

Abstract
Obstructive sleep apnea (OSA) is a highly prevalent respiratory disorder of sleep, and associated with chronic intermittent hypoxia (CIH). Experimental evidence indicates that CIH is a unique physiological state with potentially “adaptive” and “maladaptive” consequences for cardio-respiratory homeostasis. CIH is also a critical element accounting for most of cardiovascular complications of OSA. Cardiac response to CIH is time-dependent, showing a transition from cardiac compensative (such as hypertrophy) to decompensating changes (such as failure). CIH-provoked mild and transient oxidative stress can induce adaptation, but severe and persistent oxidative stress may provoke maladaptation. Hydrogen peroxide as one of major reactive oxygen species plays an important role in the transition of adaptive to maladaptive response to OSA-associated CIH. This may account for the fact that although oxidative stress has been recognized as a driver of cardiac disease progression, clinical interventions with antioxidants have had little or no impact on heart disease and progression. Here we focus on the role of hydrogen peroxide in CIH and OSA, trying to outline the potential of antioxidative therapy in preventing CIH-induced cardiac damage.
1. Introduction
Obstructive sleep apnea (OSA) is a highly prevalent respiratory disorder of sleep, characterized by recurrent episodes of complete or partial upper airway obstruction. It may develop at different ages from the premature infant to the elderly, with a prevalence related to age and sex, increasing significantly in the elderly (5–9%) [24]. The prevalence of OSA in children is generally 1–3% [25] and even higher (e.g., 5-6%) in certain conditions [26]. The prevalence of OSA in the general population of adult men and women is 3–7% and 2–5%, respectively [27, 28] and keeps increasing in the elders [24]. OSA was often found in the patients with diabetes and obesity since it is involved in the development of insulin resistance, glucose intolerance, type 2 diabetes, and metabolic syndrome independently of adiposity [29–32]. OSA was also considered as a causal factor in cardiovascular diseases [33, 34].
Collapsibility of the upper airway during sleep can be increased by underlying anatomical alteration and/or disturbances in upper airway neuromuscular control, both of which play key roles in the pathogenesis of OSA [35]. OSA-induced recurrent episodes of complete or partial collapse of the upper airway during sleep are associated with reductions in ventilation, which often leads to repetitive apneas and hypopneas. Each of these obstructive respiratory events results in recurrent episode of hypoxia. Reoxygenation occurs when the episode is terminated by an arousal that restores the airway patency. The recurrence of these hypoxia and reoxygenation episodes produces a characteristic pattern of nocturnal intermittent hypoxia that is unique to OSA. Generally, both apneas and hypopneas events can produce the same pattern of chronic intermittent hypoxia (CIH), sleep arousals also called sleep fragmentation, hemodynamic changes [36–38], and symptoms of disruptive snoring and daytime sleepiness. Clinical consequences of the disorder cover a wide spectrum, including daytime hypersomnolence, neurocognitive dysfunction, cardiovascular disease, metabolic dysfunction, and corpulmonale [37].
It has been recognized that OSA is a potential life-threatening condition and has become one of the most common public health problems [39, 40]. Since OSA is emerging as a cardiovascular risk factor, it became an important target for public health interventions aiming at reducing cardiovascular diseases [41].
Mammalian heart is an obligate aerobic organ and a constant supply of oxygen is indispensable for cardiac viability and function [42]. The heart as a contractile pump has the highest O2 consumption among all body organs, which can be increased eight-fold or more under maximal workload conditions [43]. Consequently, cardiac tissue is susceptible to lack of O2 and also highly dependent on oxidative metabolism to maintain its normal function [44]. Since CIH is one of the main events of OSA, OSA has been considered as an independent risk factor for cardiovascular morbidity [41, 45–47], such cardiovascular diseases including hypertension [45–53], stroke [54], and even glucose metabolic abnormality [55], and so forth. For instance, it has been reported that OSA can increase the risk of heart failure (HF) by 140%, stroke by 60%, and coronary heart disease by 30% [56].
As outlined in Figure 1, three major components of OSA associated with cardiovascular events are large swings in intrathoracic pressure, postapneic arousals, and CIH [57]. A body of experimental evidence has indicated that CIH is a unique physiological state with a profile of biological consequences distinct from other types of hypoxia [46, 47, 51]. In Figure 1, the negative cardiovascular consequences of OSA are illustrated, all of which were considered critically related to the unique pattern of CIH [38, 52, 53, 58]. In this paper, therefore, we would like to summarize the several features of CIH-induced cardiac changes at the early and later stages, based on both epidemiological and experimental animal information.
Figure 1.
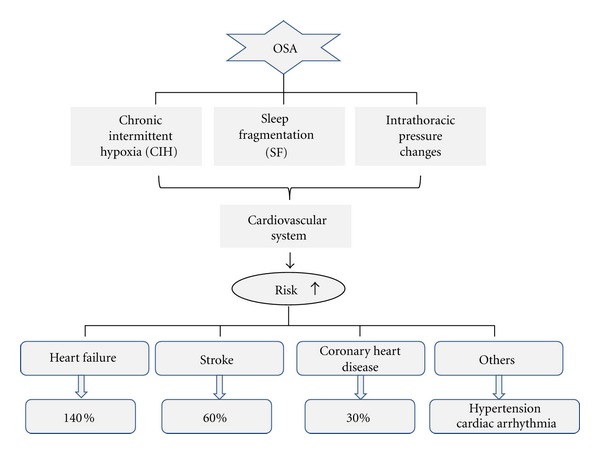
OSA-induced cardiovascular diseases. OSA increases the risk of cardiovascular diseases through three main pathological processes related with OSA: chronic intermittent hypoxia (CIH), sleep fragmentation (SF), and intrathoracic pressure changes. It can increase the risk of heart failure by 140%, stroke by 60%, and coronary heart disease by 30%. It can also increase the risk of other cardiovascular diseases such as hypertension and cardiac arrhythmia.
2. Cardiac Response to CIH: A Transition from Adaptation to Maladaptation
It has been reported that innate defense systems (adaptation) that are induced by exposure to repeated and relatively brief episodes of hypoxia act as a powerful temporal protective phenomenon [59]. Adaptation to chronic hypoxia was also reported to increase cardiac tolerance to all major deleterious consequences of acute oxygen deprivation such as myocardial infarction, contractile dysfunction, and ventricular arrhythmias [60]. However, long-term exposure to CIH may be detrimental to cardiac function (maladaptation). OSA exposes the cardiovascular system to CIH, oxidative stress, and systemic inflammation. With the extension of exposure duration to CIH, these factors can compel the heart to induce a transition from cardiac compensative (such as cardiac hypertrophy) to decompensative change (such as HF), that is, a transition from cardiac adaptation to cardiac maladaptation. OSA-related CIH has been suggested to play the main role in the development of left ventricular remodeling including cardiac hypertrophy, cardiac fibrosis, and cardiac dysfunction [61, 62]. This transition can impair myocardial contractility and cause development and progression of HF [63].
2.1. Cardiac Adaptation to CIH
2.1.1. CIH Effectively Protects the Heart against Ischemia/Reperfusion or Hypoxia/Reoxygenation-Induced Injury
It is reported that CIH can induce preconditioning-like effect to effectively protect the heart against ischemia/reperfusion (I/R) or hypoxia/reoxygenation-induced injury [1–6, 8–11, 64, 65], including the prevention of I/R-induced cardiac apoptosis and necrosis [3, 5] arrhythmias [6]. CIH also can improve postischemic recovery of cardiac function [1].
