

Abstract
Cholinergic transmission at muscarinic acetylcholine receptors (mAChR) has been implicated in higher brain functions such as learning and memory, and loss of synapses may contribute to the symptoms of Alzheimer disease. A heterogeneous family of five genetically distinct mAChR subtypes differentially modulate a variety of intracellular signaling systems as well as the processing of key molecules involved in the pathology of the disease. Although many muscarinic effects have been identified in memory circuits, including a diversity of pre- and post-synaptic actions in hippocampus, the identities of the molecular subtypes responsible for any given function remain elusive. All five mAChR genes are expressed in hippocampus, and subtype-specific antibodies have enabled identification, quantification, and localization of the encoded proteins. The m1, m2, and m4 mAChR proteins are most abundant in forebrain regions and they have distinct cellular and subcellular localizations suggestive of various pre- and postsynaptic functions in cholinergic circuits. The subtypes are also differentially altered in postmortem brain samples from Alzheimer disease cases. Further understanding of the molecular pharmacology of failing synapses in Alzheimer disease, together with the development of new subtype-selective drugs, may provide more specific and effective treatments for the disease.
Alzheimer disease (AD) is the most common cause of memory loss and dementia. Retarding or arresting disease progression is an important therapeutic goal, which at present is not possible because of limited understanding of the cause(s) of the disease. An alternative therapeutic goal of ameliorating some of the cognitive and behavioral problems may be closer to realization. Many recent advances in understanding the molecular pharmacology of the vulnerable transmitter systems in AD have provided the opportunity to develop improved pharmacological agents. Loss of synapses appears to be one of the most critical aspects of the final common pathway that leads to the dementia (1, 2), and neurochemical studies suggest that synapses containing acetylcholine (ACh), glutamate, and serotonin in neocortex and hippocampus are predominantly affected (3, 4). This brief review focuses on the cholinergic system and, in particular, the muscarinic ACh receptor (mAChR) molecules involved in modulation of memory-related synapses in the basal forebrain and hippocampus, and the therapeutic implications for AD.
Cholinergic Memory Circuits and AD
The basal forebrain contains a well characterized group of magnocellular cholinergic neurons extending from the medial septal region through the nucleus basalis of Meynert, Ch1–Ch4 (5), which provide the majority of cholinergic innervation to the hippocampus and neocortex. The septo-hippocampal component of this system is the best studied and derives from observations of the crucial role of the hippocampus in learning and memory, the early and pervasive loss of memory in AD, as well as the early and extensive hippocampal pathology in the disease (6). In fact, synapse loss in hippocampus and pathology of cholinergic basal forebrain neurons are the best predictors of memory impairment in AD (2, 7).
Several lines of evidence implicate impaired cholinergic neurotransmission at mAChR as contributing to the dementia symptoms in AD, including: (i) consistent depletion of choline acetyltransferase in neocortex and hippocampus in patients (3, 8), including both early and late onset forms of AD (e.g., see ref. 9); (ii) basal forebrain neurons, which provide the majority of cholinergic innervation of neocortex and hippocampus, are reduced in number in AD (10, 11); (iii) correlation of choline acetyltransferase levels (3, 8) and numbers of basal forebrain neurons (12, 13) with the severity of dementia; and (iv) lesions of basal forebrain neurons and pharmacological blockade of mAChR impair cognition in animals (14, 15) and humans (16, 17). That ACh plays a necessary role in learning and memory and that it is sufficient to restore these functions in lesioned animals has been recently demonstrated using cholinergic-specific lesioning methods and genetically modified grafts (15, 18). Moreover, the hypothesis has been further tested in humans by recent clinical therapeutic trials with cholinomimetics. Tacrine, an acetylcholinesterase inhibitor, yields dose-related significant improvements in several measures of cognitive performance and quality of life (19, 20), substantiating a cholinergic role in the pathophysiology of the disease. Yet the overall clinical benefits of this drug are disappointing and may be related to its indirect mechanism of action, which depends on the synthesis, storage, and release of ACh by surviving cholinergic neurons. In addition, side effects resulting from the non-specific activation of cholinergic synapses throughout the body limit the tolerability of the drug and may prevent optimum drug levels from reaching the failing synapses subserving memory and other higher brain functions. However, new insights into the molecular basis for cholinergic neurotransmission, including differential expression of mAChR subtypes at various synapses in memory circuits and surprising findings about how some of these molecules are altered in the disease, together with the development of more specific drugs targeted to receptor subtypes, offer the possibility of improved therapeutic strategies for AD.
