

The distribution and dynamics of phosphatidylserine are studied in the plasma membrane and in organellar membranes of live cells using two novel fluorescent probes in combination with various biophysical techniques, including fluorescence correlation spectroscopy and single-particle tracking.
Abstract
Much has been learned about the role of exofacial phosphatidylserine (PS) in apoptosis and blood clotting using annexin V. However, because annexins are impermeant and unable to bind PS at low calcium concentration, they are unsuitable for intracellular use. Thus little is known about the topology and dynamics of PS in the endomembranes of normal cells. We used two new probes—green fluorescent protein (GFP)–LactC2, a genetically encoded fluorescent PS biosensor, and 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phospho-l-serine (TopFluor-PS), a synthetic fluorescent PS analogue—to examine PS distribution and dynamics inside live cells. The mobility of PS was assessed by a combination of advanced optical methods, including single-particle tracking and fluorescence correlation spectroscopy. Our results reveal the existence of a sizable fraction of PS with limited mobility, with cortical actin contributing to the confinement of PS in the plasma membrane. We were also able to measure the dynamics of PS in endomembrane organelles. By targeting GFP-LactC2 to the secretory pathway, we detected the presence of PS in the luminal leaflet of the endoplasmic reticulum. Our data provide new insights into properties of PS inside cells and suggest mechanisms to account for the subcellular distribution and function of this phospholipid.
INTRODUCTION
Phosphatidylserine (PS)—the most abundant anionic phospholipid—accounts for 3–10% of the total cellular lipid. As is the case for other lipids, PS is not present evenly throughout all cellular membranes, nor is it equally distributed between the leaflets of every membrane bilayer (van Meer et al., 2008; Leventis and Grinstein, 2010). In resting cells plasmalemmal PS is found exclusively on the inner (cytoplasmic-facing) monolayer, due to the action of ATP-dependent aminophospholipid flippases (Daleke, 2007). Remarkably, most studies of PS function have analyzed its role under atypical circumstances, when PS becomes exposed on the outside-facing (extracellular) leaflet of the plasma membrane. Thus the appearance of PS on the surface of apoptotic cells and its role in signaling phagocytosis has been studied extensively (Williamson and Schlegel, 2002; Wu et al., 2006), as has the exposure of PS in activated blood platelets, which prompts the binding and activation of clotting factors (Zwaal et al., 1998; Lentz, 2003).
By comparison, the intracellular role of PS has been studied in much less detail, despite the realization that a number of important intracellular proteins require PS for their proper localization and/or activation (Cho and Stahelin, 2006). The essential role of PS is highlighted by the observation that, in mice, deletion of both PS synthase isoforms is lethal (Arikketh et al., 2008). The paucity of information regarding intracellular PS is due, at least in part, to the lack of suitable probes to analyze the distribution and dynamics of this key phospholipid in intact cells. Annexin V, the probe used most extensively to date to monitor PS appearance on the cell surface, is not suitable for intracellular use, as it requires levels of calcium that far exceed the prevailing cytosolic concentration (Tait and Gibson, 1992). The fluorescently labeled PS analogue 1-palmitoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]lauroyl}-sn-glycero-3-phospho-l-serine (NBD-PS), which until recently was the preferred alternative approach, is not a reliable mimic of the native phospholipid. The polar nature of the NBD moiety distorts the alignment of the acyl chain and alters the solubility of the conjugate, making it prone to spontaneous intermembrane translocation (Martin and Pagano, 1987; Elvington and Nichols, 2007).
We report here the characterization of the dynamics of PS in the membranes of live mammalian cells, using two new probes: green fluorescent protein (GFP)–LactC2, a genetically encoded, fluorescent PS-binding protein (Yeung et al., 2008), and 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phospho-l-serine (TopFluor-PS), a new and previously uncharacterized fluorescently labeled analogue of PS. After characterization of these probes in artificial bilayers, we used fluorescence recovery after photobleaching (FRAP), total internal reflection fluorescence microscopy (TIRFM), fluorescence correlation spectroscopy (FCS), and single-particle tracking (SPT) to study the behavior of PS in live cells. Molecular targeting enabled us to study PS not only on the cytoplasmic-facing leaflet of organellar membranes, but also in the luminal leaflet of the endoplasmic reticulum (ER).
RESULTS
TopFluor-PS as a fluorescent analogue of PS
NBD and, to a lesser extent, BODIPY, have been used to label PS at one of its acyl chains to visualize the lipid by fluorescence microscopy. NBD labeling grossly alters the lipid structure, as the hydrophilic character of the fluorescent moiety causes the acyl chain to loop back to the membrane–water interface (Chattopadhyay and London, 1987; Abrams and London, 1993). BODIPY-labeled analogues more closely mirror natural lipids (Pagano et al., 1991; Martin and Pagano, 1994; te Vruchte et al., 2004; Golebiewska et al., 2008), even though the BODIPY moiety does not fully penetrate the lipid bilayer (Kaiser and London, 1998). Of importance, BODIPY-PS is not commercially available. We therefore commissioned the synthesis of a PS conjugate labeled with a more hydrophobic derivative of BODIPY (TopFluor) at the end of a saturated 11-carbon acyl chain, referred to hereafter as TopFluor-PS (Figure 1A). Figure 1B shows the uncorrected emission spectrum of TopFluor-PS (with excitation at 470 ± 2.5 nm) and also illustrates that the fluorescence of the probe, which was incorporated into liposomes consisting of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), is practically insensitive to pH in the physiological range. The accessibility of the TopFluor-PS head group was analyzed using annexin V labeled with a far-red fluorophore. As shown in Figure 1C, when analyzed by flow cytometry, beads coated with 5% TopFluor-PS (plus 95% DOPC) bound Alexa 647–conjugated annexin V, implying that the phosphoserine head groups were correctly positioned, exposed to the aqueous milieu.
FIGURE 1:

Spectral properties and dynamics of TopFluor-PS in lipid bilayers. (A) The structure of TopFluor-PS. The fluorescent moiety is at the end of an 11-carbon acyl chain on sn-2 of the glycerophospholipid analogue. (B) Emission spectra of liposomes containing 95% DOPC and 5% TopFluor-PS excited at 470 nm. The emission peaks at 510 nm and is minimally affected by changes in pH. (C) FACS analysis of annexin V–Alexa 647 binding to 3-μm Nucleosil C18 beads coated with 100% DOPC (0% TopFluor-PS; red) or 95% DOPC, 5% TopFluor-PS (blue). (D) Quenching of the fluorescence of liposomes containing 95% DOPC and either 5% NBD-PS or 5% TopFluor-PS by varying concentrations of iodide. Data are plotted according to the Stern–Volmer relationship, where F0 is the initial fluorescence and F is the fluorescence in the presence of the quenching agent. (E, F) Emission profiles of liposomes containing TopFluor-PS (E) or NBD-PS (F) and the indicated fluorescence-quenching spin-labeled lipids (5% fluorescent lipid, 10% spin-labeled lipid, and 85% DOPC). (G) Representative FRAP recovery curve of TopFluor-PS in a supported lipid bilayer. Average Fm of all supported bilayers tested (n = 36) was 0.88 ± 0.05. (H) Diffusion coefficients calculated by FRAP analysis of TopFluor-PS in supported lipid bilayers containing varying concentrations of cholesterol. Data are means ± SEM of 9–31 independent determinations from three experiments. n.s., not significantly different from 0% cholesterol.
The positioning of the TopFluor moiety in the bilayer was analyzed next, using two separate fluorescence-quenching methods. Iodide can exert collisional quenching of a variety of fluorophores, including BODIPY and NBD derivatives. Effective quenching occurs only when the fluorescent moieties are exposed to the aqueous medium (Johnson et al., 1991). When incorporated into liposomes, TopFluor-PS was found to be much less susceptible to quenching by iodide than was NBD-PS, suggesting that the TopFluor moiety resides deeper within the hydrophobic core of the lipid bilayer (Figure 1D). This conclusion was validated using quenching of fluorescence by spin-labeled phospholipids to probe and compare the depths of the TopFluor and NBD fluorophores within the bilayer. Quenching of TopFluor-PS fluorescence was greatest when using 16-doxyl phosphocoline (PC), a lipid that places the spin-labeled moiety closest to the center of the bilayer (Figure 1E). In contrast, the much shorter 1-palmitoyl-2-stearoyl-(5-doxyl)-sn-glycero-3-phosphocholine (5-doxyl-PC) produced the greatest quenching in the case of NBD-PS (Figure 1F). Using the quenching data obtained for both fluorescent PS probes, we performed parallax analysis, which compares the quenching efficiency of multiple phospholipids having spin-labeled quenching moieties at known depths within the bilayer (Figure 1, E and F) and then uses these varying efficiencies to calculate an approximate depth at which the fluorescent moiety resides (London, 1982; Chattopadhyay and London, 1987; Abrams and London, 1993). This analysis indicated that in the case of labeled PS the NBD fluorophore mainly resides 19.6 Å from the center of the bilayer (Lc; Table 1), a value comparable to the 19.8 Å reported by Abrams and London (1993) for NBD-PC. This is consistent with the notion that the NBD moiety forces the acyl chain to which it is attached to loop back toward the interface, not aligning properly within the bilayer. In contrast, the calculations—with correction for transleaflet quenching by the deep 16-doxyl PC (Chattopadhyay and London, 1987)—indicate that TopFluor-PS sits 8.2 Å from the center of the bilayer (Table 1), implying that the fluorophore remains buried within the hydrophobic layer, deeper even than BODIPY, which was tested in other lipids (Kaiser and London, 1998). We concluded that TopFluor-PS is a more suitable mimic of PS than previously available probes, inasmuch as its head group is properly exposed to the aqueous medium, whereas the acyl chains extend into the hydrophobic core of the bilayer.
