google Scholar]
- 48.Introini-Collison I B, Miyazaki B, McGaugh J L. Psychopharmacology. 1991;104:541–544. doi: 10.1007/BF02245663. [DOI] [PubMed] [Google Scholar]
- 49.Introini-Collison I B, Dalmaz C, McGaugh J L. Neurobiol Learn Mem. 1996;65:57–64. doi: 10.1006/nlme.1996.0006. [DOI] [PubMed] [Google Scholar]
- 50.Bohus B. In: The Memory System of the Brain. Delacour J, editor. Vol. 4. Teaneck, NJ: World Scientific; 1994. pp. 337–364. [Google Scholar]
- 51.McEwen B S, Sapolsky R M. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 52.Roozendaal B, McGaugh J L. Brain Res. 1996;709:243–250. doi: 10.1016/0006-8993(95)01305-9. [DOI] [PubMed] [Google Scholar]
- 53.Roozendaal B, McGaugh J L. Neurobiol Learn Mem. 1996;65:1–8. doi: 10.1006/nlme.1996.0001. [DOI] [PubMed] [Google Scholar]
- 54.Quirarte G L, Roozendaal B, McGaugh J L. Soc Neurosci Abstr. 1996;22:1869. [Google Scholar]
- 55.McGaugh, J. L. & Cahill, L. (1996) Behav. Brain Res., in press. [DOI] [PubMed]
- 56.Izquierdo I, Dias R D. Psychoneuroendocrinology. 1983;8:81–87. doi: 10.1016/0306-4530(83)90043-4. [DOI] [PubMed] [Google Scholar]
- 57.Izquierdo I, Souza D O, Carrasco M A, Dias R D, Perry M L, Eisinger S, Elisabetsky E, Vendite D A. Psychopharmacology. 1980;70:173–177. doi: 10.1007/BF00435310. [DOI] [PubMed] [Google Scholar]
- 58.McGaugh J L, Introini-Collison I B, Castellano C. In: Handbook of Experimental Pharmacology, Opioids. Herz A, Akil H, Simon E J, editors. Heidelberg: Springer; 1993. Part 2104429447. [Google Scholar]
- 59.Gallagher M, Kapp B S, Pascoe J P, Rapp P R. In: The Amygdaloid Complex. Ben-Ari Y, editor. Amsterdam: Elsevier/North–Holland; 1981. pp. 343–354. [Google Scholar]
- 60.Introini-Collison I B, Nagahara A H, McGaugh J L. Brain Res. 1989;476:94–101. doi: 10.1016/0006-8993(89)91540-0. [DOI] [PubMed] [Google Scholar]
- 61.Introini-Collison I B, Ford L, McGaugh J L. Neurobiol Learn Mem. 1995;63:200–205. doi: 10.1006/nlme.1995.1021. [DOI] [PubMed] [Google Scholar]
- 62.Arbilla S, Langer S Z. Nature (London) 1978;271:559–561. doi: 10.1038/271559a0. [DOI] [PubMed] [Google Scholar]
- 63.Walker J M, Kahachaturian H, Watson S J. In: Norepinephrine. Ziegler M G, Lake C R, editors. Baltimore: Williams & Wilkins; 1984. pp. 74–91. [Google Scholar]
- 64.McGaugh J L, Introini-Collison I B, Nagahara A H. Brain Res. 1988;446:37–49. doi: 10.1016/0006-8993(88)91294-2. [DOI] [PubMed] [Google Scholar]
- 65.Castellano C, Brioni J D, McGaugh J L. In: Biology of Memory. Squire L, Lindenlaub E, editors. Stuttgart: Schattauer; 1990. pp. 361–378. [Google Scholar]
- 66.Brioni J D, McGaugh J L. Psychopharmacology. 1988;96:505–510. doi: 10.1007/BF02180032. [DOI] [PubMed] [Google Scholar]
- 67.Brioni J D, Nagahara A H, McGaugh J L. Brain Res. 1989;487:105–112. doi: 10.1016/0006-8993(89)90945-1. [DOI] [PubMed] [Google Scholar]
- 68.Ammassari-Teule M, Pavone F, Castellano C, McGaugh J L. Brain Res. 1991;551:104–109. doi: 10.1016/0006-8993(91)90919-m. [DOI] [PubMed] [Google Scholar]
- 69.Lister R G. Neurosci Biobehav Rev. 1985;9:87–93. doi: 10.1016/0149-7634(85)90034-x. [DOI] [PubMed] [Google Scholar]
- 70.Izquierdo I, Da Cunha C, Medina J. Neurosci Biobehav Rev. 1990;14:419–424. doi: 10.1016/s0149-7634(05)80064-8. [DOI] [PubMed] [Google Scholar]