The Molecular Diversity of Muscarinic Receptors
Cholinergic neurotransmission is mediated by two classes of receptors, the G-protein coupled muscarinic family and the ligand-gated ion channel nicotinic family. Most studies have focused on mAChR subtypes because this family has more established roles in central cholinergic transmission and functions such as learning and memory (3, 21). The molecular diversity of mAChRs became evident with cloning of a family of five genes, m1–m5, encoding highly related but distinct receptor subtypes (22, 23). The lowercase letters “m1–m5” are used to designate the five genes and their products (mRNA and protein), whereas uppercase letters “M1–M4” refer to mAChR subtypes identified in tissues using conventional pharmacological methods, such as differences in binding affinities for various compounds. Each mAChR consists of a single protein, which when stimulated by agonists such as ACh, activate GTP-binding proteins (G-protein) and evoke typically slow, modulatory second messenger responses (24, 25). In transfected cell lines individually expressing each gene, the subtypes differentially couple to intracellular G-proteins and modulate various signaling systems, including phospholipase C, phospholipase D, adenylyl cyclase, nitric oxide, and many ion channels (25, 26). Interestingly, the m1, m3, and m5 receptors also selectively influence the processing of the amyloid precursor protein, such that receptor activation increases the secretion of non-amyloidogenic peptides (27). In addition, m1 stimulation dephosphorylates tau in PC12 cells, suggesting that receptor subtypes could potentially alter the hyperphosphorylation of tau proteins and neurofibrillary pathology in AD (28). The heterogeneity of receptors and effectors suggests that the responsiveness of any cell or tissue to ACh will in part be dictated by the subtype(s) expressed.
While a great diversity of behavioral, physiological, and biochemical effects mediated by mAChR have been identified in brain, the identities of the molecular subtypes responsible for any given neural function remain elusive. The complex pharmacology of the mAChR subtypes, together with the lack of drugs having molecular specificity, has made it difficult, if not impossible, to determine the individual roles of m1–m5 receptors in brain. For example, pirenzepine is used to operationally define pharmacological “M1” receptors, although this antagonist has less than a 10-fold difference in affinity between m1 and m4 receptor proteins (29). Similarly, the “M2” pharmacological class is usually defined by AF-DX 384 or related antagonists, compounds that have virtually identical affinities for m2 and m4 receptor proteins (29). In addition, multiple subtypes are undoubtedly involved in various cholinergic responses, whether at a behavioral or cellular level. For example, performance on memory tests in animals is sensitive to a variety of drugs with preference for either M1 (30, 31, 32, 33, 34) or M2 receptors (35), suggesting that multiple subtypes are involved. Furthermore, the diversity of muscarinic effects and the presence of each of the molecular subtypes in hippocampus alone suggests that each of the receptors play special roles in memory and other functions involving this structure.
A Diversity of Muscarinic Receptor Actions in Hippocampus
The mAChR subtypes mediate a diversity of pre- and post-synaptic actions in hippocampus. Presynaptic mAChRs depress inhibitory and excitatory responses in hippocampus (36, 37, 38), with some evidence that different subtypes inhibit release of glutamate, aspartate, γ-aminobutyric acid, and ACh (39, 40). Autoreceptors that inhibit ACh release in hippocampus have been described variously as M2 (39, 41), M2-cardiac like (42), M2-noncardiac like (43), and as M4 (44). The identity of this subtype could be important therapeutically in AD, since antagonists at this site might enhance ACh release from surviving cholinergic terminals (41, 45). Physiological studies also suggest that mAChR are precisely localized at distinct excitatory synapses in hippocampus, where they profoundly influence neurotransmission. Glutamatergic systems are responsible for the majority of excitatory transmission in brain, including the excitatory feed-forward synapses that are the backbone of hippocampal circuitry, i.e., the “tri-synaptic pathway,” and are critical for memory and other hippocampal functions. ACh depresses evoked responses at each excitatory synapse. For instance, stimulation of a presynaptic mAChR depresses excitatory transmission at Schaffer collateral synapses in CA1 and at mossy fiber synapses in CA3 (36, 37, 46, 47). Postsynaptic mAChR subtypes also modulate excitatory synaptic neurotransmission in hippocampus (37). An example of this modulation in the CA1 region is enhanced responsiveness of N-methyl-d-aspartate receptors by activation of M1 receptors (48). The perforant pathway is also modulated by an mAChR selectively localized in the middle third of the dentate gyrus (49). Thus, although a diversity of pre- and postsynaptic ACh actions in memory-related circuits have been described, which exhibit a high degree of spatial- and subtype-selectivity, determination of the precise identities of the molecular subtypes has not been possible using conventional pharmacological approaches. To gain further insights into the roles of mAChR family, molecular and immunological approaches have been used to study the expression and regulation of the m1–m5 subtypes.