For instance, Park and Suzuki exposed C57BL/6 mice to CIH for 1, 2, and 4 weeks and then observed the effect on I/R injury. Results demonstrated that CIH exposure for 1-2 weeks resulted in increased susceptibility of the heart to I/R injury, while the heart appeared to adapt to CIH for 4 weeks by normalizing its susceptibility to I/R injury [7]. Their results imply that heart also has the ability to adapt to the condition of the CIH-mediated enhancement of the susceptibility to I/R. Guo et al. found that although cardiac hypertrophy may not occur in the right and left ventricles, the adaptation of guinea pigs to CIH significantly increased cardiac tolerance to I/R, shown by an improved recovery of contractile function, an increased coronary flow and a reduced level of reactive oxygen species (ROS) in cardiomyocytes [9]. These results are consistent with those observed in dogs and rats [4, 10, 11]. These data clearly show that CIH-induced cardiac protection against I/R-induced injury universally exists in different strains of animal models. CIH can promote recovery of cardiac contractile function from I/R, limiting cardiac infarction and arrhythmia caused by I/R [10, 12, 13]. Table 1 presents a summary of the protective effect of CIH on cardiac I/R-induced injuries. It seems that extensive cardiac protection by short-term CIH (less than 6 weeks) may be very similar to ischemic preconditioning, most likely sharing common signaling pathways [66].
Table 1.
The effects of CIH on cardiac I/R-induced injuries.
| Authors | Strains | Exposure time | Effects | Reference |
|---|---|---|---|---|
| Wang et al. | Male SD rats | 4 wks | Protective effects | [1] |
| Ding et al | Male SD rats | 42 days | Protective effects | [2–4] |
| Neckar et al. | Male Wistar rats | 8 h/day, 5 days/wks; | Protective effects | [5] |
| Asemu et al. | Male Wistar rats | 2 wks and 5 wks | Protective effects | [6] |
| Park and Suzuki | C57BL/6 mice | 1, 2 wks and 4 wks | Protective effects (4 wks) Deleterious effects (1, 2 wks) | [7] |
| Guo et al. | male guinea pigs | 28 days | Protective effects | [8] |
| Guo et al. | Male guinea pigs | 28 days | Protective effects | [9] |
| Zong et al. | Dogs | 20 days | Protective effects | [10] |
| Wang et al. | Male SD rats | 14, 28 and 42 days | Protective effects | [11–13] |
| Joyeux-Faure et al. | Male Wistar rats | 7 wks | Deleterious effects | [14] |
*wks: weeks.
2.1.2. CIH Induces Compensatory Increase of Cardiac Function
Naghshin et al. [15] exposed C57BL/6J mice to CIH for 4 weeks and then assessed cardiac function by echocardiography and pressure-volume loop analyses. They found that left ventricular (LV) ejection fraction (LVEF) and other measures of LV contractility were increased in CIH-exposed animals compared to controls. There was no change in contractile proteins, atrial natriuretic peptide levels, LV posterior wall thickness, or heart weight following the exposure to CIH. These results indicated that there was a compensatory increase in LV cardiac contractility that occurred independent of ventricular hypertrophy in the mouse model of CIH. It has been proposed that this adaptation, at least in part, results from activation of cardiac β-adrenergic pathways [15]. Lee et al. [16] showed that short-term IH (for 1 and 4 days) has protective effects on the heart. Campen et al. [17] found that CIH has produced a 26% increase in right ventricle weight and 10% increase in LV + septum weight, resulting in a significant increase by14% of the right ventricle/LV + septum ratio. Table 2 summarized previous studies available in the literature, which showed that cardiac hypertrophy may initially represent an adaptive response, but with the extent of exposure time, the hypertrophy ultimately is responsible for ventricular dilatation and HF. These results suggest that there is transition for cardiac response to CIH from adaptation to maladaptation.
Table 2.
Effect of CIH on cardiac functions.
| Authors | Strains | Exposure time | Effects | Reference |
|---|---|---|---|---|
| Naghshin et al. | C57BL/6J mice | 4 wks | Protective effects | [15] |
| Lee et al. | SD rats | 1, 4 days and 1, 2 wks | 1, 4 days: protective effects 1, 2 wks: deleterious effects | [16] |
| Campen et al. | C57BL/6J mice | Deleterious effects | [17] | |
| Chen et al. | Male SD rats | 5 wks | Deleterious effects | [18, 19] |
| Williams et al. | Male SD rats | 5 wks, 6 wks | Deleterious effects | [20] |
| Chen et al. | Male SD rats | 10 days | Deleterious effects | [21] |
| Yin et al. | FVB mice | 4 wks and 8 wks | Deleterious effects | [22] |
| Yang et al. | Patients | 4 wks and 8 wks | Deleterious effects | [23] |
*wks: weeks.
2.2. Cardiac Maladaptation to CIH
2.2.1. CIH Enhances Cardiac Susceptibility to I/R-Induced Injury
While short-term CIH was found to elicit a preconditioning-like events [2, 3, 67], Joyeux-Faure et al. [14] reported that CIH increased the heart susceptibility to I/R-induced injury. They exposed Wistar male rats to CIH for 7 weeks and then exposed the heart to ischemia and reperfusion for 30 and 120 min. Cardiac infarct sizes were found to be significantly higher in CIH group in comparison to the control group. This was the first study to show that CIH makes the heart more sensitive to I/R injury and suggested that although short-period exposure to CIH can stimulate protective mechanism to reduce subsequent lethal I/R-induced injury as discussed above, prolonged CIH beyond protective periods might turn to a condition with increased susceptibility to oxidative stress such as I/R-induced cardiac injury (Table 1).
2.2.2. CIH-Induced Cardiac Dysfunction
Similar to hypoxia-provoked maladaptive consequences on cardiorespiratory homeostasis [68–70], long-term exposure to CIH may decrease cardiac functions [18, 19]. OSA-induced CIH and hypercapnia are related to continuous changes in pulmonary volume, intrathoracic pressure, and microarousals. These repetitions of respiratory events and rapid changes in alertness are the main reasons why OSA induces acute hemodynamic modifications, including the heart rate, blood pressure, and cardiac output. In addition, CIH also contributes, in the long run, to an increase in autonomous nervous system drive, generation of ROS, endothelial dysfunction, and metabolic abnormalities, which in turn increase both the blood pressure and the cardiovascular risks [71]. Short-term changes of hemodynamic parameters include hypoxia, hypercapnia, negative intrathoracic pressure, and microarousal, while long-term changes of hemodynamic parameters include sympathetic activity, metabolic and hormonal changes, oxidative stress, inflammation, endothelial dysfunction, hypercoagulability, and genetic effects [71].