TABLE 1:
Determination of fluorophore depth in liposomes.
| Fluorescent lipid | Spin-labeled quencher | F/F0 ± SEM | Calculation pair | Lc (Å) ± SEM |
|---|---|---|---|---|
| TopFluor-PS | 16-doxyl | 0.30 ± 0.10 | 16 : 12 | 8.25 ± 0.06 |
| 12-doxyl | 0.59 ± 0.05 | |||
| 5-doxyl | 0.46 ± 0.16 | |||
| Tempo-PC | 0.49 ± 0.01 | |||
| NBD-PS | 16-doxyl | 0.62 ± 0.02 | 5 : Tempo | 19.62 ± 0.04 |
| 12-doxyl | 0.79 ± 0.01 | |||
| 5-doxyl | 0.47 ± 0.02 | |||
| Tempo-PC | 0.59 ± 0.04 |
PS dynamics in supported lipid bilayers
We next analyzed the behavior of TopFluor-PS in supported lipid bilayers using both FRAP and a combination of TIRFM and SPT. Figure 1G shows a representative fluorescence recovery curve after bleaching of TopFluor-PS in a bilayer containing 45% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE)/27% DOPC/15% 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS)/0.5% TopFluor-PS/2.5% DOPI/10% cholesterol/0.25% l-α-phosphatidylethanolamine-N-lissamine rhodamine B sulfonyl (Rhod-PE; see Table 5 later in the paper). In 37 similar determinations the mobile fraction (Fm) of TopFluor-PS averaged 0.88. The immobile component is likely to reflect binding of a fraction of the PS to the supporting glass; accordingly, it was unaffected when the cholesterol content of the bilayer was altered between 0 and 40%. The diffusion coefficient (D), however, was clearly affected by the cholesterol content, dropping from 1.25 μm2/s in the absence of cholesterol to 0.55 μm2/s when cholesterol was 40 mol % (Figure 1H and Table 2), as found for other lipids (Korlach et al., 1999; Filippov et al., 2003).
TABLE 5:
Liposome compositions used in this study.
| Experiment | Components | Composition (mol %) |
|---|---|---|
| TopFluor-PS self-quenching | TopFluor-PS/DOPC | 2.5–50/50–97.5 |
| TopFluor-PS spin-label quenching | TopFluor-PC/DOPC/spin-labeled PC | 5/80/15 |
| GST-LactC2/dansyl FRET | DOPS/DOPC/dansyl-PE | 20/77.5/2.5 |
| TopFluor-PS FRAP | POPE/DOPC/DOPS/TopFluor-PS/DOPI/cholesterol/Rhod-PE | 30–50/12–32/15/0.5/2.5/0–40/0.25 |
| TopFluor-PS SPT | POPE/DOPC/DOPS/TopFluor-PS/DOPI/cholesterol/Rhod-PE | 30–50/12–32/15/0.05/2.5/0–40/0.25 |
| GFP-LactC2 FRAP and SPT | POPE/DOPC/DOPS/DOPI/cholesterol/Rhod-PE | 30–50/12–32/15/2.5/0–40/0.25 |
TABLE 2:
Calculated diffusion coefficients of PS in supported bilayers (μm2/s ± SEM).
| Cholesterol (%) | TopFluor-PS FRAP (n) | GFP-LactC2 FRAP (n) |
|---|---|---|
| 0 | 1.25 ± 0.15 (10) | 1.29 ± 0.29 (13) |
| 40 | 0.55 ± 0.03 (15) | 0.45 ± 0.12 (8) |
To circumvent the complications introduced by the covalent attachment of a fluorophore to the acyl chain, we also analyzed the dynamics of PS in model membranes using the discoidin-type C2 domain of lactadherin (LactC2). Constructs containing this specific PS-binding domain were used to reveal the distribution of endogenous PS in live cells (Yeung et al., 2008; Fairn et al., 2011a). We generated a recombinant glutathione S-transferase (GST)–tagged construct to measure the affinity and validate the selectivity of the C2 domain for PS, using large unilamellar liposomes. Binding was detected by fluorescence resonance energy transfer (FRET) between tryptophan residues of the protein and dansyl-labeled PE in the liposomes, as described in Gilbert et al. (1990). As shown by the isotherms in Figure 2A, binding of the wild-type LactC2 to liposomes was stringently dependent on the presence of PS. Moreover, mutation of residues required for stable binding to PS precluded association with the liposomes (see AAA mutant in Figure 2A). Unlike the binding of annexins to PS, the interaction of LactC2 is insensitive to the ionic strength and independent of calcium. As illustrated in Figure 2B, the apparent affinity of the construct for PS was similar whether the concentration of NaCl was 15 or 150 mM or when 100 μM CaCl2 was added (the calculated Kd was 329 ± 69, 351 ± 72, and 288 ± 42 nM, respectively). Furthermore, varying the cholesterol content had no discernible effect on the ability of LactC2 to bind PS (Supplemental Figure S1). These findings confirm the notion that binding is not merely electrostatic, but stereospecific.
FIGURE 2:

PS-binding properties and bilayer dynamics of recombinant LactC2. (A) Binding isotherms of recombinant GST-LactC2 or GST-LactC2(AAA) to egg-PC/dansyl-PE (2.5 mol %) liposomes with or without 20% PS in 150 mM NaCl-containing buffer. The term (F/Fb) − 1 (y-axis) refers to the observed FRET fluorescence resulting from the close proximity of dansyl-PE in the liposome and tryptophan residues in GST-LactC2. The traces are nonlinear least-squares curves fit to a one-site binding hyperbola (Gilbert et al., 1990), yielding Kd for LactC2 PS binding of 351 ± 72 nM. (B) Effects of calcium and of changes in ionic strength on LactC2 binding to PS-containing liposomes. Ionic strength was reduced by lowering NaCl concentration to 15 mM. Where indicated, 100 μM CaCl2 was added. (C) Diagram of strategy for purification of GFP-LactC2. GST-GFP-LactC2 construct in pGEX-6p was expressed in E. coli, lysates were bound to glutathione–Sepharose, and GFP-LactC2 was cleaved on-column from GST with PreScission protease. (D) Sample Gaussian best fits of fluorescence intensity profiles obtained during FRAP of purified GFP-LactC2 bound to a supported lipid bilayer containing 15% DOPS (see Hammond et al., 2009, for details). (E, F) Diffusion coefficients (E) and τ (F) calculated for GFP-LactC2 association with supported lipid bilayers containing 15% DOPS and varying mol % cholesterol. Data are means ± SEM of 7–13 independent determinations from two experiments.
Recombinant GFP-LactC2, generated by cleavage of GST from purified GST-GFP-LactC2 (Figure 2C), was used to measure PS dynamics in supported lipid bilayers by FRAP. Because GFP-LactC2 binds to PS reversibly (Andersen et al., 2000; Shi et al., 2004), dissociation and reassociation from the membrane contribute to the recovery of fluorescence after photobleaching. Deconvolution of the lateral mobility from the rate of dissociation/reassociation was accomplished by using the procedure introduced by Hammond et al. (2009). Briefly, Gaussian curves were fit to profiles of the fluorescence intensity of the bleached membrane throughout the recovery phase (Figure 2C), which were used to determine D of GFP-LactC2-associated PS, as well as the apparent membrane dissociation time of the complex (τ), which is inversely proportional to the dissociation rate constant (Hammond et al., 2009). Using this modified FRAP analysis, we obtained a D of 1.29 μm2/s for PS in bilayers without cholesterol and 0.45 μm2/s when cholesterol was 40% (Figure 2E and Table 2). The value of τ was similar under both conditions (Figure 2F), implying that the mobility of the PS–LactC2 complex, and not its stability, is altered by cholesterol.
We also determined the mobility of PS in supported bilayers by SPT using TIRFM. By using high acquisition rates, we could obtain sufficiently long tracks (≥20 frames) to analyze molecular motion, despite the comparatively rapid dissociation of GFP-LactC2 (τ ≈ 4 s; see Figure 2F). Analysis of such tracks using the moment-scaling spectrum method (Jaqaman et al., 2008) yielded an average D of 0.33 μm2/s. Thus multiple methods of measuring PS dynamics with these two probes yielded similar results, even though some variation in the absolute values was observed, depending on the measurement technique used. These differences are considered further in the Discussion.
TopFluor-PS association with cells
Because it associates reversibly with biological membranes, NBD-PS can be rapidly removed (backwashed) from the outer leaflet of cells. This feature has facilitated the study of transbilayer movement by amino-lipid flippases (Martin and Pagano, 1987; Pomorski et al., 2003; Zhou and Graham, 2009). On the other hand, after flipping, the instability of NBD-PS in membranes causes unphysiological redistribution between intracellular organelles in a vesicular transport–independent manner (Martin and Pagano, 1987; Kobayashi and Arakawa, 1991; Pomorski et al., 2004), precluding reliable analysis of PS dynamics in intact cells. Because BODIPY-labeled lipids have considerably slower spontaneous intermembrane transfer rates (Bai and Pagano, 1997; Elvington and Nichols, 2007), we anticipated the related TopFluor-PS to be a more suitable probe for PS in cells. This was validated, as shown in Figure 3, A and B, where HeLa cells were loaded with either NBD-PS or TopFluor-PS at 4°C and maintained at low temperature to minimize flipping during subsequent backwashes with defatted albumin. We found that although >75% of NBD-PS was removed by washing with albumin, the vast majority of the TopFluor-PS remained associated with the cells (Figure 3C). Note also that the residual TopFluor-PS fluorescence is largely associated with the plasmalemma, whereas much of the detectable residual NBD-PS is associated with endomembranes.
FIGURE 3:

TopFluor-PS is poorly extracted from cellular membranes. (A, B) Cells were loaded for 10 min at 4°C with either NBD-PS (A) or TopFluor-PS (B) and images acquired by confocal microscopy before or immediately after backwashing with 1% BSA for 10 min; identical acquisition settings were maintained throughout for each fluorophore. Scale bars, 10 μm. (C) Quantification of membrane-associated fluorescence before and after BSA backwashing. Data are means ± SEM of three experiments.
When cells were rewarmed to 37°C after loading with the fluorescent PS analogues, NBD-PS and TopFluor-PS displayed divergent behavior. As reported (Martin and Pagano, 1987; Pomorski et al., 2004), NBD-PS redistributed rapidly and was seen in membranes throughout the cell after only 2 min at 37°C (Figure 4A). In contrast, TopFluor-PS was seen on intracellular membranes only after ≥5 min. After 15 min at 37°C TopFluor-PS was found mainly at the plasma membrane and in vesicles identified as components of the endocytic pathway by their colocalization with fluorescent dextran, used as a marker of fluid-phase uptake (Figure 4, B and C). Of note, labeling with TopFluor-PS had no detectable toxic effect on the cells; neither the mitochondrial potential (measured using Mitotracker) nor the permeability of the plasma membrane (assessed with propidium iodide) was altered (Supplemental Figure S2).
