

Abstract
This article reviews recent studies of memory systems in humans and nonhuman primates. Three major conclusions from recent work are that (i) the capacity for nondeclarative (nonconscious) learning can now be studied in a broad array of tasks that assess classification learning, perceptuomotor skill learning, artificial grammar learning, and prototype abstraction; (ii) cortical areas adjacent to the hippocampal formation, including entorhinal, perirhinal, and parahippocampal cortices, are an essential part of the medial temporal lobe memory system that supports declarative (conscious) memory; and (iii) in humans, bilateral damage limited to the hippocampal formation is nevertheless sufficient to produce severe anterograde amnesia and temporally graded retrograde amnesia covering as much as 25 years.
This article considers two topics relevant to the organization of memory and brain systems. The first topic is the fundamental idea that memory is not a single entity but consists of several separate entities that depend on different brain systems. The key distinction is between the capacity for conscious recollection of facts and events (declarative memory) and a heterogeneous collection of nonconscious learning capacities (nondeclarative memory) that are expressed through performance and that do not afford access to any conscious memory content. Some of the best evidence for distinguishing between kinds of memory has come from the study of amnesic patients who have sustained bilateral damage to medial temporal lobe or midline diencephalic brain structures. Amnesic patients are severely impaired on conventional memory tests that assess declarative memory—i.e., tests that assess recall or recognition of places, lists, faces, melodies, and other material. However, the same patients perform as well as normal subjects on many other tasks of learning and memory that assess, for example, the capacity for skill and habit learning and the phenomenon of priming. The implication is that the kinds of learning and memory that are intact in amnesia depend on different brain systems than those damaged in amnesia (Fig. 1). Recent studies have expanded the list of learning and memory abilities that are intact in amnesia.
Figure 1.
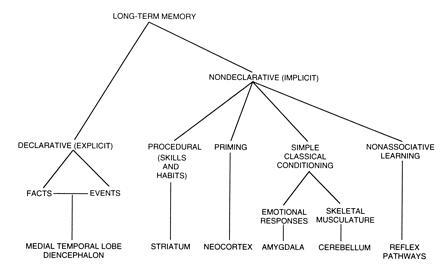
A taxonomy of long-term memory systems together with specific brain structures involved in each system (adapted from ref. 66).
The second topic concerns which structures in the medial temporal lobe are important for declarative memory. Some of this work is based on studies of an animal model of human amnesia in the monkey. Other information comes from detailed neuropathological study of the brains of amnesic patients, for whom extensive neurobehavioral data are available. One major finding, based largely on work with monkeys, is that cortical areas adjacent to the hippocampal formation, including entorhinal, perirhinal, and parahippocampal cortices, are an essential part of the medial temporal lobe memory system. A second major finding, based largely on human material, is that bilateral damage limited to the hippocampal formation (hippocampus proper, dentate gyrus, subicular complex, and entorhinal cortex) is sufficient to produce severe anterograde amnesia together with severe, temporally graded retrograde amnesia covering as much as 25 years.
Multiple Memory Systems in the Brain
Amnesic patients perform normally on a wide variety of tasks: probabilistic classification learning, perceptuomotor skill learning, and tasks that assess the ability to acquire knowledge about categories when given a series of exemplars, as in artificial grammar learning and prototype learning. These tasks depend on brain systems that are intact in amnesia and that are important for various forms of nondeclarative memory. Recent work with these tasks is beginning to provide a property list describing the operating characteristics of declarative and nondeclarative memory, and it suggests what brain systems are important for some forms of nondeclarative memory.
Probabilistic Classification Learning.
This kind of learning is analogous to the habit learning tasks studied in experimental animals. Subjects attempt to learn a set of associations. The associations are not obvious, and they are difficult to memorize because of the probabilistic structure of the task. As a result, information from a single trial is not as reliable or useful as information accrued across many trials. In one such task, the subject plays the role of weather forecaster and on each trial tries to predict from cues that are presented whether the outcome will be rain or sunshine (Fig. 2). Amnesic patients can learn gradually to predict the correct weather outcome, and they learn at the same rate as normal subjects, improving from 50% correct (chance performance) to ≈65% correct during 50 training trials (1). The same result has been obtained in three different versions of the same task. Despite the intact performance of the amnesic patients on the prediction task, they were markedly impaired at answering explicit factual questions about the training episode.
Figure 2.

Probabilistic classification learning: the weather prediction task (2). Subjects decide on each trial which of two weather outcomes (rain or sunshine) will occur based on a set of one, two, or three cues (out of four possible cues) that appear on a computer screen. Each cue is independently associated to a weather outcome with a fixed probability, and the two outcomes occur equally often. There are four possible cue–outcome association strengths: throughout training, a cue is associated either 75%, 57%, 43%, or 25% (approximately) with sunshine. During each trial, one, two, or three of the four possible cues are presented. Subjects respond by pressing a key to predict the weather outcome, and feedback is given immediately after each choice (correct or incorrect).
More recently, patients with Huntington disease and nondemented patients with Parkinson disease were found to be severely impaired in this same probabilistic learning task. Both diseases are associated with prominent pathology in the caudate nucleus. During 50 training trials, they performed no better than 53% correct in any 10-trial block (2, 3). These findings support the idea that the probabilistic classification task is akin to habit learning, which in experimental animals appears to depend on the integrity of the caudate nucleus (4, 5, 6). The important point is that the learning deficits in Huntington disease and Parkinson disease patients are not limited to learning motor programs, and the caudate nucleus is important for some kinds of habit learning, even when motor skill learning is not required (see also refs. 7 and 8).
Memory systems that are distinct from each other (i.e., declarative and nondeclarative memory systems) should be separable on the basis of multiple criteria (9, 10, 11, 12). Thus, it should be possible to distinguish declarative and nondeclarative memory systems not only in terms of anatomy, but also in terms of operating characteristics, the kind of information processed, and the purpose served by each system (13). Studies of experimental animals have suggested that declarative memory is more flexible than nondeclarative memory (14, 15, 16). To explore this issue in humans, we asked how well subjects who had learned the probabilistic classification task could use their task knowledge in a flexible way (17). Although amnesic patients and control subjects learned the classification task to the same level of proficiency, the two groups differed in their performance on transfer tests that asked for judgments about the associative strengths of the test cues. Amnesic patients performed more poorly than control subjects on the transfer tests. Importantly, they were impaired on the same test questions that were indicated by independent raters to be the most indirect and to require the most flexible use of task knowledge. These findings show that declarative and nondeclarative memory differ with respect to the flexibility of the knowledge acquired by each system. Declarative knowledge is accessible to multiple response systems. Nondeclarative memory is more encapsulated and has less access to systems not involved in the initial learning.