Localization of mAChR Gene Products in Brain
Identification of the mAChR subtypes in brain has been accomplished using in situ hybridization to localize the mRNAs (50, 51) and highly selective antibodies to directly quantify (52, 53) and localize the proteins (52). Surprisingly, all of the subtypes appear to be present in brain, albeit in different distributions and relative abundance, as summarized in Table 1. Quantitative immunoprecipitation studies performed by independent laboratories using different antibodies have shown close agreement in the composition of subtypes in various regions of rat (52, 53) and human brain (54). In the forebrain regions of interest for AD, the m1, m2, and m4 proteins are the most abundant subtypes. For example, in hippocampus and several regions of neocortex in human brain, m1 ranges from 35–60% of all mAChR binding sites, whereas m2 and m4 each account for about 15–25% of receptors in the same areas (54). In contrast, m2 is the predominant subtype in the basal forebrain, and m4 is the most abundant mAChR in the caudate and putamen. These findings suggest that m2 may play a role as autoreceptor on cholinergic neurons (see below), whereas m4 may play an important role in motor control and perhaps motor learning. The m3 and m5 receptors are expressed only at very low levels in brain.
Table 1.
Localization of mAChR subtypes in brain
| Molecular subtype | Regional abundance | Cellular localization | Synaptic localization |
|---|---|---|---|
| m1 | Abundant in forebrain (neocortex, | Pyramidal neurons | Post- ≫ Presynaptic |
| hippocampus neostriatum) | Striatal spiny neurons | ||
| m2 | Moderately abundant throughout brain | Cholinergic neurons, nonpyramidal neurons in cortex and hippocampus | Pre- ≫ Postsynaptic |
| m3 | Low levels throughout brain | Neuronal | ? |
| m4 | Abundant in neostriatum, moderate levels | Striatal spiny neurons | Pre- and postsynaptic |
| in hippocampus and cortex | Associational and commissural hippocampal projections | ||
| m5 | Low levels in hippocampus, substantia nigra | Pyramidal neurons, substantia nigra pars compacta, microglia | ? |
?, Unknown.
Immunocytochemical methods have enabled high resolution localization of the mAChR family of proteins. Light microscopic mapping studies reveal that the proteins (52, 55, 56, 57), like the mRNAs (50, 51), are differentially expressed in brain. In fact, all five receptor mRNAs and at least four of the encoded proteins are present in different populations of forebrain neurons in the rat. As shown in Fig. 1, m1–m4 receptors are all expressed in medial temporal lobe structures in non-human primates, with substantial differences in the regional and laminar patterns of immunoreactivity for each subtype. This finding suggests that the receptor proteins are differentially expressed by various neuronal populations and/or differentially transported to pre- and post-synaptic locations. However, as yet there is only limited information available about the precise synaptic distributions of the subtypes.
Figure 1.
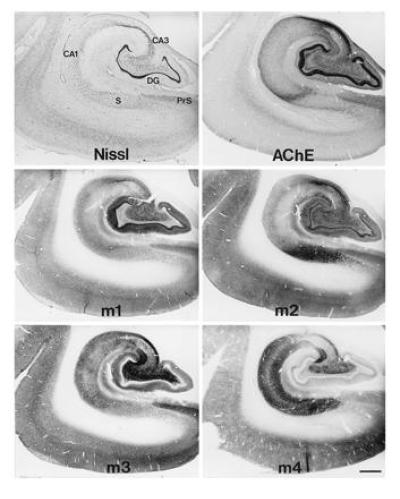
Immunocytochemical localization of m1–m4 mAChR subtypes in the medial temporal lobe of a non-human primate. Adjacent sections processed for Nissl stain to show the cytoarchitecture and acetylcholinesterase (AChE) histochemistry are shown for comparison. Note the regional and laminar differences in the distributions of the receptors, as well as the highly complementary patterns of expression. (Bar = 1.0 mm.) CA1 and CA3, fields of Ammon’s horn; DG, dentate gyrus; PrS, presubiculum; S, subiculum.