FIGURE 4:

Distribution and stability of fluorescent PS analogues in cells. (A, B) HeLa cells were loaded for 10 min at 4°C with either NBD-PS (A) or TopFluor-PS (B), transferred to a microscope with a heated stage, and confocal images acquired at the indicated times of incubation at 37°C. (C) Colocalization of intracellular TopFluor-PS with internalized dextran. After loading with TopFluor-PS to cells at 4°C, cells were transferred to medium at 37°C containing the fluid-phase marker rhodamine-dextran for 10 min, followed by imaging. All bars, 10 μm. (D) After loading with TopFluor-PS for 30 min at 4°C, cells were incubated at 37°C for the indicated times; total lipids were then extracted and analyzed by TLC. The amount of intact TopFluor-PS extracted from cells is shown. Data are means ± SEM of seven independent experiments. (E) After loading with TopFluor-PS as in D, cells were incubated at 37°C for 30 min in the presence or absence of the indicated phospholipase inhibitors, and the amount of TopFluor-PS extracted from cells was normalized to the amount extracted without incubation at 37°C. (F) RAW cells were loaded with TopFluor-PS as in D and then allowed to recover at 37°C for the indicated time before staining with annexin V–Alexa 647. Annexin V staining was determined by flow cytometry after gating for live cells.
We next determined the stability of TopFluor-PS in cells. To this end, HeLa cells were initially loaded with the probe at 4°C, followed by incubation for various times at 37°C. Total lipid extracts were then analyzed by TLC. As illustrated in Figure 4D, the amount of TopFluor-PS extractable from cells decreased slightly over time, without obvious conversion to other phospholipid species (Supplemental Figure S3). We tested a series of well-established inhibitors to assess the possible contribution of phospholipases to the disappearance of TopFluor-PS. As shown in Figure 4E, bromoenol lactone, 4-fluoro-N-(2-(4-(5-fluoro-1H-indol-1-yl)piperidin-1-yl)ethyl)benzamide (FIPI), and 1-[6-({17beta-3-methoxyestra-1,3,5(10)-trien-17-yl}amino)hexyl]-1H-pyrrole-2,5-dione (U73122), which inhibit phospholipase A, D, and C, respectively, all failed to alter the disappearance of TopFluor-PS. In summary, although caution must be exerted when interpreting results following long (≥30 min) incubations, at earlier times the bulk of the fluorescence is emitted by bona fide TopFluor-PS. Thus the probe can be used as a reliable indicator of PS for periods of many minutes.
Transmembrane flipping of TopFluor-PS
As noted, TopFluor-PS associates firmly with biological membranes and is refractory to extraction by albumin. Although this more closely reflects the behavior of native PS, it precludes the use of traditional backwashing techniques to determine the rate of transmembrane flipping. We chose instead to use annexin V, a probe commonly used to quantify exofacial PS, to monitor the disappearance of TopFluor-PS from the outer leaflet. As shown in Figure 4F, Alexa 647–tagged annexin V was readily able to detect TopFluor-PS on the exofacial leaflet of RAW cells soon after incubation with the fluorescent phospholipid (Figure 4F). The amount of exofacial PS detected by annexin V decreased rapidly when the cells were incubated at 37°C, becoming undetectable at ≥15 min. Because at comparable (or longer) times of incubation at 37°C TopFluor-PS fluorescence was still visible at the plasma membrane (Figure 4B), we concluded that the tagged lipid was indeed being flipped to the cytosolic leaflet of the plasmalemma.
TopFluor-PS quenching
While performing the flow cytometric analyses of annexin V staining, which required comparatively high loading with TopFluor-PS to generate adequate signals, we noticed that the cell-associated fluorescence emitted by TopFluor-PS increased over time (Figure 5A). This increase was not due to continued incorporation of the probe, which was absent from the medium during the incubation at 37°C. It is well known that BODIPY is able to self-quench and undergo spectral changes at high concentrations (Marks et al., 2008) not only in vitro, but also when added to cells (Chen et al., 1997). We therefore tested whether TopFluor-labeled PS similarly undergoes self-quenching, by incorporating increasing amounts of the lipid into liposomes (Figure 5B). When excited at 485 nm, the fluorescence emission was red-shifted and quenched at mole ratios >10%. Unlike BODIPY (Marks et al., 2008), however, TopFluor-labeled PS did not display a second emission peak.
FIGURE 5:

TopFluor-PS self-quenches in the plasmalemma. (A) TopFluor-PS self-quenches when added to cells at high concentrations and dequenches upon redistribution. TopFluor-PS fluorescence detected by flow cytometry in cells loaded for 30 min at 4°C with 10 nmol of TopFluor-PS complexed to BSA per ml of PBS, followed by extensive washing and incubation at 37°C for the indicated times. The emission (scattering) from unlabeled cells is shown for reference. (B) Fluorescence emission spectra of liposomes containing increasing concentrations of TopFluor-PS (in mol %). (C) HeLa cells were loaded as in A (left, 0 min), followed by warming to 37°C for 30 min (right). Scale bar,10 μm. (D, E) HeLa (D) or RAW (E) cells were loaded at 4°C with TopFluor-PS and then warmed for the indicated times. Fluorescence intensity at the plasma membrane and inside the cells was quantified. Where indicated, the cells were fixed with paraformaldehyde before loading. Data are means ± SEM of four independent experiments. (F) Cells were loaded at 4°C with TopFluor-PS and treated with or without 20 μM Dyngo-4a. Fluorescence intensity at the plasma membrane was quantified over time. (G) HeLa cells were loaded with TopFluor-PS or TopFluor-PC at 4°C; the plasmalemmal fluorescence intensity was quantified during subsequent incubation at either 37 or 15°C.
We hypothesized that dequenching of the probe was responsible for the progressive fluorescence increase and proceeded to investigate the underlying mechanism. The outer leaflet of the membrane of HeLa cells was labeled heavily by incubating cells with TopFluor-PS. Under these conditions fluorescence emission was comparatively low, likely as a result of self-quenching. The fluorescence intensity increased progressively upon warming to 37°C, despite extensive washing to remove the extracellular TopFluor-PS not associated with the cells (Figure 5C). Intensification of the plasmalemmal fluorescence was accompanied by appearance of intracellular emission; these components were quantified separately. As shown in Figure 5D, the plasma membrane fluorescence increased rapidly, attaining a plateau within 20 min, whereas the endomembrane intensity increased more slowly and continuously. Very similar behavior was noted when using RAW264.7 cells, a murine monocyte/macrophage line (Figure 5E). Of note, TopFluor-PS dequenching did not occur when the probe was added to cells that were previously fixed using paraformaldehyde (which were loaded with similar amounts of TopFluor-PS; Supplemental Figure S4), indicating the involvement of active transport in the dequenching process.
Two active processes could have contributed to TopFluor-PS dequenching: transbilayer flipping, resulting in dilution of the fluorophore in the inner leaflet, or endocytosis, in which case dilution would have resulted from fusion with unlabeled endomembranes. To assess the contribution of endocytosis we treated cells with Dyngo-4a, a potent inhibitor of dynamin (Hill et al., 2009; Harper et al., 2011), or cooled the cells to 15°C; at this temperature endocytosis is largely inhibited but PS flippases are not (Martin and Pagano, 1987). As illustrated in Figure 5, F and G, both of these conditions greatly reduced the rate and extent of fluorescence dequenching, implying that endocytosis is primarily responsible for dilution of the probe. This conclusion was validated using 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phosphocholine (TopFluor-PC); unlike PS, PC and its analogues do not undergo active flipping at the plasma membrane (van Meer et al., 2008). As shown in Figure 5G, the fluorescence of cells loaded with TopFluor-PC in the cold also increased upon warming to 37°C, although to a lesser extent than seen for TopFluor-PS. The increase in TopFluor-PC was blunted when incubation was at 15°C, consistent with involvement of endocytosis. Because the relative fluorescence increase seen at 37°C was greater for TopFluor-PS than for TopFluor-PC, and because a fraction of the TopFluor-PS dequenching persisted at 15°C, we believe that flipping and dilution with inner leaflet lipids contribute to the fluorescence increase, although this contribution is comparatively small.
PS dynamics in the plasma membrane
We next examined PS dynamics in cellular membranes using TopFluor-PS and LactC2-GFP by FRAP, TIRFM/SPT, and FCS. The behavior of PS in the plasma membranes was first analyzed by loading cells with TopFluor-PS, followed by incubation at 37°C for ∼15 min to allow for transmembrane flipping while minimizing chemical conversion of the probe (Figure 4). A representative FRAP experiment is shown in Figure 6A, and a corresponding recovery curve is depicted in Figure 6B. In the membrane of HeLa cells TopFluor-PS had an average D of 0.49 μm2/s (Table 3). Strikingly, the Fm was only 0.43, suggesting that a considerable subpopulation of the plasmalemmal PS (or at least of TopFluor-PS) is associated within complexes that curtail its lateral diffusion, at least within the time scale of our measurements.
FIGURE 6:

Movement of TopFluor-PS in the plasma membrane. (A) The mobility of TopFluor-PS was analyzed by FRAP in HeLa cells at 37°C. Representative images are shown, with magnifications of the indicated area shown in insets. Scale bar, 10 μm. (B) Sample FRAP recovery curve of TopFluor-PS obtained as in A. The average Fm of TopFluor-PS obtained from 13 independent determinations was 0.43 ± 0.06. (C, D) Fm and D of various fluorophores in the intact plasma membrane and in membrane blebs. HeLa cells were transfected with PM-GFP or tK-Ras-GFP or loaded with TopFluor-PS. Where indicated, blebs were induced using jasplakinolide. Fm and D were measured using FRAP. Data are means ± SEM of at least 10 independent determinations from two experiments. (E) Induction of membrane blebs by jasplakinolide. HeLa cells were treated with 1 μM jasplakinolide for 10 min and imaged by differential interference contrast. Size bar, 10 μm. (F) Representative image (i) and tracks (ii) of single TopFluor-PS particles in the ventral membrane of HeLa cells detected by TIRFM using an electron-multiplied charge-coupled device camera. Tracks are color coded: particles displaying free Brownian diffusion are green, and those classified as confined are red. Graph in (iii) shows classification breakdown obtained for 1237 single-particle tracks like those in (i). Scale bar, 2 μm.