Perceptuomotor Skill Learning.
Another important distinction between declarative and nondeclarative memory is that declarative memory supports conscious recollections, whereas nondeclarative memory does not afford awareness of any memory content. The issue of awareness has been addressed particularly well in the serial reaction time task, a test of perceptuomotor skill learning (Fig. 3; refs. 18, 19, 20, 21). In this task, subjects respond as rapidly as possible with a key press to a cue, which can appear in any one of four locations. The location of the cue follows a repeating sequence of 10 cued locations for 400 training trials. In one recent study (22), amnesic patients and control subjects exhibited equivalent learning of the repeating sequence, as demonstrated by gradually improving reaction times and by an increase in reaction times when the repeating sequence was replaced by a random sequence. Reaction times did not improve when subjects were given a random sequence.
Figure 3.
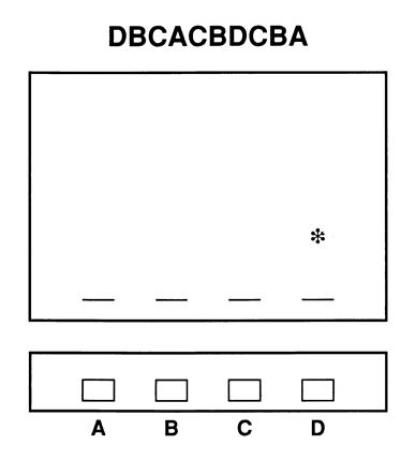
The serial reaction time task as presented on a computer (22). Four dashes appear continuously at the bottom of the screen to denote four possible locations of an asterisk (A, B, C, or D). During training, the asterisk appears sequentially, moving from one to another of the four locations. Subjects respond to each appearance of the asterisk as rapidly as possible by pressing a key directly beneath the cue. Five hundred milliseconds after each response, the asterisk appears at a new location. Unbeknownst to the subject, a sequence of 10 locations (e.g., DBCACBDCBA) repeats every 10 trials throughout 400 training trials—i.e., there are 40 repetitions of a 10-trial sequence. Learning is demonstrated by gradually improving reaction times when the asterisk appears in the repeating sequence of locations, as compared with reaction times when a random sequence of locations is presented.
In contrast to their good performance on the repeating sequence, four different tests of declarative knowledge indicated that the amnesic patients were unaware of what they had learned. It appears that control subjects, by virtue of their intact medial temporal lobe/diencephalic memory system, are able to acquire some declarative knowledge about what they are learning as they work on the repeating sequence. This knowledge is not needed to learn the sequence and does not contribute to fast reaction times. It is acquired concomitantly to the learning that is measured by improved reaction times (22).
Artificial Grammar Learning.
In tasks of artificial grammar learning, subjects see a series of letter strings generated by a rule system (Fig. 4). Even though subjects are not told that the formation of the letter strings is governed by rules until after they are all presented, subjects are nevertheless able to classify new letter strings as either grammatical or nongrammatical. Amnesic patients learn artificial grammars as well as normal subjects (23, 24, 25). Yet, despite their excellent classification performance, amnesic patients are markedly impaired at recognizing the particular letter strings, or the particular letter string fragments, that appeared during training.
Figure 4.
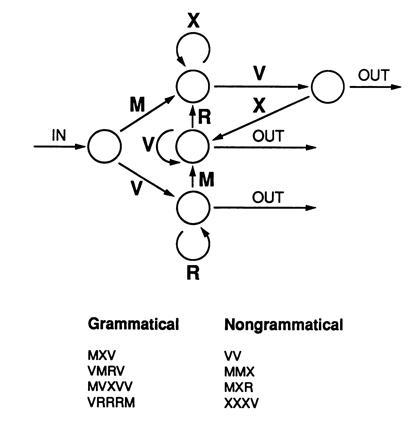
Artificial grammar learning (24). Letter strings are generated from a finite state rule system. Grammatical letter strings can be formed by traversing the diagram from the in arrow to the out arrows, adding a letter at each transition from one node to the next. In a typical experiment, 23 grammatical items are used for training, and a different 23 items are used for testing. An additional 23 nongrammatical test items are also used as foils for testing. These are generated by introducing an error in each of 23 different grammatical items. Subjects first study 23 grammatical letter strings one at a time. Five minutes later, they are informed for the first time that the letter strings they have just seen were formed by a set of rules. They are told that their task is to classify new letter strings according to whether they appear to conform to these rules. The 46 test items are then displayed one at a time, and subjects judge the item to be correct or incorrect (grammatical or nongrammatical).
It appears that artificial grammar learning is nondeclarative. It seems to be based on both abstract, rule-based knowledge and on more concrete, exemplar-specific knowledge—for example, knowledge of which bigrams and trigrams appeared frequently in the training set (25). Artificial grammar learning has some similarities to other kinds of habit and skill learning in that information is extracted across training items by a process of invariance detection. Yet, patients with Huntington disease performed as well as normal subjects at artificial grammar learning when they were given sufficient time to encode the training strings (3). Another possibility is that artificial grammar learning depends on changes in neocortex similar to what is thought to occur in perceptual priming.
Category Learning and Prototype Abstraction.
After inspecting a series of 40 training stimuli, amnesic patients are as good as normal subjects at classifying novel stimuli according to whether they do or do not belong to the same category as the training stimuli (Fig. 5; refs. 26 and 27). In contrast, they are impaired at recognizing the particular training stimuli that were presented. One severely amnesic patient could classify novel stimuli after inspecting 40 different training stimuli but could not recognize a single training stimulus as familiar even after it was presented 40 times in succession (Fig. 6).
Figure 5.
