

Abstract
Stabilization of amorphous state is a focal area for formulators to reap benefits related with solubility and consequently bioavailability of poorly soluble drugs. In the present work, an attempt has been made to explore the potential of moringa coagulant as an amorphous state stabilizer by investigating its role in stabilization of spray-dried (amorphous) ibuprofen, meloxicam and felodipine. Thermal studies like glass forming ability, glass transition temperature, hot stage microscopy and DSC were carried out for understanding thermodynamic stabilization of drugs. PXRD and dissolution studies were performed to support contribution of moringa coagulant. Studies showed that hydrogen bonding and electrostatic interactions between drug and moringa coagulant are responsible for amorphous state stabilization as explored by ATR-FTIR and molecular docking. Especially, H-bonding was found to be predominant mechanism for drug stabilization. Therein, arginine (basic amino acid in coagulant) exhibited various interactions and played important role in stabilization of aforesaid amorphous drugs.
KEY WORDS: amorphous state, arginine, drug–coagulant interactions, moringa coagulant, stabilization
INTRODUCTION
Recent days have seen tremendous strides in the dissolution enhancement of poorly soluble drugs. Amorphous form of the solids is a high-energy state responsible for dissolution enhancement; but from thermodynamic view point, amorphous form of drug is a metastable state and tends to recrystallize on storage over a period of time (1–3). So, formulation of stable amorphous form for successful drug delivery is becoming crucial step for formulation scientist. Stabilization of amorphous form of drug can be achieved by various methods like storage below the glass transition temperature (Tg); (4,5) dilution by carrier; antiplasticization (6), or allowing hydrogen bonding and/or electrostatic interactions between drug and polymer at molecular level (7–11).
Exhaustive research has been carried out on stabilization of amorphous state solids using different polymers. Polyvinyl pyrrolidone (PVP), hydroxypropyl cellulose (HPC), gelucire and other carriers have been investigated to check their potential as stabilizer for amorphous form of drugs. PVP has been used to prevent devitrification of amorphous ketoconazole (12). The significance of hydrogen bonding in stabilization of amorphous valdecoxib using PVP and HPC has been explored earlier (13). Reports are available on comparative study of stabilization efficiency of gelucire 50/13 and PVP and investigation of mechanism involved (14). Nanoporous silica xerogel, kaolin, and Mg–Al–hydrocalcite have been used in stabilization of amorphous nifedipine (15), ibuprofen (16), and celecoxib (17) respectively.
Moringa oleifera is a tropical plant belonging to the family Moringaceae. It is non-toxic (18) and contains active coagulant characterized as dimeric cationic polypeptide having a molecular weight of 6–13 kDa and an isoelectric pH value in the range of 10–11 (19,20). Traditionally, moringa coagulant (MC) was used for water purification purposes. It acts as a surfactant (21), solubility enhancer (22), and stabilizer in amorphous formulations (23). It has been reported that MC contains sequence of 60 amino acids with more amount of glutamine, arginine, and glycine (20). The literature has revealed that exhaustive work has been contributed on arginine, an amino acid, which carries out dissolution enhancement; alone or in combination with hydroxypropyl-β-cyclodextrin (24–26). Its interaction with gluten, coumarin, octyl gallate (27), alkyl gallate (28,29), amorphous silica (30), indomethacin (31), lornoxicam (32) have demonstrated increased dissolution. These studies confirm that, structural components of amino acids responsible for dissolution enhancement may play vital role in establishing various interactions with amorphous drugs also, consequently stabilize them thermodynamically.
In the present study, attempt has been made to investigate potential of biopolymer MC in stabilization of amorphous state poorly soluble drugs. Spray-dried solid dispersions of ibuprofen; meloxicam and felodipine with MC were prepared. In order to understand the mechanism contributing for stability of the amorphous solid dispersions, various physicochemical characterizations like glass forming ability (GFA), glass transition temperature (Tg), hot stage microscopy (HSM), powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), and in vitro dissolution studies were carried out. Interactions taking place at molecular level were studied by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR), molecular docking and interaction intensity, etc.