TABLE 3:
Calculated diffusion coefficients of PS in the plasma membrane of HeLa cells (μm2/s ± SEM).
| TopFluor-PS FRAP (n) | GFP-LactC2 FRAP (n) | TopFluor-PS SPT (n) [n tracks] | GFP-LactC2 SPT (n) [n tracks] | TopFluor-PS FCS Fast D Slow D Percent slow (n)a | GFP-LactC2 FCS Fast D Slow D Percent slow (n) | GFP-LactC2(AAA) FCS Fast D Slow D Percent slow (n) |
|---|---|---|---|---|---|---|
| 0.49 ± 0.09 | 0.33 ± 0.05 | 0.22 ± 0.03 | 0.44 ± 0.04 | 22.41 ± 3.72 | 26.43 ± 1.40 | 22.63 ± 1.52 |
| (13) | (40) | (24) | (34) | 1.52 ± 0.09 | 0.46 ± 0.03 | 1.28 ± 0.16 |
| [1237] | [29,007] | 77 ± 2 | 60 ± 1 | 18 ± 1 | ||
| (29) | (31) | (34) |
aFor TopFluor-PS in 25 of 85 measurements, performed on 29 different cells, a fast-diffusing particle was detected. The slow diffusion was detected in all 85 measurements.
Because the mobile fraction of PS in the plasmalemma was unexpectedly low (considerably lower than that estimated on pure lipid bilayers; see earlier discussion), we investigated the underlying mechanism. As shown in Figure 6C, the mobile fraction of PS is markedly lower than that of tK-Ras-GFP, a farnesylated construct that associates electrostatically with the inner aspect of the plasma membrane. The mobile fraction of PM-GFP, a lipid-anchored construct that partitions into rafts on the cytosolic leaflet, was also much greater. We therefore concluded that neither rafts nor partitioning into anionic domains account for the constrained mobility of PS. Instead, we hypothesized that specific association with immobile protein complexes may be involved. Moreover, we assumed that the actin cytoskeleton contributes to this immobilization. This hypothesis was tested by generating plasma membrane blebs detached from the cortical cytoskeleton. This was accomplished by using jasplakinolide, an actin stabilizer that induces the formation of blebs as the skeleton contracts (Figure 6E). The contraction is mediated by myosin II, since it is prevented by blebbistatin (Flannagan et al., 2010). FRAP measurements of TopFluor-PS in such blebs yielded an average Fm of 0.93, more than double the value in untreated membranes (Figure 6C). The diffusion coefficient of the mobile fraction did not increase significantly (Figure 6D). These findings are consistent with the notion that a considerable fraction of PS associates with (protein) complexes that are immobilized by the actin cytoskeleton.
We also analyzed the mobility of TopFluor-PS in the membrane of HeLa cells by SPT in TIRFM mode. A typical experiment is illustrated in Figure 6F. The diffusion coefficient estimated from SPT averaged 0.22 μm2/s (Table 3). In agreement with the FRAP data, only a fraction of the molecules displayed free diffusion (29%), whereas others (71%) diffused within a confined space with an average radius of 111 ± 4 nm.
Last, we also investigated the properties of TopFluor-PS in the membrane of HeLa cells by FCS. In most experiments (60 of 85) a single population was identified, with D = 1.75 ± 0.09 μm2/s. In a smaller fraction of experiments 77% of the detected molecules showed slow diffusion of the same order as before (D = 1.08 ± 0.11 μm2/s; Table 3), whereas 23% displayed much faster diffusion (D = 22.41 ± 3.72 μm2/s). The latter may reflect the time-dependent generation of soluble products of degradation of TopFluor-PS, as implied in Figure 4D. Accordingly, the observed diffusion coefficient is very similar to that of fluorophores diffusing freely in the cytosol, such as enhanced GFP (in HeLa cells D = 19.84 ± 1.83 μm2/s).
FRAP, SPT, and FCS analyses were also performed on HeLa cells expressing GFP-LactC2 (Figure 7). As reported for other cells (Yeung et al., 2008), GFP-LactC2 detects PS on the cytosolic aspect of the plasmalemma and of endocytic organelles (Figure 7A). Fluorescence at the membrane was photobleached and the diffusion coefficient calculated using Gaussian fitting (Figure 7B) of the recovery curves, as before. As found for the supported lipid bilayers, the interaction of GFP-LactC2 with cellular PS was reversible. The dissociation time of GFP-LactC2 from the membrane averaged 12.5 s (Figure 7C), somewhat longer than that of PH-PLCδ-GFP, the reversible phosphatidylinositol (4,5)-bisphosphate-binding domain used by Hammond et al. (2009) but orders of magnitude shorter than that of a diacylated probe (PM-GFP, a GFP-tagged form of the membrane-targeting sequence of Lyn kinase). FRAP analysis of GFP-LactC2 in the membrane of HeLa cells yielded an average D of 0.33 μm2/s (Table 3 and Figure 7D). This value is significantly lower than the coefficients calculated for PM-GFP and PH-PLCδ-GFP (D = 1.35 and 1.21 μm2/s, respectively; Figure 7D) and is consistent with the notion that (a fraction of) PS associates with complexes of reduced mobility.
FIGURE 7:

Movement of PS-associated GFP-LactC2 in the plasma membrane. (A) The mobility of PS-associated GFP-LactC2 was analyzed by FRAP in HeLa cells at 37°C. Representative images are shown, with magnifications of the indicated area shown in insets. Scale bar, 10 μm. (B) Sample Gaussian best fits of fluorescence intensity profiles obtained during FRAP of GFP-LactC2 in HeLa cells. (C, D) HeLa cells were transfected with PM-GFP, PLCδ-GFP, or GFP-LactC2 and subjected to FRAP, followed by analysis as described in Hammond et al. (2009). Average values of τ and D are shown in C and D, respectively. Data are means ± SEM of 18–39 independent determinations. (E) Representative image (i) and tracks (ii) of single PS-associated GFP-LactC2 particles in the ventral membrane of HeLa cells detected by TIRFM. Scale bar, 2 μm. Graph in (iii) shows classification breakdown obtained for 29,007 single-particle tracks like those in (i). (F) Normalized FCS curves representing the following constructs: red, GFP; blue, GFP-LactC2; green, GFP-LactC2(AAA). See Supplementary Methods for a description of the autocorrelation function (G(τ)). Dashed lines are measured, whereas the full lines are fitted curves. (G) Fraction of slow components as measured by FCS for GFP-LactC2 (blue) and GFP-LactC2(AAA) (green). The lines indicate the mean value of the fraction.
The mobility of PS in the membrane was also assessed by tracking single GFP-LactC2 molecules monitored by TIRFM in HeLa cells (Figure 7E). These studies indicated that 78% of the detected molecules displayed free diffusion, whereas 22% were confined within an average area of 360 ± 3 nm. Using this approach, we estimated D to average 0.44 μm2/s (Table 3). Finally, we used also FCS to investigate the behavior of PS in the membrane of cells expressing GFP-LactC2. Because the focal plane of FCS exceeds the thickness of the membrane, and because GFP-LactC2 is a reversible probe that coexists bound to membranes and in solution, we additionally analyzed the AAA mutant of GFP-LactC2, which is not well retained by the membrane, as well as soluble GFP. Typical autocorrelation curves are shown in Figure 7F. Two diffusion coefficients were detected for GFP-LactC2: D = 0.46 ± 0.03 μm2/s for ∼60% of the molecules and 26.43 ± 1.40 μm2/s for the remainder (Figure 7, F and G). The lower value is similar to that obtained for TopFluor-PS using FCS, suggesting that it reflects LactC2 bound to plasmalemmal PS. The higher diffusion coefficient likely represents soluble LactC2, since it resembles the D calculated for free GFP (19.21 ± 1.83 μm2/s). In accordance with this interpretation, the vast majority (82%) of the AAA mutant LactC2 displayed a high diffusion coefficient (22.63 ± 1.52 μm2/s), with only a small fraction (Figure 7G) existing in a seemingly membrane-bound form (D = 1.28 ± 0.16 μm2/s).
Cholesterol affects PS mobility in the plasmalemma
Cholesterol affects the diffusion rates of a variety of lipids and membrane-anchored proteins (Hao et al., 2001; Klein et al., 2003; Kenworthy et al., 2004; Vrljic et al., 2005; Nishimura et al., 2006; Pucadyil and Chattopadhyay, 2006; Shrivastava et al., 2010). We therefore tested the effects of varying the cholesterol content of HeLa cells on PS dynamics. Cholesterol was removed using methyl-β-cyclodextrin (MβCD) or increased by incubation with excess cholesterol loaded onto MβCD. The effectiveness of these procedures was validated by measuring the cellular cholesterol content using cholesterol oxidase and Amplex Red (Figure 8A). FRAP of cells expressing GFP-LactC2, followed by analysis by the method of Hammond et al. (2009), indicated that the diffusion coefficient of PS in the plasmalemma decreased significantly (from 0.33 to 0.10 μm2/s) when cholesterol content was increased (Figure 8B), as found for supported lipid bilayers. Remarkably, diffusion was also retarded (D = 0.15 μm2/s) when cholesterol was depleted (Figure 8B). The reduction in diffusion coefficient of PS in cells with decreased cholesterol was confirmed by FRAP measurements of TopFluor-PS mobility in cells extracted with MβCD (Figure 8C). A similar paradoxical effect has been reported for several membrane-associated proteins (Hao et al., 2001; Kenworthy et al., 2004; Vrljic et al., 2005; Nishimura et al., 2006).