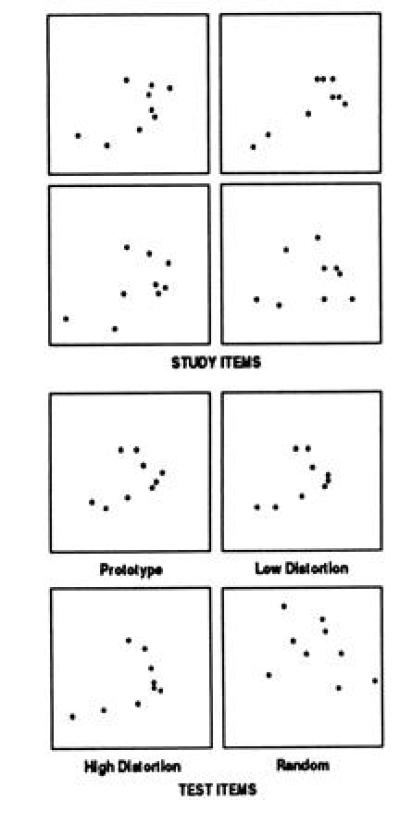
Prototype learning (27). Examples of the 40 study items and 84 test items used to study prototype learning. The study items are all distortions of a prototype (average) dot pattern that is never presented. For training, the 40 study patterns are presented for 5 sec each, and the subject points to the dot closest to the center of the pattern (to guarantee attention). Five minutes later, subjects are instructed that the dot patterns they have just seen all belong to a single category of patterns in the same sense that, if a series of different dogs had been presented, they would all belong to the category “dog.” Then for each of 84 new dot patterns subjects judge (yes/no) whether or not it belongs to the same category as the training patterns. The test items consist of four repetitions of the prototype, 20 new “low” distortions of the prototype, 20 new “high” distortions of the prototype, and 40 random dot patterns.
Figure 6.
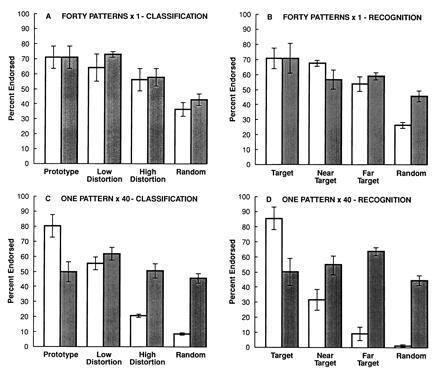
Each panel shows the results for four control subjects tested six times each (open bars) and the results for the profoundly amnesic patient EP (average of six tests; filled bars). (A) Classification of 84 novel dot patterns after studying 40 different training patterns (see Fig. 5 caption). Control subjects and EP performed similarly, endorsing the test items as a function of how closely they resembled the prototype of the training category (27). (B) Exactly the same task as in A but now with instructions to recognize the dot patterns that had been presented before (i.e., subjects made yes/no judgments). Actually, none of the 40 training patterns appeared on the test. Instead, the 84 test patterns varied in their resemblance to the training patterns as in A. (C) Classification of 84 novel dot patterns after studying a single dot pattern presented 40 times in succession. The training pattern was a prototype dot pattern, and the test patterns consisted of four repetitions of the prototype, 40 low distortions of the prototype, 20 high distortions of the prototype, and 40 random dot patterns. The instructions were as in A. (D) Exactly the same task was presented as in C, but now with instructions to recognize the dot patterns that had been presented before, as in B. Actually, only one dot pattern had been presented during training. The test items consisted of four repetitions of this same pattern and 80 other patterns that varied in their resemblance to the training pattern. A four-way ANOVA (EP versus controls, classification versus recognition instructions, 40 different study items versus one study item, and four types of test item) revealed significant effects of group, type of study item, and type of study item (Fs > 17.0, Ps < 0.002), but the effect of instructions fell short of significance [F(1,103) = 4.7, P = 0.06]. EP performed entirely normally at classification after seeing 40 different training patterns (A), but he performed significantly worse and at chance when he had to recognize a single pattern presented 40 times in succession (D). When asked to recognize 40 stimuli that had been presented once each (B), both EP and control subjects tended to use a classification strategy. When asked to classify after seeing only one pattern 40 times (C), normal subjects tended to rely on declarative memory, but EP could not perform the task. Subjects were more influenced by the kinds of material they studied than by the instructions given at test. Classification learning can proceed nondeclaratively when there is some variability in the training stimuli (A and B).
These results suggest that category-level knowledge can develop independently of and in the absence of normal declarative memory for the items presented during learning. Thus, experience with a succession of items appears to lead to two parallel consequences. First, declarative memory can be acquired and retained about each of the training items, and this ability depends on the medial temporal lobe and diencephalic structures that are damaged in amnesia and that are essential for declarative memory. Second, repeated experience leads to knowledge about the category to which the training items belong. Category-level knowledge might be acquired by abstracting information across encounters with specific training examples. Alternatively, category-level knowledge could depend on specific item information that is stored in a distributed fashion, as commonly proposed in theoretical models. In either case, the information supporting classification learning must be distinct from declarative information about the specific items presented for training. Models in which classification judgments derive from, or in any way depend on, long-term declarative memory do not account for the finding that amnesic patients can acquire category knowledge as well as normal subjects.
If classification learning does not depend on the limbic or diencephalic structures damaged in amnesia, which brain systems could be involved? One clue comes from the parallel between classification learning and the learning of skills and habits; namely, knowledge of a specific trial is not crucial. Rather, subjects detect invariance in the stimulus environment across many trials, independently of declarative memory. It is therefore possible that corticostriatal systems are involved in category learning, as has been suggested for habit learning. Alternatively, the learning could reflect gradual changes intrinsic to neocortex, as is thought to occur in the case of priming, (28, 29) such that the neocortex gradually accrues knowledge independently of the hippocampus and related structures.
The Medial Temporal Lobe Memory System Important for Declarative Memory: Studies in Monkeys
During the last two decades, work with nonhuman primates has been successful in establishing an animal model of human amnesia and in identifying a system of structures in the medial temporal lobe essential for declarative memory (30, 31). This system (Fig. 7) includes the hippocampal formation (i.e., the hippocampus proper, the dentate gyrus, the subicular complex, and the entorhinal cortex) and the adjacent perirhinal and parahippocampal cortices.
Figure 7.
