

Abstract
The vitamin D receptor (VDR) is a nuclear hormone receptor that regulates cell proliferation, cell differentiation, and calcium homeostasis. The receptor is activated by vitamin D analogs that induce the disruption of VDR-corepressor binding and promote VDR-coactivator interactions. The interactions between VDR and coregulators are essential for VDR-mediated transcription. Small molecule inhibition of VDR–coregulator binding represents an alternative method to the traditional ligand-based approach in order to modulate the expression of VDR target genes. A high throughput fluorescence polarization screen that quantifies the inhibition of binding between VDR and a fluorescently labeled steroid receptor coactivator 2 peptide was applied to discover the new small molecule VDR–coactivator inhibitors, 3-indolyl-methanamines. Structure-activity relationship studies with 3-indolyl-methanamine analogs were used to determine their mode of VDR-binding and to produce the first VDR-selective and irreversible VDR–coactivator inhibitors with the ability to regulate the transcription of the human VDR target gene, TRPV6.
Keywords: Vitamin D receptor, steroid receptor coactivator, fluorescence polarization, high throughput screening, 3-indolyl-methanamines, TRPV6
Introduction
The vitamin D receptor (VDR) is a ligand-activated transcription factor that belongs to the nuclear receptor (NR) superfamily. VDR binds to its endogenous ligand, 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3), with high affinity,1 mediating the modulation of genes responsible for cell differentiation, proliferation, and calcium homeostasis.2 Based on its biological function, the VDR has been identified as an important pharmaceutical target for the treatment of metabolic disorders, skin diseases, cancer, autoimmune diseases, and cardiovascular diseases.3 The receptor contains several functional domains, including a DNA binding domain (VDR-DBD) and a ligand-binding domain (VDR-LBD), which mediates ligand-dependent gene regulation.4 VDR binds DNA as a heterodimer with the retinoid X receptor (RXR).5 In the unliganded state, VDR is associated with corepressor proteins, which repress transcription of VDR target genes.6 In the presence of 1,25-(OH)2D3, the VDR-LBD undergoes a conformational change. This conformational change prevents corepressor binding and permits interactions with coactivator proteins, resulting in the formation of a multi-protein complex that activates VDR-mediated transcription.7
VDR ligand agonists have been developed to treat metabolic bone diseases and proliferative skin disorders.3 In contrast to the large number of reported VDR agonist, only a limited number of VDR ligand antagonists have been described with the ability to allosterically inhibit the interactions between VDR and its coactivators.8 All of these antagonists are based on the secosteroid scaffold of 1,25-(OH)2D3. A different approach to modulate gene regulation that is mediated by VDR represents the disruption of VDR–coregulator binding in the presence of 1,25-(OH)2D3. Small molecule NR–coactivator inhibitors have been discovered for the estrogen receptor, thyroid receptor, androgen receptor, and pregnane X receptor using rational design and high throughput screening (HTS).9 Mita et al. introduced the first reversible small molecule VDR–coactivator inhibitors.10 These compounds inhibited both VDR-mediated and estrogen receptor β-mediated transcription. Thus, highly potent and selective VDR–coactivator inhibitors are still missing. It is expected that these compounds can be applied as molecular probes to identify the biological functions of VDR–coactivator interactions. Herein, we describe the first HTS campaign that identified small molecule VDR–coactivator inhibitors and their evaluation using carefully selected secondary assays. One class of inhibitors were investigated in regard to their mode of VDR binding, selectivity, and SARs that resulted in the identification of the first irreversible and highly selective VDR–coactivator inhibitor with the ability to modulate VDRmediated transcription.
Results
The HTS was carried out using 384 well black polystyrene plates with 20 μl of assay reagent per well. This assay reagent included VDR-LBD (1 μM), LG190178 (5 μM), Alexa Fluor 647 labeled SRC2-3 (7.5 nM), and a selected small molecule from the screening library (30 μM). FP was measured after 3 hours of incubation. Recently, we quantified the binding affinities between VDR-LBD and different coregulator peptides using the same technique, and discovered that the third nuclear interaction domain (NID) of steroid receptor coactivator 2 (SRC2), called SRC2-3, has the strongest interaction with VDR among other coregulator peptides tested.11 The study also showed that the binding affinities of theses peptides were similar in the presence of either 1,25-(OH)2D3 or synthetic agonist LG190178.12 Three hundred different compounds were investigated per plate, which contained also the negative control, dimethyl sulfoxide (DMSO), and the positive control compound, 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one, which has been reported to inhibit the interaction between coactivator SRC2 and the thyroid receptor β13 and which inhibit the interaction between VDR and SRC2-3 (see supplement data). A far-red fluorescent label (Alexa Fluor 647, 630/685nm) was used for SRC2-3 to minimize fluorescent interference by fluorescence quenching and aggregation of screening compounds. Two hundred and seventy-five thousand compounds were tested in the primary screening campaign at a single dosage of 30 μM. Based on the FP signal, 589 compounds exhibited more than 40 percent inhibition of the VDR–SRC2-3 interaction at that concentration and were less likely to bear structure elements of promiscuous aggregating molecules14 or electrophilic compounds.15 HTS evaluation of the frozen stock solutions of these compounds confirmed that 140 compounds exhibited a dose-dependent response with IC50 values of less than 40 μM. Further analyses of these stock solutions were performed in HEK293 cells, which express a fusion protein of VDR-LBD and GAL4-DBD. It was determined that these cells induced the transcription of a β-lactamase reporter gene in the presence of 1,25-(OH)2D3, (GeneBLAzer, Invitrogen). Quantification of β-lactamase was accomplished by detecting the decrease of fluorescence resonance energy transfer (FRET) caused by the enzymatic cleavage of the β-lactam containing substrate, which was added after an incubation time of 24 hours. The quantity of uncleaved substrate, which was determined by measuring the fluorescence emission at 447 nm, revealed that 48 of the 140 active compounds were able to regulate the VDR-mediated transcription of β-lactamase. Additionally, the abilities of these compounds to inhibit the interaction between VDR and 1,25-(OH)2D3 were determined to exclude allosteric VDR–coactivator binding disruption through VDR ligand antagonism. The application of a VDR PolarScreen (Invitrogen) confirmed that none of the active compounds was able to replace labeled 1,25-(OH)2D3.
The 48 compounds were then purchased as solids, dissolved in DMSO as a 10 mM solution, and analyzed by liquid chromatography-mass spectroscopy (LC-MS) to determine purity and identity (Table 1). A subsequent dose-response analysis using the described FP assay determined that 29 of the 48 purchased compounds exhibited IC50 values of less than 40 μM. These compounds were divided into 6 groups, based on their scaffold similarity, and depicted in Figure 1 with their determined FP IC50 values.
Table 1.
Summary of biophysical and biochemical properties of validated VDR–SRC2-3 inhibitors.