In neocortical regions and hippocampus, m1 receptor is expressed in virtually all pyramidal neurons, where it is localized in somatodendritic regions. By immunoelectron microscopy, m1 immunoreactivity has been found to be primarily postsynaptic, and quite specifically enriched at certain synapses (Fig. 2). Although ACh is likely to be released at some of these synapses, no studies have identified the cholinergic terminals directly together with the receptor subtypes. However, many m1-positive synapses appear to receive additional innervation from terminals containing excitatory amino acids (58). As described above, cholinergic modulation of glutamatergic synapses is well established and m1 at such synapses may provide part of the anatomical and molecular basis for this interaction, e.g., as the postsynaptic M1-like receptor underlying the cholinergic potentiation of glutamate N-methyl-d-aspartate receptor-mediated neurotransmission (48). Given the failure of glutamatergic synapses in AD, drugs acting at these postsynaptic m1 receptors might augment these crucial memory circuits in the disease as well. The m1 receptor has a similar postsynaptic distribution at excitatory synapses in striatum (59), suggesting that this subtype may play a general role in cholinergic modulation of glutamatergic transmission.
Figure 2.
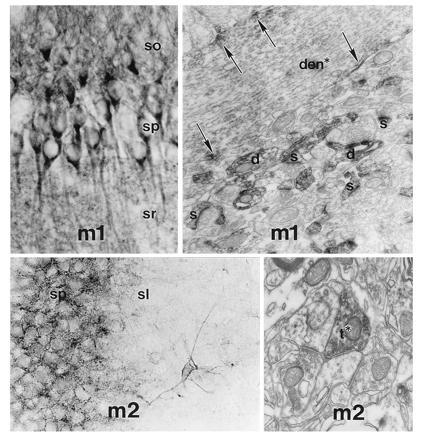
Light and electron microscopic localization of m1 and m2 immunoreactivity in rat hippocampus. (Upper Left) A light photomicrograph showing m1 immunoreactivity in pyramidal neurons in CA1. There is also abundant immunoreactivity in the neuropil in the stratum oriens (so) and stratum radiatum (sr). (Upper Right) The stratum radiatum at the electron microscopic level, with m1 immunoreactivity primarily at postsynaptic sites (arrows) along the membrane of a dendrite (den*) of a pyramidal neuron, and with much of the neuropil consisting of immunoreactive dendritic spines (s). In contrast, m2 is primarily localized in nonpyramidal neurons in hippocampus (Lower Left). An isolated neuron with several dendrites is seen in stratum lucidum (sl). Also note the dense neuropil immunoreactivity surrounding neurons in the stratum pyramidale (sp). At the electron microscopic level (Lower Right), much of the m2 immunoreactivity is presynaptically located within axon terminals (t*).
The m2 mAChR subtype has also been of considerable interest for AD, with the assumption that this molecular subtype is a presynaptic autoreceptor that inhibits ACh release (45). For drug development in AD, an antagonist acting at such a receptor would be predicted to increase ACh release from surviving cholinergic terminals. In addition, the effectiveness of subtype-nonselective agonists or cholinomimetics such as tacrine and other cholinesterase inhibitors might be diminished if the presynaptic autoreceptors are stimulated. While some pharmacological evidence has accumulated for the role of m2 as the cholinergic autoreceptor in cortex and hippocampus (45), this issue has been controversial. Recent molecular and immunocytochemical approaches have provided the first direct assessment of the cellular and synaptic localization of m2 in animal and human brain. In the basal forebrain, the m2 subtype is expressed at high levels in the cholinergic neurons, but it is also present in the admixed populations of neurons that are noncholinergic and which also project to cortex and hippocampus (56). In fact, lesions of the cholinergic neurons that spare the noncholinergic neurons have little apparent effect on m2 receptor expression, indicating that most m2 receptors in this region are located on noncholinergic structures (56). In neocortex and hippocampus, m2 receptors are found in discrete lamina in the neuropil, as well as in certain populations of nonpyramidal neurons (52, 57). As shown in Fig. 2, electron microscopic analysis reveals that many m2 receptors in the hippocampus are present in axons and axon terminals. The m2 receptor is also presynaptic in other regions of the brain, including neocortex (58), basal forebrain (56), and striatum (59). Although ACh may be contained within some of these terminals, many terminals have morphological features suggesting that other transmitters may be contained within. In neocortex and hippocampus most of the presynaptic m2 receptors are probably derived from noncholinergic neurons intrinsic to the cortex and hippocampus (57), because virtually complete lesions of the cholinergic projection neurons have little effect on the abundance or distribution of m2 receptors in the terminal fields. However, the m2 positive terminals in striatum (59), and perhaps a minority of those in cortex and hippocampus, are cholinergic, where this subtype is believed to be an autoreceptor.