FIGURE 8:

Cholesterol influences PS movement in the plasma membrane. (A) Verification of cholesterol depletion and enrichment. HeLa cells were either left untreated (control) or were exposed for 30 min to 10 mM MβCD in the absence or presence of excess (50 μg/ml) cholesterol. The cholesterol content of the cells was then measured using cholesterol oxidase-Amplex Red, as described in Materials and Methods. Data are means ± SEM of four independent experiments. (B) Diffusion coefficients of PS in HeLa cells before and after cholesterol depletion or enrichment as obtained by FRAP analysis of GFP-LactC2 (C) Diffusion coefficients of TopFluor-PS in HeLa cells before and after cholesterol depletion accomplished by 30-min incubation with 10 mM MβCD. Data are means ± SEM of 21–40 independent determinations.
Diffusion of PS on the cytosolic leaflet of intracellular membranes
In addition to the plasmalemma, PS is also present in various organellar membranes. Most of these, however, are too small to analyze by FRAP. To analyze the behavior of PS in an endomembrane compartment, we chose the phagosome. Phagosomes suitable for FRAP determinations can be generated by exposing macrophages to large particles, such as sheep red blood cells (4–5 μm in diameter; Figure 9A). Phagosomes generated by RAW264.7 cells transfected with GFP-LactC2 were subjected to photobleaching between 15 and 45 min after particle ingestion. Determinations of fluorescence recovery indicated that PS diffuses at a similar rate in this endocytic organelle (0.13 ± 0.05 μm2/s) as found for the plasma membrane of the same cells (0.18 ± 0.03 μm2/s; Figure 9B). Note that PS diffusion in general was somewhat slower in RAW264.7 macrophages compared with HeLa cells, estimated using the same method (compare Figures 7D and 9B).
FIGURE 9:

Measurements of PS in intracellular membranes. (A, B) Measurement of the diffusion coefficient of PS in phagosomes by FRAP analysis of GFP-LactC2. (A) Example of phagosomes used to perform FRAP analysis in RAW macrophages. Photobleached area is encircled. Scale bar, 5 μm. (B) Comparison of diffusion coefficients calculated for the plasma membrane and phagosomal membrane. Data are means ± SEM of 19–32 independent determinations. (C) Schematic structures for the constructs used for detection of fluorescence in the ER: ss-LactC2-GFP-KDEL and ss-GFP-KDEL. (D) Spinning-disk confocal images of HeLa cells transfected with ss-LactC2-GFP-KDEL, ss-LactC2(AAA)-GFP-KDEL, or ss-GFP-KDEL and immunostained for protein disulfide isomerase (PDI). Scale bars, 10 μm. (E) Normalized FCS curves for the following constructs: red, ss-GFP-KDEL; blue, ss-LactC2-GFP-KDEL; green, ss-LactC2(AAA)-GFP-KDEL. Dashed lines are measured, whereas the solid lines are fitted curves. (F) Fraction of slow components measured by FCS in each cell for different proteins. The lines indicate the mean value of the fraction. (G) Comparison of the fraction of total molecules assigned to the slow component for the different constructs, calculated by FCS. Data are means ± SEM of 29–41 independent determinations.
Diffusion of PS on the luminal leaflet of intracellular membranes
PS is synthesized on the cytosolic leaflet of the ER and can be subsequently translocated across the bilayer (Stone and Vance, 2000; Leventis and Grinstein, 2010). Although the precise transmembrane distribution of PS in the ER has not been definitively established, several studies suggested that the majority of the PS is on the inner (luminal) leaflet (Bollen and Higgins, 1980; Boon and Smith, 2002). This afforded us the opportunity to study the behavior of PS in the luminal aspect of an organellar membrane.
To this end, we selectively targeted GFP-LactC2 to the ER by generating a construct that included the signal sequence of preprolactin, as well as an ER retention motif (KDEL) at the C-terminus (Figure 9C). We verified that this conformation of the probe did not interfere with PS binding; the modified LactC2 domain bound effectively to PS-coated Nucleosil beads (Supplemental Figure S5). When transfected into HeLa cells, the resulting construct (ss-LactC2-GFP-KDEL) displayed a reticular appearance and was found lining the nuclear membrane, consistent with ER localization. Indeed, ss-LactC2-GFP-KDEL colocalized extensively with protein disulfide isomerase, an ER-resident enzyme (Figure 9D; Pearson's coefficient, 0.65 ± 0.03). We initially used FRAP to assess the mobility of ss-LactC2-GFP-KDEL. Because, like the parental molecule, ss-LactC2-GFP-KDEL interacts reversibly with PS, FRAP analysis requires Gaussian fittings to line scans across the bleached area. Because the convolutions of the cytoplasmic ER make such analysis impossible, we bleached only regions of the nuclear membrane, which showed the most distinct linear membranous structures (Figure 9D). We calculated an average D of 0.79 ± 0.08 μm2/s (Table 4) and an average τ of 5.05 ± 0.70 s.
TABLE 4:
Diffusion coefficients of GFP constructs in the ER estimated using FRAP and FCS (μm2/s ± SEM).
| GFP-LactC2 FRAP (n) | ss-GFP-KDEL FCS (n) | ss-LactC2-GFP-KDEL FCS (n) | ss-LactC2(AAA)-GFP-KDEL FCS (n) | |
|---|---|---|---|---|
| D of the fast component | n.a. | 23.62 ± 1.86 (41) | 18.52 ± 1.51 (30) | 18.54 ±1.51 (35) |
| D of the slow component | 0.79 ± 0.08 (15) | 0.78 ± 0.09 (41) | 0.57 ± 0.04 (30) | 0.53 ± 0.05 (35) |
| Fraction of slow component | n.a. | 32% ± 1% (41) | 48% ± 2% (30) | 33% ± 2% (35) |
We were concerned that protein crowding within the lumen of the ER might have affected the estimation of the diffusion coefficient, reducing the mobility of the probe in a manner independent of PS. This possibility was tested by generating an ER-targeted form of the AAA mutant of LactC2 (ss-LactC2(AAA)-GFP-KDEL) that binds poorly to PS. We also expressed ER-targeted GFP lacking the LactC2 domain (ss-GFP-KDEL; Figure 9C). Both of these probes were found to recover rapidly after photobleaching, with recovery half-times of 2.7 s (95% confidence interval [CI], 2.4–3.1 s) and 3.0 s (95% CI, 2.7–3.4 s), respectively. These recoveries were significantly faster than ss-LactC2-GFP-KDEL (4.3 s; 95% CI, 4.0–4.5 s). Moreover, the recovery patterns of the soluble ER probes did not fit Gaussian patterns, indicating that lateral diffusion of membrane-bound probe did not contribute significantly to the fluorescence recovery. Together, these observations imply that crowding of luminal molecules did not alter measurably the mobility of the wild-type ss-LactC2-GFP-KDEL bound to PS.
We also measured PS dynamics in the ER using FCS. This method enabled us to assess the properties of PS in regions other than the nuclear membrane, which may have unique properties, and provided a more reliable comparison of ss-LactC2-GFP-KDEL with the soluble ss-LactC2(AAA)-GFP-KDEL and ss-GFP-KDEL. Analysis of the autocorrelation curves for ss-LactC2-GFP-KDEL fluorescence revealed the existence of two components with distinct diffusion coefficients. The fast-diffusing component (D = 18.52 ± 1.51 μm2/s; n = 30) likely represents unbound ss-LactC2-GFP-KDEL, as well as some GFP-containing degradation products, which are detectable by immunoblotting (Supplemental Figure S6). The slow-diffusing fraction (D = 0.57 ± 0.04 μm2/s) reflects the diffusion of PS but likely also includes some ss-LactC2-GFP-KDEL bound to the KDEL receptor and/or protein nonspecifically associated with the ER membrane (∼30%; Figure 9F). The latter was concluded from parallel FCS analyses of ss-LactC2(AAA)-GFP-KDEL and ss-GFP-KDEL. As expected, these constructs displayed a large rapidly diffusing fraction (18.54 ± 1.51, n = 35; and 23.62 ± 1.86, n = 41; respectively). However, they also showed a slower-diffusing component. Although significantly smaller than that noted for the wild-type ss-LactC2-GFP-KDEL, this smaller component was not insignificant (Figure 9, F and G). The uncertainty introduced by this component precludes accurate estimation of the diffusion of PS in the luminal aspect of the ER. On the other hand, the greater magnitude of the slow component of ss-LactC2-GFP-KDEL compared with ss-LactC2(AAA)-GFP-KDEL and ss-GFP-KDEL validates the existence of a pool of luminal PS that diffuses in the plane of the membrane.
Concentration of PS in the ER
The FCS measurements not only confirmed the presence of luminal PS in the ER of HeLa cells, but they also enabled us to calculate the minimum concentration of the phospholipid in the ER. In transiently transfected cells ss-LactC2-GFP-KDEL is expressed at varying levels, ranging from ∼2 to 200 molecules in the observation volume of 0.75 fl (see Supplementary Material). When calibrated with a concentration standard, this corresponds to concentrations of 5 nM to 0.5 μM. Of note, the slowly diffusing fraction changed little despite the wide range (two orders of magnitude) of concentrations analyzed. Because on average ∼35% of the construct is cleaved (as quantified by immunoblotting), this implies that most of the intact ss-LactC2-GFP-KDEL is membrane bound at any time. Making use of the Kd for the ss-LactC2-GFP-KDEL/PS complex estimated earlier (∼300 nM; Figure 2), we can estimate a lower limit for the concentration of PS in the lumen of the ER as follows: the bound fraction of molecules y in FCS is given by
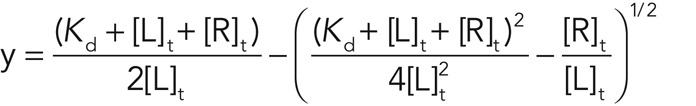 |
where [L]t is the total ligand concentration (LactC2) and [R]t is the total receptor (PS) concentration (Wohland et al., 1999). For the bound fraction y, assuming close to full binding, we use values between 0.85 and 0.99. Solving the equation for [R]t, we estimate the PS concentration in the ER to be in the range of 2–30 μM. This is a lower limit, since the estimated PS concentration steadily increases with the value of y used. Note that 30 μM in a volume of 1 fl (= 1 μm3) would correspond to about 18,000 molecules.