| Compound | % Puritya | Solubility (μM)b | Permeabilit yc Log(Pe) (cm/s) | VDR–SRC2-3 Inhibition IC50 (μM)d | Inhibition of Transcription (%)e | Toxicity (%)g | ||
|---|---|---|---|---|---|---|---|---|
| Conc. 62.5 μM | Conc. 20.8 μM | Conc. 62.5 μM | Conc. 20.8 μM | |||||
| 1 | 93.0 | 0.1 | −7.86 | 3.3 | 6.0f | 3.1f | ||
| 2 | 89.8 | 273.3 | −6.55 | 4.0 | 5.5f | 4.7f | ||
| 3 | 89.2 | 0.7 | −8.22 | 5.9 | 5.7f | 3.6f | ||
| 4 | 94.7 | 0.2 | −7.93 | 8.6 | 11.5f | 7.6f | ||
| 5 | 88.6 | 218.7 | −7.70 | 13.8 | 6.8f | 5.5f | ||
| 7 | 95.2 | 21.9 | −6.60 | 7.1 | 1.6f | 2.4f | ||
| 8 | 96.1 | 49.9 | −6.67 | 8.1 | 5.3f | 12.5f | ||
| 9 | 94.5 | 17.6 | −6.78 | 6.9 | 6.2f | 16.7f | ||
| 10 | 99.6 | 34.2 | −6.36 | 22.3 | 2.6f | 7.7f | ||
| 11 | 42.8 | 342.0 | −6.25 | 9.6 | 100 | 90 | 100 | 30 |
| 12 | 58.0 | 422.6 | −6.10 | 27.7 | 100 | 10 | 90 | 0 |
| 13 | 65.6 | 403.3 | −6.12 | 30.3 | 100 | 0 | 60 | 0 |
| 14 | 64.9 | 132.5 | −6.11 | 14.1 | 100 | 10 | 100 | 10 |
| 15 | 84.1 | 84.6 | −6.01 | 17.3 | 100 | 90 | 100 | 40 |
| 16 | 78.5 | 468.7 | −6.05 | 28.9 | 100 | 10 | 100 | 0 |
| 17 | 42.4 | 201.4 | −6.12 | 15.4 | 9.1f | 18.0f | ||
| 18 | 81.6 | 277.7 | −6.01 | 25.1 | 100 | 0 | 100 | 0 |
| 19 | 79.1 | 427.8 | −5.99 | 22.2 | 80 | 0 | 60 | 0 |
| 20 | 58.1 | 466.8 | −8.71 | 21.6 | 100 | 60 | 50 | 25 |
| 21 | 41.6 | 453.9 | −7.84 | 37.7 | 100 | 30 | 50 | 25 |
| 22 | 57.6 | 462.0 | −7.83 | 10.5 | 10.6f | 13.5f (partial 50%) | ||
| 23 | 99.2 | 576.0 | −8.28 | 20.5 | 50 | 15 | 20 | 0 |
| 24 | 84.1 | 42.1 | −6.29 | 28.0 | 80 | 20 | 20 | 0 |
| 25 | 87.1 | 15.3 | −6.52 | 14.3 | 100 | 60 | 60 | 10 |
| 26 | 67.6 | 493.0 | −7.48 | 13.7 | 0 | 0 | 0 | 0 |
| 27 | 93.2 | 338.8 | −7.28 | 2.0 | 30 | 0 | 30 | 0 |
| 28 | 98.9 | 487.0 | −6.78 | 10.5 | 100 | 10 | 100 | 10 |
| 29 | 34.4 | 377.4 | −5.95 | 25.1 | 65 | 0 | 50 | 0 |
Purities were determined by high pressure liquid chromatography using a photodiode array and identity was confirmed by mass spectrometry;
Solubilities were determined in phosphate-buffered saline at pH 7.4;
Permeabilities were measured using the parallel artificial membrane permeation assay (PAMPA) at neutral pH (pH = 7.4). The following permeability standards (logPe) were used: Ranitidine (−8.02 ± 0.074 cm/s) low permeability, carbamazepine (−6.81 ± 0.0011 cm/s) medium permeability, and verapamil (−5.93 ± 0.015 cm/s) high permeability. The solubility and permeability assay conditions reflect conditions required for activity in cell-based assays;
A fluorescence polarization competition assay was carried out using VDR-LBD (1μM), Alexa Fluor-labeled peptide SRC2-3 (7 nM), VDR-agonist LG190178 (5 μM), and serially diluted small molecules. IC50 values were obtained by fitting data to the following equation: Y=Bottom + (Top- Bottom)/(1+10^((LogIC50-X)*HillSlope)) using three independent experiments in quadruplet;
VDR-mediated inhibition of transcription was carried out using a commercially available GeneBLAzer (Invitrogen) assay. Data was normalized by signals observed for inactive and activated VDR (± 1,25(OH)2D3);
IC50/LD50 values (μM) are given instead of percentages for highly active compounds using the following non-linear regression equation: Y=Bottom + (Top- Bottom)/(1+10^((LogIC50-X)*HillSlope)) using three independent experiments in quadruplet;
Toxicity was determined using CellTiter-Glo (Promega) and data was normalized by signal observed for living and dead cells (± 100 μM 3- dibutylamino-1-(4-hexyl-phenyl)-propan-1-one).
Figure 1.

Hit structures from HTS for inhibitors of the interaction between VDR-LBD and fluorescently labeled peptide SRC2-3. Structures of validated hits are shown, grouped by chemotype, and annotated with IC50 values that were determined using a fluorescence polarization assay that employed VDR-LBD and fluorescently labeled peptide SRC2-3.
Further characterizations were carried out, which include the determination of small molecule aqueous solubility, permeability using PAMPA (Parallel Artificial Membrane Permeability), toxicity in HEK293T cells, and their ability to inhibit VDR-mediated transcription using the described GeneBLAzer assay. The results are summarized in Table 1.
The 3-indolyl-methanamines grouped in C showed the most promising characteristics, which included good to excellent solubility, high permeability, inhibition of VDR-SRC2-3 interaction at low micromolar concentrations, and the ability to inhibit VDR-mediated transcription in the range of 62.5–20.8 μM.
Selected analogs of 3-indolyl-methanamines were regenerated in our laboratory in regard of the low purities and limited commercial availability of these compounds. In order to analyze the electronic effects of both aromatic substituents, we substituted the pyridine ring of the compounds depicted in Figure 1 with substituted phenyl groups as shown in Figure 2. For synthesis details, see supplemental data. Three series of 3-indolyl-methanamines were generated, including those bearing electron-donating and accepting aromatic substituents (Figure 2).
Figure 2.

Structures of synthesized 3-indolyl-methanamines.
The compounds were characterized using carefully selected biophysical and biochemical assays that are summarized in Table 2. The 2-chloro phenyl substitutent was chosen for series 31, because 31a exhibit a higher activity (IC50) and higher rate constant (k) in comparison with others members of group 30.
Table 2.
Summary of biophysical and biochemical properties of 3-indolyl-methanamines.
| Compound | Solubilit y (μM)a | Permeabilityb Log(Pe) (cm/s) | VDR–SRC2-3 Inhibition IC50 (μM)c | Rate constant k for the dissociation of SRC2-3 from VDR (10−5)d | Transcription Inhibition IC50 (μM)e | Toxicity LC50 (μM)f |
|---|---|---|---|---|---|---|
| 30a | 252.9 | −6.00 | 30.2±4.8 | 45.1 ± 1.6 | 10.9±2.8 | 15.3±2.9 |
| 30b | 93.9 | −6.03 | 31.7±4.3 | 34.2 ± 1.0 | 8.1±1.7 | 14.6±3.4 |
| 30c | 237.5 | −6.10 | 31.2±3.2 | 88.0 ± 4.7 | 12.1±1.8 | 17.2±2.5 |
| 30d | 175.1 | −6.34 | 43.6±7.8 | 35.7 ± 1.4 | 14.6±2.5 | 21.7±4.2 |
| 30e | 157.7 | −6.88 | n.s. | 2.0 ± 0.16 | 13.5±1.3 | 25.8±6.3 |
| 30f | 117.6 | −6.08 | 28.5±4.5 | 6.7 ± 0.12 | 12.2±2.4 | 16.2±2.5 |
| 30g | 189.0 | −6.40 | 104.8±15.2 | 1.8 ± 0.15 | 20.1±5.2 | 37.4±7.7 |
| 30h | 67.3 | −6.30 | 29.6±3.1 | 14.2 ± 0.28 | 15.0±2.4 | 20.8±3.5 |
| 30i | 503.4 | −6.83 | 58.6±8.1 | 2.3 ± 0.21 | 13.1±2.5 | 31.4±8.2 |
| 31a | 31.6 | −6.41 | 29.8±4.5 | 38.6 ± 0.89 | 8.5±1.8 | 12.6±2.2 |
| 31b | 68.0 | −6.24 | 36.7±5.1 | 4.5 ± 0.59 | 4.2±1.9 | 11.6±1.7 |
| 31c | 84.2 | −6.30 | 26.5±3.4 | 39.1 ± 1.3 | 5.8±2.1 | 12.1±2.2 |
| 31d | 35.9 | −6.45 | 17.7±3.2 | 23.7 ± 0.35 | 8.2±1.5 | 12.7±3.0 |
| 31e | 21.0 | −7.28 | n.s. | 0.87 ± 0.13 | 8.7±1.5 | 19.4±3.8 |
| 31f | 4.9 | −7.60 | n.s. | 1.2 ± 0.16 | 4.4±1.1 | 11.0±2.1 |
| 31g | n.d. | n.d. | n.s. | 2.5 ± 0.07 | 24.0±5.7 | 43.1±9.1 |
| 31 h | 59.7 | −6.22 | 24.3±4.4 | 25.2 ± 0.36 | 8.6±1.1 | 12.1±2.7 |
| 32a | 15.3 | −6.58 | n.o. | n.o. | >70 | >70 |
| 32b | 52.3 | −6.51 | 20.3±4.5 (partial) | 1.4 ± 0.12 | 21.6±3.3 | >70 |
| 32c | 11.8 | −6.89 | n.o. | n.o. | >70 | >70 |
Solubilities were determined in phosphate-buffered saline at pH 7.4;
Permeabilities were measured using the parallel artificial membrane permeation assay (PAMPA) at neutral pH (pH = 7.4). The following permeability standards were used (logPe): Ranitidine (−8.02 ± 0.074 cm/s) low permeability, Carbamazepine (−6.81 ± 0.0011 cm/s) medium permeability, and Verapamil (−5.93 ± 0.015 cm/s) high permeability. The solubility and permeability assay conditions reflect conditions required for activity in cell-based assays;
A fluorescence polarization competition assay was carried out using VDR-LBD (1μM) , Alexa Fluor-labeled peptide SRC2-3 (7 nM), VDR-agonist LG190178 (5 μM), and serial diluted small molecules. IC50 values were obtained by fitting data obtained after 3 hours to the following equation: Y=Bottom + (Top- Bottom)/(1+10^((LogIC50-X)*HillSlope)) using three independent experiments in quadruplet;
A fluorescence polarization assay described under c was monitored over time. Dissociation rate constants were obtained by linear fitting of ln(mP) (fluorescence polarization) against time (first order kinetics);
HEK293T cells were transfected with CMV-VDR, a CYP24A1 promoter driven luciferase expression vector, and a Renilla luciferase control vector. The data was normalized to Renilla luciferase activity and by signals observed for inactive and activated VDR (±1,25(OH)2D3);
Toxicity was determined based on signal observed for Renilla luciferase activity and normalized to signal observed for living and dead cells (± 100 μM of 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one, CBT358); n.d. = not determined; n.s. = no saturation of signal at higher small molecule concentration (no reliable non-linear fitting possible); n.o. = not observed.