Much less is known about the precise localization of the other mAChR in forebrain circuits relevant to memory and cognition. The m3 receptors are present in various neuronal populations throughout the brain (55). The m4 subtype is fairly abundant in cortex and hippocampus, although it is most enriched in striatum. At the light microscopic level m4 receptors, like m2, appear mostly in the neuropil (57). Although no ultrastructural information has been reported for m4, this subtype is present in several key pathways, including the corpus callosum, hippocampal commissure, and fimbria–fornix. This pattern of localization, together with the laminar distribution of m4, suggests that these receptors may be presynaptically located on associational and commissural projection pathways of the hippocampus. If so, this receptor might be important in the regulation of glutamate release. Finally, m5 is the only receptor protein that has yet to be localized by immunocytochemistry, although the mRNA has been reported in hippocampal pyramidal neurons, dopamine-containing neurons in the substantia nigra, and few other regions (51, 60).
Muscarinic Receptor Subtypes in AD
The ultimate application of basic advances in the molecular neurobiology of mAChR to human conditions, such as AD, depends on knowledge about the expression of the mAChR family in human brain and possible alterations in the receptors by the disease process. Pharmacologically defined binding sites have been analyzed in detail in control and in AD postmortem brain (for review see ref. 61). It has been suggested that “postsynaptic” receptors are largely unchanged in number in AD (45, 57) but may not be functional (62). There has been less consensus regarding “presynaptic” receptors, but some studies have found these are reduced in AD (45). However, because the ligands used in these studies do not have the specificity necessary to distinguish among the individual mAChR proteins or to spatially resolve their pre- and postsynaptic locations, it is difficult to interpret these earlier findings vis a vis the molecular subtypes.
Only recently has any information been obtained regarding the molecular subtypes in AD. A solution hybridization study revealed a significant decrease of m1 mRNA in temporal cortex of six AD patients, with no change in the levels of m2, m3, or m4 (63). No changes in any subtypes were found in other brain regions tested. This finding is at odds with one other study of the mAChR mRNAs, which described an almost 3-fold increase in m1 mRNA in temporal cortex using in situ hybridization (64). A recent immunoprecipitation study has provided the first direct analysis of the family of receptor proteins in AD, with assay of several brain regions from 13 AD cases and 11 age-matched controls (54). The results yielded several surprising findings. First, the m1 protein was decreased throughout cortex and hippocampus despite unchanged levels of the M1 binding sites in the same tissues. This finding is in conflict with current dogma, since it suggests that m1, the predominant postsynaptic receptor in cholinergic terminal fields, may be reduced in diseased brain, perhaps as a result of shrinkage or degeneration of pyramidal neurons and their dendrites and spines. Alternatively, the receptor may lose the epitopes recognized by antibodies yet retain ligand binding ability. In addition, there were marked increases in m4 receptors in AD, which occurred only in cortical regions and hippocampus and not in putamen. Because the ligand binding preferences of m1 and m4 overlap (both are “M1”-like), the opposing directions of change in the levels of these two receptors could also reasonably account for the findings of previous studies in which “M1” receptor binding sites were unchanged. Although the cellular basis for the increase in m4 is presently unknown, this subtype might be an interesting target for novel cholinergic therapies. Other subtype changes of note in AD brain include a decrease in the levels of m2 receptor protein. As discussed above, because only a minority of the m2 receptors are present in cholinergic terminals, the reduced levels of this subtype probably reflect changes in other neuron populations, which are intrinsic to the neocortex and hippocampus.