DISCUSSION
The appearance and properties of PS on the outer leaflet of activated or apoptotic cells have been studied extensively, mostly using annexin V. By contrast, the analysis of endomembrane PS has been severely limited by the paucity of suitable intracellular probes. Here we analyzed several aspects of the behavior of intracellular PS using two novel fluorescent probes and multiple biophysical approaches. TopFluor-PS is a better mimic of PS than the NBD-conjugated form; the hydrophobicity of the TopFluor moiety produces less distortion of the molecular structure, which is reflected in its greater retention by (lower extractability from) the bilayer. Moreover, TopFluor is more photostable than NBD.
Although inserted deeply in the bilayer (Table 1), TopFluor is nevertheless bulkier and unlikely to replicate entirely the behavior of the native acyl chain of PS. In addition, TopFluor-PS undergoes time-dependent degradation, which limits its usefulness to a window of ∼30 min at 37°C. Both of these shortcomings are obviated by LactC2-GFP, which interacts with endogenous PS. The comparatively large probe does not hinder the mobility of the complex significantly, since the viscosity of the hydrophobic bilayer is much greater than that of the aqueous milieu. Accordingly, the diffusion of free LactC2-GFP in the cytosol, deduced from the behavior of the AAA mutant, is more than an order of magnitude faster than that of the membrane-associated LactC2-GFP/PS complex. A second concern—scavenging of PS that may result in altered cell function—is also unlikely to be significant. PS is abundant—the average cellular concentration is estimated to be well in excess of 100 μM (Blom et al., 2001; Junqueira et al., 1957; Kay and Grinstein, 2011)—whereas GFP-tagged fluorescent probes like LactC2-GFP are generally expressed at low micromolar concentrations (Botelho et al., 2000). Nevertheless, there are notable limitations to the use of LactC2-GFP as a probe for PS. First, PS molecules whose head groups are engaged by endogenous proteins or obscured by other lipids may not be detected by LactC2-GFP. Second, cytosolically expressed LactC2-GFP has no access to, and therefore fails to detect, PS in the lumen of intracellular organelles. Because of the limitations of the individual probes, we believe that a combined approach using both TopFluor-PS and LactC2-GFP yields a more accurate reflection of the behavior of PS.
That LactC2-GFP and TopFluor-PS are suitable and complementary is suggested by the similarity of the results obtained with the two probes. Comparable diffusion coefficients were estimated for supported lipid bilayers using the two probes (Table 2). In fact, the diffusion coefficients varied less between probes than they did when the same probe was assessed by two different methods (Table 3). Differences in the calculated diffusion coefficient between measurement techniques are not unprecedented (Saxton and Jacobson, 1997; Bates et al., 2006; Guo et al., 2008; Owen et al., 2009) and can be rationalized: FRAP measurements are dominated by those fluorophores that travel comparatively long distances, whereas SPT determinations are influenced more by molecules that diffuse within confined areas (Bates et al., 2006; Rayan et al., 2010). There is also a bias toward the detection of slower-moving probes by TIRFM/SPT because identification of the faster molecules is less reliable; the ability of the software to adjust Gaussian fits to their fluorescence pattern often fails if they undergo significant displacement within the period used for image acquisition (32 ms in our experiments). Furthermore, although in SPT one measures the behavior of a single particle over a relatively large area, in FCS one observes many particles passing through a very small area (Guo et al., 2008). However, the FCS experiments clearly show that there are membrane-bound TopFluor-PS molecules with diffusion coefficients in the expected range and possibly some soluble degradation products with much faster diffusion in a smaller subset of the measurements.
The experiments described here yielded two salient findings. First, using FRAP and SPT, we detected a sizable fraction of PS that was immobile or poorly mobile (confined). Although interaction of PS with cholesterol in microdomains could in principle account for this observation, we found that D was reduced both by increasing and by depleting cholesterol from the membrane (Figure 8). Instead, we believe that the reduced mobility is more likely due to association of PS with protein complexes that become trapped in picket fences established by the cortical actin cytoskeleton. This is suggested by observations made in membrane blebs devoid of cytoskeleton, where the fractional mobility of PS increased to >0.9 (Figure 6). That PS forms part of comparatively stable macromolecular complexes is consistent with a recent report that, when visualized by immunogold, PS in the inner leaflet was not homogeneously dispersed (Ripley's K function >1; Fairn et al., 2011b). As indicated, SPT gave two sizes of apparent corralling, depending on the probe used, 111 ± 4 nm for TopFluor-PS and 360 ± 3 nm for GFP-LactC2, as well as vastly differing fractions of the probe confined in those corrals (compare Figures 6F and 7E). This difference is likely a result of the nature of the probe, with TopFluor-PS having a free phosphoserine head group able to interact with endogenous proteins to form part of macromolecular complexes. GFP-LactC2, in contrast, is able to interact with endogenous PS, but only if it has a free head group for binding LactC2, preferentially reporting on PS that is not part of larger complexes. This again highlights the importance of using two different probes for PS to gain a deeper understanding of its dynamics.
The second finding of note was the observation that PS is present in the luminal membrane of the ER in intact cells. This conclusion was based on both FRAP and FCS determinations of the mobility of ss-LactC2-GFP-KDEL, a genetically encoded probe targeted to the lumen of the ER by a signal sequence. Escape of the soluble probe via the secretory pathway is minimized by introduction of a C-terminal KDEL sequence, which promotes retrieval of the protein from the more acidic Golgi complex by KDEL receptors. At the neutral pH prevailing in the ER lumen, however, the interaction between the receptor and the KDEL-tagged probe is weak, and the majority of the probe is released into the lumen. The small fraction that remains bound to the receptors could be distinguished from that associated to PS by comparing ss-LactC2-GFP-KDEL with ss-LactC2(AAA)-GFP-KDEL and ss-GFP-KDEL, which bind poorly, if at all, to PS. The larger fraction of slowly diffusible ss-LactC2-GFP-KDEL is a reliable indicator of the presence of PS in the lumen of the ER. The existence of a fraction of the probe that is confined by binding to KDEL and possibly by other nonspecific interactions complicates the quantification of the concentration of PS. Nevertheless, by subtraction of this component we estimate that the concentration of PS in the ER lumen is at least 2–30 μM.
Previous determinations of the sidedness of PS in the ER were made in fractionated cells and yielded conflicting results. Some researchers believe that, like other lipids, PS is distributed more or less symmetrically across the bilayer by a scramblase intended to deliver newly synthesized lipids to the luminal leaflet, thereby minimizing membrane curvature (Buton et al., 1996; Pomorski et al., 2001; Kol et al., 2004). Other studies, however, showed that the bulk of the PS is latent inside ER-derived vesicles (Higgins and Dawson, 1977; Bollen and Higgins, 1980; Vale, 1977), implying that directional flipping, not random scrambling, takes place. Our results cannot unambiguously distinguish between these proposals. The failure to detect PS in the ER of cells expressing cytosolic LactC2-GFP, which was also reported earlier (Yeung et al., 2008), is not necessarily an indication of asymmetry; because PS is abundant in the cytosolic leaflet of the plasma and endocytic membranes (van Meer et al., 2008), most of the cytosolically expressed probe could be preferentially recruited to these compartments, leaving little available to bind the lower amounts of PS on the cytosolic aspect of the ER. On the other hand, recent evidence using on-section labeling of frozen sections analyzed by electron microscopy (Fairn et al., 2011b) revealed considerable labeling of ER membranes with recombinant LactC2. When considered together with these findings, our data make a compelling case for asymmetrical PS distribution in the ER.
How and where PS becomes concentrated en route from the ER to the plasma membrane are also unclear. Our observations suggest one possible mechanism: PS located in the lumen of the ER may travel to the trans-Golgi network, where phosphoaminolipid flippases translocate it to the cytosolic monolayer. We propose that PS is accumulated in subdomains of the trans-Golgi network, possibly by coat-forming proteins with polycationic motifs. By selectively budding these subdomains, vesicles highly enriched in PS would be preferentially delivered to the plasma membrane.
In summary, we present the first reliable determinations of the dynamics of PS in the cytosolic leaflet of the plasma membrane and of an endocytic organelle, as well as in the luminal aspect of the ER, using a variety of approaches. Some of these could be modified and extended for use in other organelles if suitable targeting signals are incorporated when designing a new generation of probes.
MATERIALS AND METHODS
Lipids and reagents
The lipids TopFluor-PS, NBD-PS, TopFluor-PC, 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phosphoethanolamine (TopFluor-PE), DOPC, DOPS, POPE, 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol) (DOPI), 5-doxyl-PC, 1-palmitoyl-2-stearoyl-(12-doxyl)-sn-glycero-3-phosphocholine (12-doxyl-PC), 1-palmitoyl-2-stearoyl-(16-doxyl)-sn-glycero-3-phosphocholine (16-doxyl-PC), 1,2-dipalmitoyl-sn-glycero-3-phospho(tempo)choline (Tempo-PC), egg-transphosphatidylated Rhod-PE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl) (dansyl-PE), egg-PC, and brain-PS were obtained from Avanti Polar Lipids (Alabaster, AL). Dyngo-4a was a kind gift from Phillip Robinson (Children's Medical Research Institute, Westmead, Australia). Glutathione Sepharose and PreScission protease were from GE Healthcare (Little Chalfont, United Kingdom), and cholesterol and all other chemicals were from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Liposomes and supported bilayers
The liposome compositions used in this study are listed in Table 5. Liposomes were made by adding the required amount of stock lipids (in chloroform) into a glass vial and drying under a stream of N2. The lipid was resuspended in liposome buffer (140 mM KCl, 0.5 mM MgCl2, 15 mM NaCl, and 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5) by vortexing and sonication. Uniform large unilamellar liposomes were made by passing through a LiposoFast extruder with a filter of 100 nm pore size (Avestin, Ottawa, Canada). To make supported lipid bilayers, liposomes were mixed with 1 M NaCl on piranha solution–washed glass coverslips (Marienfeld, Lauda-Königshofen, Germany) as described previously (Knight and Falke, 2009) and incubated for 1 h at 37°C. After extensive washing with Milli-Q water, supported bilayers were imaged in phosphate-buffered saline (PBS).
Spectroscopic measurements and parallax calculations
Spectroscopy was performed with an F-2500 fluorescence spectrometer (Hitachi High-Tech, Tokyo, Japan) using 0.75 mM total lipids in liposome buffer. Emission scans were made in 2.5-nm steps using a 5-nm slit; excitation wavelength was 470 nm for NBD-PS and 485 nm for TopFluor-PS. Parallax calculations were performed as described by Chattopadhyay and London (1987) and Kaiser and London (1998). For calculations involving 16-doxyl-PC quenching of TopFluor-PS, special care was taken to account for transleaflet quenching, using the calculations described by Chattopadhyay and London (1987).
