

Abstract
Osteosarcoma is a primary bone malignancy with a particularly high incidence rate in children and adolescents relative to other age groups. The etiology of this often aggressive cancer is currently unknown, because complicated structural and numeric genomic rearrangements in cancer cells preclude understanding of tumour development. In addition, few consistent genetic changes that may indicate effective molecular therapeutic targets have been reported. However, high-resolution techniques continue to improve knowledge of distinct areas of the genome that are more commonly associated with osteosarcomas. Copy number gains at chromosomes 1p, 1q, 6p, 8q, and 17p as well as copy number losses at chromosomes 3q, 6q, 9, 10, 13, 17p, and 18q have been detected by numerous groups, but definitive oncogenes or tumour suppressor genes remain elusive with respect to many loci. In this paper, we examine studies of the genetics of osteosarcoma to comprehensively describe the heterogeneity and complexity of this cancer.
1. Introduction
Osteosarcoma is the most common primary bone malignancy, with a high incidence rate in children and adolescents compared to other age groups. Tumours most often arise in the long bones from osteoid-producing neoplastic cells adjacent to the growth plates, occurring less commonly in the axial skeleton and other nonlong bones [1]. Survival rates for osteosarcoma have remained at 60–70% for localised disease for decades despite ongoing studies [2]. Unlike many sarcomas which are characterised by specific chromosome translocations, complex genomic rearrangements involving any chromosome characterise individual osteosarcoma cells. Because of this few consistent genetic changes that may indicate effective molecular targets for treatment have been reported.
Decades' worth of molecular cytogenetics studies and genomic analyses of osteosarcomas have been completed through karyotyping, comparative genomic hybridisation (CGH), fluorescence in situ hybridisation, quantitative PCR, and single-strand conformation polymorphism analysis, among others. Genome-wide association studies utilising single-nucleotide polymorphisms (SNPs) have been used more recently to learn more broadly about osteosarcoma genomics [3]. Resolution of alterations has increased from visualisation at the chromosome level to point mutations, but the genetic etiology of osteosarcoma is still unknown. One consistent finding, however, is the higher incidence of osteosarcoma relative to the general population in individuals with familial Li-Fraumeni syndrome (germline TP53 inactivation), hereditary retinoblastoma (germline RB1 inactivation), Rothmund-Thomson syndrome (germline RECQL4 inactivation), or Bloom or Werner syndrome (germline BLM or WRN inactivation, resp.) [4–8]. The genes associated with all of these familial syndromes encode protein products necessary to stabilise the genome, and their impairment can manifest in defective maintenance of DNA.
In this paper, we have collected studies of the genetics of osteosarcoma to illustrate the heterogeneity and complexity of this tumour type at the level of the chromosome and gene. Osteosarcoma-specific epigenetic changes, mRNA and protein level aberrations, and changes to microRNA (miRNA) will not be described extensively in this paper. Other publications on these topics exist and offer more thorough descriptions of the epigenetic [9], expression [10, 11], and miRNA profiling [12, 13] of osteosarcoma. To understand the molecular dynamics of this disease at any level, it is important to first recognize the fundamental role of the disruption of cellular mechanisms intended to maintain genomic instability.
2. Genomic Instability in Osteosarcoma
Osteosarcoma is characterised by a high level of genomic instability, in particular one subcategory of instability known as chromosomal instability (CIN) [14, 15]. Microsatellite instability (MIN) and CpG island methylator phenotype (CIMP) are two other forms of genomic instability, and they have been described extensively and predominantly in colorectal cancer [16, 17]. CIN is the elevated rate of gain or loss of entire chromosomes or sections of chromosomes [16, 18], and it appears to be significant in the pathogenesis of osteosarcoma tumours, resulting in complicated structural and numerical aberrations and wide variability between cells [19].
CIN is categorised in two subtypes, numerical CIN (N-CIN) and structural CIN (S-CIN). Processes underlying N-CIN are those leading to copy number alterations. N-CIN is manifested in polyploidy, caused by errors in mitosis, aneuploidy, segmental amplifications, or deletions, and unbalanced translocations. S-CIN can result from ineffective DNA damage response mechanisms following exogenous insults or replication errors, leading to aberrant genomic rearrangements, chromosomal breakages, and usually, but not necessarily, gene copy number alterations [20]. Karyotypic complexity in tumours, an end product of CIN, is correlated with higher expression of survival- and tissue invasion-related genes and lower expression of those involved in checking cell cycle regulation and ensuring DNA repair [21].
Mutations or deregulation of genes important for mitotic checkpoints is thought to be the underlying cause of CIN [22]. For example, inactivation of the tumour suppressor proteins p53 and pRB cause CIN in vivo [23, 24]. Additionally, mutation of TP53 is significantly correlated with high levels of genomic instability in osteosarcoma [25], while mutation of RB1 contributes to mitotic missegregation and loss of heterozygosity (LOH) in mice [26]. In a study of 18 osteosarcomas, an association was made between overexpression of RECQL4, a gene which encodes a DNA helicase, and S-CIN [27]. Whether mutator mutations are in fact required to induce carcinogenesis by increasing the rate of genetic change is still in question [28].
Telomere maintenance, or lack thereof, is another potential source of the instability typical of osteosarcoma, in addition to reducing the likelihood of favourable outcome in patients with the disease. Telomerase activation is a mechanism by which human cells can bypass their theoretical life span defined by the number of cell divisions required to critically deplete telomere length (the Hayflick limit), thereby avoiding senescence [29]. Rather than activation of the telomerase subunit genes, the alternative lengthening of telomeres' (ALTs) mechanism of preserving telomeres is more frequently observed in sarcomas [30]. Telomerase activation and ALT both contribute to telomere maintenance in osteosarcoma, but ALT seems to be the predominant process [31, 32]. Interestingly, ALT is more common in sarcomas not associated with specific translocations [33] and therefore may be associated with more complex chromosomal aberrations in some tumours [34, 35], including osteosarcomas [36, 37]. In females, shorter telomere length is associated with increased risk of osteosarcoma [38]. Additionally, cellular telomere maintenance is associated with poor outcome for osteosarcoma patients [39], but enzymes facilitating ALT may have potential as therapeutic targets [40].
3. Genetic Alterations by Osteosarcoma Subtype
The vast majority of studies have been descriptions of osteosarcomas focused on the conventional, high-grade subtypes including the chondroblastic, fibroblastic, and osteoblastic variants. These are the most frequently occurring types of osteosarcoma. The rarer subtypes include telangiectatic, small cell, periosteal, high-grade surface, and low-grade osteosarcoma. These forms often present with distinguishing genetic features infrequent in conventional tumours.
3.1. Conventional Osteosarcoma
Complex and largely inconsistent genetic alterations are typical of conventional osteosarcoma. Overall, some frequent genetic alterations in conventional osteosarcoma are losses of portions of chromosomes 3q, 6q, 9, 10, 13, 17p, and 18q and gains of portions of chromosomes 1p, 1q, 6p, 8q, and 17p (Table 1; Figure 1). In general, regions in which known tumour suppressor genes are located undergo deletion and mutation events, while those possessing established oncogenes are gained or amplified in cells. Unfortunately, for many of the alterations described in this paper there exist wide ranges of observed frequencies among published reports. These can be due to inconsistencies between materials and methodology used by groups, including differences in the resolution of cytogenetic techniques and platforms, variation between tumour cohorts with respect to staging, histological subtype, and sample size, and whether specimens have been exposed to chemotherapy (chemotherapy drugs may induce DNA damage). The low incidence rate of osteosarcoma exacerbates the limitations on genetic studies of this disease because it lowers the availability of samples. Furthermore, a high level of chromosomal instability is thought to cause the profound intra- and intertumoural heterogeneity observed in and among specimens, in which abnormalities such as heterogeneously staining regions, double-minute chromosomes, and dicentric chromosomes are not uncommon.