Conclusions
Impaired neurotransmission at muscarinic cholinergic synapses may contribute to the devastating loss of memory and other cognitive abilities in AD. Identification of a family of five mAChR genes encoding highly related receptor subtypes with markedly different cellular and synaptic distributions in brain, raises the exciting possibility that individual receptors may be targets for improved therapies. Presently used cholinergic compounds suffer from a lack of subtype-selectivity and potency, which favor negative peripheral side effects and may limit cognitive effects because of weak and/or opposing actions in brain. In contrast, the high levels and selective expression of several of the molecular subtypes, including m1, m2, and m4, in memory-related forebrain circuits, provides an opportunity for “magic bullet” therapies to be targeted to precise pre- and postsynaptic sites. However, the complexity of cholinergic transmission, together with alterations in these receptors in AD brain, makes predictions about the behavioral and therapeutic relevance of these receptors uncertain. Progress in the development of many selective compounds will likely clarify the ultimate therapeutic implications of the mAChR family for AD in the near future.
Acknowledgments
I am deeply grateful to C. Heilman, S. Hersch, S. Taylor-Rouse, H. Rees, S. Edmunds, D. Rye, A. Serbenescu, M. Wakai, E. Mufson, D. Mash, and D. Flynn for their invaluable contributions and helpful discussions. This work was supported by U.S. Public Health Service Grants NS30454, NS31937, and AG10130.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: ACh, acetylcholine; AD, Alzheimer disease; mAChR, muscarinic acetylcholine receptors.
References
- 1.DeKosky S T, Scheff S W. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 2.Terry R D, Masliah E, Salmon D P, Butters N, DeTeresa R, Hill R, Hansen L A, Katzman R. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 3.Coyle J R, Price D L, DeLong M R. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 4.Bowen D M, Francis P T. Semin Neurosci. 1990;2:101–108. [Google Scholar]
- 5.Mesulam M-M, Mufson E J, Levey A I, Wainer B H. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 6.Morris J C, McKeel D W, Jr, Storandt M, Rubin E H, Price J L, Grant E A, Ball M J, Berg L. Neurology. 1991;41:469–478. doi: 10.1212/wnl.41.4.469. [DOI] [PubMed] [Google Scholar]
- 7.Samuel W, Terry R D, DeTeresa R, Butters N, Masliah E. Arch Neurol. 1994;51:772–778. doi: 10.1001/archneur.1994.00540200048015. [DOI] [PubMed] [Google Scholar]
- 8.Perry E K, Tomlinson B E, Blessed G, Bergmann K, Gibson P H, Perry R H. Br Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Etienne P, Robitaille Y, Wood P, Gauthier S, Nair N P, Quirion R. Neuroscience. 1986;19:1279–1291. doi: 10.1016/0306-4522(86)90142-9. [DOI] [PubMed] [Google Scholar]
- 10.Whitehouse P J, Price D L, Clark A W, Coyle J T, DeLong M R. Ann Neurol. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 11.Arendt T, Bigl V, Arendt A, Tennstedt A. Acta Neuropathol. 1983;61:101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- 12.Doucette R, Fisman M, Hachinski V C, Mersky H. Can J Neurol Sci. 1986;13:435–440. doi: 10.1017/s0317167100037070. [DOI] [PubMed] [Google Scholar]
- 13.Lehericy S, Hirsch E C, Cervera-Pierot P, Hersh L B, Bakchine S, Piette F, Duyckaerts C, Hauw J, Javoy-Agid F, Agid Y. J Comp Neurol. 1993;330:15–31. doi: 10.1002/cne.903300103. [DOI] [PubMed] [Google Scholar]