Rodent models of CIH exposure lasting from days to weeks have exhibited multiple adverse outcomes, including hypertension [17, 72], insulin resistance [73, 74], hyperlipidemia [75], atherosclerosis [76], and increased size of experimentally induced cardiac infarction [7, 14]. In rats, CIH leads to several cardiovascular consequences that are also observed in human OSA, including blood pressure elevation, biventricular hypertrophy, and LV contractile dysfunction [18–20, 53]. These data suggest that CIH exposure is detrimental to many physiological processes. Accumulating data indicate that CIH can induce cardiac dysfunction as its maladaptation [18, 19, 21]. Several studies have focused on the point that CIH induces cardiac dysfunction, but conclusions were inconsistent. Lee et al. [16] showed that short-term CIH exposure of rats for 1 and 4 days appeared to exert protective effects on the hearts, whereas long-term CIH exposure for 1 and 2 weeks appeared to exert deleterious effects. Williams et al. also used rats to confirm that after exposure to CIH for 10 days, cardiac function was decreased [20]. Fagan reported that treatment of CB57BL/6J mice with 2 min cycles of 10% alternating with 21% oxygen for 8 h per day for 4 weeks increased right ventricular systolic pressure, right ventricular mass, and neovascularization of distal pulmonary vessels, but had no effect on LV mass [77]. Campen and coworkers [17] reported that CB57BL/6J mice exposed to a 5-week regimen of 60-second cycles of 21% alternating with 5% oxygen for 12 h per day did develop significant increases in both systemic and pulmonary vascular pressures showing more prominent right than LV hypertrophy. Chen's group exposed male Sprague-Dawley rats to CIH for 4 and 8 weeks and found that abnormal myocardial architecture and increased interstitial space were observed after 4 weeks and became more obvious at 8 weeks [21]. In addition, a decreased ratio of wall thickness to cavity diameter and cardiac dilatation were observed after 8 weeks. They have proposed that a longer duration of CIH could induce abnormal myocardial architecture and even lead to the transition from eccentric cardiac hypertrophy to dilated cardiac hypertrophy. In our own study, we used 8–10-week-old FVB mice for exposure to CIH for 4 and 8 weeks. Exposure to CIH for 4 weeks induced cardiac hypertrophy, cardiac fibrosis, cardiac inflammation, and even cardiac dysfunction. Beside the above changes, exposure to CIH for 8 weeks induced cardiac apoptosis, oxidative stress and damage along with cardiac dysfunction, showing progressive pathological changes [22].
The diversity of observations among different research groups implies that the difference of cardiac responses to CIH may be related to many factors: (1) different resistance to hypoxia of different animals [15, 16, 21, 77]; (2) age of the animals that were used for starting the experiments; (3) the different protocols of CIH (including oxygen level, cycle length, number of hypoxic episodes per day, and number of exposure days, etc.). For example, Zhang et al. [78] have exposed postnatal male Sprague-Dawley rats to intermittent hypobaric hypoxia (IHH) at 3000 m high-altitude (P B = 525 mm Hg, P O2 = 108.8 mm Hg) or at 5000 m high-altitude (P B = 404 mm Hg, P O2 = 84 mm Hg), and sham control, and then perfused the isolated hearts in the Langendorff system with 30 min. global ischemia and 60 min. reperfusion. They found that the recovery of cardiac function was enhanced, coronary flow was increased and lactate dehydrogenase activity was decreased in 3000 m-IHH group at 60 min after I/R. In addition, cardiac function restored better in 3000 m-IHH group after 42 days of IHH than that after 28 days of IHH. In contrast to the above, the recovery of cardiac function was lower, coronary flow was decreased, and lactate dehydrogenase activity was increased in 5000 m-IHH group. The results suggest that the effect of CIH on I/R could be affected by the modes of IH exposure. In addition, the duration of CIH was the key determinant for the different effects on the I/R-induced cardiac damages and functional changes [15, 17, 21].
Although there are some differences of CIH-induced cardiac functional changes among animal models, Yang et al. [23] have examined the cardiac function of OSA patients with echocardiogram and found that the LVEF, fractional shortening (FS), and the ratio of early-to-late diastolic filling in patients with severe OSA was lower than in those with moderate OSA and in healthy controls. The inner diameters of the main pulmonary artery and right ventricle as well as the thickness of anterior wall of the right ventricle were increased in patients with severe OSA compared to those with moderate disease, which was worsened as a function of time with disease. The tissue Doppler imaging-derived Tei index and pulmonary artery systolic pressure were also increased along with the severity of OSA. LVEF and FS in patients who had suffered from OSA over 10 years were decreased compared to those suffering from OSA for a shorter time. LVEF and FS in patients with secondary hypertension were decreased significantly relative to nonhypertensive OSA patients and healthy controls. The ratio of early-to-late diastolic filling was decreased in OSA patients whether they had secondary hypertension or not. These results suggest that OSA affects the LV diastolic function in the early stage of the disease. Extended exposure to OSA resulted in LV systolic dysfunction with increased hypertension. Right ventricle dysfunction and abnormalities became more severe as the disease progressed. The results confirm the conclusion from clinical data that the duration of OSA is the key factor for OSA-induced cardiac damage.
HF is frequently observed in OSA patients, with a prevalence of 11–37% [79, 80]. Data obtained in the Sleep Heart Health Study have shown a 2.38 times increased likelihood of having HF in association with OSA, independent of other risk factors. OSA is not only a consequence of HF but indeed represents a risk factor for this condition [81], independent of hypertension [82]. Several grades of cardiac alterations have been reported in OSA patients, from silent or subclinical echocardiographic LV abnormalities to symptomatic systolic dysfunction (Table 2).
Based on the above discussion, we propose that the longer exposure to OSA-relevant CIH, the worse of the cardiac dysfunction. With the extension of exposure time to CIH, cardiac functions transit from compensative to decompensative phase as from cardiac hypertrophy to cardiac dilation even cardiac failure at the end. The development of contractile dysfunction is an important feature of cardiac decompensation (maladaptation) during CIH exposure to stress and also a hallmark of adverse cardiac remodeling [83].
3. Oxidative Stress Is the Predominant Mechanism of Cardiac Response to CIH: Role of Hydrogen Peroxide
Although OSA has been implicated in the pathogenesis of various cardiovascular diseases, mechanisms by which OSA affects the cardiovascular system are largely unknown. Oxidative stress, endothelial dysfunction, and inflammation are long-term consequences that mediate cardiovascular disease in patients with OSA [38]. Now, numerous studies have shown that oxidative stress is the main mechanism of cardiac I/R injury [84–86]. Because there is resemblance between the patterns of CIH associated with OSA and I/R injury, potential mechanisms of oxidative stress in OSA have been postulated to be related directly to CIH in a manner similar to I/R injury or indirectly via inflammatory response. The increased sympathetic tone and elevated catecholamine level might also be associated with increased ROS production [38]. Most recent studies in patients with OSA and animal models of CIH confirm that OSA is associated with oxidative stress, which generally correlates with the severity of sleep apnea, and improves with treatment [38, 87].
Oxidative stress represents an imbalance between the production of ROS and the antioxidant capacity of a biological system to buffer ROS. On the other hand, ROS are involved in signaling cascades, so that subphysiological ROS production may lead to reductive stress which has recently also been suggested to be detrimental in certain cardiac conditions [43, 88]. A fine balance between redox state and metabolism is more important than oxidative stress itself, and an imbalance in either the oxidative or the reductive direction could be detrimental [43]. In the past, ROS were considered solely injurious, but now it is generally accepted that they may exert both deleterious and beneficial actions [89]. In the last decade, they were consistently described as regulators of signal transduction and as second messengers in many signaling pathways in all cells that mediate cardioprotection [90–93]. The data of Kolar and Ostadel suggest that ROS not only contribute to I/R injury in normoxic rat hearts, but also are involved in the protective mechanisms induced by CIH [60]. During adaptation to CIH, repetitive cycles of hypoxia and reoxygenation (sublethal stresses) may lead to the production of ROS in the hypoxic heart, and the latter trigger a cascade of events that lead to increased antioxidant enzyme activity. These findings indicate that the ROS produced in cardiac tissue during sub-lethal stress may participate and activate signal transduction pathways [94] that form a positive feedback loop consisting of ROS and transcriptional factors [95]. This kind of strictly regulated generation of ROS at low levels can mediate physiological functions, such as increased level of antioxidative protection (enzymatic and nonenzymatic), growth, differentiation, and metabolism in cardiomyocytes [42, 96]. It may be involved in potentially adaptive processes such as adaptation to hypoxia and modulation of excitation—contraction coupling. On the other hand, the generation of higher levels of ROS and/or more potent oxidants such as hydroxyl radical may result in pathological changes as a result of macromolecular damage as well as inadequate signaling [43]. It is important to note that in addition to ROS, reactive nitrogen species, such as nitric oxide, play crucial roles in the regulation of cardiac functions. The metabolism of reactive nitrogen species is intertwined with ROS.