- 71.Izquierdo I, Da Cunha C, Huang C H, Walz R, Wolfman C, Medina J H. Behav Neural Biol. 1990;54:105–109. doi: 10.1016/0163-1047(90)91282-g. [DOI] [PubMed] [Google Scholar]
- 72.Tomaz C, Dickinson-Anson H, McGaugh J L. Proc Natl Acad Sci USA. 1992;89:3615–3619. doi: 10.1073/pnas.89.8.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dickinson-Anson H, Mesches M H, Coleman K, McGaugh J L. Behav Neural Biol. 1993;60:1–4. doi: 10.1016/0163-1047(93)90638-x. [DOI] [PubMed] [Google Scholar]
- 74.Dickinson-Anson H, McGaugh J L. Behav Neural Biol. 1993;60:84–87. doi: 10.1016/0163-1047(93)90781-c. [DOI] [PubMed] [Google Scholar]
- 75.de Souza-Silva M A, Tomaz C. Neuropsychobiology. 1995;32:31–36. doi: 10.1159/000119209. [DOI] [PubMed] [Google Scholar]
- 76.Hunter B, Zornetzer S F, Jarvik M E, McGaugh J L. In: Handbook of Psychopharmacology. Iversen L, Iversen S, Snyder S, editors. Vol. 8. New York: Plenum; 1977. pp. 531–577. [Google Scholar]
- 77.Packard M G, Williams C L, Cahill L, McGaugh J L. In: Neurobehavioral Plasticity: Learning, Development and Response to Brain Insults. Spear N E, Spear L, Woodruff M, editors. Hillsdale, NJ: Lawrence Erlbaum; 1995. pp. 149–184. [Google Scholar]
- 78.Chapman P F, Kairiss E W, Keenan C L, Brown T H. Synapse. 1990;6:271–278. doi: 10.1002/syn.890060306. [DOI] [PubMed] [Google Scholar]
- 79.Clugnet M C, LeDoux J E. J Neurosci. 1990;10:1055–1061. doi: 10.1523/JNEUROSCI.10-04-01055.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maren S, Fanselow M S. J Neurosci. 1995;15:7548–7564. doi: 10.1523/JNEUROSCI.15-11-07548.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang K C, McGaugh J L. Brain Res. 1983;274:309–318. doi: 10.1016/0006-8993(83)90709-6. [DOI] [PubMed] [Google Scholar]
- 82.Introini-Collison I B, McGaugh J L. Psychopharmacology. 1988;94:379–385. doi: 10.1007/BF00174693. [DOI] [PubMed] [Google Scholar]
- 83.Salinas J A, Packard M G, McGaugh J L. Behav Brain Res. 1993;59:153–159. doi: 10.1016/0166-4328(93)90162-j. [DOI] [PubMed] [Google Scholar]
- 84.Salinas J A, McGaugh J L. Behav Brain Res. 1996;80:87–98. doi: 10.1016/0166-4328(96)00023-x. [DOI] [PubMed] [Google Scholar]
- 85.Packard M G, Cahill L, McGaugh J L. Proc Natl Acad Sci USA. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Packard M G, Hirsh R, White N M. J Neurosci. 1989;9:1465–1472. doi: 10.1523/JNEUROSCI.09-05-01465.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Packard M G, White N M. Behav Neurosci. 1991;105:295–306. doi: 10.1037//0735-7044.105.2.295. [DOI] [PubMed] [Google Scholar]
- 88.Packard M G, McGaugh J L. Behav Neurosci. 1992;106:439–446. doi: 10.1037//0735-7044.106.3.439. [DOI] [PubMed] [Google Scholar]
- 89.Kesner R P, Bolland B L, Dakis M. Exp Brain Res. 1993;93:462–470. doi: 10.1007/BF00229361. [DOI] [PubMed] [Google Scholar]
- 90.McDonald R J, White N M. Behav Neurosci. 1993;107:3–22. doi: 10.1037//0735-7044.107.1.3. [DOI] [PubMed] [Google Scholar]
- 91.Packard M G, McGaugh J L. Neurobiol Learn Mem. 1996;65:65–72. doi: 10.1006/nlme.1996.0007. [DOI] [PubMed] [Google Scholar]
- 92.Roozendaal, B. & McGaugh, J. L. (1996) Eur. J. Neurosci., in press.