Table 1.
Frequent genetic alterations in sporadic conventional osteosarcoma.
| Genomic region | Event | Frequency | Effected genes | References | |
|---|---|---|---|---|---|
| Tumour suppressor gene(s) | Oncogene(s) | ||||
| 1q10-q12, 1q21-q31 | Amp | 6–59% | [50, 55, 77, 82–84] | ||
| 3q13.31 | Del, LOH |
6–80% | LSAMP | [44, 45, 49–55] | |
| 5q21 | LOH | 62% | APC | [109] | |
| 6p12-p21 | Gain, Amp |
16–75% | RUNX2, CDC5L, VEGFA, PIM1 | [9, 20, 45, 49, 65, 77, 78, 82, 89–92] | |
| 6p22.3 | Gain, Amp |
60% | E2F3 | [92] | |
| 7p21 7q31 |
Del Amp Del Amp |
36% 14% 41% 9% |
TWIST
MET |
[109] | |
| 8q24.21 | Amp | 7–67% | MYC | [20, 45, 49, 55, 71, 78, 81–83] | |
| 8q24.4 | Mut | <5% | RECQL4 | [85] | |
| Gain | 33% | RECQL4 | [27] | ||
| 9p21 | Del | 5–21% |
p16/INK4A,
p14/ARF, p15/INK4B |
[48, 60–63] | |
| 10q26 | LOH | 60% |
BUB3,
FGFR2 |
[106] | |
| 12q13 | Amp | 41% | PRIM1 | [58] | |
| 12q14 | Amp | 10% | CDK4 | [45, 57] | |
| 12q15 | Amp | 3–25% | MDM2 | [47, 48, 57, 72, 73] | |
| 13q14.2 | LOH | 19–67% | RB1 | [43–55] | |
| Mut | 25–35% | RB1 | [46, 56] | ||
| 16q23.1-q23.2 | Del | 30% | WWOX | [107] | |
| 17p11.2-p12 | Amp | 20–78% | COPS3, PMP22, MAPK7 | [20, 49, 52, 55, 65, 68, 70, 75–80] | |
| 17p13.1 | Del, LOH |
29–42% | TP53 | [44, 62, 65] | |
| Mut | 10–39% | TP53 | [25, 44, 47, 48, 56, 62, 67–71] | ||
| 18q (MCR 18q21-q23) | Del | 31–64% | [44, 53, 110, 114] | ||
MCR, minimal common region; Del, deletion; Amp, amplification; Mut, mutation.
Figure 1.
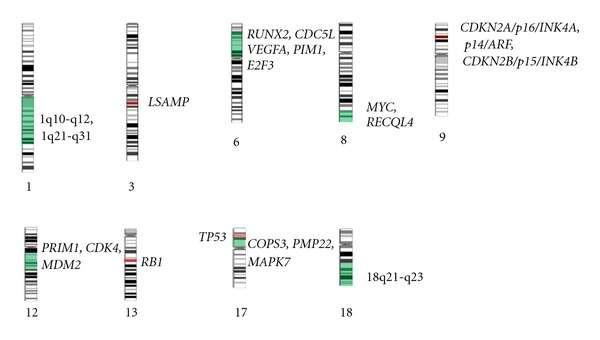
Frequent chromosomal aberrations in sporadic conventional osteosarcoma. Green highlighted areas represent minimal common regions of gain and amplification, or cytobands containing frequently gained and amplified genes. Red highlighted areas represent minimal common regions of loss, or cytobands containing genes frequently lost. Refer to Table 1 and the text for more details regarding minimal common regions and the presence of genetic mutations in some areas of the genome. Chromosome images adapted from the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman/. Accessed January 25, 2012).
Inactivation of RB1, located at chromosome 13q14.2, is frequent in sporadic osteosarcoma, and when it occurs due to germline mutation, osteosarcoma incidence significantly increases [41]. RB1 encodes the tumour suppressor protein pRB which is essential in preventing cell cycle progression through G1/S following DNA damage. Mechanistically, the protein inhibits members of the E2F transcription factor family, a process that requires strict regulation of the cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors (CDKNs), to promote stability of the genome [42]. LOH or deletion of the RB1 locus has been detected in 19–67% of tumours [43–55], and RB1 mutations have been detected in about 25–35% of cases [56]. Either type of alteration is associated with inactivation of RB1 expression in about 50% of tumours [46].
Loss of cellular control of other components of the pRB pathway is often deduced upon observing genetic alterations in osteosarcoma tumours. As such, pRB-independent mechanisms of pRB pathway deregulation may be present in addition to pRB inactivation. Amplification of the cyclin-dependent kinase gene CDK4 (chromosome 12q13-14) has been detected in approximately 10% of tumours [45, 57]. Approximately 41% of tumours possess amplification of the DNA primase gene PRIM1, which is also at chromosome 12q13 [58]. Both PRIM1 and CDK4 are involved in different aspects of the cell cycle phase transition from G1 to S, but the consequences of increased copy number of both genes are unknown. On the other hand, genomic losses of the CDKN genes, all of which encode tumour suppressor proteins that inactivate the CDK proteins, are also frequent. The genes CDKN2A/p16/INK4A, p14/ARF, and CDKN2B/p15/INK4B are located at chromosome 9p21, and CDKN2A/p16 alteration has been implicated in osteosarcoma development [59]. Chromosome 9p21 undergoes deletion in 5–21% of osteosarcomas [48, 60–63].
Deregulation of TP53 is also thought to be significant in the development of osteosarcoma and occurs due to mutations of the gene or gross changes to the gene locus at 17p13.1. Like pRB, the p53 protein is a tumour suppressor that is activated upon DNA damage recognition and can induce cellular quiescence, senescence, or apoptosis. However, p53 is by far the more commonly inactivated protein in human cancer [64]. Individuals with the Li-Fraumeni syndrome, the manifestation of germline TP53 mutations, have an increased incidence of osteosarcoma [4, 6]. LOH and deletions of the 17p13.1 locus have been detected in 29–42% of sporadic osteosarcomas [44, 62, 65, 66]. Mutations of TP53 are present in 10–39% of cases [25, 44, 47, 48, 56, 62, 67–71].
Direct inactivation of TP53 expression is only one mechanism by which the p53 pathway can be disrupted. Functional inactivation of p53 at the posttranslational level can also occur through regulation by tumourigenic proteins. The oncoprotein MDM2 is a well-described inhibitor of p53, functioning both in the promotion of p53 degradation and the downregulation of its transcription. Amplification of MDM2 (chromosome 12q15) is a relatively infrequent event in primary osteosarcoma, occurring in 3–25% of tumours [47, 48, 57, 72, 73] but appears to be considerably more frequent in metastases and recurrences [47, 73, 74]. Nearby the TP53 locus at chromosome 17p11.2-p12 is another focus of amplification that leads to increased copy number of COPS3, PMP22, and MAPK7, among other genes. Amplification of chromosome 17p11.2-p12 is more frequent than that of chromosome 12q15, at a range of 20% to 78% of tumours [20, 49, 52, 55, 65, 68, 70, 75–80]. COPS3 is strongly suspected to be the amplicon target because, like MDM2, it has an important role in promoting proteasome-mediated degradation of p53.
Instability of chromosome 8q has been described by many laboratories, with MYC (cytoband 8q24.21, also known as c-MYC) being gained at varying frequencies. An early report sets the frequency of amplification at 7%, and those events only occurred in tumours from adult patients [81]. Other groups have reported frequencies of gain and amplification of MYC at 14–67% [20, 45, 49, 55, 71, 78, 82, 83]. However, other regions of 8q, including 8q23-qter, 8q21.3-8q23, and 8q21 commonly undergo copy number increases as well [20, 49, 55, 77–79, 83, 84], suggesting that other oncogenes located within these bands could have roles in osteosarcoma pathogenesis [82].