MATERIALS
Ibuprofen, meloxicam and felodipine were obtained as a gift sample from Cipla pharmaceuticals (Mumbai, India), Lupin Ltd. (Pune, India) and Mistair health and hygiene Pvt. Ltd. (Kolhapur, India) respectively. Moringa seed powder was supplied by Veg India Export (Tamil Nadu, India). All other chemicals were of analytical grade.
METHODS
Separately spray-dried drugs (ibuprofen (SDI), meloxicam (SDM) and felodipine (SDF)) and spray-dried solid dispersions (SDs) thereof (ibuprofen (SDIM), meloxicam (SDMM) and felodipine (SDFM)) in combination with MC in drug/MC ratio (1:2, 1:4, 1:6, 1:8, 1:10, w/w) were separately dissolved in suitable solvent system (ibuprofen and its SDs, ethanol 95%; meloxicam and its SDs, ethanol 95% and dichloromethane (DCM) mixture; felodipine and its SDs, methanol). The fine dispersion was spray-dried using a spray dryer (Advanced Labultima, LU-222, Mumbai, India) under set of conditions described in Table I. Spray-dried pure drugs and their solid dispersions with MC (SDI, SDM, SDF, SDIM, SDMM, and SDFM) were further dried in oven at 40°C for 24 h to remove moisture and residual solvent. SDs containing different drugs subjected to different evaluation studies.
Table I.
Operational Parameters for Spray Drying
| Parameters | SDI/SDIM | SDM/SDMM | SDF/SDFM |
|---|---|---|---|
| Solvent system | Ethanol (95%) | Ethanol (95%) and Dichloromethane (70:30) | Methanol |
| Inlet temperature (°C) | 50 | 50 | 40 |
| Outlet temperature (°C) | 70 | 70 | 60 |
| Aspirate speed | 50 | 50 | 60 |
| Vaccum applied (mmWc) | 260 | 240–250 | 250 |
| Feed flow rate (ml/min) | 5 | 7 | 5 |
SDI spray-dried pure ibuprofen, SDM spray-dried pure meloxicam, SDF spray-dried pure felodipine, SDIM SD of ibuprofen and MC, SDMM SD of meloxicam and MC and SDFM SD of felodipine and MC
Drug Content
Accurately weighed 20 mg of SDIM, SDMM and SDFM SDs was dissolved in aforementioned solvents for spray drying viz, ethanol 95%, ethanol–DCM mixture, and methanol, respectively, in which drug is completely soluble. Subsequently, the solution was shaken, sonicated, and filtered through Whatman filter paper no. 45. The filtrate was assayed spectrophotometrically (V-530, JASCO, Japan) at λmax 221, 362, 238 nm for ibuprofen, meloxicam, and felodipine, respectively.
Glass Transition Temperature (Tg)
Tg has been used as an indicator of phase behavior/miscibility of drugs and carriers. For SDs (1:6, drug/MC) it was predicted using the Gordon–Taylor equation.
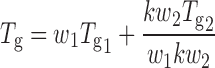 |
1 |
Where w1 and w2 are weight fractions of each component and Tg1 and Tg2 are their corresponding Tg values. k = δ1Tg1/δ2Tg2 where δ1 and δ2 are the density of drug/s and MC, respectively (12).
Glass Forming Ability
Diverse theories have been anticipated to understand vitrification phenomenon of solids (33). GFA is a measure of tendency of amorphous drugs to remain in an amorphous state. The GFA describes the relative ability of compounds to form a glassy state upon supercooling of the melt. Good glass formers are ones for whom the probability of germinating a crystal rather than forming a glassy solid during cooling at normal rates is so small that crystallization does not take place. They have very low nucleation probabilities at temperatures between Tg and Tm (melting temperature). On the contrary, poor glass formers require exceeding of a critical cooling rate for successful formation of glass (34). Means, if difference between Tg and Tm is more, probability of nucleation and consequently crystallization is more, thus resulting in less GFA. Turnbull (35) has proposed a classical parameter based on the assumption that the nucleation frequency in an undercooled melt is inversely proportional to its viscosity. The ratio of Tg to Tm called as GFA, is a criterion for glass formation as the avoidance of a single nucleation event,
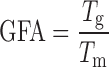 |
2 |
Hot Stage Microscopy
At thermomicroscope, drug crystals display prismatic morphology that progressively converts into liquid vesicles where melting occurs. Hot stage microscopy of SDs (1:6, drug/MC) was carried out by Leica DMLP polarized microscope and Leica LMV hot stage (Leica, Germany). Photographs were acquired using Leica DC 300 camera and analyzed using Leica IM50, version 1.20 release 19, image manager software. Hot plate was gradually heated at 20°C min−1 according to Tg or melting point of samples (36).