These assays included a VDR transcription assay that employed luminescence instead of FRET. The assay is based on the VDR-mediated transcription of a luciferase gene under control of the CYP24A1 gene promoter 16. The gene product of CYP24A1 is 24(R)-hydroxylase, which is responsible for the catabolism of vitamin D analogs into their 24-hydroxylated species 17. CYP24A1 is highly and directly regulated by VDR.
The solubility of compounds from series 30 is excellent, except those bearing a 2-chloro-aryl or 2-naphthalenyl substituent (Table 2, compounds 31a and 30h). Molecules from series 31 and 32, which bear a 2-chloro-aryl substituent, also have lower solubility, ranging from 4.9–85 μM. The permeability of all series 30–32 range from medium to high in comparison with drug standards, carbamazepine (logPe = −6.81 cm/s, medium) and verapamil (logPe = −5.93 cm/s, high). Determination of the ability of 3-indolyl-methanamines to inhibit the interaction between VDR and SRC2-3 resulted in similar IC50 values, ranging from 27–44 μM, for the majority of compounds after 3 hours. Significantly higher IC50 values were observed for compound 30g, bearing a methyl group (IC50 = 104 μM) instead of an aryl group, and compounds 30e, 31e, 31f, and 31g, bearing electron-withdrawing aromatic substitutents. For the last 4 compounds, non-linear fitting resulted in IC50 values with high standard deviation caused by lack of saturation at higher compound concentrations (Table 2). Additionally, we observed loss of activity for 3-indolyl-methanamines with the alkylation of the indole nitrogen (32a) or nitrogen-sulfur substitution (32c). Compound 32b, missing the 2-methyl indole substituents, inhibited only 50% of the interaction between VDR and SRC2-3. The FP analysis of the VDR–coactivator inhibition reaction at different time points identified significant changes of inhibition in time. This prompted us to determine each compound’s rate constant by fitting the data to first order dissociation kinetics (see supplemental data). Small standard deviations support the application of this model and enabled us to identify large reactivity differences between the 3-indolyl-methanamines tested. As expected, we observed smaller rate constants for 3-indolyl-methanamines with higher IC50 values of more than 44 μM (30e, 30g, 30i, 31e, 31f, and 31g). Four 3-indolyl-methanamines (30f, 30h, 31b, and 32b) exhibited IC50 values in the range of 27–44 μM, but showed relative small reaction rates. Interestingly, compound 31d, which has the lowest IC50 values, does not have the highest reaction rate. The ability of 3-indolyl-methanamines to displace 1,25-(OH)2D3 from VDR was determined by a commercially available FP assay (Polarscreen, Invitrogen) and excludes VDR ligand displacement by 3-indolyl-methanamines, which can cause allosteric disruption of the VDR–coactivator interaction. None of the synthesized 3-indolyl-methanamines were able inhibit the interaction between labeled 1,25-(OH)2D3 and VDR, except compounds 30e and 30f, which exhibited weak inhibition at higher concentrations (see supplemental data). Almost all 3-indolyl-methanamines were able to inhibit the VDR-mediated transcription at lower micromolar concentrations, except those that were not able to inhibit the interaction between VDR and SRC2-3 (Table 2, compounds 32a and 32c). We also observed significant cell toxicity caused by 3-indolyl-methanamines at higher concentrations. The small difference between transcriptional inhibition and toxicity prompted us to use rt-PCR to determine the modulation of gene regulation in the presence of 3-indolyl-methanamines.
The different reaction rates of the 3-indolyl-methanamines and similar IC50 values indicate that these compounds are likely to react with VDR or SRC2-3 in an irreversible fashion. Indeed, it was reported that, especially under acidic conditions or elevated temperature, 3-indolyl-methanamines underwent elimination reactions by breaking the carbon-nitrogen bonds and forming the corresponding azafulvenium salts (Scheme 1).18
Scheme 1.

Proposed mechanism of action for 3-indolyl-methanamines.
To discriminate which of the binding partners (VDR or SRC2-3) is alkylated by 3-indolyl-methanamines, we incubated 31b for 3 hours with either VDR or SRC2-3 followed by the addition of the other interaction partner that was SRC2-3 or VDR, respectively (Figure 3). The binding isotherm of each condition was different. Pre-incubation of 31b with SRC2-3 followed by the addition of VDR did not result in an alkylation reaction because the FP signal did not change with higher concentration of 31b. In contrast, pre-incubation of VDR with different 31b concentrations followed by the addition of SRC2-3 did result in a change of FP signal. The corresponding isotherm was similar to the inhibition observed for combining all reagents at the same time and measuring FP after 3 hours.
Figure 3.
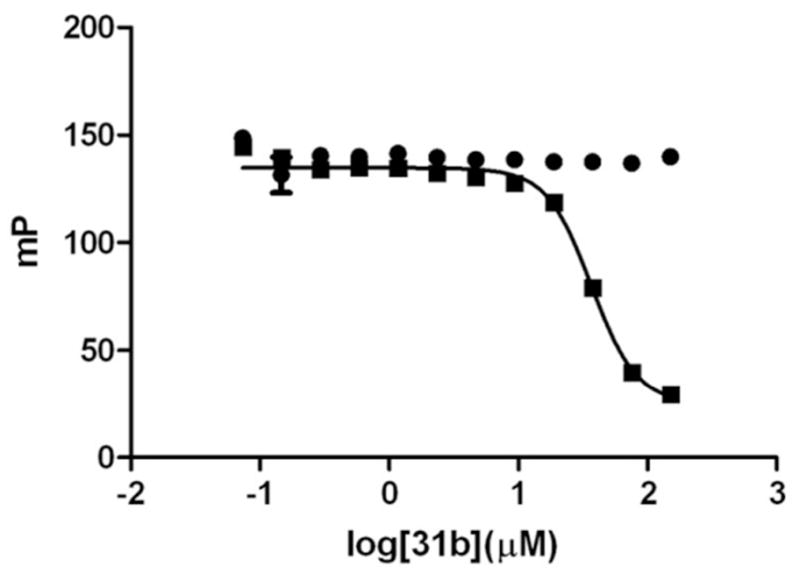
Identification of 31b’s reaction partner. ● Alexa Fluor-labeled SRC2-3 peptide (7 nM) was incubated for 3 hours with different concentration of 31b followed by the addition of VDR-LBD (1 μM) and LG190178 (5 μM). Fluorescence polarization was detected after 5 minutes. ■ VDR-LBD (1 μM) and LG190178 (5 μM) was incubated for 3 hours with different concentration of 31b followed by the addition of Alexa Fluor-labeled SRC2-3 peptide (7 nM). Fluorescence polarization was detected after 5 minutes.
The azafulvenium salts are reactive electrophilies that can undergo reactions with natural occurring nucleophilies such as cysteine residues of proteins. The wide range of reaction rates of the differently substituted 3-indolyl-methanamines and the fact that these compounds are likely to react with VDR irreversibly, prompted us to investigate the possibility of a linear free energy relationship between the alkylation of VDR measured by disruption of VDR-SRC2-3 binding and the electronic nature of different aromatic 3-indolyl-methanamine substituents; therefore, log(kx/k0) was plotted against Hammett σ-values for the compounds of series 30 and 31 (Figure 4).19
Figure 4.

Linear free energy relationship between VDR-SRC2-3 inhibition and the electronic nature of 3-indolyl-methanamines substitutents. A Series 30; B Series 31; log(kx/k0) values were calculated using rate constants given in Table 2. K0 being the non-substituted compounds 30a and 31a. σ-values were obtained from Ritchie et al. ρ-values represent the slopes of the linear regressions with the corresponding r2 values.
A strong correlation was found for both series (r2 values are 0.93) with significant negative ρ-values, supporting the proposed mechanism that during the rate determining step, a positive charge is building up. Additionally, we observed that this reaction is less sensitive to substituents of compound series 30 (ρ = −1.1), than to substituents of compound series 31 (ρ = −1.5). This supports the fact that substituents of series 30 have a majorly inductive stabilizing effect, resulting in a smaller absolute ρ-value than substituents of compound series 31, which can stabilize the positive charge via resonance.
In water at a neutral pH, 3-indolyl-methanamines were stable for 24 hours, but reaction occurred when high concentrations of 2-mercaptoethanol (5 mM) were added. To determine the selectivity of 3-indolyl-methanamine (31b) towards different nucleophiles, an FP assay of different concentrations 2-mercaptoethanol was carried out (Figure 5).