Sources of ROS and RNS in cardiomyocytes include mitochondria [97, 98], NADPH oxidase [99, 100], xanthine oxidase [101, 102] and uncoupled nitric oxide synthases [103, 104]. ROS include superoxide radical anion, hydroxyl radical, and hydrogen peroxide (H2O2). The superoxide is a relatively nonreactive, particularly targeting metalloproteins. It can be dismutated to H2O2 by superoxide dismutase. ROS exerts biological effects either by causing nonspecific oxidative damage to DNA, proteins, lipids, and macromolecules or through specific modulation of cellular signaling pathways (redox signaling). Cellular levels of ROS and their effects are regulated by a variety of specific and antioxidant systems (e.g., catalase, superoxide dismutase, glutathione peroxidase, peroxiredoxin, thioredoxin, and various vitamins) [105].
H2O2 is membrane permeable and diffusible, less reactive and longer-lived than hydroxyl radical or superoxide radical anion, and it is best suited for intra- and even inter-cellular signaling [106]. The physiological range of intracellular H2O2 concentrations appears to be remarkably conserved in different forms of life [107]. Among ROS, H2O2 is the only species that is generated and removed by several specific enzymes, which suggests that the intracellular concentration of H2O2 is tightly regulated and may serve specific cellular functions. Superoxide dismutase can catalyze the dismutation of the superoxide radical to H2O2 and molecular oxygen. Then it can be converted to H2O by catalase and glutathione peroxidase (Figure 2). H2O2 may react with transition metals, such as iron or copper to produce the highly reactive hydroxyl radical. In living organisms, besides its well-known cytotoxic effects, H2O2 also plays an essential role as a signaling molecule in regulating diverse biological processes such as immune cell activation, vascular remodeling, and apoptosis [108–110].
Figure 2.
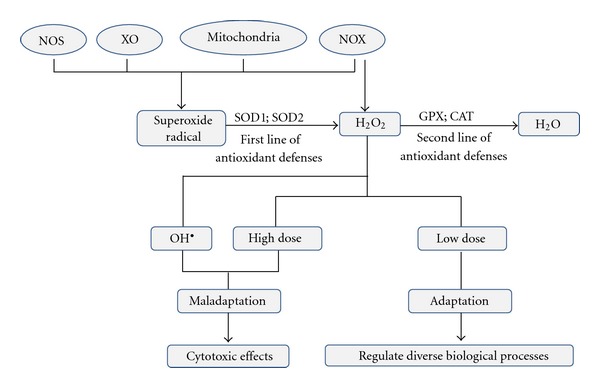
The schematic diagram of H2O2 source and effects. SOD can catalyze the dismutation of the superoxide radical to H2O2 and molecular oxygen. Then it can be converted to H2O by CAT and GPX. H2O2 also can be converted either to water or OH•. Low dose of H2O2 can induce adaptation, regulating diverse biological processes. High dose of H2O2 together with its conversion to OH• can induce maladaptation and cause cytotoxic effects. NOX: NADPH oxidase; NOS: uncoupled NO synthases; XO: xanthine oxidase; H2O2: Hydrogen peroxide; OH•: hydroxyl radical; SOD: superoxide dismutase; CAT: catalase; GPX: glutathione peroxidase.
Although H2O2 can contribute to I/R injury, it appears to play the part of activator in processes in which CIH upregulates the antioxidant enzymes. Using a cultured chick embryonic cardiomyocyte H/R model, Zhang et al. demonstrated that H2O2 is involved in ischemic preconditioning [111]. Preconditioning protects ischemic cardiomyocytes through H2O2 by opening mitochondrial K-ATP channels via activating PKC-ε pathway [111]. Park and Suzuki presented evidence that a redox regulator, thioredoxin, which can scavenge H2O2, is upregulated in adapted hearts in response to I/R and downregulated in the heart showing increased susceptibility to I/R following 2 weeks of CIH [7]. Their results suggested that thioredoxin plays a role in this adaptive mechanism as a scavenger of H2O2 and also represent a proof about H2O2's role in cardiac maladaptation. The Janus character of H2O2 as a mediator of growth and apoptosis suggests specificity of its biological activity. A number of studies have illustrated that, at concentrations in the high physiological range, H2O2 induces adaptive changes, increasing resistance of biological systems not only to oxidative stress but also to many other stimuli [43, 106]. The capability of H2O2 to induce a large number of protein syntheses and to provide cross-resistance implies that living systems may “intentionally” produce H2O2 as a component of adaptation in response to different fluctuations and perturbations shifting the system away from homeostasis. However, if the concentration of H2O2 exceeds the physiological concentration or the stress persists for a longer period of time, it can induce cardiac maladaptation. Thus, H2O2 is an important ROS in the transition of cardiac response to CIH from the adaptive phase to maladaptive phase or from cardiac hypertrophy to cardiac failure.
As H2O2 is generated by many compartmentalized enzymes, local variations in the concentration of H2O2 could also be crucial for the activation of specific targets [110]. Treatment of different primary cells with increasing doses of exogenous H2O2 induces proliferation, senescence, or apoptosis. Studies using other experimental models also showed H2O2 as a ROS having a flag potential for adjustments related to hypertrophy and/or cell death depending on its intracellular concentration [112, 113].
4. Effects of Antioxidants on the CIH-Induced Cardiac Response
OSA is recognized as an oxidative stress-related disorder [114]. In addition, the concept that oxidative stress resulting from an imbalance between increased ROS generation and inadequate endogenous antioxidant pools contributing to HF is well established [43]. Therefore, agents able to abolish oxidative stress are attractive for prophylactic and therapeutic intervention of OSA. Antioxidant pharmacotherapy for OSA is considered as a viable and attractive clinical option, especially in the light of poor compliance of continuous positive airway pressure—the gold standard treatment for OSA.
Understanding of complex actions of CIH, which can exert both protective and detrimental effects, may reveal important information for developing therapeutic strategies for a better management for OSA patients [7]. Cardiac response to CIH should help developing therapeutic strategies to prevent and/or treat OSA-induced cardiovascular complications. A number of studies have been conducted using CIH-treated animal models to explore the effects of antioxidants, but the outcomes of these studies are controversial.