Accelerated Stability Studies
Stability study of pure drugs and SDs was performed by packing them in polyethylene laminated aluminum foils and storing under ICH specified accelerated stability conditions at temperature 40°C and 75% relative humidity. Periodically, samples were withdrawn and characterized using following techniques.
Powder X-ray Diffraction
PXRD patterns of pure drugs, spray-dried drugs, and SDs were obtained by Philips X-ray diffractometer (PW-3710, Holland) using Cu Kα radiation (λ = 1.5405 Å) at voltage of 40 kV, and 30 mA current. The data recorded over a range of 10° to 50° at a scanning rate of 5 × 103 cps using a chart speed of 5 mm/2°.
Differential Scanning Calorimetry
DSC thermograms of pure drugs, spray-dried drugs and SDs were obtained using DSC 821e (Mettler Toledo, Switzerland) instrument, operating with STARe software, version 5.1, equipped with an intracooler. The samples (3–5 mg) were heated at 10°C to 350°C at rate 10°C/min. under dry nitrogen purge (80 ml/min) in crimped and pin-holed aluminum pans. The DSC instrument was calibrated for temperature and heat flow using high purity standards of gallium, indium, and tin.
In Vitro Dissolution Studies
In vitro dissolution studies of the pure drugs and SDs was carried out each drug in United States Pharmacopeia Type-II dissolution test apparatus (Electrolab, TDT 08L, Mumbai, India). The dissolution medium was 900 ml of phosphate buffer maintained at 37.5 ± 0.5°C and stirred by paddles at 100 rpm. Aliquots, 5 ml were withdrawn at predefined time intervals and analyzed by UV spectroscopy at the λmax of 221, 362, 238 nm for pH of 7.2, 7.4, 6.8 for ibuprofen, meloxicam, and felodipine, respectively. The same amount of fresh phosphate buffer was used to replace at each time as amount withdrawn for respective dissolution media. Analysis of data was done using PCP-Disso V3 software (BVCP, Pune, India).
Interaction Studies
-
Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy (ATR-FTIR)
ATR-FTIR is an infrared spectroscopic method as potential tool for analysis of drug–carrier interactions. The ATR-FTIR utilizing a high refractive-index crystals, such as Ge, ZnSe, or diamond has to study various interactions between drug and carrier. ATR spectra of pure drugs and SDs were recorded at resolution of 4 cm−1 with 32 scans on a Nicolet Nexus Smart 470 ARK FTIR spectrometer (Thermo Nicolet Corp, Madison, WI) equipped with a DTGS-KBr detector and a zinc selenide trapezoidal IRE ATR crystal (37).
-
Molecular Docking
Receiving molecule, MC, (host) was obtained from the PDB code 2DS2. All the complementary partner (guest) molecule, i.e., ibuprofen, meloxicam and felodipine were designed on Molecular Operating Environment (MOE.2008, version 10, Canada) and geometries were optimized by MMFF94 Forcefield until the gradient of 0.0001 was achieved. Chiral geometry was retained during optimization. Molecular modeling database was created for the docking of guest with the host. The dummies obtained via Site Finder were used as default complementary molecule.
-
Interaction Intensity
A quantitative evaluation of interactions between MC and drug/s was investigated using UV–Visible double beam spectrophotometer (Jasco V-530, Japan). All the proportions of SDs were dissolved in suitable solvent and spectra were recorded in the 200–450 nm range. Interaction intensity (F) was calculated according to the equation: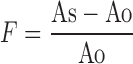
3 Where As and Ao represent the absorbance of the drug/MC composite and free drug at respective λmax (38).
RESULTS AND DISCUSSION
Drug content of SDIM, SDMM and SDFM was in the range of 90.31 to 95.37% (w/w), 95.30 to 98.78% (w/w) and 91.71 to 98.94% (w/w), respectively. SDs of ibuprofen, meloxicam and felodipine, at 1:6 proportions showed maximum, i.e., 85.85%, 93.25%, and 87.10% (w/v) drug dissolution (Table II). It has been observed that significant increase (P < 0.05) in % drug dissolution with increase in concentration of MC takes place in SDs up to 1:6 proportions, above which there was no significant dissolution enhancement. It may be due to saturation of all functionalities present in the drug with MC. As 1:6 proportions of SDIM, SDMM, and SDFM gives highest dissolution rate, they were used for further evaluation.