Figure 5.

Influence of 2-mercaptoethanol for the inhibition of the VDR-SRC2-3 binding in the presence of 3-indolyl-methanamine, 31b. VDR-LBD (1 μM), LG190178 (5 μM), and Alexa Fluorlabeled SRC2-3 peptide (7 nM) were incubated in the presence of different concentrations of 31b and in the absence and presence of different concentrations of 2-mercaptoethanol. Interactions between VDR and SRC2-3 were determined by fluorescence polarization. ○ DMSO (negative control), □ 3- dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (positive control), 2-mercaptoethanol concentrations (31b IC50 values): ● 100 mM (82.2 ± 7.3 μM), ■ 10 mM (54.5 ± 3.1 μM), ▲ 1 mM (45.4 ± 2.0 μM), ▼ 0.1 mM (37.5 ± 1.1 μM), ◆ 0.01 mM (36.8 ± 0.5 μM).
Two effects were observed. First, the IC50 values for 31b under identical conditions increased with the amount of 2-mercaptoethanol from 36.8 μM (0.01 mM 2-mercaptoethanol) to 82.2 μM (100 mM 2-mercaptoethanol). Secondly, the efficacy of each isotherm decreased with increasing amount of 2-mercaptoethanol. These results show that VDR–SRC2-3 binding inhibition by 31b was only changed in the presence of more than a 1000-fold excess of an alternative nucleophile, such as 2-mercaptoethanol.
Small molecule target selectivity is very important, especially towards different NR–coactivator interactions. To determine the NR-selectivity of 3-indolyl-methanamines, five additional (NR)–coactivator interactions were investigated: androgen receptor (AR)–SRC2-3, thyroid receptor (TRα)–SRC2-2 and (TRβ)–SRC2-2, estrogen receptor β (ERβ)–SRC2-2, and peroxisome proliferator-activated receptor γ (PPARγ)–DRIP2 (VDR-interacting protein 205). The quantification of these NR–coactivator interactions has been reported previously.20 All 3-indolyl-methanamines exclusively disrupted the VDR–SRC2-3 interaction, as depicted for compound 31b in Figure 6. The binding isotherms for all 3-indolyl-methanamines are presented in the supplemental material.
Figure 6.

Nuclear receptor–coactivator binding studies in the presence of 3-indolyl-methanamine 31b using fluorescence polarization. FP was detected at an excitation/emission wavelength of 595/615 nm. The conditions for different NRs are as follows: AR: androgen receptor LBD (5 μM), Texas Redlabeled SRC2-3 (7 nM), and dihydrotestosterone (5 μM) were incubated with small molecule for 3h; TRα: thyroid receptor α LBD (2 μM), Texas Red-labeled SRC2-2 (7 nM), and triiodothyronine (1 μM) were incubated with small molecule for 3h; TRβ: thyroid receptor β LBD (0.8 μM), Texas Red-labeled SRC2-2 (7 nM), and triiodothyronine (1 μM) were incubated with small molecule for 3h; estrogen receptor β (3 μM), Texas Red-labeled SRC2-2 (5 nM), and estradiol (0.1 μM) were incubated with small molecule for 3h; peroxisome proliferator-activated receptor γ (5 μM), Texas Red-labeled DRIP2 (7 nM), and rosiglitazone (5 μM) were incubated with small molecule for 3h; VDR: vitamin D receptor LBD (1 μM), Texas Red-labeled SRC2-3 (7 nM), and LG190178 (5 μM) were incubated with small molecule for 3h.
We also determined the abilities of 3-indolyl-methanamines to inhibit different VDR–coregulator interactions. The quantification of interactions between VDR and coregulator peptides was reported recently.11 Herein, we focused on three different coregulators, which include SRC221, DRIP20522, and Hairless [Hr]23. The results employing compound 31b are summarized in Figure 7.
Figure 7.

VDR-coactivator interactions in the presence of 31b using FP. A VDR-LBD (1 μM), LG190178 (5 μM), and different Texas Red-labeled coregulator peptides (7 nM) were incubated for 3h in the presence of different concentrations of compound 31b.
Inhibition of binding was observed in the presence of 31b for VDR-SRC2-2, VDR-SRC2-3, and VDR-DRIP2 (Figure 7). The efficacy of these isotherms is dissimilar because of the different binding constants between VDR and SRC2-2 (Kd = 1.7±0.2 μM), VDR and SRC2-3 (Kd = 0.93±0.17 μM), and VDR and DRIP-2 (Kd = 1.6±0.2 μM). In comparison, the binding constant between VDR and SRC2-1 and VDR and Hr1 is greater than 5 μM and no inhibition by compound 31b was observed under these conditions. Interestingly, the IC50-values of 31b and for the VDR-SRC2-2, VDR-SRC2-3, and VDRDRIP-2 interactions is very similar (IC50 = 25.4±6.5 μM, IC50 = 28.5±6.9 μM, and IC50 = 33.1±3.4 μM, respectively).
The ability of 31b to partially inhibit the interaction between VDR and SRC2, containing all three SRC2 NIDs was tested using a pull down assay (Figure 8).
Figure 8.

Western Blot of in vitro binding reactions between SRC2 bearing all three NIDs and VDRLBD in the presence of 31b. Lane 1-3 different concentrations of 31b in the presence of VDR, SRC2 and 1,25(OH)2D3, lane 4 VDR, SRC2 and 1,25(OH)2D3, lane 5 no ligand (1,25(OH)2D3), no coregulator (SRC2).
Control experiments indicate that SRC2 bound to VDR in the presence of VDR ligand 1,25(OH)2D3 (Figure 8, lane 4) but not in the absence of 1,25(OH)2D3 (Figure 8, lane 5). The VDR–SRC2 interaction was blocked in a dose dependent manner by 31b (Figure 8, lane 1–3). Although significant inhibition of the VDR–SRC2 interaction was observed at a concentration of 50 μM and 100 μM of 31b, a residual interaction between VDR and SRC2 could still be detected. Thus, the inhibition of the interaction between VDR and full length SRC2 by 31b exhibit dose response dependence, similar to the SRC2-2 peptide binding study described above.
Additionally, we investigated the interaction between VDR and SRC2 in the presence of 3-indolylmethanamine or vehicle prior to the pull-down assay, to discriminate between 31b–VDR and 31b–SRC2 binding, respectively. VDR–SRC2 interactions could be verified for all reaction conditions, although 31b, in contrast to 32a, was able to inhibit the interaction between VDR and SRC2 (Table 2 and Figure 8). Thus, pre-incubation of 31b with SRC2, in contrast to pre-incubation of 31b with VDR (Figure 8), did not result in an alkylation reaction and therefore VDR–SRC2 binding was observed.
To examine the influence of VDR–coactivator inhibition by 31b in respect to VDR-mediated transcription, we investigated the expression levels of the transient receptor potential vanilloid type 6 gene (TRPV6). The gene product of TRPV6 (ECaC2 or CaT1) is a membrane Ca2+ ion channel, which is highly expressed in advanced prostate cancer24 and was reported to be directly regulated by VDR in the presence of 1,25-(OH)2D325. The expression levels of TRPV6 in the prostate cancer cell line DU145 in the presence and absence of 1,25-(OH)2D3 and compound 31b are depicted in Figure 10.
Figure 10.

Modulation of expression of TRPV6 in the presence and absence of 1,25-(OH)2D3 and increasing concentrations of small molecule 31b. DU145 cells were cultured in six-well plates and treated with 1,25-(OH)2D3 (20 nM) and/or small molecule 31b. TRPV6 expression levels were determined by semi-quantitative RT-PCR and normalized to GAPDH transcript level and to DMSO control condition. The ΔΔCt method was used to measure the fold change in expression of genes. Standard deviations were calculated from three biological independent experiments performed in triplicates.
In the presence of 1,25-(OH)2D3, TRPV6 was up-regulated in DU145 cells. The single treatment of cells with 3-indolyl-methanamine 31b at 20 μM showed no regulation of TRPV6. For 31b concentrations higher than 20 μM an increased cytotoxicity was observed (see supplemental material). In the contrary, in the presence of 20 nM 1,25-(OH)2D3 and different concentrations of 31b, TRPV6 transcription was reduced in a dose dependent manner confirming that 31b is modulating TRPV6 expression by interacting with VDR.
Discussion
It was demonstrated that the application of HTS was successful in identifying the first irreversible inhibitors of the VDR–coactivator interactions. The screening campaign utilized a FP-based primary assay to quantify small molecule inhibition of the VDR–coactivator peptide binding. The original hit rate of 1.3 percent was reduced to 0.32 percent through the application of different scaffold filters to eliminate highly reactive molecules from the hit compound selection. These reactive molecules included enones, thiols, imines, and thioisocyanates. Rescreening of the remaining 579 compounds resulted in 140 confirmed hit compounds. Further evaluation of these molecules using secondary cell-based assays (transcription and toxicity assays) confirmed 48 compounds that exhibit biochemical and cellular activity. The purchase of these compounds as solids and screening the freshly dissolved quality-controlled compounds using the HTS assay confirmed only 29 compounds of the initial 48 compounds. We hypothesize that the low hit reproducibility is based on at least two reasons. Firstly, the deviation of compound activities for single point HTS assays, and secondly, the limited stability of some screening compounds in DMSO.