- 93.Ikegaya Y, Saito H, Abe K. Brain Res. 1994;656:157–164. doi: 10.1016/0006-8993(94)91377-3. [DOI] [PubMed] [Google Scholar]
- 94.Ikegaya Y, Saito H, Abe K. Brain Res. 1995;671:351–354. doi: 10.1016/0006-8993(94)01403-5. [DOI] [PubMed] [Google Scholar]
- 95.Ikegaya Y, Saito H, Abe K. Neurosci Res. 1995;22:203–207. doi: 10.1016/0168-0102(95)00894-7. [DOI] [PubMed] [Google Scholar]
- 96.Cahill L, McGaugh J L. Behav Neurosci. 1990;104:532–543. doi: 10.1037//0735-7044.104.4.532. [DOI] [PubMed] [Google Scholar]
- 97.Parent M B, Tomaz C, McGaugh J L. Behav Neurosci. 1992;106:791–799. doi: 10.1037//0735-7044.106.5.789. [DOI] [PubMed] [Google Scholar]
- 98.Parent M B, West M, McGaugh J L. Behav Neurosci. 1994;6:1080–1087. doi: 10.1037//0735-7044.108.6.1080. [DOI] [PubMed] [Google Scholar]
- 99.Parent M B, Avila E, McGaugh J L. Brain Res. 1995;676:235–244. doi: 10.1016/0006-8993(95)00095-8. [DOI] [PubMed] [Google Scholar]
- 100.Parent M B, Quirarte G L, Cahill L, McGaugh J L. Behav Neurosci. 1995;109:803–807. doi: 10.1037//0735-7044.109.4.803. [DOI] [PubMed] [Google Scholar]
- 101.Coleman-Mesches K, Salinas J A, McGaugh J L. Behav Brain Res. 1996;77:175–180. doi: 10.1016/0166-4328(95)00231-6. [DOI] [PubMed] [Google Scholar]
- 102.Izquierdo I, Bianchin M, Silva M, Zanatta M S, Walz R, Ruschel A C, Da Silva R C, Paczko N, Medina J. Behav Neural Biol. 1993;59:1–4. doi: 10.1016/0163-1047(93)91061-q. [DOI] [PubMed] [Google Scholar]
- 103.Liang K C. Soc Neurosci Abstr. 1991;17:486. [Google Scholar]
- 104.Kim M, Campeau S, Falls W A, Davis M. Behav Neural Biol. 1993;59:5–8. doi: 10.1016/0163-1047(93)91075-x. [DOI] [PubMed] [Google Scholar]
- 105.Collingridge G L, Bashir Z I, Blake J F, Davies C H, Davies S N, Irvining A J, Randall A D, Schofield J G. In: The Biology of Memory. Squire L R, Lindenlaub E, editors. Stuttgart: Schattauer; 1990. pp. 233–250. [Google Scholar]
- 106.Mesches, M. H., Bianchin, M. & McGaugh, J. L. (1996) Neurobiol. Learn. Mem., in press. [DOI] [PubMed]
- 107.Goldstein L E, Rasmusson A M, Bunney B S, Roth R H. J Neurosci. 1996;16:4787–4798. doi: 10.1523/JNEUROSCI.16-15-04787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Young B J, Leaton R N. Behav Neurosci. 1996;110:228–237. doi: 10.1037//0735-7044.110.2.228. [DOI] [PubMed] [Google Scholar]
- 109.Nielson K A, Jensen R A. Behav Neural Biol. 1994;62:190–200. doi: 10.1016/s0163-1047(05)80017-2. [DOI] [PubMed] [Google Scholar]
- 110.Cahill L, Roozendaal B, McGaugh J L. In: The Functional Behaviorism of Robert C. Bolles: Learning, Motivation and Cognition. Bouton M E, Fanselow M S, editors. Washington, DC: Am. Psychol. Assoc.; 1996. in press. [Google Scholar]