Table II.
In vitro Drug Dissolution Study of API, Spray-Dried API, SDIM, SDMM, and SDFM at Specified Period of Time
| Proportions | 0th day | 15th day | 30th day | 45th day | 60th day | 75th day | 90th day | |
|---|---|---|---|---|---|---|---|---|
| SDIM | Ibuprofen API | 21.65 ± 2.15 | 20.02 ± 1.56 | 19.69 ± 2.04 | 18.36 ± 1.27 | 17.38 ± 1.21 | 16.58 ± 1.84 | 15.95 ± 2.34 |
| SDI | 96.62 ± 1.68 | 22.04 ± 2.07 | 20.38 ± 1.85 | 18.40 ± 1.08 | 17.52 ± 2.01 | 16.60 ± 2.56 | 16.02 ± 1.36 | |
| 1:2 | 83.15 ± 2.28 | 83.09 ± 1.17 | 82.98 ± 1.27 | 82.87 ± 2.01 | 82.79 ± 2.0 | 82.65 ± 2.12 | 82.07 ± 1.17 | |
| 1:4 | 87.30 ± 2.12 | 87.21 ± 1.47 | 87.03 ± 2.08 | 86.94 ± 2.17 | 86.84 ± 2.09 | 86.72 ± 1.90 | 86.22 ± 1.87 | |
| 1:6 | 94.85 ± 1.44 | 94.64 ± 1.77 | 94.58 ± 2.12 | 94.46 ± 2.11 | 94.20 ± 1.27 | 93.56 ± 1.44 | 92.77 ± 2.09 | |
| 1:8 | 93.38 ± 1.48 | 93.01 ± 2.06 | 92.84 ± 2.18 | 92.68 ± 2.01 | 92.51 ± 1.77 | 92.35 ± 2.09 | 92.3 ± 2.17 | |
| 1:10 | 92.44 ± 2.18 | 92.15 ± 1.99 | 91.95 ± 1.88 | 91.75 ± 1.80 | 91.55 ± 2.12 | 91.15 ± 2.00 | 90.36 ± 2.34 | |
| SDMM | Meloxicam API | 13.06 ± 1.62 | 12.36 ± 2.08 | 11.78 ± 1.45 | 10.89 ± 2.54 | 10.08 ± 1.40 | 9.36 ± 1.32 | 8.59 ± 0.96 |
| SDM | 98.65 ± 2.30 | 14.95 ± 1.96 | 12.54 ± 1.52 | 11.34 ± 1.20 | 10.68 ± 1.68 | 9.68 ± 1.85 | 8.97 ± 2.08 | |
| 1:2 | 83.87 ± 0.84 | 83.58 ± 1.22 | 83.39 ± 0.55 | 83.01 ± 0.67 | 82.89 ± 1.23 | 82.82 ± 0.87 | 82.71 ± 1.35 | |
| 1:4 | 97.89 ± 1.47 | 97.55 ± 0.80 | 97.33 ± 0.27 | 97.21 ± 1.11 | 97.16 ± 1.45 | 96.96 ± 0.67 | 96.73 ± 1.44 | |
| 1:6 | 98.25 ± 1.24 | 98.07 ± 1.23 | 97.87 ± 1.45 | 97.67 ± 1.29 | 97.56 ± 0.99 | 97.37 ± 1.45 | 97.05 ± 1.56 | |
| 1:8 | 97.02 ± 1.84 | 96.89 ± 1.11 | 96.76 ± 1.56 | 96.53 ± 1.23 | 96.23 ± 0.78 | 95.97 ± 1.56 | 95.86 ± 1.67 | |
| 1:10 | 95.90 ± 0.92 | 95.64 ± 0.52 | 95.37 ± 0.88 | 95.25 ± 0.99 | 95.04 ± 0.89 | 94.96 ± 1.23 | 94.74 ± 0.99 | |
| SDFM | Felodipine API | 17.09 ± 1.64 | 16.64 ± 1.84 | 15.48 ± 1.48 | 14.89 ± 1.84 | 13.67 ± 1.20 | 12.75 ± 1.84 | 11.71 ± 1.05 |
| SDF | 96.71 ± 2.08 | 18.37 ± 1.24 | 16.28 ± 2.36 | 15.34 ± 1.08 | 13.51 ± 1.54 | 12.84 ± 1.42 | 11.87 ± 1.27 | |
| 1:2 | 81.89 ± 0.84 | 81.72 ± 0.80 | 81.57 ± 1.45 | 81.45 ± 0.88 | 81.22 ± 1.11 | 80.85 ± 1.09 | 80.03 ± 0.56 | |
| 1:4 | 84.47 ± 1.47 | 84.28 ± 1.21 | 84.04 ± 0.79 | 84.76 ± 1.45 | 84.56 ± 0.45 | 84.12 ± 0.77 | 83.61 ± 0.34 | |
| 1:6 | 98.10 ± 1.29 | 98.05 ± 1.11 | 97.60 ± 0.78 | 97.52 ± 1.22 | 97.43 ± 0.55 | 96.85 ± 1.54 | 96.24 ± 1.22 | |
| 1:8 | 96.76 ± 1.84 | 96.62 ± 1.45 | 96.21 ± 1.22 | 95.76 ± 0.76 | 95.73 ± 0.76 | 95.28 ± 1.02 | 94.9 ± 0.44 | |