In the study of Skelly et al. [115], tempol (1 mM, superoxide dismutase mimetic) was applied in the treatment of rats exposed to CIH. It was found that antioxidant treatment may be beneficial as an adjunct OSA therapy. Doehner et al. found that allopurinol, a xanthine oxidase inhibitor, free radical scavenger, and lipid peroxidation inhibitor [116], was associated with the improvement of CIH-related oxidant stress, cardiac dysfunction, and apoptosis in rats [20]. However, Kolor and coworkers demonstrated that the pretreatment of antioxidant N-acetylcysteine completely prevented the development of cardioprotection in CIH rats although the infarct size was reduced [117]. This clearly implies the dual roles, adaptive and maladaptive, of ROS in CIH. The suppression of ROS production prior to hypoxia prevented the adaptive responses to be activated, but on the other hand, on the long run, it showed positive effects on cardiac function. Inamoto et al. [118] confirmed in their study using CIH-treated mice model that pitavastatin preserved, at least partially, the morphological structure of the LV myocardium in lean mice exposed to CIH, through its antioxidant effect. Unlike in the acute experiment, a chronic antioxidative treatment with N-acetylcysteine during the adaptation to hypoxia led to a significant attenuation of the improvement in tolerance to lethal myocardial injury. Generally, the dual effects might, at least partially, explain why clinical trials with antioxidants failed to confirm promising data obtained in a number of animal studies. It is obvious that beneficial consequences of antioxidant supplementation in normal healthy heart cannot be used to predict an outcome in adapted or diseased hearts [117].
In clinical studies, the results of antioxidants treatments were also inconsistent. From a clinical perspective, much attention has focused on the concept that oxidative stress may be a driver of cardiac disease progression (e.g., HF), but clinical interventions with antioxidants have had little or no impact on heart disease risk and progression [43]. Results of relatively few controlled clinical trials with antioxidants such as vitamin C, α-tocopherol (vitamin E), and coenzyme Q10 are also controversial [119, 120]. Oral N-acetylcysteine administration appears to have a therapeutic potential in the treatment of OSA. It is proposed that long-term treatment with N-acetylcysteine in patients with OSA may reduce their dependency on continuous positive airway pressure therapy [121].
5. Prospective
Excessive oxidative stress undoubtedly exerts toxic and detrimental effects through modification of biomolecules or triggering damaging signals and also plays important roles in the pathogenesis of certain oxidative stress-related diseases, such as OSA-elicited cardiac dysfunction. Thus, supplementation of exogenous antioxidants may be used to balance cellular redox status. However, low levels of ROS such as H2O2 also function as signal molecules and induce adaptive response to protect the cells. Under such condition, we may not supply exogenous antioxidants since they may attenuate or completely block the adaptive signals, as proposed in a recent review [122]. Antioxidants may be considered at higher H2O2 concentrations related to pronounced oxidative damage, but if applied too early they may interfere with adaptive processes. However, the threshold of H2O2 or other ROS associated with a switch from mild/transient to severe/persistent oxidative stress has not yet been identified in OSA patients or OSA-relevant CIH models with cardiac dysfunction, which could be a good molecular diagnosis and/or monitoring in the development and progression of this disorder. Nevertheless, the concept of cardiac response to chronic intermittent hypoxia with a transition from adaptation to maladaptation will help us to understand the pathogenic mechanisms of OSA-induced cardiac damage and shed light on potential prevention and therapy. Since OSA is considered as a life-threatening condition and as an independent risk factor of cardiovascular diseases, we anticipate an intervention of effective exogenous antioxidants and/or induction of endogenous antioxidants in heart at the right time and proper dose may have protective effect against OSA-induced cardiac damage.
Acknowledgments
The data cited from the laboratories of the authors were supported in part by grants from American Diabetes Association (1-11-BS-17 to L. Cai) and Sleep Research Society Foundation/J. Christian Gillin M.D. Research Grant (001GN09 to J. Cai).
Abbreviations
- OSA:
Obstructive sleep apnea
- CIH:
Chronic intermittent hypoxia
- HF:
Heart failure
- I/R:
Ischemia/reperfusion
- LV:
Left ventricular
- LVEF:
Left ventricular ejection fraction
- IHH:
Intermittent hypobaric hypoxia
- FS:
Fractional shortening
- ROS:
Reactive oxygen species
- H2O2:
Hydrogen peroxide.
References
- 1.Wang ZH, Chen YX, Zhang CM, et al. Intermittent hypobaric hypoxia improves postischemic recovery of myocardial contractile function via redox signaling during early reperfusion. American Journal of Physiology. 2011;301(4):H1695–H1705. doi: 10.1152/ajpheart.00276.2011. [DOI] [PubMed] [Google Scholar]
- 2.Ding HL, Zhu HF, Dong JW, Zhu WZ, Zhou ZN. Intermittent hypoxia protects the rat heart against ischemia/reperfusion injury by activating protein kinase C. Life Sciences. 2004;75(21):2587–2603. doi: 10.1016/j.lfs.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Dong JW, Zhu HF, Zhu WZ, Ding HL, Ma TM, Zhou ZN. Intermittent hypoxia attenuates ischemia/reperfusion induced apoptosis in cardiac myocytes via regulating Bcl-2/Bax expression. Cell Research. 2003;13(5):385–391. doi: 10.1038/sj.cr.7290184. [DOI] [PubMed] [Google Scholar]
- 4.Zhu HF, Dong JW, Zhu WZ, Ding HL, Zhou ZN. ATP-dependent potassium channels involved in the cardiac protection induced by intermittent hypoxia against ischemia/reperfusion injury. Life Sciences. 2003;73(10):1275–1287. doi: 10.1016/s0024-3205(03)00429-6. [DOI] [PubMed] [Google Scholar]
- 5.Neckar J, Szarszoi O, Koten L, et al. Effects of mitochondrial K(ATP) modulators on cardioprotection induced by chronic high altitude hypoxia in rats. Cardiovascular Research. 2002;55(3):567–575. doi: 10.1016/s0008-6363(02)00456-x. [DOI] [PubMed] [Google Scholar]
- 6.Asemu G, Papousek F, Ostadal B, Kolar F. Adaptation to high altitude hypoxia protects the rat heart against ischemia-induced arrhythmias. Involvement of mitochondrial K(ATP) channel. Journal of Molecular and Cellular Cardiology. 1999;31(10):1821–1831. doi: 10.1006/jmcc.1999.1013. [DOI] [PubMed] [Google Scholar]
- 7.Park AM, Suzuki YJ. Effects of intermittent hypoxia on oxidative stress-induced myocardial damage in mice. Journal of Applied Physiology. 2007;102(5):1806–1814. doi: 10.1152/japplphysiol.01291.2006. [DOI] [PubMed] [Google Scholar]
- 8.Guo HC, Guo F, Zhang LN, et al. Enhancement of Na/K pump activity by chronic intermittent hypobaric hypoxia protected against reperfusion injury. American Journal of Physiology. 2011;300(6):H2280–H2287. doi: 10.1152/ajpheart.01164.2010. [DOI] [PubMed] [Google Scholar]
- 9.Guo HC, Zhang Z, Zhang LN, et al. Chronic intermittent hypobaric hypoxia protects the heart against ischemia/reperfusion injury through upregulation of antioxidant enzymes in adult guinea pigs. Acta Pharmacologica Sinica. 2009;30(7):947–955. doi: 10.1038/aps.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zong P, Setty S, Sun W, et al. Intermittent hypoxic training protects canine myocardium from infarction. Experimental Biology and Medicine. 2004;229(8):806–812. doi: 10.1177/153537020422900813. [DOI] [PubMed] [Google Scholar]
- 11.Wang YP, Cui F, Zhang LP, et al. Effect of chronic intermittent hypobaric hypoxia on alpha(1)-adrenergic receptor of myocardium participates in the cardioprotection. Sheng Li Xue Bao. 2009;61(1):21–36. [PubMed] [Google Scholar]