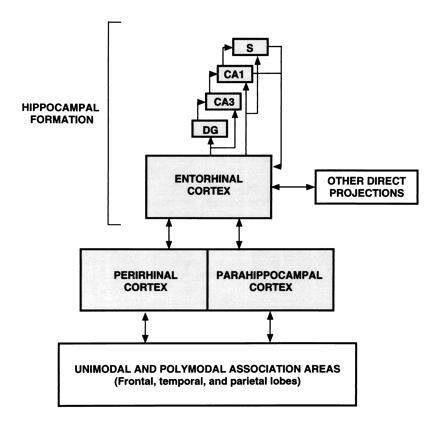
Schematic view of the medial temporal lobe memory system. The entorhinal cortex is a major source of projections to the hippocampal region (which includes the dentate gyrus, the cell fields of the hippocampus, and the subicular complex). Nearly two-thirds of the cortical input to entorhinal cortex originates in the adjacent perirhinal and parahippocampal cortices, which in turn receive projections from unimodal and polymodal areas in the frontal, temporal, and parietal lobes. The entorhinal cortex also receives other direct inputs from orbital frontal cortex, insular cortex, and superior temporal gyrus. All these projections are reciprocal.
Here, we consider the hippocampal formation to consist of two components, the hippocampal region and the entorhinal cortex. The hippocampal region consists of the cell fields of the hippocampus proper, the dentate gyrus, and the subicular complex. The entorhinal cortex is the major source of cortical projections to the hippocampal region. The entorhinal cortex in turn receives nearly two-thirds of its cortical input from the adjacent perirhinal and parahippocampal cortices. The entorhinal cortex also receives other direct inputs from the olfactory bulb, orbital frontal cortex, insular cortex, cingulate cortex, and superior temporal gyrus. The following sections summarize recent findings from monkeys that illuminate the contribution of medial temporal lobe structures to memory.
The Effects on Memory of Damage to the Hippocampal Region.
Two techniques have been used to damage the hippocampal region: global ischemia and stereotaxic lesions. For ischemia, a noninvasive technique involving carotid occlusion and pharmacologically induced hypotension was used to produce 15 min of reversible ischemia in monkeys (the ISC lesion; ref. 32). The monkeys sustained significant bilateral cell loss in fields CA1 and CA2 of the hippocampus as well as loss of somatostatin-immunoreactive cells in the dentate gyrus. This damage was associated with significant and long-lasting memory impairment. For example, performance was impaired on the delayed nonmatching to sample task, a standard task of visual recognition memory (normal group, 79% correct; ISC group, 61% correct; P < 0.05). Across several tasks used to assess memory functions, the memory impairment of monkeys with ISC lesions was less severe than in monkeys with more extensive damage to the hippocampal formation or other components of the medial temporal lobe memory system. For example, monkeys with ISC lesions were less impaired than monkeys with surgical lesions that involved nearly all the hippocampal region bilaterally as well as the parahippocampal cortex and posterior entorhinal cortex (the H+ lesion; in the notation used here, H refers to the hippocampal region and + refers to adjacent cortex; ref. 32). Similarly, monkeys with ISC lesions were less impaired than monkeys with combined lesions of the perirhinal and parahippocampal cortices (the PRPH lesion; refs. 33 and 34).
An important issue about ischemic damage is whether the damage identifiable in histopathological examination provides an accurate estimate of the neural damage responsible for the memory impairment. The concern is that additional (“covert”) damage might be present, which is sufficient to disrupt neuronal function in areas important for memory and sufficient to impair behavioral performance, but not sufficient to progress to cell death and to be detectable in histopathology. It is an important issue because if covert damage commonly occurs, then the findings from monkeys with ischemic damage (as well as the findings from humans with memory impairment in association with ischemic damage; ref. 35) cannot be interpreted with confidence and cannot provide reliable anatomical information about the neuroanatomy of memory impairment. Although few studies have addressed this issue directly, the available experimental data in rats and monkeys suggest that covert damage is not a serious concern (for detailed discussion, see ref. 36). However, additional experimental work, especially in rats, will be needed to settle the issue of how to compare the behavioral effects of ischemic and neurosurgical lesions.
The second technique that has been used to make lesions of the hippocampal region involves the use of stereotaxic neurosurgery combined with magnetic resonance imaging (37). Magnetic resonance imaging is used to create a brain atlas for each monkey, and the atlas is then used to derive coordinates for making stereotaxic lesions limited to the hippocampal region. Monkeys with bilateral radio-frequency lesions of the hippocampal region made in this way (the H lesion; ref. 38) exhibited significant impairment on the delayed nonmatching to sample task (performance on the delayed nonmatching task at the 10-min delay: normal group, 79% correct; H group, 68% correct; P < 0.05). Overall, the performance of the H and the ISC groups was similar, and in both groups performance remained impaired 6–9 months after surgery.
These findings from monkeys with ISC and H lesions are consistent with a preliminary report of impaired memory in monkeys with lesions of the hippocampal region that were made with ibotenic acid (39). Another preliminary report found no impairment on the delayed nonmatching to sample task following ibotenate lesions of the hippocampal region (40). However, in this study, the delay intervals used for the delayed nonmatching task were relatively short (<3.5 min), and the monkeys were trained on the task before surgery, which is known to attenuate postoperative performance deficits (41, 42). Thus, impairment may not have been detected because the task, as it was administered, was insufficiently sensitive to memory impairment.
The Effects on Memory of Damage to the Entorhinal Cortex.
Most of the sensory information to the hippocampal formation enters via the entorhinal cortex, and the entorhinal cortex receives a substantial portion of its cortical input from the perirhinal and parahippocampal cortices. An earlier study concluded that damage limited to the entorhinal cortex produced only a mild memory impairment (43). We also found that monkeys with bilateral lesions limited to entorhinal cortex (the E lesion; ref. 44) were only mildly impaired on the delayed nonmatching to sample task at the 10-min delay (normal group, 79% correct; E group, 63% correct; P < 0.05). In addition, we found that when the delayed nonmatching task was readministered 6–13 months later, the E animals performed normally at all delay intervals (performance at the 10-min delay: normal group, 77% correct; E group, 79% correct). The finding of behavioral recovery following the E lesion shows that the entorhinal cortex itself, unlike the remainder of the hippocampal formation, is not essential for memory, at least as measured by the several tasks that have to date been given to monkeys with the E lesion. One way to understand the behavioral recovery is to suppose that the hippocampal region itself is eventually able to support memory performance. Interestingly, the perirhinal cortex originates a projection directly to the hippocampus, and, in association with behavioral recovery, the transverse length of the perirhinal terminal field in CA1 increased ≈70% (44).