| 1:10 | 96.62 ± 0.92 | 96.53 ± 0.78 | 96.16 ± 1.23 | 96.09 ± 1.23 | 96.01 ± 1.22 | 95.92 ± 0.44 | 95.76 ± 1.07 |
Glass Transition Temperature
Most current approaches on stability prediction involve evaluation of Tg (39). Generally, in an amorphous solid solution, the drug is molecularly distributed in an amorphous carrier. This type of solid dispersion is homogeneous on a molecular level. Therefore, only one phase is present, i.e., drug and carrier completely miscible and Tg will appear somewhere between the Tg of the carrier and drug (40). From the DSC spectra (Fig. 1) SDIM, SDMM, and SDFM shows there was only a single Tg over the entire temperature range of DSC measurements. This indicated complete miscibility of drugs and MC in the SDs. Gordon–Taylor equation used to predict physical stability of amorphous solid dispersion. If the drug and the carrier bind more strongly to each other, the Tg will be higher than expected, because the stronger binding lowers chain mobility and vice-versa. Here, it was observed that Tg value is greater than predicted Tg by Gordon–Taylor equation. At the same Tg of respective SD was also greater than Tg of pure drugs and MC (ibuprofen 105.41°C, meloxicam 201.38°C, felodipine 63.28°C, and MC 72.88°C). From the above observation, it was concluded that there may be strong binding between drugs and MC which ultimately lowers chain mobility. Decreased molecular mobility is important parameter for physical stability of amorphous SD and greater molecular mobility has been regarded as the key factor responsible for their physical instability.
Fig. 1.

Glass transition temperature (T g) of SDIM, SDMM, and SDFM
GFA
Drugs or carriers can be divided into poor (fragile) glass formers and good (strong) glass formers. In case of poor or fragile glass formers GFA ≤ 0.7 and good or strong glass formers GFA ≥ 0.7. Good glass formers exhibit minimal molecular mobility changes at Tg, and hence the shift in heat capacity tends to be small which exhibited a relatively stable amorphous form. GFA ≥ 0.7 indicates slow crystallization rates and ultimately increased tendency for an amorphous drugs to remain in amorphous state. GFA of ibuprofen, meloxicam, felodipine, and MC was found to be 0.82, 0.71, 0.72, and 0.83. So, all the selected candidates are good glass former and they are good candidates for amorphous solid dispersions.
HSM
The thermomicroscopic pictures of SDIM, SDFM and SDMM in Fig. 2 revealed that ibuprofen, felodipine and meloxicam crystals were dissolved in the MC. HSM images showed characteristic images before Tg and SDs exhibited supercooled liquid like appearance after Tg.