GST- and GFP-LactC2 purification
A construct for the expression of GST-GFP-LactC2 was engineered with pGEX6P (GE Healthcare), which introduces a recognition site for cleavage by PreScission protease. Purification of the resultant protein was performed according to manufacturer's instructions. Briefly, GST-GFP-LactC2 expression was induced in Escherichia coli, and the cell lysate was loaded onto a glutathione–Sepharose column. PreScission protease was used to cleave GST from the construct, and the resultant GFP-LactC2 was collected and concentrated.
GST-LactC2 WT or AAA for in vitro FRET analysis were also produced in E. coli, which were pelleted and lysed in 20 ml of B-PER (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Cell lysate supernatants were bound to a glutathione–Sepharose column. The slurry was washed with PBS, and the protein was eluted with 25 mM glutathione in PBS, pH 7.4, in 500-μl fractions. The fractions were analyzed for GST-LactC2 by Coomassie staining of SDS–PAGE gels, the positive fractions were pooled and dialyzed in PBS at 4°C overnight to remove excess glutathione.
Cell culture, transfections, drug treatments, cholesterol measurements, lipid loading, and extraction
Cells were maintained at 37°C with 5% CO2 in αMEM (Wisent, St. Bruno, Canada) supplemented with 10% heat-inactivated serum (HeLa cells) or RPMI (Wisent) with 10% serum (RAW264.7 cells). For plasmid transfections, 2 μg of DNA and 3 μl of Fugene 6 (Roche, Basel, Switzerland; HeLa cells) or 5 μl of Fugene HD (RAW cells) per ml of cell medium were premixed and added to cells. After 4–15 h (depending on expression level required) cells were imaged.
Cholesterol was removed from cells by incubating with 10 mM MβCD for 30 min in serum-free medium, and cholesterol was added to cells by incubating cells for 30 min with 50 μg/ml cholesterol complexed with 1.5 mg/ml MβCD (Blom et al., 2001). Cholesterol determinations were performed with an Amplex Red Cholesterol Assay Kit (Invitrogen, Carlsbad, CA), following the manufacturer's protocol.
To add TopFluor-PS and NBD-PS to cells, the required amount of stock lipid in chloroform was dried under N2, resuspended in methanol, and injected into a vortex PBS containing 3 mg of bovine serum albumin (BSA). Generally, 10 nmol of lipid per ml of PBS:BSA was used. The lipid:BSA:PBS mixture was incubated with the cells on ice for 5–30 min, followed by washing three times with PBS before placing the cells in serum-free medium for subsequent analysis.
The Folch method of lipid extraction was used (Folch et al., 1957), and TLC was performed on 250-μm silica gel TLC plates (Whatman, Maidstone, United Kingdom) with a resolving solution of CHCl3, MeOH, CH3COOH, and H2O (25:15:4:1). TopFluor-PS was visualized with a Storm 840 chemiluminescence imager system (Molecular Dynamics, Sunnyvale, CA).
Where indicated, the phospholipase inhibitors bromoenol lactone, FIPI, and U73122 were used at final concentrations of 20 μM, 750 nM, and 10 μM, respectively.
LactC2 Kd measurements by fluorescence resonance energy transfer
Binding of GST-LactC2 to liposomes was measured via FRET from intrinsic tryptophan residues to dansyl linked to PE head groups as described previously (Gilbert et al., 1990). Experiments were performed in a 5 × 5 × 30 mm quartz cuvette in a Hitachi F-2500 fluorescence spectrophotometer at room temperature. Excitation and emission wavelengths were 280 and 510 nm, respectively, with 5-nm slit widths. The total lipid concentration for the measurement was adjusted to 10 μM, GST-LactC2 was added in increments, and the fluorescence was recorded and corrected for dilution. Data were analyzed as previously described (Gilbert et al., 1990).
Preparation of Nucleosil C18 silica beads for ss-LactC2-GFP-KDEL binding assay
We coated 0.5 mg of Nucleosil C18 silica beads (Macherey-Nagel, Düren, Germany) with 1 μmol of lipids (100% egg-PC or 95% egg-PC/5% brain-PS) by suspending them in chloroform, drying the mixture under N2, and rehydrating them in 20 mM Tris-HCl, pH 7.0. The beads were bath sonicated and washed several times before use.
FCS setup
FCS measurements were conducted on an Olympus FV 300 confocal microscope (Olympus, Tokyo, Japan). Fluorescence excitation was achieved with the 488-nm line of an argon ion laser (Melles Griot, Albuquerque, NM). The laser beam was focused into the back aperture of a water-immersion objective (60×, numerical aperture [NA] 1.2; Olympus). The emitted light passed a 3× magnification system and was spatially filtered by a 150-μm pinhole. The emitted fluorescence of the proteins was collected by the same objective, filtered by a band-pass filter (510AF23; Omega Optical, Brattleboro, VT), and detected by an avalanche photodiode (SPCM-AQR-14-FC; Pacer, Berkshire, United Kingdom). The autocorrelation curves were computed online by a hardware correlator (Flex02-01D; Correlator.com, Bridgewater, NJ). The laser power was set to 10 μW, as measured before the microscope objective.
Confocal microscopy, FRAP, TIRF, and SPT
Confocal microscopy and FRAP were performed on a WaveFX spinning-disk microscopy system (Quorum Technologies, Guelph, Canada) equipped with a Mosaic digital diaphragm for photobleaching and operated with Volocity software, version 4.4 (PerkinElmer-Cetus, Waltham, MA). TIRF microscopy was performed on a cellˆTIRF system (Olympus) equipped with 50-mW 491- and 532-nm lasers, a 150×, 1.45 NA lens and operated with MetaMorph Advanced, version 7.7 (Molecular Devices, Sunnyvale, CA). Image analysis was performed with Volocity and ImageJ (National Institutes of Health, Bethesda, MD). FRAP analysis of TopFluor-PS was performed with traditional calculations (Axelrod et al., 1976), and FRAP analysis of the GFP-LactC2 and other GFP-tagged probes was performed as described (Hammond et al., 2009). For SPT, raw images were cropped in ImageJ, and tracking was performed with Matlab (MathWorks, Natick, MA), detecting local intensity maxima and then fitting two-dimensional Gaussian kernels approximating the point-spread function of the microscope as described previously (Jaqaman et al., 2008). We did not perform time averaging of our images, to improve our ability to detect fast-moving molecules. SPT diffusion analysis and type classification were performed using a moment scaling spectrum analysis (Ewers et al., 2005; Ferrari and Manfroi, 2001) as described previously (Flannagan et al., 2010).
Additional method details are included in the Supplementary Material.
Supplementary Material
Acknowledgments
We thank R. Flannagan for his help. This work was supported by Canadian Institutes of Health Research Grants MOP7075 and MOP93634. S.G. is the holder of the Pitblado Chair in Cell Biology at The Hospital for Sick Children and is cross-appointed to the Department of Biochemistry, University of Toronto, Toronto, Canada.
Abbreviations used:
- D
diffusion coefficient
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPI
1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol)
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- FCS
fluorescence correlation spectroscopy
- Fm
mobile fraction
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescence resonance energy transfer
- NBD-PS
1-palmitoyl-2-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]lauroyl}-sn-glycero-3-phospho-l-serine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- PS
phosphatidylserine
- SPT
single-particle tracking
- TIRFM
total internal reflection fluorescence microscopy
- TopFluor-PC
1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phosphocholine
- TopFluor-PS
1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phospho-l-serine
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-11-0936) on April 11, 2012.