From the selection of validated hit compounds, further characterization assays of 3-indolyl-methanamines were chosen based on their promising characteristics, which included good to excellent solubility, high permeability, inhibition of VDR–SRC2-3 interaction at low micromolar concentrations, and the ability to inhibit VDR-mediated transcription in cells. In order to identify SARs for 3-indolyl-methanamines, we synthesized structural analogs with diverse substituents at different positions of the scaffold. These compounds were evaluated with assays mentioned above for the initial HTS hit compounds. In general, 3-indolyl-methanamines with electron donating aryl substituents inhibited the interaction between VDR and SRC2-3 coactivator peptide faster than 3-indolyl-methanamines with electron withdrawing ones. The significance of this correlation was demonstrated with the application of a linear free energy relationship (LFER) equation, which indicated a strong and different relationship for series 30 and 31. The negative rho values support the hypothesized mode of binding of 3-indolyl-methanamines, which includes the formation of an azafulvenium salt. This salt is then expected to react with solvent-exposed nucleophilic residue of VDR inhibiting the interaction with SRC2. The reactivity of 3-indolyl-methanamines towards thiols was demonstrated by conducting FP assays in the presence of 2-mercaptoethanol, which diminished binding between VDR and compound 31b at higher concentrations.
Many irreversible antagonists are among FDA-approved drugs, such as dibenzyline, which is an α-adrenoceptor blocker used for hypertension. The majority of these drugs are reactive towards biological nucleophilies, but they usually exhibit high selectivities among them. An exclusive selectivity could be demonstrated for 3-indolyl-methanamines in regard to their ability to disrupt the VDR–coactivator interaction in comparison with five other NR–coactivator interactions. This is remarkable because different electrophilic inhibitors (β-aminoketones and methylsulfonylnitrobenzoates) have been developed for three of these interactions (TRα–SRC2, TRβ–SRC2, and PPARγ–DRIP2).26 The micromolar activity of 3-indolylmethanamine is comparable to the reported activity of direct irreversible and reversible inhibitors of other NR-coactivator interactions9, 26 as well as to the binding affinities reported for native coactivator peptides.11 Additionally, a selectivity of 3-indolyl-methanamines towards the interactions between VDR–SRC2-3, VDR-SRC2-2, and VDR-DIP2 was observed among other NR–coregulator interactions tested. This is consistent with the fact that coactivator SRCs and DRIP are binding VDR at the same interaction site.27
Problematic is the quantification of transcriptional inhibition for 3-indolylmethanamines because of their ability to induce cell death at a similar concentration. This resulting narrow therapeutic window is of concern for the development of non-toxic VDR-coactivator inhibitor. We hypothesize that the inhibition of transcription by itself has the potential to induce cell death including apoptosis, which has been demonstrated for TRPV6 expression by using siRNA-TRPV6.28 Similar, 3-indolylmenthanamine 31b, reduced the expression of TRPV6 in a dose-depended manner as determined by rt-PCR and induced cell death at similar concentrations. Unfortunately, this anti-proliferative behavior of 3-indolylmenthanamines complicates the identification of transcriptional inhibition using the described transcriptions assays due to their sensitivity towards cell toxicity.
The regulation of VDR target genes by small molecules, which includes the inhibition of VDR–coactivator interactions, represents a new strategy to develop new drug candidates for diseases related to 1,25-(OH)2D3 and VDR. For prostate cancer cells, for example LNCaP cells, it was reported that 1,25-(OH)2D3 can induce proliferation or antiproliferation depending on the amount of supplemental serum used.28 Additionally, it has been shown that the expression of TRPV6, induced by 1,25-(OH)2D3, is responsible for cell proliferation and apoptosis resistance.29 We hypothesize that especially for prostate cancer with 1,25-(OH)2D3-independent proliferation (DU145), VDR antagonists such as 3-indolyl-methanamines represents a new anti cancer approach because of their ability to downregulate the transcription of TRPV6 and to induce cell death.
Conclusion
This study reports a successful HTS strategy to identify small molecule inhibitors of VDR–coregulator interactions. To date, these are the first irreversible inhibitors of VDR–coactivator interactions that have been reported. This new class of VDR antagonists exhibits excellent selectivity among different NR–coregulator interactions and inhibit the interaction between VDR and different coactivators. This inhibition regulated the expression of VDR target genes such as TRPV6, which has been shown to be up-regulated in last stage prostate cancers. The application of 3-indolyl-methanamines such as 31b is expected to be part of new therapy to reduce prostate cancer cell growth and enables investigations towards the function of VDR–coactivator interactions during gene regulation. 3-indolyl-methanamines have also a great potential as novel drug candidates for human diseases associated with the overproduction of 1,25-(OH)2D3, such as sarcoidosis 30 or Crohn’s disease 31. We are currently developing more selective and more potent 3-indolyl-methanamines in order to study their modulation of VDR-mediated transcription and their biological activity in vivo. These future studies will include prostate cancer xenographs that enable the determination of efficacy of 3-indolylmethanamines by monitoring the expression of VDR target genes in tumors as well as the expected tumor regression. Simultaneously, any signs of acute and chronic toxicity will be determined by observation and necropsy.
Experimental Section
Chemistry
All materials were obtained from commercial suppliers and used without further purification. All solvents used were dried using an aluminum oxide column. Thin-layer chromatography was performed on precoated silica gel 60 F254 plates. Purification of compounds was carried out by normal phase column chromatography (SP1 [Biotage], Silica gel 230–400 mesh) followed by evaporation. Purity determination were performed by element analysis (EA1110, CarloErba) or using a LC-MS (Surveyor&MSQ) with a C18 column. The total flow rate was 1.0 mL/min and gradient program started at 90% A (0.1% formic acid in H2O), changed to 95% B (0.1% formic acid in methanol), and then to 90% A. The mass spectrometer was operated in positive-ion mode with electrospray ionization. All compounds presented were confirmed at 95% purity or better using either method. NMR spectra are recorded on a Bruker 400 MHz and referenced internally to the residual resonance in CDCl3 (δ) 7.26 ppm) for hydrogen and (δ) 77 ppm) for carbon atoms.
General procedure for the aza-Friedel-Crafts reaction
In a dry flask, aniline (2 mmol) and aldehyde (2 mmol) were dissolved in toluene (2 ml) and stirred for 1h. Then indole (2 mmol) and decanoic acid (0.2 mmol, 10 mol%)32 were added slowly as a solution in toluene (2 ml). The reaction mixture was stirred at room temperature and monitored by TLC. After the reaction was completed, saturated aqu. NaHCO3 (6 ml) was added. The mixture was extracted with dichloromethane (3 × 10 ml). The organic layer was combined, washed with brine (10 mL), and dried over anhydrous Na2SO4. The solvents were removed under reduced pressure and the residue was purified by recrystallization or chromatography through Biotage SP1 flash system.
Example: N-((2-methyl-1H-indol-3-yl)(phenyl)methyl)aniline (30a)
Rf = 0.3 (EtOAc/hexanes = ¼). 230 mg white solid, 37% yield. 1H NMR (300 MHz, CDCl3, TMS): δ 2.34 (s, 3H), 4.34 (s, 1H), 5.76 (s, 1H), 6.59 (dd, J = 1.8, 4.8 Hz, 2H), 6.69–6.73 (m, 1H), 6.69–7.05 (m, 1H), 7.08–7.18 (m, 4H), 7.26–7.29 (m, 3H), 7.38 (d, J = 7.5 Hz, 2H), 7.49 (d, J = 7.8 Hz, 1H), 7.82 (s, 1H). 13C NMR (300 MHz, CDCl3, TMS): 12.35, 54.40, 110.8, 112.45, 113.28, 116.12, 118.76, 119.10, 120.43, 126.79, 128.58, 143.9, 144.1, 148.88, 148.96; Anal. Calcd. for C22H20N2: C 84.58, H 6.45, N 8.97 found C 84.2027, H 6.72, N 8.66.
Reagents
1,25-(OH)2D3 (calcitriol) was purchased from Endotherm, Germany; LG190178 was synthesized using a published procedure 12.
Labeled Coregulator Peptides
Peptides, such as SRC2-3 (CLQEKHRILHKLLQNGNSPA), 11 were purchased and labeled with cysteine-reactive fluorophores, such as Texas-Red maleimides and Alexa Fluor 647 maleimides, in DMF/PBS 50:50. After purification by HPLC, the corresponding labeled peptides were dissolved in DMSO and stored at −20°C.