Fig. 2.

SDIM, SDFM and SDMM before T g a, b, and c; and after T g d, e, and f
Accelerated Stability Studies
SDs are partially or fully amorphous in nature and are thermodynamically unstable and have the tendency to change to a more stable state due to devitrification during storage. To predict the long-term stability of amorphous SD periodically samples were characterized for dissolution along with DSC and PXRD studies.
PXRD
Crystalline ibuprofen, meloxicam and felodipine exhibit characteristic peaks at 23.95, 24.13, 25.73, 27.21, 27.71, 29.45, 32.93, 35.75, 36.97, 37.29 2θ values (41), 12.97, 18.69, 21.57, 27.05, 38.07, 38.91, 43.81 2θ values (42), and 10.20, 10.80, 16.50, 20.50 23.30, 24.50, 25.40, 26.50, 29.50, and 30.20 2θ values (43), respectively. MC being amorphous in nature, spray drying of crystalline drugs or its SDs with MC achieves amorphous state (SDI, SDM, SDF, SDIM, SDMM and SDFM) and it is confirmed by the intensity (counts) at respective 2θ values as shown in Fig. 3. Stability samples of amorphous drugs after 1 month showed complete devitrification (SDI1, SDM1 and SDF1); but its SDs with MC still stabilizing after 3 month and it clearly shown in SDIM3, SDMM3, and SDFM3 spectra.
Fig. 3.

PXRD spectra of pure drugs, spray-dried drugs and stability study of SDs
DSC
Thermal analysis of ibuprofen, meloxicam, and felodipine (Fig. 4) showed melting endotherm at 76.93°C, 257.8°C, and 145.85°C respectively. SDI, SDM, and SDF showed change in heat capacity indicating Tg at 74.29°C, 201.3°C, and 138.9°C, respectively, which clearly indicates existence of amorphous state of the drugs. After 1 month, SDI1, SDM1 and SDF1 showed melting peak at 75.42°C, 257.1°C, and 144.28°C which indicates physical instability of amorphous state; it is due to conversion of high free energy (amorphous state) to less energetic (crystalline state). Spray-dried solid dispersions (SDIM, SDMM, and SDFM) showed shifting to lower temperature and broadening of melting endotherm at 73.84°C, 256.2°C, and 141.74°C and also 15.36%, 13.22%, and 16.02% crystallinity, respectively, indicates interactions and amorphization of the crystalline drugs. From the study of 3 month stability (SDIM3, SDMM3, and SDFM3) it was clear that after aging MC still stabilizing amorphous form of SDs. The mechanism behind stabilization is either alone or combination of reasons like dilution effect of the MC and/or interactions occurring at molecular level, like hydrogen bonding or any other weak electrostatic bonding between the two components of SD.
Fig. 4.

DSC thermograms of pure drugs, spray-dried drugs and stability study of SDs
In Vitro—Dissolution Study
From dissolution data of pure drugs and SDs shown in Table II, amongst the SDs prepared, 1:6 ratio of SDMM shows highest dissolution and above that there was decreased dissolution rate. The drug release is influenced not only by concentration of MC, but also by the coagulation behavior of the MC, and the interactions between the drugs and the MC. 100% dissolution could not be achieved even at 6 h dissolution time at their respective phosphate buffer. The drug release was greatly slowed down after 1:6 concentration ie. The MC increases dissolution up to 1:6 ratio due to its polarity and wetting effect, but in higher concentrations its coagulation property may come to the forefront and it may slowdown the dissolution of the drug.
It has been observed that MC improves the dissolution and stabilization of meloxicam, to a greater extent than ibuprofen and felodipine. It can be attributed to five hydrogen acceptors and two hydrogen donors of meloxicam, unlike other drugs.