The Effects on Memory of Damage to Structures Adjacent to the Hippocampal Formation.
A major finding that has emerged from studies of the primate medial temporal lobe memory system is that structures adjacent to the hippocampal formation play a significant role in memory function (30, 31). For example, PRPH lesions, which did not directly damage the hippocampal formation, impaired memory performance nearly as severely as larger medial temporal lobe lesions, which included the perirhinal and parahippocampal cortices, the hippocampal formation, and the amygdala. Thus, at the 10-min delay in the delayed nonmatching to sample task, five monkeys with the PRPH lesion scored 60% correct, and nine normal monkeys scored 77% correct (P < 0.02; refs. 33 and 34). In addition, monkeys with bilateral lesions involving the hippocampal formation plus the perirhinal cortex (the H++ lesion) were severely impaired on the delayed nonmatching to sample task (45). Four H++ monkeys scored 56% correct at the 10-min delay, while seven normal monkeys scored 78% correct.
Importantly, bilateral stereotaxic lesions limited to the amygdaloid complex (the A lesion) did not affect memory on the same tasks that were sensitive to removal of the hippocampal formation or the adjacent cortical areas (46). Monkeys with the A lesion scored 77% correct on the 10-min delay of the delayed nonmatching to sample task; normal monkeys in this study scored 80% correct. Thus the amygdaloid complex does not appear to be a part of the medial temporal lobe memory system.
When the available studies on monkeys are considered together, it appears that memory impairment becomes more severe as more components of the medial temporal lobe memory system are damaged (47). Thus, damage limited to the hippocampal region (H) caused significant memory impairment. More severe memory impairment occurred following hippocampal region damage that was combined with damage to the adjacent entorhinal and parahippocampal cortex (H+). Finally, the severity of memory impairment was still greater following damage that included the hippocampal region plus all the adjacent cortical regions—i.e., the perirhinal, entorhinal, and parahippocampal cortices (H++).
The Effects of Memory of Selective Damage to Perirhinal or Parahippocampal Cortex.
The finding that large medial temporal lobe lesions impair memory more severely than small lesions does not mean that lesion size is the only determinant of memory impairment. It is also possible that different components of the system contribute to memory in specific ways. This possibility follows from anatomical findings that information from neocortex enters the medial temporal lobe memory system at different points. For example, perirhinal cortex receives stronger visual projections from unimodal visual areas than parahippocampal cortex (48, 49). Accordingly, perirhinal cortex might be expected to play a greater role than parahippocampal cortex in visual memory. Preliminary findings directly comparing the separate effects of perirhinal cortex and parahippocampal cortex lesions are consistent with this idea. First, monkeys with bilateral lesions limited to the perirhinal cortex (the PR lesion) perform poorly on memory tasks (43, 50, 51). On the 10-min delay of the delayed nonmatching to sample task monkeys with PR lesions scored 67% correct, and normal monkeys scored 79% correct (P < 0.05). Across several different tasks, damage to the perirhinal cortex produces more severe memory impairment than damage to any other single component of the medial temporal lobe memory system (Fig. 8). By contrast, preliminary findings from monkeys with bilateral parahippocampal cortex lesions (the PH lesion) suggest that at least some kinds of memory are not affected (performance on the delayed nonmatching task at the 10-min delay: normal group, 79% correct; PH group, 77% correct; P > 0.10; ref. 51; but see ref. 54).
Figure 8.
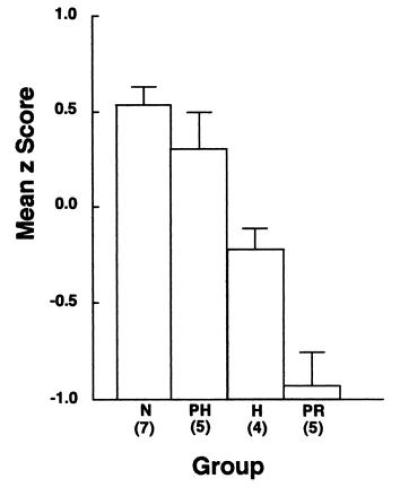
Mean z scores based on data from four measures of memory (47) for ten normal monkeys (N), five monkeys with surgical aspiration lesions of the parahippocampal cortex (the PH lesion; cortical areas TH and TF; ref. 52), four monkeys with surgical stereotaxic damage limited to the hippocampal region (H; dentate gyrus, the cell fields of the hippocampus, and the subicular complex), and five monkeys with surgical aspiration lesions of perirhinal cortex (PR; cortical areas 35 and 36; ref. 53). The PR group was impaired relative to the N, PH, and H groups. The PH group was not impaired (51). Error bars indicate standard errors of the mean.
In view of the substantial anatomical projection to parahippocampal cortex from area 7 of the parietal cortex (49), the parahippocampal cortex may be involved in spatial memory. Consistent with this idea is the finding that monkeys with lesions involving the parahippocampal cortex, hippocampus, and posterior entorhinal cortex exhibited a more severe spatial memory impairment than monkeys with lesions involving the rostral perirhinal cortex, rostral entorhinal cortex, and the amygdala (55). The effect of selective parahippocampal cortical lesions on spatial memory tasks has not yet been evaluated.
The Effects on Memory of Damage to Inferotemporal Cortex Area TE.
Inferotemporal cortex (area TE; ref. 52) is a unimodal visual area situated immediately adjacent and lateral to the perirhinal cortex. Previous studies of the behavioral effects of lesions of area TE have reported impaired visual perception (as measured, for example, by pattern discrimination learning) as well as impaired recognition memory (as measured by performance on the delayed nonmatching to sample task; ref. 56). However, virtually all previous studies of TE lesions have included damage to what is now known to be perirhinal cortex (57). If area TE is involved in visual perception and is also a repository of long-term visual memories, (56) and if perirhinal cortex is involved in the formation of visual memory, the question naturally arises as to whether damage to area TE and perirhinal cortex can be distinguished within the visual modality. A preliminary study compared performance of monkeys with perirhinal lesions (PR) and monkeys with TE lesions that did not include perirhinal cortex (the TE lesion; ref. 58). Both the PR and TE groups were similarly impaired on the delayed nonmatching to sample task (performance at the 10-min delay: normal group, 79% correct; PR group, 67% correct; TE group, 65%).