- 12.Zhang Y, Zhong N, Zhou ZN. Effects of intermittent hypoxia on action potential and contraction in non-ischemic and ischemic rat papillary muscle. Life Sciences. 2000;67(20):2465–2471. doi: 10.1016/s0024-3205(00)00832-8. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Zhong N, Zhu HF, Zhou ZN. Antiarrhythmic and antioxidative effects of intermittent hypoxia exposure on rat myocardium. Acta Physiologica Sinica. 2000;52(2):89–92. [PubMed] [Google Scholar]
- 14.Joyeux-Faure M, Stanke-Labesque F, Lefebvre B, et al. Chronic intermittent hypoxia increases infarction in the isolated rat heart. Journal of Applied Physiology. 2005;98(5):1691–1696. doi: 10.1152/japplphysiol.01146.2004. [DOI] [PubMed] [Google Scholar]
- 15.Naghshin J, McGaffin KR, Witham WG, et al. Chronic intermittent hypoxia increases left ventricular contractility in C57BL/6J mice. Journal of Applied Physiology. 2009;107(3):787–793. doi: 10.1152/japplphysiol.91256.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SD, Kuo WW, Wu CH, et al. Effects of short- and long-term hypobaric hypoxia on Bcl2 family in rat heart. International Journal of Cardiology. 2006;108(3):376–384. doi: 10.1016/j.ijcard.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 17.Campen MJ, Shimoda LA, O’Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. Journal of Applied Physiology. 2005;99(5):2028–2035. doi: 10.1152/japplphysiol.00411.2005. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Einbinder E, Zhang Q, Hasday J, Balke CW, Scharf SM. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. American Journal of Respiratory and Critical Care Medicine. 2005;172(7):915–920. doi: 10.1164/rccm.200504-560OC. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Zhang J, Gan TX, et al. Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. Journal of Applied Physiology. 2008;104(1):218–223. doi: 10.1152/japplphysiol.00301.2007. [DOI] [PubMed] [Google Scholar]
- 20.Williams AL, Chen L, Scharf SM. Effects of allopurinol on cardiac function and oxidant stress in chronic intermittent hypoxia. Sleep and Breathing. 2010;14(1):51–57. doi: 10.1007/s11325-009-0279-x. [DOI] [PubMed] [Google Scholar]
- 21.Chen LM, Kuo WW, Yang JJ, et al. Eccentric cardiac hypertrophy was induced by long-term intermittent hypoxia in rats. Experimental Physiology. 2007;92(2):409–416. doi: 10.1113/expphysiol.2006.036590. [DOI] [PubMed] [Google Scholar]
- 22.Yin X, Tan Y, Wang B, Zheng Y, Cai J, Cai L. Metallothionein protects intermittent hypoxia-induced cardiomyopathy by inhibition of cardiac oxidative damage, apoptosis and inflammation. In: Proceedings of the 51st Annual Meeting and ToxExpoTM, vol. 126; 2012; p. 327. [Google Scholar]
- 23.Yang SQ, Han LL, Dong XL, et al. Mal-effects of obstructive sleep apnea on the heart. doi: 10.1007/s11325-011-0566-1. Sleep and Breathing. In press. [DOI] [PubMed] [Google Scholar]
- 24.Ancoli-Israel S, Kripke DF, Klauber MR, Mason WJ, Fell R, Kaplan O. Sleep-disordered breathing in community-dwelling elderly. Sleep. 1991;14(6):486–495. doi: 10.1093/sleep/14.6.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newacheck PW, Taylor WR. Childhood chronic illness: prevalence, severity, and impact. American Journal of Public Health. 1992;82(3):364–371. doi: 10.2105/ajph.82.3.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guilleminault C, Ji HL, Chan A. Pediatric obstructive sleep apnea syndrome. Archives of Pediatrics and Adolescent Medicine. 2005;159(8):775–785. doi: 10.1001/archpedi.159.8.775. [DOI] [PubMed] [Google Scholar]
- 27.Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proceedings of the American Thoracic Society. 2008;5(2):136–143. doi: 10.1513/pats.200709-155MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. The New England Journal of Medicine. 1993;328(17):1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 29.Botros N, Concato J, Mohsenin V, Selim B, Doctor K, Yaggi HK. Obstructive sleep apnea as a risk factor for type 2 diabetes. American Journal of Medicine. 2009;122(12):1122–1127. doi: 10.1016/j.amjmed.2009.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw JE, Punjabi NM, Wilding JP, Alberti KGMM, Zimmet PZ. Sleep-disordered breathing and type 2 diabetes. A report from the International Diabetes Federation Taskforce on Epidemiology and Prevention. Diabetes Research and Clinical Practice. 2008;81(1):2–12. doi: 10.1016/j.diabres.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 31.Keckeis M, Lattova Z, Maurovich-Horvat E, et al. Impaired glucose tolerance in sleep disorders. PLoS One. 2010;5(3) doi: 10.1371/journal.pone.0009444. Article ID e9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punjabi NM, Beamer BA. Alterations in glucose disposal in sleep-disordered breathing. American Journal of Respiratory and Critical Care Medicine. 2009;179(3):235–240. doi: 10.1164/rccm.200809-1392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das AM, Khayat R. Hypertension in obstructive sleep apnea: risk and therapy. Expert Review of Cardiovascular Therapy. 2009;7(6):619–626. doi: 10.1586/erc.09.25. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Jimenez F, Cortes-Bergoderi M. Update: systemic diseases and the cardiovascular system (i): obesity and the heart. Revista Española de Cardiología. 2011;64(2):140–149. doi: 10.1016/j.recesp.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Lam JC, Mak JC, Ip MS. Obesity, obstructive sleep apnoea and metabolic syndrome. Respirology. 2012;17(2):223–236. doi: 10.1111/j.1440-1843.2011.02081.x. [DOI] [PubMed] [Google Scholar]
- 36.Pinto JMB, Garpestad E, Weiss JW, Bergau DM, Kirby DA. Hemodynamic changes associated with obstructive sleep apnea followed by arousal in a porcine model. Journal of Applied Physiology. 1993;75(4):1439–1443. doi: 10.1152/jappl.1993.75.4.1439. [DOI] [PubMed] [Google Scholar]
- 37.Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest. 2007;132(1):325–337. doi: 10.1378/chest.07-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khayat R, Patt B, Hayes D., Jr. Obstructive sleep apnea: the new cardiovascular disease. Part I: obstructive sleep apnea and the pathogenesis of vascular disease. Heart Failure Reviews. 2009;14(3):143–153. doi: 10.1007/s10741-008-9112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. American Journal of Respiratory and Critical Care Medicine. 2002;165(9):1217–1239. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 40.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. Journal of the American Medical Association. 2006;295(13):1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 41.Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics-2008 update: a report from the American heart association statistics committee and stroke statistics subcommittee. Circulation. 2008;117(4):e25–e146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 42.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. Journal of Clinical Investigation. 2005;115(3):500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos CXC, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radical Biology and Medicine. 2011;50(7):777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiological Reviews. 2005;85(3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 45.Steiner S, Schueller PO, Hennersdorf MG, Behrendt D, Strauer BE. Impact of obstructive sleep apnea on the occurrence of restenosis after elective percutaneous coronary intervention in ischemic heart disease. Respiratory Research. 2008;9, article 50 doi: 10.1186/1465-9921-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prabhakar NR, Fields RD, Baker T, Fletcher EC. Intermittent hypoxia: cell to system. American Journal of Physiology. 2001;281(3):L524–L528. doi: 10.1152/ajplung.2001.281.3.L524. [DOI] [PubMed] [Google Scholar]