Aberrations of the RECQL4 gene (8q24.4) are also associated with osteosarcoma development. Loss of RECQL4 function via truncating mutation in individuals with the autosomal recessive familial Rothmund-Thomson syndrome results in significantly higher risk of osteosarcoma [7], but in sporadic osteosarcoma the rate of RECQL4 mutation is less than 5% [85]. However, increased copy number and increased protein expression of RECQL4 have been reported as a frequent event in sporadic osteosarcoma [27]. Bloom syndrome and Werner syndrome are two additional autosomal recessive syndromes that predispose affected individuals to osteosarcoma [86, 87]. Both syndromes result from genomic instability caused by hereditary mutation of a RECQL family DNA helicase gene [8]: Bloom syndrome due to mutation of BLM (RECQL3) located at chromosome 15q26.1 and Werner syndrome due to mutation of WRN (RECQL2) located at chromosome 8p12. Distinct regions of chromosome arms 15q and 8p are prone to inconsistent rearrangements and copy number alterations in sporadic osteosarcoma, frequently as amplicons within 15q and loss of 8p regions [50, 75, 78, 88].
Amplifications within the short arm of chromosome 6, with a minimal common region at 6p12-p21, have been frequently observed at rates of 16–75% in conventional osteosarcoma tumour specimens [9, 20, 45, 49, 65, 77, 78, 82, 89–92], including those from biopsy, surgical resection, and metastases [89]. Data obtained using 10 osteosarcoma patient samples indicate amplification-related overexpression of genes within the 6p12-p21 region [93]. Notably, in addition to this, a conditional mouse model of osteosarcoma demonstrated overexpression of genes within mouse genomic regions homologous to human 6p12-p21 [94], consistent with observations of 6p deregulation in human osteosarcoma.
A number of genes with oncogenic potential lie within chromosome 6p12-p21 and in close proximity to this region. E2F3 (6p22.3) is gained or amplified in approximately 60% of osteosarcomas [92] and encodes the E2F3 transcription factor. An increased level of E2F3 is associated with the accumulation of DNA damage [95] and increased proliferation rate in cancer [96, 97]. PIM1 is a protooncogene located at 6p21.2 that encodes a serine/threonine-protein kinase and whose overexpression is associated with high-grade prostate cancer [98]. VEGFA (6p21.1) is amplified in 25% of a cohort of osteosarcoma specimens [99], and its protein product promotes angiogenesis and blood vessel permeability in cancer [100]. Also at cytoband 6p21.1 is the human cyclin D3 gene CCND3, which is commonly amplified in other cancers [101, 102], CDC5L (cell division cycle 5-like), and RUNX2 (runt-related transcription factor 2). CDC5L encodes a cell cycle regulator which may function in human osteosarcoma [89], and its overexpression may promote mitotic entry and shorten the G2 phase [103]. RUNX2 encodes a transcription factor important in osteogenesis [104] and has been expressed in up to 87% of tumour specimens, including biopsy samples, implying that alteration of 6p12-p21 may be an early event in the disease [45, 89]. In another report, gain-related overexpression of RUNX2 was observed in 60% of the analysed osteosarcoma tumours [9], and overexpression of RUNX2 is correlated with poor response to chemotherapy [93].
Other genomic regions frequently altered in copy number but whose potential gene targets are less well characterised in osteosarcoma have been abundantly described. Amplifications of chromosome 1q, at minimal regions including 1q10-q12 and 1q21-q31, occur in 6–59% of tumours in addition to other rearrangements of 1q [50, 55, 77, 82–84]. Portions of chromosome 17q undergo mixed duplication and deletion events in osteosarcoma [65, 75], and LOH of the tumour suppressor gene BRCA1 (17q21.31) has been detected [66].
Loss of chromosome 3q, with a minimal common region at 3q13.31, has been observed in 6–80% of tumours [44, 45, 49–55]. The presence of a novel gene, limbic system-associated membrane protein (LSAMP), at 3q13.31 is suggested to have a significant tumour suppressive role in osteosarcoma [49]. Another group has suggested the presence of an osteosarcoma tumour suppressor gene at locus 3q26.2-q26.3 based on findings of frequent LOH of this region [105].
LOH at chromosome 10q26 has been reported in 60% of a cohort, and the genes BUB3 and fibroblast growth factor receptor 2 (FGFR2) are suspected to be of importance in this region. BUB3 encodes a mitotic checkpoint protein and could have a role in maintaining genomic stability, while FGFR2 is involved in skeletal formation [106]. Deletion of WWOX (chromosome 16q23.1-q23.2) has been reported in 30% of osteosarcomas [107], but, perhaps more importantly, reduction of its expression occurs in up to 58% of specimens and is associated with elevated RUNX2 expression [108]. In a study of 91 osteosarcomas, LOH of the tumour-suppressor gene APC (chromosome 5q21) was detected in 62% of cases, while both amplification and deletion (the latter the more frequent event) of TWIST (chromosome 7p21) and MET (chromosome 7q31) were detected [109] (Table 1).
As at chromosome 3q, LOH at chromosome 18q has been frequently observed, but no studies have defined a distinct tumour suppressor gene important in osteosarcoma. Chromosome 18q is lost in 31–64% of specimens [44, 53, 110]. This portion of chromosome 18 contains a locus of susceptibility near 18q21-q23 that is linked to the Paget disease of bone (PDB) [111, 112]. PDB is a disorder of older adults which leads to osteosarcoma in about 1% of pagetic patients, particularly in the case of familial PDB [113]. A minimal common region of loss in osteosarcoma has been identified as overlapping the locus associated with PDB [110, 114]. This region excludes previously identified candidate genes including TNFRSF11A, which encodes RANK [114], and deleted in colorectal cancer (DCC), which nonetheless is frequently reduced in expression in osteosarcoma [115].
3.2. Telangiectatic Osteosarcoma
Telangiectatic osteosarcoma is a rare subtype of the disease, accounting for between 2 and 12% of cases [116]. Few cytogenetic studies of this subtype have been published, and most of the published observations have described individual cases. Tumour cells from two female patients with telangiectatic osteosarcoma were predominantly normal genomically (46xx), though one tumour possessed cells with trisomy 3 and the other possessed pseudotetraploidy and telomeric associations in a few cells [117]. One group has reported a TP53 mutation in a single case which otherwise had normal RB1 and no copy number change in MDM2 [56]. Other studies have reported a constitutional inversion at chromosome 9p11-9q12 in a patient, along with non-clonal balanced translocations in the tumour [20], and a familial occurrence of telangiectatic osteosarcoma in cousins, but without any apparent hereditary components [118]. Gains of chromosomes 6p12-p21, 8q, 12q13-q15, and 14q, along with loss of 2q24-qter, have been observed in one tumour [50]. Overall, however, reported cases of telangiectatic osteosarcoma appear to have relatively few structural and numeric chromosomal alterations in comparison to the other subtypes of the disease [50, 91].
3.3. Small Cell Osteosarcoma
Histologically, small cell osteosarcoma can be mistaken for Ewing's sarcoma, but cytogenetically they lack any consistent genetic alteration. The t(11; 22)(q24; q12) translocation, typical of Ewing's sarcoma, has been reported in one case of small cell osteosarcoma tumour [119]. These results have not been replicated in subsequent studies [120, 121], but a EWSR1-CREB3L1 fusion transcript was detected in a small cell osteosarcoma tumour [122]. Complex structural and numerical rearrangements of multiple chromosomes have been found in two cases of this subtype studied by different labs [75, 88], one of which possessed amplification of 6p12-p21. A study of MDM2 copy number and TP53 and RB1 mutations in a single small cell osteosarcoma specimen reported normal TP53, RB1, and MDM2 [56]. Another study found complex structural rearrangements of chromosomes 6, 16, and 17 and monoallelic deletion of TP53 in one tumour [123].