Performance on two other visual tasks, however, did distinguish the behavioral effects of the two lesions. Monkeys with TE lesions were impaired on the concurrent learning task, where eight different two-choice object discriminations must be learned concurrently, but they were unimpaired at learning and retaining single-object discriminations. By contrast, monkeys with PR lesions showed the opposite pattern: they performed well at concurrent discrimination learning but poorly at single object discriminations, and there was a significant interaction of group × task (P < 0.05). One way to understand these results is to suppose that the concurrent discrimination task places a heavy burden on visual perceptual functions but does not necessarily depend on declarative memory (47). By contrast, the perceptual demands of the single object discrimination task may not require area TE, because task performance can be supported by more posterior visual areas, but the task does depend on declarative memory (59).
The Medial Temporal Lobe Memory System Important for Declarative Memory: Studies in Humans
Several cases of circumscribed human amnesia have become available in recent years, which confirm and extend the findings with monkeys. These cases make three important points. First, bilateral damage limited to the CA1 region of the hippocampal formation is sufficient to produce moderately severe anterograde memory impairment. Second, bilateral damage beyond the CA1 region, but still limited to the hippocampal formation, can produce more severe anterograde memory impairment. Finally, bilateral damage limited to the hippocampal formation can produce extensive, temporally graded retrograde amnesia covering >15 years.
The first reported case of human amnesia in which there was detailed neuropsychological information and detailed postmortem neuropathological analysis demonstrated that damage limited to the CA1 region of the hippocampus is sufficient to produce anterograde memory impairment (patient RB; ref. 35). Recently, neuropsychological and postmortem neuropathological findings have been described for an additional amnesic patient who became amnesic as the result of an ischemic episode and who, like RB, sustained bilateral damage limited to the CA1 region (patient GD; Table 1; ref. 60). In addition, neuropsychological and postmortem findings have been described for two other patients who sustained bilateral damage to the CA1 region of the hippocampus together with other damage in the hippocampal formation including entorhinal cortex (patient LM; and entorhinal cortex together with the subiculum, patient WH; Table 1; ref. 60).
Table 1.
Summary of neuropsychological and neuropathological findings from four patients with bilateral damage to the hippocampal formation
| Patient | Anterograde amnesia | Retrograde amnesia | Damage to the hippocampal formation |
|---|---|---|---|
| RB | Moderate | Minimal | CA1 field |
| GD | Moderate | Minimal (?) | CA1 field |
| LM | Moderate | Extensive | CA1, CA2, CA3 fields |
| Dentate gyrus | |||
| Entorhinal cortex | |||
| WH | Severe | Extensive | CA1, CA2, CA3 fields |
| Dentate gyrus, subiculum | |||
| Entorhinal cortex |
Anterograde and retrograde memory impairment in four patients with damage to the hippocampal formation (35, 60). Patients GD and RB had damage restricted primarily to the CA1 field of the hippocampus bilaterally. GD and RB additionally had cell loss in the region of the CA1/subiculum border. Patient LM, in addition to substantial cell loss in the dentate gyrus and all the cell fields of the hippocampus, evidenced some loss of cells in entorhinal cortex. Patient WH sustained more extensive damage to the hippocampal region overall than the other patients, including considerable loss of cells in the subiculum, complete loss of polymorphic cells in the dentate gyrus and dispersion of the granule cell layer in the dentate gyrus. In addition, WH sustained cell loss in entorhinal cortex. The severity of memory impairment correlated with the extent of damage to the hippocampal formation. Thus, RB and GD had moderate anterograde amnesia and minimal retrograde amnesia. (The question mark for GD indicates that some uncertainty remains about the interpretation of his performance on remote memory tests.) LM had moderate anterograde amnesia, together with extensive, temporally graded retrograde amnesia covering at least 15 years. WH had severe anterograde amnesia together with extensive, temporally graded retrograde amnesia covering as much as 25 years.
For this group of four patients, the severity of anterograde amnesia was generally related to the extent of damage. It was not possible to rank order with confidence the severity of anterograde memory impairment in the three patients RB, GD, and LM. GD had the least education and was not always interested in testing, and he had a low WAIS-R IQ [92 for GD versus 103 for RB (estimated from his WAIS score) and 109 for LM]. Accordingly, GD’s test performance might well have reflected in part these other factors, and he may have been less amnesic than the other patients. Nevertheless, of the patients listed in Table 1, WH had the most extensive damage to the hippocampal formation and was also the most severely amnesic. Thus, damage limited to the CA1 region, as in RB and GD, produced a moderate level of anterograde amnesia. More severe anterograde memory impairment can occur when there is bilateral damage beyond the CA1 region including the subiculum and the adjacent entorhinal cortex (patient WH; Table l). Finally, the severely amnesic patient HM (61), who sustained a bilateral resection of the medial temporal lobe in l953, provides a useful point of reference. Patient HM is more severely amnesic than any of the patients listed in Table 1, and HM has extensive damage to the perirhinal cortex as well as to the hippocampal formation (62).
The severity of retrograde amnesia is also related to the nature and extent of damage in the hippocampal formation. Damage limited to the CA1 field of the hippocampal formation (patients RB and GD) caused minimal retrograde amnesia, involving perhaps 1 or 2 years. More extensive damage, involving more of the hippocampal region and the entorhinal cortex (patients LM and WH) caused extensive, temporally graded retrograde amnesia covering at least 15 years. LM and WH appear to be the first reported cases of memory impairment showing that extensive and temporally graded retrograde amnesia can occur following bilateral damage limited to the hippocampal formation. An earlier view was that retrograde amnesia in amnesic patients, even severely amnesic patients like HM, is typically quite limited and involves perhaps a few years or less. This issue has been reexamined in some detail (60) with the conclusion that, when formal tests are given to severely amnesic patients, retrograde amnesia is typically graded and extensive covering a decade or more (see also ref. 63).
Taken together, the findings from patients suggest that, in humans as in monkeys, damage limited to the hippocampal region produces significant memory impairment. Moreover, as more extensive damage occurs, involving the entorhinal cortex (patients LM and WH) or the entorhinal and perirhinal cortices (patient HM), the memory impairment becomes increasingly severe. It seems reasonable to suppose that each of these medial temporal lobe structures should contribute uniquely to memory function. If so, it should be possible in principle to identify some function that is as impaired in patients RB and GD as in patient HM. All three patients have CA1 hippocampal damage. HM additionally has extensive damage to other medial temporal lobe structures. However, RB and GD always appeared to have been simply mild versions of patient HM. No task was ever identified that RB and GD performed as poorly as HM. Accordingly, the possibility must be kept in mind that the several components of the medial temporal lobe memory system all contribute similarly to behavior. They may contribute different subfunctions, but these may be difficult with behavioral measures.