Protein Expression and Purification
The VDR-LBDmt DNA was kindly provided by D. Moras 33 and cloned into pMAL-c2X vector (New England Biolabs). For a detailed expression and purification protocol, see 11. For detailed expression and purification of protocols of PPARγ-LBD, TRα-LBD, TRβ-LBD, and AR-LBD, see.26b
High Throughput FP Assay
The HTS was carried out at St. Jude Children’s Research Hospital. The small molecule collection consisted of 275,000 unique compounds from commercial sources (ChemDiv, ChemBridge, and Life Chemicals). The FP assay was conducted in 384 well black polystyrene microplates (Corning, #3573). The assay solution contained buffer (25 mM PIPES (pH 6.75) 50 mM NaCl, 0.01% NP-40, 2% DMSO, VDR-LBD protein (1 μM), LG190178 (5 μM), and Alexa Fluor 647-labeled SRC2-3 (7.5 nM). Small molecule transfer into 20 μl assay solution was accomplished using a 50H hydrophobic coated pin tool (V&P Scientific), delivering 60 nl of a 10 mM compound solution, which resulted in a final concentration of 30 μM. Inhibition of binding was detected using FP performed on an EnVision multi-label plate reader (GE) with a 620 nm excitation filter, a 688 nm S polarized emission filter, a 688 nm R polarized emission filter, and a Cy5 FP dichroic mirror. Automation was realized using a system developed by high resolution engineering, which uses a Staubli T60 robot arm to transfer plates from instrument to instrument. The assay solution was dispensed in bulk into empty plates using Matrix Wellmates (Matrix Technologies), followed by compound addition, centrifugation using a Vspin plate centrifuge (Velocity11), and incubation for 3 hours at room temperature. The positive control (3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one 26a) and negative control (DMSO) were measured within each plate to determine the assay plate quality and to enable data normalization.
Fluorescence Resonance Energy Transfer (FRET) Transcription Assay
A GeneBLAzer (Invitrogen) assay was used to identify VDR–coactivator inhibitors that regulate VDR-mediated transcription. The provided HEK293 cells of this assay expressed a fusion protein of VDR-LBD and the GAL4-DBD, which was activated by 1,25-(OH)2D3, and induced transcription of a β-lactamase reporter gene. Quantification of β-lactamase was accomplished by detecting the decrease in FRET caused by the enzymatic cleavage of the β-lactam-containing substrate, which was added after an incubation time of 24 hours. The cleaved substrate concentration was quantified by measuring the fluorescence emission at 447 nm. Controls for this assay were 1,25-(OH)2D3 and LG190178 (positives) and DMSO (negative). Toxicity was determined by luminescence using Cell-Titer Glo (Promega), which was added to the plates after recording the FRET signal. Controls for cell viability were 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (100 μM in DMSO) (positive) and DMSO (negative). Two independent experiments were conducted in triplet.
CYP24A1 Promoter Transcription Assay
This assay was used to determine the regulation of transcription of the VDR-target gene, CYP24A1, in the presence of small molecules. Briefly, HEK 293T cells (ATTC) were cultured in 75 cm2 flasks using MEM/EBSS (Hyclone) with L-glutamine (2 mM), glucose (1 mM), non-essential amino acids, sodium pyruvate (1 mM), penicillin and streptomycin, and 10 percent heat inactivated FBS (Hyclone). At 50–70 percent confluency, cell media was changed to phenol red free MEM/EBSS with L-glutamine (2 mM), glucose (1 mM), non-essential amino acids, sodium pyruvate (1 mM), penicillin and streptomycin, and 10 percent dialyzed and heat inactivated FBS (Invitrogen), followed by the addition of 2 ml of untreated MEM/EBSS containing 1.56 μg of a VDRpRc/ CMV plasmid, 16 μg of a luciferase reporter gene plasmid containing a rat 24-hydroxylase gene promoter (−1399 to +76), 17.4 μg of a Renilla luciferase control vector (Promega), Lipofectamine™ LTX (75 μl), and PLUS™ reagent (25 μl). After 16 hours, the cells were harvested and plated in sterile cell culture treated black 384 well plates with optical bottom (Nunc 142761) at a concentration of 15,000 cells per well. After 2 hours, plated cells were treated with small molecules in vehicle DMSO, followed by a 16 hours incubation time. Transcription was determined using a Dual-LuciferaseR Reporter Assay (Promega). Cell viability was determined using the Renilla luciferase signal. IC50 values and standard errors were calculated based on two independent experiments performed in quadruplet. Controls for this assay were 1,25-(OH)2D3 and LG190178 (positives) and DMSO (negative). Controls for cell viability were 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (100 μM in DMSO) (positive) and DMSO (negative). Two independent experiments were conducted in quadruplets.
VDR Ligand Competition Assay
Ligand antagonism was determined by using a FP assay (PolarScreen, Invitrogen), which employs a fluorescently labeled 1,25-(OH)2D3 analog. Two independent experiments were conducted in quadruplet and data was analyzed using nonlinear regression with variable slope (GraphPrism).
Solubility Assay
In a 384 UV plate (Corning #3675), 16 compounds were serial diluted in quadruplet starting from a 10 mM compound stock solution in DMSO. Therefore, buffer (90 mM ethanolamine, 90 mM KH2PO4, 90 mM potassium acetate, and 30 mM KCl (pH 7.4)) containing 20 percent acetonitrile was used. The plate was sealed (Corning #6570), sonicated for 1 minute, and agitated for an additional 5 minutes before scanning from 230–800 nm at 5 minutes increments. A calibration plot was prepared for each compound for the maximal absorbance using background-subtracted values. A 384 well filter plate (Pall # 5037) was pre-wetted with 20 percent acetonitrile/buffer, and filled with buffer (47.5 μl) and 10 mM of compound in DMSO (2.5 μl). The final DMSO concentration was 5 percent. After sonication (1 minute) and agitation (12 hours), the mixtures were filtered and 30 μl of each well was transferred into a 384 well UV plate, together with the addition of 20 μl of acetonitrile. The plate was agitated for 5 minutes and scanned from 230–800 nm at 5 minutes increments. The solubility was determined using background-subtracted values and the following equation: sol=absorbance at λmax/slope*(5/3). Each plate had the following solubility standards: 4,5-diphenylimidazole (67.3 ± 3.7 μM), β-estradiol (43.0 ± 2.3 μM), diethylstilbestrol (108.3 ± 5.4 μM), ketoconazole (134.5 ± 2.4 μM), and 3-phenylazo-2,6-diaminopyridine (357.7 ± 7.0 μM). All experiments were conducted in quadruplet.
Permeability Assay
This assay was carried out using Millipore’s Multiscreen™ protocol, AN1725EN00. Each plate had the following standards with the following permeability values (logPe): Ranitidine (−8.02 ± 0.074 cm/s) represents low permeability, carbamazepine (−6.81 ± 0.0011 cm/s) represents medium permeability, and verapamil (−5.93 ± 0.015 cm/s) represents high permeability. All experiments were conducted in triplet.
NR–Coactivator Binding Studies in the Presence of 3-Indolyl-methamines
These assays were conducted in 384 well black polystyrene microplates (Corning) using a buffer (20 mM TRIS (pH 7.50), 100 mM NaCl, 0.01% NP-40, 2% DMSO) and analyzed with a M1000 reader (Tecan) to detect FP at an excitation/emission wavelength of 595/615 nm. For the androgen receptor, AR-LBD (5 μM): Texas Red-labeled SRC2-3 (7 nM) and dihydrotestosterone (5 μM) were incubated in buffer with small molecule for 3 hours; for the thyroid receptor α, TRα-LBD (2 μM): Texas Red-labeled SRC2-2 (7 nM) and triiodothyronine (1 μM) were incubated with small molecule for 3 hours; for the thyroid receptor β, TRβ-LBD (0.8 μM): Texas Red-labeled SRC2-2 (7 nM) and triiodothyronine (1 μM) were incubated with small molecule for 3 hours; for the peroxisome proliferator-activated receptor γ, PPARγ-LBD (5 μM): Texas Red-labeled DRIP2 (7 nM) and rosiglitazone (5 μM) were incubated with small molecule for 3 hours; for the VDR, VDR-LBD (1 μM): Texas Red-labeled SRC2-3 (7 nM) and LG190178 (5 μM) were incubated with small molecule for 3 hours. Two independent experiments were carried out in quadruplet and data was analyzed using nonlinear regression with variable slope (GraphPrism).
VDR–Coactivator Binding Studies in the presence of 3-Indolyl-methamine 31b
These assays were conducted in 384 well black polystyrene microplates (Corning) using a buffer (25 mM PIPES (pH 6.75) 50 mM NaCl, 0.01% NP-40, 2% DMSO) and analyzed with a M1000 reader (Tecan) to detect FP at an excitation/emission wavelength of 595/615 nm. VDR-LBD protein (1 μM), LG190178 (5 μM), and 7.5 nM of Texas-Red labeled SRC1-3 [CESKDHQLLRYLLDKDEKDL], Texas-Red labeled SRC2-3 [CLQEKHRILHKLLQNGNSPA], Texas-Red labeled SRC3-3 [CKKENNALLRYLLDRDDPSD], or Texas-Red labeled DRIP2 [CNTKNHPMLMNLLKDNPAQD] were incubated with different concentration of 31b. Two independent experiments were carried out in quadruplet and data was analyzed using nonlinear regression with variable slope (GraphPrism).