- 47.Prabhakar NR, Kline DD. Ventilatory changes during intermittent hypoxia: importance of pattern and duration. High Altitude Medicine and Biology. 2002;3(2):195–204. doi: 10.1089/15270290260131920. [DOI] [PubMed] [Google Scholar]
- 48.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. The New England Journal of Medicine. 2000;342(19):1378–1384. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi M, Fujimoto K, Urushibata K, Uchikawa SI, Imamura H, Kubo K. Nocturnal oxygen desaturation correlates with the severity of coronary atherosclerosis in coronary artery disease. Chest. 2003;124(3):936–941. doi: 10.1378/chest.124.3.936. [DOI] [PubMed] [Google Scholar]
- 50.Milleron O, Pilliere R, Foucher A, et al. Benefits of obstructive sleep apnoea treatment in coronary artery disease: a long-term follow-up study. European Heart Journal. 2004;25(9):728–734. doi: 10.1016/j.ehj.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. Journal of Applied Physiology. 2003;94(6):2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- 52.Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia—influence of chemoreceptors and sympathetic nervous system. Journal of Hypertension. 1997;15(12) part 2:1593–1603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- 53.Fletcher EC. Physiological and genomic consequences of intermittent hypoxia: invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. Journal of Applied Physiology. 2001;90(4):1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- 54.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. The New England Journal of Medicine. 2005;353(19):2034–2041. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 55.Punjabi NM, Polotsky VY. Disorders of glucose metabolism in sleep apnea. Journal of Applied Physiology. 2005;99(5):1998–2007. doi: 10.1152/japplphysiol.00695.2005. [DOI] [PubMed] [Google Scholar]
- 56.Rosamond W, Flegal K, Friday G, et al. Heart disease and stroke statistics—2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115(5):e69–e171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 57.Lavie L. Sleep-disordered breathing and cerebrovascular disease: a mechanistic approach. Neurologic Clinics. 2005;23(4):1059–1075. doi: 10.1016/j.ncl.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 58.Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, Phillipson EA. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. Journal of Clinical Investigation. 1997;99(1):106–109. doi: 10.1172/JCI119120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 60.Kolar F, Ostadal B. Molecular mechanisms of cardiac protection by adaptation to chronic hypoxia. Physiological Research. 2004;53(supplement 1):S3–S13. [PubMed] [Google Scholar]
- 61.Alchanatis M, Tourkohoriti G, Kosmas EN, et al. Evidence for left ventricular dysfunction in patients with obstructive sleep apnoea syndrome. European Respiratory Journal. 2002;20(5):1239–1245. doi: 10.1183/09031936.02.00278002. [DOI] [PubMed] [Google Scholar]
- 62.Nakashima H, Katayama T, Takagi C, et al. Obstructive sleep apnoea inhibits the recovery of left ventricular function in patients with acute myocardial infarction. European Heart Journal. 2006;27(19):2317–2322. doi: 10.1093/eurheartj/ehl219. [DOI] [PubMed] [Google Scholar]
- 63.Kasai T, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. Journal of the American College of Cardiology. 2011;57(2):119–127. doi: 10.1016/j.jacc.2010.08.627. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Y, Yang HT, Zhou ZN. The cardioprotection of intermittent hypoxic adaptation. Sheng Li Xue Bao. 2007;59(5):601–613. [PubMed] [Google Scholar]
- 65.Zhuang JG, Zhou ZN. Protective effects of intermittent hypoxic adaptation on myocardium and its mechanisms. Biological Signals and Receptors. 1999;8(4-5):316–322. doi: 10.1159/000014602. [DOI] [PubMed] [Google Scholar]
- 66.Neckar J, Papousek F, Novakova O, Ost’adal B, Kolar F. Cardioprotective effects of chronic hypoxia and ischaemic preconditioning are not additive. Basic Research in Cardiology. 2002;97(2):161–167. doi: 10.1007/s003950200007. [DOI] [PubMed] [Google Scholar]
- 67.Cai Z, Manalo DJ, Wei G, et al. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108(1):79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 68.Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr., Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. Journal of Neuroscience. 2001;21(14):5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitchell GS, Baker TL, Nanda SA, et al. Physiological and genomic consequences of intermittent hypoxia: Invited review: Intermittent hypoxia and respiratory plasticity. Journal of Applied Physiology. 2001;90(6):2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- 70.Mateika JH, Narwani G. Intermittent hypoxia and respiratory plasticity in humans and other animals: does exposure to intermittent hypoxia promote or mitigate sleep apnoea? Experimental Physiology. 2009;94(3):279–296. doi: 10.1113/expphysiol.2008.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fava C, Montagnana M, Favaloro EJ, Guidi GC, Lippi G. Obstructive sleep apnea syndrome and cardiovascular diseases. Seminars in Thrombosis and Hemostasis. 2011;37(3):280–297. doi: 10.1055/s-0031-1273092. [DOI] [PubMed] [Google Scholar]
- 72.Fletcher EC, Lesske J, Qian W, Miller CC, III, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992;19(6):555–561. doi: 10.1161/01.hyp.19.6.555. [DOI] [PubMed] [Google Scholar]
- 73.Iiyori N, Alonso LC, Li J, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. American Journal of Respiratory and Critical Care Medicine. 2007;175(8):851–857. doi: 10.1164/rccm.200610-1527OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polotsky VY, Li J, Punjabi NM, et al. Intermittent hypoxia increases insulin resistance in genetically obese mice. Journal of Physiology. 2003;552(1):253–264. doi: 10.1113/jphysiol.2003.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Savransky V, Nanayakkara A, Smith PL, O’Donnell CP, Polotsky VY. Hyperlipidemia and lipid peroxidation are dependent on the severity of chronic intermittent hypoxia. Journal of Applied Physiology. 2007;102(2):557–563. doi: 10.1152/japplphysiol.01081.2006. [DOI] [PubMed] [Google Scholar]
- 76.Savransky V, Nanayakkara A, Li J, et al. Chronic intermittent hypoxia induces atherosclerosis. American Journal of Respiratory and Critical Care Medicine. 2007;175(12):1290–1297. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fagan KA. Physiological and genomic consequences of intermittent hypoxia selected contribution: pulmonary hypertension in mice following intermittent hypoxia. Journal of Applied Physiology. 2001;90(6):2502–2507. doi: 10.1152/jappl.2001.90.6.2502. [DOI] [PubMed] [Google Scholar]
- 78.Zhang H, Yang CY, Wang YP, et al. Effects of different modes of intermittent hypobaric hypoxia on ischemia/reperfusion injury in developing rat hearts. Sheng Li Xue Bao. 2007;59(5):660–666. [PubMed] [Google Scholar]
- 79.Javaheri S, Parker TJ, Liming JD, et al. Sleep apnea in 81 ambulatory male patients with stable heart failure: types and their prevalences, consequences, and presentations. Circulation. 1998;97(21):2154–2159. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- 80.Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. American Journal of Respiratory and Critical Care Medicine. 1999;160(4):1101–1106. doi: 10.1164/ajrccm.160.4.9903020. [DOI] [PubMed] [Google Scholar]
- 81.Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health study. American Journal of Respiratory and Critical Care Medicine. 2001;163(1):19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- 82.Hedner J, Ejnell H, Caidahl K. Left ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoea. Journal of Hypertension. 1990;8(10):941–946. doi: 10.1097/00004872-199010000-00009. [DOI] [PubMed] [Google Scholar]