- 14.Dunnett S B. Psychopharmacology. 1985;87:357–363. doi: 10.1007/BF00432721. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson O G, Leanza G, Rosenblad C, Lappi D A, Wiley R G, Bjorklund A. NeuroReport. 1992;3:1005–1008. doi: 10.1097/00001756-199211000-00015. [DOI] [PubMed] [Google Scholar]
- 16.Drachman D A, Leavitt J. Arch Neurol. 1974;30:113–121. doi: 10.1001/archneur.1974.00490320001001. [DOI] [PubMed] [Google Scholar]
- 17.Damasio A R, Graff-Radford N R, Eslinger P J, Damasio H, Kassell N. Arch Neurol. 1985;42:263–271. doi: 10.1001/archneur.1985.04060030081013. [DOI] [PubMed] [Google Scholar]
- 18.Winkler J, Suhr S, Gage F, Thal L, Fisher L. Nature (London) 1995;375:484–487. doi: 10.1038/375484a0. [DOI] [PubMed] [Google Scholar]
- 19.Davis K L, Thal L J, Gamzu E R, Davis C S, Woolson R F, Gracon S I, Drachman D A, Schneider L S, Whitehouse P J, Hoover T M, Morris J C, Kawas C H, Knopman D S, Earl N L, Kumar V, Doody R S, Group T C S. N Engl J Med. 1992;327:1253–1259. doi: 10.1056/NEJM199210293271801. [DOI] [PubMed] [Google Scholar]
- 20.Farlow M, Gracon S I, Hershey L A, Lewis K W, Sadowsky C H, Dolan-Ureno J. J Am Med Assoc. 1992;268:2523–2529. [PubMed] [Google Scholar]
- 21.Bartus R T, Dean R L, Beer B, Lippa A S. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 22.Bonner T I, Buckley N J, Young A C, Brann M R. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 23.Peralta E G, Ashkenazi A, Winslow J W, Smith D H, Ramachandran J, Capon D J. EMBO J. 1987;6:3923–3929. doi: 10.1002/j.1460-2075.1987.tb02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hulme E C, Birdsall N J M, Buckley N J. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 25.Caulfield M P. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- 26.McKinney M. Prog Brain Res. 1993;98:333–340. doi: 10.1016/s0079-6123(08)62416-4. [DOI] [PubMed] [Google Scholar]
- 27.Farber S A, Nitsch R M, Schulz J G, Wurtman R J. J Neurosci. 1995;15:7442–7451. doi: 10.1523/JNEUROSCI.15-11-07442.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadot E, Gurwitz D, Barg J, Behar L, Ginzburg I, Fisher A. J Neurochem. 1996;66:877–880. doi: 10.1046/j.1471-4159.1996.66020877.x. [DOI] [PubMed] [Google Scholar]
- 29.Dorje F, Wess J, Lambrecht G, Tacke R, Mutschler E, Brann M R. J Pharmacol Exp Ther. 1991;256:727–733. [PubMed] [Google Scholar]
- 30.Caulfield M, Higgins G, Straughan D. J Pharm Pharmacol. 1983;35:131–132. doi: 10.1111/j.2042-7158.1983.tb04290.x. [DOI] [PubMed] [Google Scholar]
- 31.Hagan J J, Jansen J H M, Broekkamp C L E. Psychopharmacology. 1987;93:470–476. doi: 10.1007/BF00207237. [DOI] [PubMed] [Google Scholar]
- 32.Messer W S, Jr, Thomas W S, Hoss W. Brain Res. 1987;407:37–45. doi: 10.1016/0006-8993(87)91217-0. [DOI] [PubMed] [Google Scholar]
- 33.Bymaster F P, Heath I, Hendrix J C, Shannon H E. J Pharmacol Exp Ther. 1993;267:16–24. [PubMed] [Google Scholar]
- 34.Dawson G, Iverson S. Behav Brain Res. 1993;57:143–153. doi: 10.1016/0166-4328(93)90130-i. [DOI] [PubMed] [Google Scholar]
- 35.Packard M G, Regenold W, Quirion R, White N M. Brain Res. 1990;524:72–76. doi: 10.1016/0006-8993(90)90493-u. [DOI] [PubMed] [Google Scholar]
- 36.Valentino R J, Dingledine R. J. Neurosci. 1981;1:784–792. doi: 10.1523/JNEUROSCI.01-07-00784.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halliwell J V. Prog Brain Res. 1990;84:255–272. doi: 10.1016/s0079-6123(08)60910-3. [DOI] [PubMed] [Google Scholar]
- 38.Krnjevic K. Prog Brain Res. 1993;98:285–292. doi: 10.1016/s0079-6123(08)62410-3. [DOI] [PubMed] [Google Scholar]