Western Blot of in vitro binding reactions between SRC2 bearing all three NIDs and VDR-LBD in the presence of 31b
GST fusion to the SRC2 bearing all three NIDs were expressed in Escherichia coli BL21. Cultures were grown to OD600 = 0.5–0.6 at 22 °C and induced with 0.5 mM isopropyl- Dthiogalactoside for 12 h. The cultures were centrifuged (1000 × g), and bacterial pellets were resuspended in 20 mM TRIS, pH 7.4, 200 mM NaCl, 1 mM NaN3, 0.5M EDTA, 1 mM DTT, protein inhibitors cocktail (Roche) and sonicated. Debris was pelleted by centrifugation (100,000 × g). The supernatant was incubated with glutathione-Sepharose 4B beads (Amersham Biosciences) and washed. Protein on bead was stored with 10% glycerol at −20°C. Each pull-down reaction was carried out in 100 μl buffer (25 mM PIPES (pH 6.75) 50 mM NaCl, 0.01% NP-40, 2% DMSO) using 100 nM calcitriol, 10 μM VDR-LBD-MPB, and 31b. After 2 hours at rt, 15 μl of SRC2-beads was added to each reaction followed by 30 minutes incubation. The reaction was filtered, washed with buffer (100 μl) and eluted from the bead using a buffer and 10 mM reduced glutathione. Separation was carried out using SDSPAGE followed western blotting using standard procedures with anti-MBP (E8032S, New England BioLabs) and anti-mouse IgG-Tr (sc-2781, Santa Cruz).
Semi-quantitative real time PCR
DU145 cells were incubated at 37 °C with 31b (20 μM) in the presence or absence of 20 nM calcitriol for 18 h. Total RNA was isolated from cells using an RNAeasy kit (Qiagen). Genomic DNA was removed and cDNA was generated using equal amounts of RNA (QuantiTect Reverse Transcription Kit, Qiagen). The cDNA reaction was then diluted 5-fold, and the QuantiFast SYBR Green PCR Kit (Qiagen) was used for the real time PCR following manufacturer’s recommendations. Primers used in these studies are as follows: GAPDH Forward Primer 5=-accacagtccatgccatcac-3=, Reverse Primer 5=-tccaccaccctgttgctgta-3=; TRPV6 FP 5=-ACTGTCATTGGGGCTATCATC-3=, RP 5=-CAGCAGAATCGCATCAGGTC-3=; Real-time rt-PCR was carried out on a Mastercycler (Eppendorf). We used the ΔΔCt method to measure the fold change in gene expression of target genes. Standard deviations were calculated from 3 biological independent experiments performed in triplicates.
Supplementary Material
Figure 9.

Western Blot of in vitro binding reactions between SRC2 bearing all three NIDs and 3- indolylmethanamines. Lane 1 pre-incubation SRC2 with 31b, Lane 2 pre-incubation with 32a, and lane 3 pre-incubation with vehicle only. After incubation, beads were washed, treated with VDR and VDR–SRC2 interactions were determined by Western blot.
Acknowledgments
We thank Taosheng Chen, Fu-Yue Zeng, Wenwei Lin, and Jimmy Cui for their assistance with the HTS at the St. Jude High Throughput Screening Center, as well as Aaron Kosinski for his assistance with protein preparation. We also thank Paul Webb and Phuong Nguyen for providing us with the SRC2 plasmid. This work was supported by the University of Wisconsin-Milwaukee [LAA], the NIH (DA031090) [LAA] and (DK58080) [RKG], the American Lebanese Syrian Associated Charities (ALSAC) [RKG], and St. Jude Children’s Research Hospital [RKG].
Abbreviations
- VDR
vitamin D receptor
- NR
nuclear hormone receptor
- FP
fluorescence polarization
- SRC2
steroid receptor coactivator 2
- SRC2-3
labeled steroid receptor coactivator 2 peptide
- SAR
structureactivity relationship
- TRPV6
transient receptor potential vanilloid type 6 gene
- 1,25-(OH)2D3
1,25-dihydroxyvitamin D3
- DBD
DNA binding domain
- LBD
ligand-binding domain
- RXR
retinoid X receptor
- HTS
high throughput screening
- DMSO
dimethyl sulfoxide
- HEK293
human embryonic kidney cells
- FRET
fluorescence resonance energy transfer
- LC-MS
liquid chromatography-mass spectroscopy
- PAMPA
parallel artificial membrane permeability
- AR
androgen receptor
- ERβ
estrogen receptor beta
- TRα
thyroid receptor alpha
- TRβ
thyroid receptor beta
- PPARγ
peroxisome proliferator-activated receptor γ
- DRIP205
VDR-interacting protein 205
- GST
glutathione-S-transferase
- DU145
human prostate cancer cells
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- LFER
linear free energy relationship
Footnotes
Supporting Information Availability. Inhibition of VDR-SRC2-3 binding in the presence of 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one, synthesis and characterization of molecules, graphical representation of rate constants, nuclear receptor-coactivator binding isotherms and VDR-ligand competition isotherms. This material is free of charge via the Internet at http://pubs.org.
References
- 1.Brumbaugh PF, Haussler MR. 1 Alpha,25-dihydroxycholecalciferol receptors in intestine. I. Association of 1 alpha,25-dihydroxycholecalciferol with intestinal mucosa chromatin. J Biol Chem. 1974;249:1251–1257. [PubMed] [Google Scholar]
- 2.Jurutka PW, Whitfield GK, Hsieh JC, Thompson PD, Haussler CA, Haussler MR. Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Rev Endocr Metab Disord. 2001;2:203–216. doi: 10.1023/a:1010062929140. [DOI] [PubMed] [Google Scholar]
- 3.Feldman D, Pike JW, Glorieux FH. Vitamin D. 2. 1–2 Elsevier; Burlington: 2005. [Google Scholar]
- 4.Yamada S, Shimizu M, Yamamoto K. Vitamin D receptor. Endocr Dev. 2003;6:50–63. doi: 10.1159/000072769. [DOI] [PubMed] [Google Scholar]
- 5.Toell A, Polly P, Carlberg C. All natural DR3-type vitamin D response elements show a similar functionality in vitro. Biochem J. 2000;352(Pt 2):301–309. [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JY, Son YL, Lee YC. Involvement of SMRT corepressor in transcriptional repression by the vitamin D receptor. Mol Endocrinol. 2009;23:251–264. doi: 10.1210/me.2008-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene. 2000;246:9–21. doi: 10.1016/s0378-1119(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 8.(a) Lamblin M, Spingarn R, Wang TT, Burger MC, Dabbas B, Moitessier N, White JH, Gleason JL. An o-aminoanilide analogue of 1alpha,25-dihydroxyvitamin D(3) functions as a strong vitamin D receptor antagonist. J Med Chem. 2010;53:7461–7465. doi: 10.1021/jm1007159. [DOI] [PubMed] [Google Scholar]; (b) Kato Y, Nakano Y, Sano H, Tanatani A, Kobayashi H, Shimazawa R, Koshino H, Hashimoto Y, Nagasawa K. Synthesis of 1alpha,25-dihydroxyvitamin D3-26,23-lactams (DLAMs), a novel series of 1 alpha,25-dihydroxyvitamin D3 antagonist. Bioorg Med Chem Lett. 2004;14:2579–2583. doi: 10.1016/j.bmcl.2004.02.076. [DOI] [PubMed] [Google Scholar]; (c) Inaba Y, Yoshimoto N, Sakamaki Y, Nakabayashi M, Ikura T, Tamamura H, Ito N, Shimizu M, Yamamoto K. A new class of vitamin D analogues that induce structural rearrangement of the ligand-binding pocket of the receptor. J Med Chem. 2009;52:1438–1449. doi: 10.1021/jm8014348. [DOI] [PubMed] [Google Scholar]; (d) Nakabayashi M, Yamada S, Yoshimoto N, Tanaka T, Igarashi M, Ikura T, Ito N, Makishima M, Tokiwa H, DeLuca HF, Shimizu M. Crystal structures of rat vitamin D receptor bound to adamantyl vitamin D analogs: structural basis for vitamin D receptor antagonism and partial agonism. J Med Chem. 2008;51:5320–5329. doi: 10.1021/jm8004477. [DOI] [PubMed] [Google Scholar]; (e) Saito N, Kittaka A. Highly potent vitamin D receptor antagonists: design, synthesis, and biological evaluation. Chembiochem. 2006;7:1479–1490. doi: 10.1002/cbic.200600054. [DOI] [PubMed] [Google Scholar]; (f) Cho K, Uneuchi F, Kato-Nakamura Y, Namekawa J, Ishizuka S, Takenouchi K, Nagasawa K. Structure-activity relationship studies on vitamin D lactam derivatives as vitamin D receptor antagonist. Bioorg Med Chem Lett. 2008;18:4287–4290. doi: 10.1016/j.bmcl.2008.06.095. [DOI] [PubMed] [Google Scholar]; (g) Herdick M, Steinmeyer A, Carlberg C. Antagonistic action of a 25-carboxylic ester analogue of 1alpha, 25-dihydroxyvitamin D3 is mediated by a lack of ligand-induced vitamin D receptor interaction with coactivators. J Biol Chem. 2000;275:16506–16512. doi: 10.1074/jbc.M910000199. [DOI] [PubMed] [Google Scholar]; (h) Ishizuka S, Kurihara N, Miura D, Takenouchi K, Cornish J, Cundy T, Reddy SV, Roodman GD. Vitamin D antagonist, TEI-9647, inhibits osteoclast formation induced by 1alpha,25-dihydroxyvitamin D3 from pagetic bone marrow cells. J Steroid Biochem Mol Biol. 2004;89–90:331–334. doi: 10.1016/j.jsbmb.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 9.Moore TW, Katzenellenbogen JA. Inhibitors of Nuclear Hormone Receptor/Coactivator Interactions. Annu Rep Med Chem. 2009;44:443–457. [Google Scholar]