3.4. Periosteal Osteosarcoma
The genetic alterations observed in this subtype have been largely inconsistent. Cells in one case were found only with an additional copy of chromosome 17 [117], in another possessed only gain of 20q12-q13.2 [50], while in a third case were the only cells in a cohort of 31 osteosarcomas of various subtypes to have no DNA copy number aberrations at all [83]. Another study of three periosteal osteosarcomas reported gains of 2q, 5p, 8q, portions of 12p and 12q, and chromosomes 14 and 21, as well as losses of chromosomes 6, 8p, and 13. The same study reported focal amplifications of 8q11-q24 in one case and of 12q11-q15 in each of the other two cases, in addition to various other amplicons [75]. Complex chromosomal alterations have been reported by others [124, 125], and point mutations in TP53 have also been detected [126].
3.5. High-Grade Surface Osteosarcoma
Amplification of the sarcoma amplified sequence (SAS) gene (located at 12q14.3-15) was reported in a single case of high-grade surface osteosarcoma and six cases of low-grade surface tumours [127]. However, there are no published observations of cytogenetic alterations in prechemotherapy biopsies of high-grade surface osteosarcomas.
3.6. Low-Grade Osteosarcoma
In one CGH study of low-grade central osteosarcoma, six of seven specimens possessed a single copy number change and there were recurrent gains at 12q13-q14, 12p, and 6p21.1-p21.3 among the cases [128]. Amplification of oncogene ERBB2 (chromosome 17q12) has been detected in 26% of low-grade tumours [129]. Other researchers assayed 21 tumours of this subtype by sequencing for TP53 and the oncogene HRAS, and no specimens possessed mutations of either gene. However, amplification of MDM2 was detected in 19% of the 21 cases [130]. A separate study described amplification of chromosome 12q13-q15 in five low-grade central osteosarcomas and amplification-related overexpression of MDM2 and CDK4 which lie within the region [131]. Both the overall lack of complex chromosomal aberrations and the low frequency of TP53 mutations differentiate this subtype from conventional high-grade osteosarcoma.
Parosteal osteosarcoma is characterised by a high rate of MDM2 amplification (chromosome 12q13-q14), in up to 83% of studied tumours [72, 132]. Chromosome 12q13-q15 amplification products have also been found within supernumerary ring chromosomes in another study that detected amplification of the region in 100% of the specimens examined [133].
4. Conclusions
Osteosarcoma is characterised by extensive and heterogeneous genetic complexity, which is reflected in the similarly complex epigenetic and expression alterations in tumours [134] and is visually apparent in the results of quantitative research (Figure 2). Mechanisms of genomic instability may be facilitated by the repetitive DNA sequences ubiquitous in the human genome, particularly low copy repeats [92, 135], but this area still requires further study. Unfortunately, even though several alterations are relatively consistent across cohorts of tumours, the accumulated knowledge of genetic changes in osteosarcoma has yet to significantly impact survival rates. Clinical markers continue to be the most reliable indicators for prognostication [136]. Overall, the multitude of genetics studies of osteosarcoma serves to illustrate the extremes to which DNA alterations in cancer can reach, but it is hoped that accurate biomarkers and targeted therapies will soon be revealed for this disease.
Figure 2.
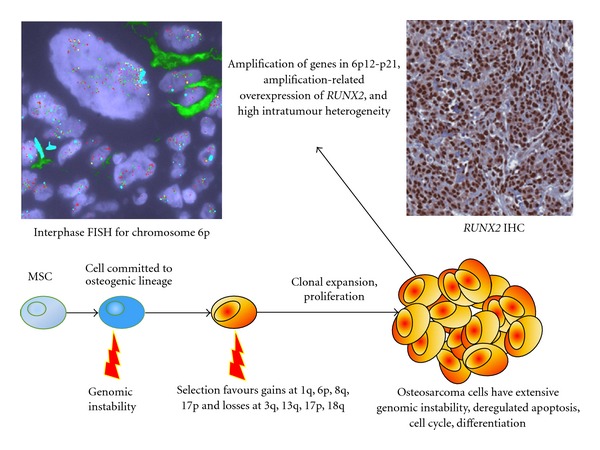
Chromosome 6p rearrangement. Chromosome 6p12-p21, which contains RUNX2, frequently undergoes complex rearrangements in osteosarcoma and is one example of a distinct genomic locus that undergoes such alterations in this cancer. In this case, there is gain and amplification of the labeled genes (with wide variation between cells) as shown in the image of interphase fluorescence in situ hybridisation (FISH) for chromosome 6p. The FISH experiment employed probes for FBXO9 (yellow), RUNX2 (orange), PIM1 (green), E2F3 (red), and the centromere of chromosome 6 (light blue). The RUNX2 immunohistochemistry (IHC) image was obtained after staining for RUNX2 protein. High levels of the protein were nearly ubiquitous in the nuclei of cells and were associated with genetic amplification of RUNX2. The FISH and IHC images were obtained via experiments performed on serial formalin-fixed paraffin-embedded sections of one osteoblastic (conventional) osteosarcoma tissue specimen. MSC, mesenchymal stem cell.
Acknowledgment
The authors are supported by the Canadian Cancer Society through Grant CCRI-020247.
References
- 1.Raymond AK, Ayala AG, Knuutila S. Conventional osteosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002. pp. 264–270. [Google Scholar]
- 2.Longhi A, Errani C, De Paolis M, Mercuri M, Bacci G. Primary bone osteosarcoma in the pediatric age: state of the art. Cancer Treatment Reviews. 2006;32(6):423–436. doi: 10.1016/j.ctrv.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:13 pages. doi: 10.1155/2011/548151. Article ID 548151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs B, Pritchard DJ. Etiology of osteosarcoma. Clinical Orthopaedics and Related Research. 2002;(397):40–52. doi: 10.1097/00003086-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Hansen MF, Koufos A, Gallie BL. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(18):6216–6220. doi: 10.1073/pnas.82.18.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 7.Wang LL, Gannavarapu A, Kozinetz CA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene Rothmund-Thomson syndrome. Journal of the National Cancer Institute. 2003;95(9):669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- 8.Mohaghegh P, Hickson ID. DNA helicase deficiencies associated with cancer predisposition and premature ageing disorders. Human Molecular Genetics. 2001;10(7):741–746. doi: 10.1093/hmg/10.7.741. [DOI] [PubMed] [Google Scholar]
- 9.Sadikovic B, Yoshimoto M, Chilton-MacNeill S, Thorner P, Squire JA, Zielenska M. Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Human Molecular Genetics. 2009;18(11):1962–1975. doi: 10.1093/hmg/ddp117. [DOI] [PubMed] [Google Scholar]
- 10.Posthumadeboer J, Witlox MA, Kaspers GJL, Van Royen BJ. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clinical and Experimental Metastasis. 2011;28(5):493–503. doi: 10.1007/s10585-011-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubista B, Klinglmueller F, Bilban M, et al. Microarray analysis identifies distinct gene expression profiles associated with histological subtype in human osteosarcoma. International Orthopaedics. 2011;35(3):401–411. doi: 10.1007/s00264-010-0996-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lulla RR, Costa FF, Bischof JM, et al. Identification of differentially expressed microRNAs in osteosarcoma. Sarcoma. 2011;2011:6 pages. doi: 10.1155/2011/732690. Article ID 732690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maire G, Martin JW, Yoshimoto M, Chilton-MacNeill S, Zielenska M, Squire JA. Analysis of miRNA-gene expression-genomic profiles reveals complex mechanisms of microRNA deregulation in osteosarcoma. Cancer Genetics. 2011;204(3):138–146. doi: 10.1016/j.cancergen.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Selvarajah S, Yoshimoto M, Maire G, et al. Identification of cryptic microaberrations in osteosarcoma by high-definition oligonucleotide array comparative genomic hybridization. Cancer Genetics and Cytogenetics. 2007;179(1):52–61. doi: 10.1016/j.cancergencyto.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Selvarajah S, Yoshimoto M, Park PC, et al. The breakage-fusion-bridge (BFB) cycle as a mechanism for generating genetic heterogeneity in osteosarcoma. Chromosoma. 2006;115(6):459–467. doi: 10.1007/s00412-006-0074-4. [DOI] [PubMed] [Google Scholar]