- 39.Raiteri M, Leardi R, Marchi M. J Pharmacol Exp Ther. 1984;228:209–214. [PubMed] [Google Scholar]
- 40.Raiteri M, Marchi M, Paudice P. Ann NY Acad Sci. 1990;604:113–129. doi: 10.1111/j.1749-6632.1990.tb31987.x. [DOI] [PubMed] [Google Scholar]
- 41.Lapchak P A, Araujo D M, Quirion R, Collier B. Brain Res. 1989;496:285–294. doi: 10.1016/0006-8993(89)91075-5. [DOI] [PubMed] [Google Scholar]
- 42.Richards M H. Br J Pharmacol. 1990;99:753–761. doi: 10.1111/j.1476-5381.1990.tb13002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marchi M, Raiteri M. J Pharmacol Exp Ther. 1989;248:1255–1260. [PubMed] [Google Scholar]
- 44.McKinney M, Miller J H, Aagaard P J. J Pharmacol Exp Ther. 1993;264:74–78. [PubMed] [Google Scholar]
- 45.Mash D C, Flynn D D, Potter L T. Science. 1985;228:1115–1117. doi: 10.1126/science.3992249. [DOI] [PubMed] [Google Scholar]
- 46.Herreras O, Solis J, Herranz A, Martin del Rio R, Lerma J. Brain Res. 1988;461:303–313. doi: 10.1016/0006-8993(88)90260-0. [DOI] [PubMed] [Google Scholar]
- 47.Williams S, Johnston D. J Neurophysiol. 1990;64:1089–1097. doi: 10.1152/jn.1990.64.4.1089. [DOI] [PubMed] [Google Scholar]
- 48.Markram H, Segal M. J Physiol (London) 1992;447:513–533. doi: 10.1113/jphysiol.1992.sp019015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kahle J S, Cotman C W. Brain Res. 1989;482:159–163. doi: 10.1016/0006-8993(89)90554-4. [DOI] [PubMed] [Google Scholar]
- 50.Buckley N J, Bonner T I, Brann M R. J Neurosci. 1988;8:4646–4652. doi: 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vilaro M T, Mengod G, Palacios J M. Prog Brain Res. 1993;98:95–101. doi: 10.1016/s0079-6123(08)62385-7. [DOI] [PubMed] [Google Scholar]
- 52.Levey A, Kitt C, Simonds W, Price D, Brann M. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yasuda R P, Ciesla W, Flores L R, Wall S J, Li M, Satkus S A, Weisstein J S, Spagnola B V, Wolfe B B. Mol Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- 54.Flynn D, Ferrari-DiLeo G, Mash D, Levey A. J Neurochem. 1995;64:1888–1891. doi: 10.1046/j.1471-4159.1995.64041888.x. [DOI] [PubMed] [Google Scholar]
- 55.Levey A I, Edmunds S M, Heilman C J, Desmond T J, Frey K A. Neuroscience. 1994;63:207–221. doi: 10.1016/0306-4522(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 56.Levey A I, Edmunds S M, Hersch S M, Wiley R G, Heilman C J. J Comp Neurol. 1995;351:339–356. doi: 10.1002/cne.903510303. [DOI] [PubMed] [Google Scholar]
- 57.Levey A I, Edmunds S M, Koliatsos V, Wiley R G, Heilman C J. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mrzljak L, Levey A I, Goldman-Rakic P S. Proc Natl Acad Sci USA. 1993;90:5194–5198. doi: 10.1073/pnas.90.11.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hersch S M, Gutekunst C A, Rees H D, Heilman C J, Levey A I. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiner D M, Levey A I, Brann M R. Proc Natl Acad Sci USA. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nordberg A. Cerebrovasc Brain Metab Rev. 1992;4:303–328. [PubMed] [Google Scholar]
- 62.Flynn D D, Weinstein D A, Mash D C. Ann Neurol. 1991;29:256–262. doi: 10.1002/ana.410290305. [DOI] [PubMed] [Google Scholar]
- 63.Wang S Z, Zhu S Z, Mash D C, El-Fakahany E E. Mol Brain Res. 1992;16:64–70. doi: 10.1016/0169-328x(92)90194-g. [DOI] [PubMed] [Google Scholar]
- 64.Harrison P J, Barton A J L, Najlerahim A, McDonald B, Pearson R C A. Mol Brain Res. 1991;9:15–21. doi: 10.1016/0169-328x(91)90125-h. [DOI] [PubMed] [Google Scholar]