The Function of the Medial Temporal Lobe Memory System
The medial temporal lobe is required to form permanent and usable long-term declarative memory. The neocortex is believed to be the permanent respository of memory. The medial temporal lobe memory system is needed at the time of learning and for a lengthy period of time afterwards. During this lengthy period, it has been proposed that medial temporal lobe structures direct consolidation in neocortex by gradually binding together the multiple, geographically separate cortical regions that together store memory for a whole event (63, 64). As a result of these gradual changes, which may involve modifications in the strengths of connections between cortical areas, perhaps dependent on morphological growth and change, storage of declarative memory and its retrieval become independent of the medial temporal lobe memory system. It may soon be possible to study consolidation directly in neocortex at different times after learning by sampling neurons in neocortex that are part of memory representation dependent on the medial temporal lobe (65).
Acknowledgments
This work was supported by the Medical Research Service of the Department of Veterans Affairs, National Institutes of Health Grant NS19063, and National Institute of Mental Health Grant MH24600.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Knowlton B J, Squire L R, Gluck M. Learn Mem. 1994;1:106–120. [PubMed] [Google Scholar]
- 2.Knowlton B J, Mangels J A, Squire L R. Science. 1996;273:1399–1402. doi: 10.1126/science.273.5280.1399. [DOI] [PubMed] [Google Scholar]
- 3.Knowlton, B. J., Squire, L. R., Paulsen, J. S., Swerdlow, N., Swenson, M. & Butters, N. (1996) Neuropsychology, in press.
- 4.Malamut B L, Saunders R C, Mishkin M. Behav Neurosci. 1984;98:759–769. doi: 10.1037//0735-7044.98.5.759. [DOI] [PubMed] [Google Scholar]
- 5.McDonald R J, White N M. Behav Neurosci. 1993;61:260–270. [Google Scholar]
- 6.Packard M G, Hirsh R, White N M. J Neurosci. 1989;9:1465–1472. doi: 10.1523/JNEUROSCI.09-05-01465.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graybiel A M. Curr Opin Neurobiol. 1995;5:733–741. doi: 10.1016/0959-4388(95)80100-6. [DOI] [PubMed] [Google Scholar]
- 8.Salmon D P, Butters N. Curr Opin Neurobiol. 1995;5:184–190. doi: 10.1016/0959-4388(95)80025-5. [DOI] [PubMed] [Google Scholar]
- 9.Sherry D F, Schacter D L. Psychol Rev. 1987;94:439–454. [Google Scholar]
- 10.Schacter D L. Am Psychol. 1992;47:559–569. doi: 10.1037//0003-066x.47.4.559. [DOI] [PubMed] [Google Scholar]
- 11.Nadel L. In: Memory Systems 1994. Schacter D L, Tulving E, editors. Cambridge, MA: MIT Press; 1994. pp. 39–64. [Google Scholar]
- 12.Weiskrantz L. In: Behavioral and Neural Aspects of Learning and Memory. Krebs J R, Horn G, editors. Oxford: Clarendon; 1991. pp. 1–10. [Google Scholar]
- 13.Tulving E. In: Memory: Organization and Locus of Change. Squire L R, Weinberger N M, Lynch G, McGaugh J L, editors. New York: Oxford Univ. Press; 1991. pp. 3–32. [Google Scholar]
- 14.Eichenbaum H, Mathews P, Cohen N J. Behav Neurosci. 1989;103:1207–1216. doi: 10.1037//0735-7044.103.6.1207. [DOI] [PubMed] [Google Scholar]
- 15.Eichenbaum H, Stewart C, Morris R G M. J Neurosci. 1990;10:3531–3542. doi: 10.1523/JNEUROSCI.10-11-03531.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders R C, Weiskrantz L. Behav Brain Res. 1989;35:85–94. doi: 10.1016/s0166-4328(89)80109-3. [DOI] [PubMed] [Google Scholar]
- 17.Reber, P. J., Knowlton, B. J. & Squire, L. R. (1996) Behav. Neurosci., in press. [DOI] [PubMed]
- 18.Cohen A, Ivry R I, Keele S W. J Exp Psychol Learn Mem Cognit. 1990;16:17–30. [Google Scholar]
- 19.Stadler M A. J Exp Psychol Learn Mem Cognit. 1989;15:1061–1069. doi: 10.1037//0278-7393.15.6.1061. [DOI] [PubMed] [Google Scholar]
- 20.Nissen M J, Bullemer P. Cognit Psychol. 1987;19:1–32. [Google Scholar]
- 21.Willingham D B, Nissen M J, Bullemer P. J Exp Psychol Learn Mem Cognit. 1989;15:1047–1060. doi: 10.1037//0278-7393.15.6.1047. [DOI] [PubMed] [Google Scholar]
- 22.Reber P J, Squire L R. Learn Mem. 1994;2:1–13. [PubMed] [Google Scholar]
- 23.Knowlton B J, Ramus S J, Squire L R. Psychol Sci. 1992;3:172–179. [Google Scholar]
- 24.Knowlton B J, Squire L R. J Exp Psychol Learn Mem Cognit. 1994;20:79–91. doi: 10.1037//0278-7393.20.1.79. [DOI] [PubMed] [Google Scholar]
- 25.Knowlton B J, Squire L R. J Exp Psychol Learn Mem Cognit. 1996;22:161–181. doi: 10.1037//0278-7393.22.1.169. [DOI] [PubMed] [Google Scholar]
- 26.Knowlton B J, Squire L R. Science. 1993;262:1747–1749. doi: 10.1126/science.8259522. [DOI] [PubMed] [Google Scholar]
- 27.Squire L R, Knowlton B J. Proc Natl Acad Sci USA. 1995;92:12470–12474. doi: 10.1073/pnas.92.26.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schacter D L, Chiu C Y, Ochsner K N. Annu Rev Neurosci. 1993;16:159–182. doi: 10.1146/annurev.ne.16.030193.001111. [DOI] [PubMed] [Google Scholar]
- 29.Squire L R, Knowlton B, Musen G. Annu Rev Psychol. 1993;44:453–495. doi: 10.1146/annurev.ps.44.020193.002321. [DOI] [PubMed] [Google Scholar]
- 30.Squire L R, Zola-Morgan S. Science. 1991;253:1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- 31.Mishkin M, Murray E A. Curr Opin Neurobiol. 1994;4:200–206. doi: 10.1016/0959-4388(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 32.Zola-Morgan S, Squire L R, Rempel N L, Clower R P, Amaral D G. J Neurosci. 1992;12:2582–2596. doi: 10.1523/JNEUROSCI.12-07-02582.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zola-Morgan S, Squire L R, Amaral D G, Suzuki W A. J Neurosci. 1989;9:4355–4370. doi: 10.1523/JNEUROSCI.09-12-04355.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki W A, Zola-Morgan S, Squire L R, Amaral D G. J Neurosci. 1993;13:2430–2451. doi: 10.1523/JNEUROSCI.13-06-02430.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zola-Morgan S, Squire L R, Amaral D G. J Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Squire, L. R. & Zola, S. M. (1996) Hippocampus, in press.