REFERENCES
- Abrams FS, London E. Extension of the parallax analysis of membrane penetration depth to the polar region of model membranes: use of fluorescence quenching by a spin-label attached to the phospholipid polar headgroup. Biochemistry. 1993;32:10826–10831. doi: 10.1021/bi00091a038. [DOI] [PubMed] [Google Scholar]
- Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT. Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry. 2000;39:6200–6206. doi: 10.1021/bi992221r. [DOI] [PubMed] [Google Scholar]
- Arikketh D, Nelson R, Vance JE. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 2008;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Pagano RE. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 1997;36:8840–8848. doi: 10.1021/bi970145r. [DOI] [PubMed] [Google Scholar]
- Bates IR, Hébert B, Luo Y, Liao J, Bachir AI, Kolin DL, Wiseman PW, Hanrahan JW. Mass spectrometric analysis reveals an increase in plasma membrane polyunsaturated phospholipid species upon cellular cholesterol loading. Biochemistry. 2001;40:14635–14644. doi: 10.1021/bi0156714. [DOI] [PubMed] [Google Scholar]
- Bollen IC, Higgins JA. Phospholipid asymmetry in rough- and smooth-endoplasmic-reticulum membranes of untreated and phenobarbital-treated rat liver. Biochem J. 1980;189:475–480. doi: 10.1042/bj1890475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon JM, Smith BD. Chemical control of phospholipid distribution across bilayer membranes. Med Res Rev. 2002;22:251–281. doi: 10.1002/med.10009. [DOI] [PubMed] [Google Scholar]
- Botelho RJ, Teruel M, Dierckman R, Anderson R, Wells A, York JD, Meyer T, Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buton X, Morrot G, Fellmann P, Seigneuret M. Ultrafast glycerophospholipid-selective transbilayer motion mediated by a protein in the endoplasmic reticulum membrane. J Biol Chem. 1996;271:6651–6657. doi: 10.1074/jbc.271.12.6651. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay A, London E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 1987;26:39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- Chen CS, Martin OC, Pagano RE. Changes in the spectral properties of a plasma membrane lipid analog during the first seconds of endocytosis in living cells. Biophys J. 1997;72:37–50. doi: 10.1016/S0006-3495(97)78645-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W, Stahelin RV. Membrane binding and subcellular targeting of C2 domains. Biochim Biophys Acta. 2006;1761:838–849. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Daleke DL. Phospholipid flippases. J Biol Chem. 2007;282:821–825. doi: 10.1074/jbc.R600035200. [DOI] [PubMed] [Google Scholar]
- Elvington SM, Nichols JW. Spontaneous, intervesicular transfer rates of fluorescent, acyl chain-labeled phosphatidylcholine analogs. Biochim Biophys Acta. 2007;1768:502–508. doi: 10.1016/j.bbamem.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers H, Smith AE, Sbalzarini IF, Lilie H, Koumoutsakos P, Helenius A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc Natl Acad Sci USA. 2005;102:15110–15115. doi: 10.1073/pnas.0504407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, Hermansson M, Somerharju P, Grinstein S. Phosphatidylserine is polarized and required for proper Cdc42 localization and for development of cell polarity. Nat Cell Biol. 2011a;13:1424–1430. doi: 10.1038/ncb2351. [DOI] [PubMed] [Google Scholar]
- Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S, Parton RG. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol. 2011b;194:257–275. doi: 10.1083/jcb.201012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Manfroi A. Nonlinear phenomena: strongly and weakly self-similar diffusion. Physica D. 2001;154:111–137. [Google Scholar]
- Filippov A, Orädd G, Lindblom G. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys J. 2003;84:3079–3086. doi: 10.1016/S0006-3495(03)70033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Harrison RE, Yip CM, Jaqaman K, Grinstein S. Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J Cell Biol. 2010;191:1205–1218. doi: 10.1083/jcb.201007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gilbert GE, Furie BC, Furie B. Binding of human factor VIII to phospholipid vesicles. J Biol Chem. 1990;265:815–822. [PubMed] [Google Scholar]
- Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol Biol Cell. 2008;19:1663–1669. doi: 10.1091/mbc.E07-12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Har JY, Sankaran J, Hong Y, Kannan B, Wohland T. Molecular diffusion measurement in lipid bilayers over wide concentration ranges: a comparative study. Chem Phys Chem. 2008;9:721–728. doi: 10.1002/cphc.200700611. [DOI] [PubMed] [Google Scholar]
- Hammond GRV, Sim Y, Lagnado L, Irvine RF. Reversible binding and rapid diffusion of proteins in complex with inositol lipids serves to coordinate free movement with spatial information. J Cell Biol. 2009;184:297–308. doi: 10.1083/jcb.200809073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M, Mukherjee S, Maxfield FR. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc Natl Acad Sci USA. 2001;98:13072–13077. doi: 10.1073/pnas.231377398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CB, et al. Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J Biol Chem. 2011;286:35966–35976. doi: 10.1074/jbc.M111.283879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JA, Dawson RM. Asymmetry of the phospholipid bilayer of rat liver endoplasmic reticulum. Biochim Biophys Acta. 1977;470:342–356. doi: 10.1016/0005-2736(77)90126-2. [DOI] [PubMed] [Google Scholar]
- Hill TA, et al. Inhibition of dynamin mediated endocytosis by the dynoles—synthesis and functional activity of a family of indoles. J Med Chem. 2009;52:3762–3773. doi: 10.1021/jm900036m. [DOI] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ID, Kang HC, Haugland RP. Fluorescent membrane probes incorporating dipyrrometheneboron difluoride fluorophores. Anal Biochem. 1991;198:228–237. doi: 10.1016/0003-2697(91)90418-s. [DOI] [PubMed] [Google Scholar]
- Junqueira L, Rothschild H, Fajer A. Protein production by the rat pancreas. Exp Cell Res. 1957;12:338–341. doi: 10.1016/0014-4827(57)90147-7. [DOI] [PubMed] [Google Scholar]
- Kaiser RD, London E. Determination of the depth of BODIPY probes in model membranes by parallax analysis of fluorescence quenching. Biochim Biophys Acta. 1998;1375:13–22. doi: 10.1016/s0005-2736(98)00127-8. [DOI] [PubMed] [Google Scholar]
- Kay J, Grinstein S. Sensing phosphatidylserine in cellular membranes. Sensors (Basel) 2011;11:1744–1755. doi: 10.3390/s110201744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Pillot T, Chambaz J, Drouet B. Determination of plasma membrane fluidity with a fluorescent analogue of sphingomyelin by FRAP measurement using a standard confocal microscope. Brain Res Brain Res Protoc. 2003;11:46–51. doi: 10.1016/s1385-299x(03)00016-3. [DOI] [PubMed] [Google Scholar]
- Knight JD, Falke JJ. Single-molecule fluorescence studies of a PH domain: new insights into the membrane docking reaction. Biophys J. 2009;96:566–582. doi: 10.1016/j.bpj.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Arakawa Y. Transport of exogenous fluorescent phosphatidylserine analogue to the Golgi apparatus in cultured fibroblasts. J Cell Biol. 1991;113:235–244. doi: 10.1083/jcb.113.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol MA, de Kroon AIPM, Killian JA, de Kruijff B. Transbilayer movement of phospholipids in biogenic membranes. Biochemistry. 2004;43:2673–2681. doi: 10.1021/bi036200f. [DOI] [PubMed] [Google Scholar]
- Korlach J, Schwille P, Webb WW, Feigenson GW. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc Natl Acad Sci USA. 1999;96:8461–8466. doi: 10.1073/pnas.96.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42:423–438. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- London E. Investigation of membrane structure using fluorescence quenching by spin-labels. A review of recent studies. Mol Cell Biochem. 1982;45:181–188. doi: 10.1007/BF00230086. [DOI] [PubMed] [Google Scholar]
- Marks DL, Bittman R, Pagano RE. Use of Bodipy-labeled sphingolipid and cholesterol analogs to examine membrane microdomains in cells. Histochem Cell Biol. 2008;130:819–832. doi: 10.1007/s00418-008-0509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OC, Pagano RE. Transbilayer movement of fluorescent analogs of phosphatidylserine and phosphatidylethanolamine at the plasma membrane of cultured cells. Evidence for a protein-mediated and ATP-dependent process(es) J Biol Chem. 1987;262:5890–5898. [PubMed] [Google Scholar]
- Martin OC, Pagano RE. Internalization and sorting of a fluorescent analogue of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanisms. J Cell Biol. 1994;125:769–781. doi: 10.1083/jcb.125.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura SY, Vrljic M, Klein LO, McConnell HM, Moerner WE. Cholesterol depletion induces solid-like regions in the plasma membrane. Biophys J. 2006;90:927–938. doi: 10.1529/biophysj.105.070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DM, Williamson D, Rentero C, Gaus K. Quantitative microscopy: protein dynamics and membrane organisation. Traffic. 2009;10:962–971. doi: 10.1111/j.1600-0854.2009.00908.x. [DOI] [PubMed] [Google Scholar]
- Pagano RE, Martin OC, Kang HC, Haugland RP. A novel fluorescent ceramide analogue for studying membrane traffic in animal cells: accumulation at the Golgi apparatus results in altered spectral properties of the sphingolipid precursor. J Cell Biol. 1991;113:1267–1279. doi: 10.1083/jcb.113.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomorski T, Holthuis JCM, Herrmann A, van Meer G. Tracking down lipid flippases and their biological functions. J Cell Sci. 2004;117:805–813. doi: 10.1242/jcs.01055. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Hrafnsdóttir S, Devaux PF, van Meer G. Lipid distribution and transport across cellular membranes. Semin Cell Biol. 2001;12:139–148. doi: 10.1006/scdb.2000.0231. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JCM. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell. 2003;14:1240–1254. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucadyil TJ, Chattopadhyay A. Effect of cholesterol on lateral diffusion of fluorescent lipid probes in native hippocampal membranes. Chem Phys Lipids. 2006;143:11–21. doi: 10.1016/j.chemphyslip.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Rayan G, Guet J-E, Taulier N, Pincet F, Urbach W. Recent applications of fluorescence recovery after photobleaching (FRAP) to membrane bio-macromolecules. Sensors (Basel) 2010;10:5927–5948. doi: 10.3390/s100605927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ, Jacobson K. Single-particle tracking: applications to membrane dynamics. Annu Rev Biophys Biomol Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- Shi J, Heegaard CW, Rasmussen JT, Gilbert GE. Lactadherin binds selectively to membranes containing phosphatidyl-L-serine and increased curvature. Biochim Biophys Acta. 2004;1667:82–90. doi: 10.1016/j.bbamem.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Shrivastava S, Pucadyil TJ, Paila YD, Ganguly S, Chattopadhyay A. Chronic cholesterol depletion using statin impairs the function and dynamics of human serotonin (1A) receptors. Biochemistry. 2010;49:5426–5435. doi: 10.1021/bi100276b. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- Tait JF, Gibson D. Phospholipid binding of annexin V: effects of calcium and membrane phosphatidylserine content. Arch Biochem Biophys. 1992;298:187–191. doi: 10.1016/0003-9861(92)90111-9. [DOI] [PubMed] [Google Scholar]
- te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DCA, Dwek RA, Platt FM, van Blitterswijk WJ, Sillence DJ. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279:26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- Vale MG. Localization of the amino phospholipids in sarcoplasmic reticulum membranes revealed by trinitrobenzenesulfonate and fluorodinitrobenzene. Biochim Biophys Acta. 1977;471:39–48. doi: 10.1016/0005-2736(77)90391-1. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrljic M, Nishimura SY, Moerner WE, McConnell HM. Cholesterol depletion suppresses the translational diffusion of class II major histocompatibility complex proteins in the plasma membrane. Biophys J. 2005;88:334–347. doi: 10.1529/biophysj.104.045989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson P, Schlegel R. Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim Biophys Acta. 2002;1585:53–63. doi: 10.1016/s1388-1981(02)00324-4. [DOI] [PubMed] [Google Scholar]
- Wohland T, Friedrich K, Hovius R, Vogel H. Study of ligand-receptor interactions by fluorescence correlation spectroscopy with different fluorophores: evidence that the homopentameric 5-hydroxytryptamine type 3As receptor binds only one ligand. Biochemistry. 1999;38:8671–8681. doi: 10.1021/bi990366s. [DOI] [PubMed] [Google Scholar]
- Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Yeung T, Gilbert G, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- Zhou X, Graham TR. Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc Natl Acad Sci USA. 2009;106:16586–16591. doi: 10.1073/pnas.0904293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal RF, Comfurius P, Bevers EM. Lipid-protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376:433–453. doi: 10.1016/s0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.