- 16.Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends in Genetics. 2008;24(2):64–69. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature Genetics. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 18.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 19.Bayani J, Selvarajah S, Maire G, et al. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Seminars in Cancer Biology. 2007;17(1):5–18. doi: 10.1016/j.semcancer.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Bayani J, Zielenska M, Pandita A, et al. Spectral karyotyping identifies recurrent complex rearrangements of chromosomes 8, 17, and 20 in osteosarcomas. Genes Chromosomes and Cancer. 2003;36(1):7–16. doi: 10.1002/gcc.10132. [DOI] [PubMed] [Google Scholar]
- 21.Roschke AV, Glebov OK, Lababidi S, Gehlhaus KS, Weinstein JN, Kirsch IR. Chromosomal instability is associated with higher expression of genes implicated in epithelial-mesenchymal transition, cancer invasiveness, and metastasis and with lower expression of genes involved in cell cycle checkpoints, DNA repair, and chromatin maintenance. Neoplasia. 2008;10(11):1222–1230. doi: 10.1593/neo.08682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392(6673):300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 23.Van Harn T, Foijer F, Van Vugt M, et al. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes and Development. 2010;24(13):1377–1388. doi: 10.1101/gad.580710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss MB, Vitolo MI, Mohseni M, et al. Deletion of p53 in human mammary epithelial cells causes chromosomal instability and altered therapeutic response. Oncogene. 2010;29(33):4715–4724. doi: 10.1038/onc.2010.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overholtzer M, Rao PH, Favis R, et al. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):11547–11552. doi: 10.1073/pnas.1934852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coschi CH, Martens AL, Ritchie K, et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes and Development. 2010;24(13):1351–1363. doi: 10.1101/gad.1917610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maire G, Yoshimoto M, Chilton-MacNeill S, Thorner PS, Zielenska M, Squire JA. Recurrent RECQL4 imbalance and increased gene expression levels are associated with structural chromosomal instability in sporadic osteosarcoma. Neoplasia. 2009;11(3):260–268. doi: 10.1593/neo.81384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckman RA, Loeb LA. Genetic instability in cancer: theory and experiment. Seminars in Cancer Biology. 2005;15(6):423–435. doi: 10.1016/j.semcancer.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Shay JW, Wright WE. Role of telomeres and telomerase in cancer. Seminars in Cancer Biology. 2011;21(6):349–353. doi: 10.1016/j.semcancer.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Royle NJ, Foxon J, Jeyapalan JN, et al. Telomere length maintenance—an ALTernative mechanism. Cytogenetic and Genome Research. 2009;122(3-4):281–291. doi: 10.1159/000167814. [DOI] [PubMed] [Google Scholar]
- 31.Ulaner GA, Hoffman AR, Otero J, et al. Divergent patterns of telomere maintenance mechanisms among human sarcomas: sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing’s sarcomas and osteosarcomas. Genes Chromosomes and Cancer. 2004;41(2):155–162. doi: 10.1002/gcc.20074. [DOI] [PubMed] [Google Scholar]
- 32.Ulaner GA, Huang HY, Otero J, et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Research. 2003;63(8):1759–1763. [PubMed] [Google Scholar]
- 33.Montgomery E, Argani P, Hicks JL, DeMarzo AM, Meeker AK. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. American Journal of Pathology. 2004;164(5):1523–1529. doi: 10.1016/S0002-9440(10)63710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berardinelli F, Antoccia A, Cherubini R, et al. Transient activation of the ALT pathway in human primary fibroblasts exposed to high-LET radiation. Radiation Research. 2010;174(5):539–549. doi: 10.1667/RR2127.1. [DOI] [PubMed] [Google Scholar]
- 35.Lundberg G, Sehic D, Länsberg J-K, et al. Alternative lengthening of telomeres—an enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes Chromosomes and Cancer. 2011;50(4):250–262. doi: 10.1002/gcc.20850. [DOI] [PubMed] [Google Scholar]
- 36.Scheel C, Schaefer KL, Jauch A, et al. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene. 2001;20(29):3835–3844. doi: 10.1038/sj.onc.1204493. [DOI] [PubMed] [Google Scholar]
- 37.Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clinical Cancer Research. 2005;11(1):217–225. [PubMed] [Google Scholar]
- 38.Mirabello L, Richards EG, Duong LM, et al. Telomere length and variation in telomere biology genes in individuals with osteosarcoma. International Journal of Molecular Epidemiology and Genetics. 2011;2(1):19–29. [PMC free article] [PubMed] [Google Scholar]
- 39.Sanders RP, Drissi R, Billups CA, Daw NC, Valentine MB, Dome JS. Telomerase expression predicts unfavorable outcome in osteosarcoma. Journal of Clinical Oncology. 2004;22(18):3790–3797. doi: 10.1200/JCO.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 40.Henson JD, Cao Y, Huschtscha LI, et al. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nature Biotechnology. 2009;27(12):1181–1185. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 41.Wong FL, Boice JD, Abramson DH, et al. Cancer incidence after retinoblastoma: radiation dose and sarcoma risk. Journal of the American Medical Association. 1997;278(15):1262–1267. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- 42.Manning AL, Dyson NJ. PRB, a tumor suppressor with a stabilizing presence. Trends in Cell Biology. 2011;21(8):433–441. doi: 10.1016/j.tcb.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinsohn S, Ulrike E, Udo ZS, Bielack S, Kabisch H. Determination of the prognostic value of loss of heterozygosity at the retinoblastoma gene in osteosarcoma. International Journal of Oncology. 2007;30(5):1205–1214. [PubMed] [Google Scholar]
- 44.Patiño-García A, Sotillo Piñeiro E, Zalacaín Díez M, Gárate Iturriagagoitia L, Antillón Klüssmann F, Sierrasesúmaga Ariznabarreta L. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. Journal of Pediatric Hematology/Oncology. 2003;25(5):362–367. doi: 10.1097/00043426-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Smida J, Baumhoer D, Rosemann M, et al. Genomic alterations and allelic imbalances are strong prognostic predictors in osteosarcoma. Clinical Cancer Research. 2010;16(16):4256–4267. doi: 10.1158/1078-0432.CCR-10-0284. [DOI] [PubMed] [Google Scholar]
- 46.Wadayama BI, Toguchida J, Shimizu T, et al. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Research. 1994;54(11):3042–3048. [PubMed] [Google Scholar]