- 37.Alvarez-Royo P, Clower R P, Zola-Morgan S, Squire L R. J Neurosci Methods. 1991;38:223–232. doi: 10.1016/0165-0270(91)90172-v. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez P, Zola-Morgan S, Squire L R. J Neurosci. 1995;15:3796–3807. doi: 10.1523/JNEUROSCI.15-05-03796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beason-Held L, Rosene D L, Moss M B. Soc Neurosci Abstr. 1993;23:438. [Google Scholar]
- 40.O’Boyle V J, Murray E A, Mishkin M. Soc Neurosci Abstr. 1993;23:438. [Google Scholar]
- 41.Ringo J L. Behav Neurosci. 1988;102:173–177. doi: 10.1037//0735-7044.102.1.173. [DOI] [PubMed] [Google Scholar]
- 42.Zola-Morgan S, Squire L R. Behav Neurosci. 1986;100:155–160. doi: 10.1037//0735-7044.100.2.155. [DOI] [PubMed] [Google Scholar]
- 43.Meunier M, Bachevalier J, Mishkin M, Murray E A. J Neurosci Abstr. 1993;13:5418–5432. doi: 10.1523/JNEUROSCI.13-12-05418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leonard B W, Amaral D G, Squire L R, Zola-Morgan S. J Neurosci. 1995;15:5637–5659. doi: 10.1523/JNEUROSCI.15-08-05637.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zola-Morgan S, Squire L R, Clower R P, Rempel N L. J Neurosci. 1993;13:251–265. doi: 10.1523/JNEUROSCI.13-01-00251.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zola-Morgan S, Squire L R, Amaral D G. J Neurosci. 1989;9:1922–1936. doi: 10.1523/JNEUROSCI.09-06-01922.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zola-Morgan S, Squire L R, Ramus S J. Hippocampus. 1994;4:483–495. doi: 10.1002/hipo.450040410. [DOI] [PubMed] [Google Scholar]
- 48.Webster M J, Ungerleider L G, Bachevalier J. J Neurosci. 1991;11:1095–1116. doi: 10.1523/JNEUROSCI.11-04-01095.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki W A, Amaral D G. J Comp Neurol. 1994;350:497–533. doi: 10.1002/cne.903500402. [DOI] [PubMed] [Google Scholar]
- 50.Horel J A, Pytko-Joiner D E, Voytko M L, Salsbury K. Behav Brain Res. 1987;23:29–42. doi: 10.1016/0166-4328(87)90240-3. [DOI] [PubMed] [Google Scholar]
- 51.Ramus S J, Zola-Morgan S, Squire L R. Soc Neurosci Abstr. 1994;20:1975. [Google Scholar]
- 52.von Bonin G, Bailey P. The Neocortex of Macaca mulatta. Urbana, IL: Univ. of Illinois Press; 1947. [Google Scholar]
- 53.Brodmann K. Vergleichende Lokalisationslehre der Grosshirnrinde. Leipzig, Germany: Barth; 1909. ; trans. GareyL. J. (1994) Localisation in the Cerebral Cortex (Smith–GordonLondon). [Google Scholar]
- 54.George P J, Horel J A, Cirillo R A. Behav Brain Res. 1989;34:163–178. doi: 10.1016/s0166-4328(89)80100-7. [DOI] [PubMed] [Google Scholar]
- 55.Parkinson J K, Murray E, Mishkin M. J Neurosci. 1988;8:4159–4167. doi: 10.1523/JNEUROSCI.08-11-04159.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishkin M. Philos Trans R Soc London B. 1982;298:85–92. doi: 10.1098/rstb.1982.0074. [DOI] [PubMed] [Google Scholar]
- 57.Buffalo E A, Zola-Morgan S, Squire L R. Soc Neurosci Abstr. 1994;20:1075. [Google Scholar]
- 58.Buffalo E A, Ramus S J, Zola-Morgan S, Squire L R. Soc Neurosci Abstr. 1995;21:1493. [Google Scholar]
- 59.Zola-Morgan S, Squire L R. J Neurosci. 1984;4:1072–1085. doi: 10.1523/JNEUROSCI.04-04-01072.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rempel-Clower N L, Zola-Morgan S, Squire L R, Amaral D G. J Neurosci. 1996;16:5233–5255. doi: 10.1523/JNEUROSCI.16-16-05233.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scoville W B, Milner B. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Corkin S, Amaral D G, Johnson K A, Hyman B T. J Neurosci. 1996;16:5233–5255. [Google Scholar]
- 63.Squire L R, Alvarez P. Curr Opin Neurobiol. 1995;5:169–177. doi: 10.1016/0959-4388(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 64.Zola-Morgan S, Squire L R. Science. 1990;250:288–290. doi: 10.1126/science.2218534. [DOI] [PubMed] [Google Scholar]
- 65.Higuchi S, Miyashita Y. Proc Natl Acad Sci USA. 1996;93:739–743. doi: 10.1073/pnas.93.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Squire L R, Knowlton B J. In: The Cognitive Neurosciences. Gazzinga M, editor. Cambridge, MA: MIT Press; 1994. [Google Scholar]