- 10.Mita Y, Dodo K, Noguchi-Yachide T, Miyachi H, Makishima M, Hashimoto Y, Ishikawa M. LXXLL peptide mimetics as inhibitors of the interaction of vitamin D receptor with coactivators. Bioorganic & Medicinal Chemistry Letters. 2010;20:1712–1717. doi: 10.1016/j.bmcl.2010.01.079. [DOI] [PubMed] [Google Scholar]
- 11.Teichert A, Arnold LA, Otieno S, Oda Y, Augustinaite I, Geistlinger TR, Kriwacki RW, Guy RK, Bikle DD. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry. 2009;48:1454–1461. doi: 10.1021/bi801874n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boehm MF, Fitzgerald P, Zou A, Elgort MG, Bischoff ED, Mere L, Mais DE, Bissonnette RP, Heyman RA, Nadzan AM, Reichman M, Allegretto EA. Novel nonsecosteroidal vitamin D mimics exert VDR-modulating activities with less calcium mobilization than 1,25-dihydroxyvitamin D3. Chem Biol. 1999;6:265–275. doi: 10.1016/S1074-5521(99)80072-6. [DOI] [PubMed] [Google Scholar]
- 13.Arnold LA, Kosinski A, Estebanez-Perpina E, Fletterick RJ, Guy RK. Inhibitors of the interaction of a thyroid hormone receptor and coactivators: preliminary structure-activity relationships. J Med Chem. 2007;50:5269–5280. doi: 10.1021/jm070556y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 15.Huth JR, Song D, Mendoza RR, Black-Schaefer CL, Mack JC, Dorwin SA, Ladror US, Severin JM, Walter KA, Bartley DM, Hajduk PJ. Toxicological evaluation of thiol-reactive compounds identified using a la assay to detect reactive molecules by nuclear magnetic resonance. Chem Res Toxicol. 2007;20:1752–1759. doi: 10.1021/tx700319t. [DOI] [PubMed] [Google Scholar]
- 16.Chen KS, DeLuca HF. Cloning of the human 1 alpha,25-dihydroxyvitamin D-3 24-hydroxylase gene promoter and identification of two vitamin D-responsive elements. Biochim Biophys Acta. 1995;1263:1–9. doi: 10.1016/0167-4781(95)00060-t. [DOI] [PubMed] [Google Scholar]
- 17.Holick MF, Kleiner-Bossaller A, Schnoes HK, Kasten PM, Boyle IT, DeLuca HF. 1,24,25-Trihydroxyvitamin D3. A metabolite of vitamin D3 effective on intestine. J Biol Chem. 1973;248:6691–6696. [PubMed] [Google Scholar]
- 18.Xie WH, Bloomfield KM, Jin YF, Dolney NY, Wang PG. Lanthanide triflates catalyzed reactions of imines with indole in protic media. Synlett. 1999:498–500. [Google Scholar]
- 19.Ritchie CD, Sager WF. An Examination of Structure-Activity Relationships. Prog Phys Org Chem. 1964;2:323–400. [Google Scholar]
- 20.(a) Estebanez-Perpina E, Moore JM, Mar E, Delgado-Rodrigues E, Nguyen P, Baxter JD, Buehrer BM, Webb P, Fletterick RJ, Guy RK. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J Biol Chem. 2005;280:8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]; (b) Moore JM, Galicia SJ, McReynolds AC, Nguyen NH, Scanlan TS, Guy RK. Quantitative proteomics of the thyroid hormone receptor-coregulator interactions. J Biol Chem. 2004;279:27584–27590. doi: 10.1074/jbc.M403453200. [DOI] [PubMed] [Google Scholar]
- 21.(a) Masuyama H, Brownfield CM, St-Arnaud R, MacDonald PN. Evidence for ligand-dependent intramolecular folding of the AF-2 domain in vitamin D receptor-activated transcription and coactivator interaction. Mol Endocrinol. 1997;11:1507–1517. doi: 10.1210/mend.11.10.9990. [DOI] [PubMed] [Google Scholar]; (b) Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. 1997;17:2735–2744. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li H, Gomes PJ, Chen JD. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc Natl Acad Sci U S A. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rachez C, Suldan Z, Ward J, Chang CP, Burakov D, Erdjument-Bromage H, Tempst P, Freedman LP. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 1998;12:1787–1800. doi: 10.1101/gad.12.12.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem. 2003;278:38665–38674. doi: 10.1074/jbc.M304886200. [DOI] [PubMed] [Google Scholar]
- 24.(a) Wissenbach U, Niemeyer B, Himmerkus N, Fixemer T, Bonkhoff H, Flockerzi V. TRPV6 and prostate cancer: cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem Biophys Res Commun. 2004;322:1359–1363. doi: 10.1016/j.bbrc.2004.08.042. [DOI] [PubMed] [Google Scholar]; (b) Nijenhuis T, Hoenderop JG, van der Kemp AW, Bindels RJ. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. J Am Soc Nephrol. 2003;14:2731–2740. doi: 10.1097/01.asn.0000094081.78893.e8. [DOI] [PubMed] [Google Scholar]
- 25.Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol. 2006;20:1447–1461. doi: 10.1210/me.2006-0031. [DOI] [PubMed] [Google Scholar]
- 26.(a) Arnold LA, Estebanez-Perpina E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280:43048–43055. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]; (b) Hwang JY, Huang W, Arnold LA, Huang R, Attia RR, Connelly M, Wichterman J, Zhu F, Augustinaite I, Austin CP, Inglese J, Johnson RL, Guy RK. Methylsulfonylnitrobenzoates, a new class of irreversible inhibitors of the interaction of the thyroid hormone receptor and its obligate coactivators that functionally antagonizes thyroid hormone. J Biol Chem. 2011;286:11895–11908. doi: 10.1074/jbc.M110.200436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Ciesielski F, Rochel N, Moras D. Adaptability of the Vitamin D nuclear receptor to the synthetic ligand Gemini: remodelling the LBP with one side chain rotation. J Steroid Biochem Mol Biol. 2007;103:235–242. doi: 10.1016/j.jsbmb.2006.12.003. [DOI] [PubMed] [Google Scholar]; (b) Vanhooke JL, Benning MM, Bauer CB, Pike JW, DeLuca HF. Molecular structure of the rat vitamin D receptor ligand binding domain complexed with 2-carbon-substituted vitamin D3 hormone analogues and a LXXLL-containing coactivator peptide. Biochemistry. 2004;43:4101–4110. doi: 10.1021/bi036056y. [DOI] [PubMed] [Google Scholar]
- 28.Lehen’kyi V, Raphael M, Oulidi A, Flourakis M, Khalimonchyk S, Kondratskyi A, Gordienko DV, Mauroy B, Bonnal JL, Skryma R, Prevarskaya N. TRPV6 determines the effect of vitamin D3 on prostate cancer cell growth. PLoS One. 2011;6:e16856. doi: 10.1371/journal.pone.0016856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehen’kyi V, Flourakis M, Skryma R, Prevarskaya N. TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene. 2007;26:7380–7385. doi: 10.1038/sj.onc.1210545. [DOI] [PubMed] [Google Scholar]
- 30.Stern PH, De Olazabal J, Bell NH. Evidence for abnormal regulation of circulating 1 alpha,25-dihydroxyvitamin D in patients with sarcoidosis and normal calcium metabolism. J Clin Invest. 1980;66:852–855. doi: 10.1172/JCI109924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bosch X. Hypercalcemia due to endogenous overproduction of 1,25-dihydroxyvitamin D in Crohn’s disease. Gastroenterology. 1998;114:1061–1065. doi: 10.1016/s0016-5085(98)70327-0. [DOI] [PubMed] [Google Scholar]
- 32.Shirakawa S, Kobayashi S. Carboxylic acid catalyzed three-component aza-Friedel-Crafts reactions in water for the synthesis of 3-substituted indoles. Organic Letters. 2006;8:4939–4942. doi: 10.1021/ol062031q. [DOI] [PubMed] [Google Scholar]
- 33.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.