- 47.Miller CW, Aslo A, Won A, Tan M, Lampkin B, Koeffler HP. Alterations of the p53, Rb and MDM2 genes in osteosarcoma. Journal of Cancer Research and Clinical Oncology. 1996;122(9):559–565. doi: 10.1007/BF01213553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.López-Guerrero JA, López-Ginés C, Pellín A, Carda C, Llombart-Bosch A. Deregulation of the G1 to S-phase cell cycle checkpoint is involved in the pathogenesis of human osteosarcoma. Diagnostic Molecular Pathology. 2004;13(2):81–91. doi: 10.1097/00019606-200406000-00004. [DOI] [PubMed] [Google Scholar]
- 49.Kresse SH, Ohnstad HO, Paulsen EB, et al. LSAMP, a novel candidate tumor suppressor gene in human osteosarcomas, identified by array comparative genomic hybridization. Genes Chromosomes and Cancer. 2009;48(8):679–693. doi: 10.1002/gcc.20675. [DOI] [PubMed] [Google Scholar]
- 50.Ozaki T, Schaefer KL, Wai D, et al. Genetic imbalances revealed by comparative genomic hybridization in osteosarcomas. International Journal of Cancer. 2002;102(4):355–365. doi: 10.1002/ijc.10709. [DOI] [PubMed] [Google Scholar]
- 51.Pasic I, Shlien A, Durbin AD, et al. Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Research. 2010;70(1):160–171. doi: 10.1158/0008-5472.CAN-09-1902. [DOI] [PubMed] [Google Scholar]
- 52.Tarkkanen M, Karhu R, Kallioniemi A, et al. Gains and losses of DNA sequences in osteosarcomas by comparative genomic hybridization. Cancer Research. 1995;55(6):1334–1338. [PubMed] [Google Scholar]
- 53.Yamaguchi T, Toguchida J, Yamamuro T, et al. Allelotype analysis in osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Research. 1992;52(9):2419–2423. [PubMed] [Google Scholar]
- 54.Yen CC, Chen WM, Chen TH, et al. Identification of chromosomal aberrations associated with disease progression and a novel 3q13.31 deletion involving LSAMP gene in osteosarcoma. International Journal of Oncology. 2009;35(4):775–788. doi: 10.3892/ijo_00000390. [DOI] [PubMed] [Google Scholar]
- 55.Zielenska M, Bayani J, Pandita A, et al. Comparative genomic hybridization analysis identifies gains of 1p35∼p36 and chromosome 19 in osteosarcoma. Cancer Genetics and Cytogenetics. 2001;130(1):14–21. doi: 10.1016/s0165-4608(01)00461-7. [DOI] [PubMed] [Google Scholar]
- 56.Pellín A, Boix-Ferrero J, Carpio D, et al. Molecular alterations of the RB1, TP53, and MDM2 genes in primary and xenografted human osteosarcomas. Diagnostic Molecular Pathology. 1997;6(6):333–341. doi: 10.1097/00019606-199712000-00005. [DOI] [PubMed] [Google Scholar]
- 57.Mejia-Guerrero S, Quejada M, Gokgoz N, et al. Characterization of the 12q15 MDM2 and 12q13-14 CDK4 amplicons and clinical correlations in osteosarcoma. Genes Chromosomes and Cancer. 2010;49(6):518–525. doi: 10.1002/gcc.20761. [DOI] [PubMed] [Google Scholar]
- 58.Yotov WV, Hamel H, Rivard GE, et al. Amplifications of DNA primase 1 (PRIM 1) in human osteosarcoma. Genes Chromosomes and Cancer. 1999;26(1):62–69. doi: 10.1002/(sici)1098-2264(199909)26:1<62::aid-gcc9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 59.Mohseny AB, Szuhai K, Romeo S, et al. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. Journal of Pathology. 2009;219(3):294–305. doi: 10.1002/path.2603. [DOI] [PubMed] [Google Scholar]
- 60.Nielsen GP, Burns KL, Rosenberg AE, Louis DN. CDKN2A gene deletions and loss of p16 expression occur in osteosarcomas that lack RB alterations. American Journal of Pathology. 1998;153(1):159–163. doi: 10.1016/S0002-9440(10)65556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohseny AB, Tieken C, van der Velden PA, et al. Small deletions but not methylation underlie CDKN2A/p16 loss of expression in conventional osteosarcoma. Genes, Chromosomes & Cancer. 2010;49(12):1095–1103. doi: 10.1002/gcc.20817. [DOI] [PubMed] [Google Scholar]
- 62.Tsuchiya T, Sekine KI, Hinohara SI, Namiki T, Nobori T, Kaneko Y. Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genetics and Cytogenetics. 2000;120(2):91–98. doi: 10.1016/s0165-4608(99)00255-1. [DOI] [PubMed] [Google Scholar]
- 63.Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP. Alterations of the p15, p16, and p18 genes in osteosarcoma. Cancer Genetics and Cytogenetics. 1996;86(2):136–142. doi: 10.1016/0165-4608(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 64.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nature Reviews Cancer. 2009;9(10):749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lau CC, Harris CP, Lu XY, et al. Frequent amplification and rearrangement of chromosomal bands 6p12-p21 and 17p11.2 in osteosarcoma. Genes Chromosomes and Cancer. 2004;39(1):11–21. doi: 10.1002/gcc.10291. [DOI] [PubMed] [Google Scholar]
- 66.Sztan M, Papai Z, Szendroi M, et al. Allelic Losses from Chromosome 17 in Human Osteosarcomas. Pathology Oncology Research. 1997;3(2):115–120. doi: 10.1007/BF02907805. [DOI] [PubMed] [Google Scholar]
- 67.Gokgoz N, Wunder JS, Mousses S, Eskandarian S, Bell RS, Andrulis IL. Comparison of p53 mutations in patients with localized osteosarcoma and metastatic osteosarcoma. Cancer. 2001;92(8):2181–2189. doi: 10.1002/1097-0142(20011015)92:8<2181::aid-cncr1561>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 68.Henriksen J, Aagesen TH, Maelandsmo GM, Lothe RA, Myklebost O, Forus A. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene. 2003;22(34):5358–5361. doi: 10.1038/sj.onc.1206671. [DOI] [PubMed] [Google Scholar]
- 69.Miller CW, Aslo A, Tsay C, et al. Frequency and structure of p53 rearrangements in human osteosarcoma. Cancer Research. 1990;50(24):7950–7954. [PubMed] [Google Scholar]
- 70.Yan T, Wunder JS, Gokgoz N, et al. COPS3 amplification and clinical outcome in osteosarcoma. Cancer. 2007;109(9):1870–1876. doi: 10.1002/cncr.22595. [DOI] [PubMed] [Google Scholar]
- 71.Pompetti F, Rizzo P, Simon RM, et al. Oncogene alterations in primary, recurrent, and metastatic human bone tumors. Journal of Cellular Biochemistry. 1996;63(1):37–50. doi: 10.1002/(SICI)1097-4644(199610)63:1%3C37::AID-JCB3%3E3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 72.Duhamel LAE, Ye H, Halai D, et al. Frequency of Mouse Double Minute 2 (MDM2) and Mouse Double Minute 4 (MDM4) amplification in parosteal and conventional osteosarcoma subtypes. Histopathology. 2012;60(2):357–359. doi: 10.1111/j.1365-2559.2011.04023.x. [DOI] [PubMed] [Google Scholar]
- 73.Lonardo F, Ueda T, Huvos AG, Healey J, Ladanyi M. p53 and MDM2 alterations in osteosarcomas: correlation with clinicopathologic features and proliferative rate. Cancer. 1997;79(8):1541–1547. [PubMed] [Google Scholar]
- 74.Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH. MDM2 gene amplification in metastatic osteosarcoma. Cancer Research. 1993;53(1):16–18. [PubMed] [Google Scholar]
- 75.Atiye J, Wolf M, Kaur S, et al. Gene amplifications in osteosarcoma-CGH microarray analysis. Genes Chromosomes and Cancer. 2005;42(2):158–163. doi: 10.1002/gcc.20120. [DOI] [PubMed] [Google Scholar]
- 76.Van Dartel M, Cornelissen PWA, Redeker S, et al. Amplification of 17p11.2∼p12, including PMP22, TOP3A, and MAPK7, in high-grade osteosarcoma. Cancer Genetics and Cytogenetics. 2002;139(2):91–96. doi: 10.1016/s0165-4608(02)00627-1. [DOI] [PubMed] [Google Scholar]