- 83.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling-concepts and clinical implications: a consensus paper from an International Forum on Cardiac Remodeling. Journal of the American College of Cardiology. 2000;35(3):569–582. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- 84.Downey JM, Cohen MV. Reducing infarct size in the setting of acute myocardial infarction. Progress in Cardiovascular Diseases. 2006;48(5):363–371. doi: 10.1016/j.pcad.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 85.Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et Biophysica Acta. 1998;1366(1-2):33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- 86.Hung LM, Chen JK, Lee RS, Liang HC, Su MJ. Beneficial effects of astringinin, a resveratrol analogue, on the ischemia and reperfusion damage in rat heart. Free Radical Biology and Medicine. 2001;30(8):877–883. doi: 10.1016/s0891-5849(01)00474-9. [DOI] [PubMed] [Google Scholar]
- 87.Laaban JP, Pascal-Sebaoun S, Bloch E, Orvoen-Frija E, Oppert JM, Huchon G. Left ventricular systolic dysfunction in patients with obstructive sleep apnea syndrome. Chest. 2002;122(4):1133–1138. doi: 10.1378/chest.122.4.1133. [DOI] [PubMed] [Google Scholar]
- 88.Rajasekaran NS, Connell P, Christians ES, et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130(3):427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovascular Research. 2004;61(3):461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 90.Das DK, Engelman RM, Maulik N. Oxygen free radical signaling in ischemic preconditioning. Annals of the New York Academy of Sciences. 1999;874:49–65. doi: 10.1111/j.1749-6632.1999.tb09224.x. [DOI] [PubMed] [Google Scholar]
- 91.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. The Journal of Biological Chemistry. 1998;273(19):11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 92.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biology and Medicine. 1996;22(1-2):269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 93.Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxidants and Redox Signaling. 2007;9(9):1397–1403. doi: 10.1089/ars.2007.1732. [DOI] [PubMed] [Google Scholar]
- 94.Marklund SL, Westman NG, Lundgren E, Roos G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Research. 1982;42(5):1955–1961. [PubMed] [Google Scholar]
- 95.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacological Research. 2007;55(6):523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 96.Finkel T. Oxidant signals and oxidative stress. Current Opinion in Cell Biology. 2003;15(2):247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 97.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 98.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 100.Brandes RP, Weissmann N, Schroder K. NADPH oxidases in cardiovascular disease. Free Radical Biology and Medicine. 2010;49(5):687–706. doi: 10.1016/j.freeradbiomed.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 101.Kelley EE, Khoo NKH, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radical Biology and Medicine. 2010;48(4):493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circulation Research. 2006;98(2):271–279. doi: 10.1161/01.RES.0000200181.59551.71. [DOI] [PubMed] [Google Scholar]
- 103.Massion PB, Balligand JL. Relevance of nitric oxide for myocardial remodeling. Current Heart Failure Reports. 2007;4(1):18–25. doi: 10.1007/s11897-007-0021-6. [DOI] [PubMed] [Google Scholar]
- 104.Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovascular Research. 2007;75(2):315–326. doi: 10.1016/j.cardiores.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 105.Kost KM. Endoscopic percutaneous dilatational tracheotomy: a prospective evaluation of 500 consecutive cases. Laryngoscope. 2005;115(10) part 2:1–30. doi: 10.1097/01.MLG.0000163744.89688.E8. [DOI] [PubMed] [Google Scholar]
- 106.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants and Redox Signaling. 2006;8(3-4):243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 107.Mueller S. Sensitive and nonenzymatic measurement of hydrogen peroxide in biological systems. Free Radical Biology and Medicine. 2000;29(5):410–415. doi: 10.1016/s0891-5849(00)00261-6. [DOI] [PubMed] [Google Scholar]
- 108.Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. The Journal of Biological Chemistry. 2004;279(50):51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- 109.Laloi C, Apel K, Danon A. Reactive oxygen signalling: the latest news. Current Opinion in Plant Biology. 2004;7(3):323–328. doi: 10.1016/j.pbi.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 110.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nature Reviews Molecular Cell Biology. 2007;8(9):722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 111.Zhang HY, McPherson BC, Liu H, Baman TS, Rock P, Yao Z. H2O2 opens mitochondrial KATP channels and inhibits GABA receptors via protein kinase C-ε in cardiomyocytes. American Journal of Physiology. 2002;282(4):H1395–H1403. doi: 10.1152/ajpheart.00683.2001. [DOI] [PubMed] [Google Scholar]
- 112.Schenkel PC, Tavares AM, Fernandes RO, et al. Redox-sensitive prosurvival and proapoptotic protein expression in the myocardial remodeling post-infarction in rats. Molecular and Cellular Biochemistry. 2010;341(1-2):1–8. doi: 10.1007/s11010-010-0431-8. [DOI] [PubMed] [Google Scholar]
- 113.da Rosa Araujo AS, Silva de Miranda MF, de Oliveira UO, et al. Increased resistance to hydrogen peroxide-induced cardiac contracture is associated with decreased myocardial oxidative stress in hypothyroid rats. Cell Biochemistry and Function. 2010;28(1):38–44. doi: 10.1002/cbf.1616. [DOI] [PubMed] [Google Scholar]
- 114.Lavie L. Obstructive sleep apnoea syndrome—an oxidative stress disorder. Sleep Medicine Reviews. 2003;7(1):35–51. doi: 10.1053/smrv.2002.0261. [DOI] [PubMed] [Google Scholar]
- 115.Skelly JR, Edge D, Shortt CM, Jones JF, Bradford A, O’Halloran KD. Tempol ameliorates pharyngeal dilator muscle dysfunction in a rodent model of chronic intermittent hypoxia. American Journal of Respiratory Cell and Molecular Biology. 2012;46(2):139–148. doi: 10.1165/rcmb.2011-0084OC. [DOI] [PubMed] [Google Scholar]
- 116.Doehner W, Schoene N, Rauchhaus M, et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105(22):2619–2624. doi: 10.1161/01.cir.0000017502.58595.ed. [DOI] [PubMed] [Google Scholar]
- 117.Kolar F, Jezkova J, Balkova P, et al. Role of oxidative stress in PKC-delta upregulation and cardioprotection induced by chronic intermittent hypoxia. American Journal of Physiology. 2007;292(1):H224–H230. doi: 10.1152/ajpheart.00689.2006. [DOI] [PubMed] [Google Scholar]
- 118.Inamoto S, Yoshioka T, Yamashita C, et al. Pitavastatin reduces oxidative stress and attenuates intermittent hypoxia-induced left ventricular remodeling in lean mice. Hypertension Research. 2010;33(6):579–586. doi: 10.1038/hr.2010.36. [DOI] [PubMed] [Google Scholar]
- 119.Ferrari R, Guardigli G, Mele D, Percoco GF, Ceconi C, Curello S. Oxidative stress during myocardial ischaemia and heart failure. Current Pharmaceutical Design. 2004;10(14):1699–1711. doi: 10.2174/1381612043384718. [DOI] [PubMed] [Google Scholar]
- 120.Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends in Pharmacological Sciences. 2005;26(6):302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sadasivam K, Patial K, Vijayan VK, Ravi K. Anti-oxidant treatment in obstructive sleep apnoea syndrome. The Indian Journal of Chest Diseases & Allied sciences. 2011;53(3):153–162. [PubMed] [Google Scholar]
- 122.Zhi-Hua Chen EN. Two faces of lipid peroxidation products: the “Yin and Yang” principles of oxidative stress. Journal of Experimental and Integrative Medicine. 2011;1(4):215–219. [Google Scholar]