- 77.Forus A, Weghuis DO, Smeets D, Fodstad O, Myklebost O, Van Kessel AG. Comparative genomic hybridization analysis of human sarcomas: II. Identification of novel amplicons at 6p and 17p in osteosarcomas. Genes Chromosomes and Cancer. 1995;14(1):15–21. doi: 10.1002/gcc.2870140104. [DOI] [PubMed] [Google Scholar]
- 78.Squire JA, Pei J, Marrano P, et al. High-resolution mapping of amplifications and deletions in pediatric osteosarcoma by use of CGH analysis of cDNA microarrays. Genes Chromosomes and Cancer. 2003;38(3):215–225. doi: 10.1002/gcc.10273. [DOI] [PubMed] [Google Scholar]
- 79.Batanian JR, Cavalli LR, Aldosari NM, et al. Evaluation of paediatric osteosarcomas by classic cytogenetic and CGH analyses. Molecular Pathology. 2002;55(6):389–393. doi: 10.1136/mp.55.6.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hulsebos TJM, Bijleveld EH, Oskam NT, et al. Malignant astrocytoma-derived region of common amplification in chromosomal band 17p12 is frequently amplified in high-grade osteosarcomas. Genes Chromosomes and Cancer. 1997;18(4):279–285. [PubMed] [Google Scholar]
- 81.Ladanyi M, Chan Kum Park, Lewis R, Jhanwar SC, Healey JH, Huvos AG. Sporadic amplification of the MYC gene in human osteosarcomas. Diagnostic Molecular Pathology. 1993;2(3):163–167. [PubMed] [Google Scholar]
- 82.Stock C, Kager L, Fink FM, Gadner H, Ambros PF. Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes and Cancer. 2000;28(3):329–336. doi: 10.1002/1098-2264(200007)28:3<329::aid-gcc11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 83.Tarkkanen M, Elomaa I, Blomqvist C, et al. DNA sequence copy number increase at 8q: a potential new prognostic marker in high-grade osteosarcoma. International Journal of Cancer. 1999;84(2):114–121. doi: 10.1002/(sici)1097-0215(19990420)84:2<114::aid-ijc4>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 84.dos Santos Aguiar S, de Jesus Girotto Zambaldi L, dos Santos AM, Pinto W, Brandalise SR. Comparative genomic hybridization analysis of abnormalities in chromosome 21 in childhood osteosarcoma. Cancer Genetics and Cytogenetics. 2007;175(1):35–40. doi: 10.1016/j.cancergencyto.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 85.Nishijo K, Nakayama T, Aoyama T, et al. Mutation analysis of the RECQL4 gene in sporadic osteosarcomas. International Journal of Cancer. 2004;111(3):367–372. doi: 10.1002/ijc.20269. [DOI] [PubMed] [Google Scholar]
- 86.Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiology, Biomarkers and Prevention. 1996;5(4):239–246. [PubMed] [Google Scholar]
- 87.German J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genetics and Cytogenetics. 1997;93(1):100–106. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- 88.Boehm AK, Neff JR, Squire JA, Bayani J, Nelson M, Bridge JA. Cytogenetic findings in 36 osteosarcoma specimens and a review of the literature. Pediatric Pathology and Molecular Medicine. 2000;19(5):359–376. [Google Scholar]
- 89.Lu XY, Lu Y, Zhao YJ, et al. Cell cycle regulator gene CDC5L, a potential target for 6p12-p21 amplicon in osteosarcoma. Molecular Cancer Research. 2008;6(6):937–946. doi: 10.1158/1541-7786.MCR-07-2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Man TK, Lu XY, Jaeweon K, et al. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer. 2004;4, article no. 45 doi: 10.1186/1471-2407-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zielenska M, Marrano P, Thorner P, et al. High-resolution cDNA microarray CGH mapping of genomic imbalances in osteosarcoma using formalin-fixed paraffin-embedded tissue. Cytogenetic and Genome Research. 2004;107(1-2):77–82. doi: 10.1159/000079574. [DOI] [PubMed] [Google Scholar]
- 92.Martin JW, Yoshimoto M, Ludkovski O, et al. Analysis of segmental duplications, mouse genome synteny and recurrent cancer-associated amplicons in human chromosome 6p21-p12. Cytogenetic and Genome Research. 2010;128(4):199–213. doi: 10.1159/000308353. [DOI] [PubMed] [Google Scholar]
- 93.Sadikovic B, Thorner P, Chilton-MacNeill S, et al. Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer. 2010;10, article no. 202 doi: 10.1186/1471-2407-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walkley CR, Qudsi R, Sankaran VG, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes and Development. 2008;22(12):1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paulson QX, Pusapati RV, Hong S, Weaks RL, Conti CJ, Johnson DG. Transgenic expression of E2F3a causes DNA damage leading to ATM-dependent apoptosis. Oncogene. 2008;27(36):4954–4961. doi: 10.1038/onc.2008.138. [DOI] [PubMed] [Google Scholar]
- 96.Hurst CD, Tomlinson DC, Williams SV, Platt FM, Knowles MA. Inactivation of the Rb pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. Oncogene. 2008;27(19):2716–2727. doi: 10.1038/sj.onc.1210934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Olsson AY, Feber A, Edwards S, et al. Role of E2F3 expression in modulating cellular proliferation rate in human bladder and prostate cancer cells. Oncogene. 2007;26(7):1028–1037. doi: 10.1038/sj.onc.1209854. [DOI] [PubMed] [Google Scholar]
- 98.Van Der Poel HG, Zevenhoven J, Bergman AM. Pim1 regulates androgen-dependent survival signaling in prostate cancer cells. Urologia Internationalis. 2010;84(2):212–220. doi: 10.1159/000277601. [DOI] [PubMed] [Google Scholar]
- 99.Yang J, Yang D, Sun Y, et al. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer. 2011;117(21):4925–4938. doi: 10.1002/cncr.26116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nature Reviews Cancer. 2008;8(11):880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kasugai Y, Tagawa H, Kameoka Y, Morishima Y, Nakamura S, Seto M. Identification of CCND3 and BYSL as candidate targets for the 6p21 amplification in diffuse large B-cell lymphoma. Clinical Cancer Research. 2005;11(23):8265–8272. doi: 10.1158/1078-0432.CCR-05-1028. [DOI] [PubMed] [Google Scholar]
- 102.Büschiges R, Weber RG, Actor B, Lichter P, Collins VP, Reifenberger G. Amplification and expression of cyclin D genes (CCND 1, CCND2 and CCND3) in human malignant gliomas. Brain Pathology. 1999;9(3):435–443. doi: 10.1111/j.1750-3639.1999.tb00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bernstein HS, Coughlin SR. A mammalian homolog of fission yeast Cdc5 regulates G2 progression and mitotic entry. Journal of Biological Chemistry. 1998;273(8):4666–4671. doi: 10.1074/jbc.273.8.4666. [DOI] [PubMed] [Google Scholar]
- 104.Lian JB, Javed A, Zaidi SK, et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Critical Reviews in Eukaryotic Gene Expression. 2004;14(1-2):1–41. [PubMed] [Google Scholar]
- 105.Kruzelock RP, Murphy EC, Strong LC, Naylor SL, Hansen MF. Localization of a novel tumor suppressor locus on human chromosome 3q important in osteosarcoma tumorigenesis. Cancer Research. 1997;57(1):106–109. [PubMed] [Google Scholar]
- 106.Mendoza S, David H, Gaylord GM, Miller CW. Allelic loss at 10q26 in osteosarcoma in the region of the BUB3 and FGFR2 genes. Cancer Genetics and Cytogenetics. 2005;158(2):142–147. doi: 10.1016/j.cancergencyto.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 107.Yang J, Cogdell D, Yang D, et al. Deletion of the WWOX gene and frequent loss of its protein expression in human osteosarcoma. Cancer Letters. 2010;291(1):31–38. doi: 10.1016/j.canlet.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 108.Kurek KC, Del Mare S, Salah Z, et al. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Research. 2010;70(13):5577–5586. doi: 10.1158/0008-5472.CAN-09-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Entz-Werle N, Lavaux T, Metzger N, et al. Involvement of MET/TWIST/APC combination or the potential role of ossification factors in pediatric high-grade osteosarcoma oncogenesis. Neoplasia. 2007;9(8):678–688. doi: 10.1593/neo.07367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nellissery MJ, Padalecki SS, Brkanac Z, et al. Evidence for a novel osteosarcoma tumor-suppressor gene in the chromosome 18 region genetically linked with Paget disease of bone. American Journal of Human Genetics. 1998;63(3):817–824. doi: 10.1086/302019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Haslam SI, Van Hul W, Morales-Piga A, et al. Paget’s disease of bone: evidence for a susceptibility locus on chromosome 18q and for genetic heterogeneity. Journal of Bone and Mineral Research. 1998;13(6):911–917. doi: 10.1359/jbmr.1998.13.6.911. [DOI] [PubMed] [Google Scholar]
- 112.Good DA, Busfield F, Fletcher BH, et al. Linkage of Paget disease of bone to a novel region on human chromosome 18q23. American Journal of Human Genetics. 2002;70(2):517–525. doi: 10.1086/338658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. Journal of Bone and Mineral Research. 2006;21:P58–P63. doi: 10.1359/jbmr.06s211. [DOI] [PubMed] [Google Scholar]
- 114.Johnson-Pais TL, Nellissery MJ, Ammerman DG, et al. Determination of a minimal region of loss of heterozygosity on chromosome 18Q21.33 in osteosarcoma. International Journal of Cancer. 2003;105(2):285–288. doi: 10.1002/ijc.11070. [DOI] [PubMed] [Google Scholar]
- 115.Horstmann MA, Pösl M, Scholz RB, et al. Frequent reduction or loss of DCC gene expression in human osteosarcoma. British Journal of Cancer. 1997;75(9):1309–1317. doi: 10.1038/bjc.1997.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weiss A, Khoury JD, Hoffer FA, et al. Telangiectatic osteosarcoma: the St. Jude Children’s Research Hospital’s experience. Cancer. 2007;109(8):1627–1637. doi: 10.1002/cncr.22574. [DOI] [PubMed] [Google Scholar]
- 117.Bridge JA, Nelson M, McComb E, et al. Cytogenetic findings in 73 osteosarcoma specimens and a review of the literature. Cancer Genetics and Cytogenetics. 1997;95(1):74–87. doi: 10.1016/s0165-4608(96)00306-8. [DOI] [PubMed] [Google Scholar]
- 118.Nishida J, Abe M, Shiraishi H, et al. Familial occurrence of telangiectatic osteosarcoma: cousin cases. Journal of Pediatric Orthopaedics. 1994;14(1):119–122. doi: 10.1097/01241398-199401000-00023. [DOI] [PubMed] [Google Scholar]
- 119.Noguera R, Navarro S, Triche TJ. Translocation (11;22) in small cell osteosarcoma. Cancer Genetics and Cytogenetics. 1990;45(1):121–124. doi: 10.1016/0165-4608(90)90074-k. [DOI] [PubMed] [Google Scholar]
- 120.Giovannini M, Selleri L, Biegel JA, Scotlandi K, Emanuel BS, Evans GA. Interphase cytogenetics for the detection of the t(11;22)(q24;q12) in small round cell tumors. Journal of Clinical Investigation. 1992;90(5):1911–1918. doi: 10.1172/JCI116068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.MacHado I, Alberghini M, Giner F, et al. Histopathological characterization of small cell osteosarcoma with immunohistochemistry and molecular genetic support. A study of 10 cases. Histopathology. 2010;57(1):162–167. doi: 10.1111/j.1365-2559.2010.03589.x. [DOI] [PubMed] [Google Scholar]
- 122.Debelenko LV, McGregor LM, Shivakumar BR, Dorfman HD, Raimondi SC. A novel EWSR1-CREB3L1 fusion transcript in a case of small cell osteosarcoma. Genes Chromosomes and Cancer. 2011;50(12):1054–1062. doi: 10.1002/gcc.20923. [DOI] [PubMed] [Google Scholar]
- 123.Nishio J, Gentry JD, Neff JR, et al. Monoallelic deletion of the p53 gene through chromosomal translocation in a small cell osteosarcoma. Virchows Archiv. 2006;448(6):852–856. doi: 10.1007/s00428-006-0181-x. [DOI] [PubMed] [Google Scholar]
- 124.Gisselsson D, Höglund M, Mertens F, Mitelman F, Mandahl N. Chromosomal organization of amplified chromosome 12 sequences in mesenchymal tumors detected by fluorescence in situ hybridization. Genes Chromosomes and Cancer. 1998;23(3):203–212. doi: 10.1002/(sici)1098-2264(199811)23:3<203::aid-gcc1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 125.Hoogerwerf WA, Hawkins AL, Perlman EJ, Griffin CA. Chromosome analysis of nine osteosarcomas. Genes Chromosomes and Cancer. 1994;9(2):88–92. doi: 10.1002/gcc.2870090203. [DOI] [PubMed] [Google Scholar]
- 126.Radig K, Schneider-Stock R, Haeckel C, Neumann W, Roessner A. p53 gene mutations in osteosarcomas of low-grade malignancy. Human Pathology. 1998;29(11):1310–1316. doi: 10.1016/s0046-8177(98)90263-5. [DOI] [PubMed] [Google Scholar]
- 127.Noble-Topham SE, Burrow SR, Eppert K, et al. SAS is amplified predominantly in surface osteosarcoma. Journal of Orthopaedic Research. 1996;14(5):700–705. doi: 10.1002/jor.1100140504. [DOI] [PubMed] [Google Scholar]
- 128.Tarkkanen M, Böhling T, Gamberi G, et al. Comparative genomic hybridization of low-grade central osteosarcoma. Modern Pathology. 1998;11(5):421–426. [PubMed] [Google Scholar]
- 129.Lee WI, Bacchni P, Bertoni F, Maeng YH, Park YK. Quantitative assessment of HER2/neu expression by real-time PCR and fluorescent in situ hybridization analysis in low-grade osteosarcoma. Oncology Reports. 2004;12(1):125–128. [PubMed] [Google Scholar]
- 130.Park HR, Won Jung W, Bertoni F, et al. Molecular analysis of p53, MDM2 and H-ras genes in low-grade central osteosarcoma. Pathology Research and Practice. 2004;200(6):439–445. doi: 10.1016/j.prp.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 131.Dujardin F, Binh MBN, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Modern Pathology. 2011;24(5):624–637. doi: 10.1038/modpathol.2010.229. [DOI] [PubMed] [Google Scholar]
- 132.Gamberi G, Ragazzini P, Benassi MS, et al. Analysis of 12q13-15 genes in parosteal osteosarcoma. Clinical Orthopaedics and Related Research. 2000;(377):195–204. doi: 10.1097/00003086-200008000-00026. [DOI] [PubMed] [Google Scholar]
- 133.Szymanska J, Mandahl N, Mertens F, Tarkkanen M, Karaharju E, Knuutila S. Ring chromosomes in parosteal osteosarcoma contain sequences from 12q13-15: a combined cytogenetic and comparative genomic hybridization study. Genes Chromosomes and Cancer. 1996;16(1):31–34. doi: 10.1002/(SICI)1098-2264(199605)16:1<31::AID-GCC4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 134.Sadikovic B, Yoshimoto M, Al-Romaih K, Maire G, Zielenska M, Squire JA. In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma. PLoS One. 2008;3(7) doi: 10.1371/journal.pone.0002834. Article ID e2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Van Dartel M, Hulsebos TJM. Amplification and overexpression of genes in 17p11.2∼p12 in osteosarcoma. Cancer Genetics and Cytogenetics. 2004;153(1):77–80. doi: 10.1016/j.cancergencyto.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 136.Bramer JAM, van Linge JH, Grimer RJ, Scholten RJPM. Prognostic factors in localized extremity osteosarcoma: a systematic review. European Journal of Surgical Oncology. 2009;35(10):1030–1036. doi: 10.1016/j.ejso.2009.01.011. [DOI] [PubMed] [Google Scholar]