ATR-FTIR Study
ATR-FTIR spectra (Fig. 5) of ibuprofen showed principal peaks at wavenumber 2,955.43 cm−1 (hydroxyl), 2,868.33 cm−1 (C–H stretching), 1,721.42 cm−1 (C═O stretching), frequency in the range of 1,230.70 cm−1 to 1,183.55 cm−1 (–C–O stretching) of alcohol and 779.49 cm−1 to 865.97 cm−1 (═C–O bending) of carboxylic acid. Meloxicam exhibit characteristic strong and narrow absorption peaks at 3,290.52 cm−1 (N–H stretching), 1,620.17 cm−1 (C═N stretching), 1,550.03 cm−1 (C–O group), 2,967.08 cm−1 (–CONH), 1164.13 cm−1 (S═O stretching) and felodipine showed peaks at 3,370.32 cm−1 (–NH stretching), 3,068.87 cm−1, 1,697.41 cm−1 (C═O stretching), 1643.56 cm−1 (N–H bending), 1,098.23 cm−1 (C–O–C stretching), 563.59 cm−1 (Cl stretching). Spectra of MC shows the peaks for N–H stretching, for C–H stretching, for amide, for C═O stretching, for C–N stretching, C═C aromatic compound stretching at wavenumber of 3,308.81 cm−1, 2,961.10 cm−1, 1,677.62 cm−1, 1,444.82 cm−1, 1,536.65 cm−1, 1,234.44 cm−1, respectively. The appearance of above peaks, confirms the presence of amino acids like tyrosine, phenylalanine, tryptophan, arginine, cystine, etc.
Fig. 5.

ATR-FTIR spectra of pure drugs, moringa coagulant, and SDs
Absence or shifting or broadening of characteristic peaks, in infrared spectrum of the SDs would indicate changes in the drug characteristics, amorphous form or possibilities of drug-MC interactions.
Ibuprofen –OH group interacts with C═O group of MC and peak get shifted to lower wavenumber 1,668.32 cm−1. A new peak at 1,732.70 cm−1 was observed in the spectra of SDIM which also confirms hydrogen bonding. Interaction of proton donors with carbonyl oxygen via hydrogen bonding is known to decrease the frequency of the C═O mode due to weakening of C═O bond. The fact that simultaneous growth of C═O band of ibuprofen shifted to higher wavenumber and C═O group of MC shifted to lower wavenumber provides strong evidence of hydrogen bonded ibuprofen with MC.
The spectrum of SDMM, significant decrease in intensities of N–H and S═O stretching vibration peaks of meloxicam at 3,289.52 cm−1 and 1,152.64 cm−1 respectively. Shift or broadening in the bands may be due to overlapping of –OH band of MC with N–H of meloxicam and also there was slight shift along with decrease in intensity of C═N stretching vibrations. It has been assumed that negative charge on oxygen atom of meloxicam and positive charge on MC interacts electrostatically. These observations indicates possibility of intermolecular electrostatic interaction and hydrogen bonding via N–H, S═O, and C═N group of meloxicam and –OH group of MC.
When hydrogen bonding occurs, bond energy at the N–H or C═O bond of felodipine decreases and peak shift to lower frequencies 3,369.56 cm−1 and 1,658.35 cm−1, respectively.
Molecular Docking
The best site of MC had a size of 221 squares with a hydrophobic index of the 37 and side residue 162. The active site has been found in chains 2, 3, and 4. The fourth chain contains most active amino acids in their binding site. The active amino acids found in the chains 2, 3, and 4 as following.
-
Residues in chain 2
Arg27, Gln28, Gln31, Gln32, Leu34, Gln35, Leu45, Phe49, Ala71.
-
Residues in chain 3
Leu2, Trp3, Cys5, Gln6, Phe9, Arg14.
-
Residues in chain 4
Pro24, Arg27, Gln28, Gln31, Gln32, Leu34, Gln35, Arg36, Leu45, Phe49, Arg70, Ala71.
As arginine is a most active (basic) amino acid, it favoring more interaction with poorly soluble drugs (Fig. 6). Ibuprofen exhibit possible hydrogen bonding interaction with ArgC14 and ArgD36 amino acids of MC and Gln D35 establish hydrogen bonding interaction with –OH group of ibuprofen by accepting hydrogen. OH and S═O group of meloxicam exhibit H-bond interaction with ArgD70 of MC. ArgD27 and ArgB27 forms H-bond interaction and arene cation interaction. Felodipine also interacts with ArgB27 and ArgD70 by H-bond and arene cation interaction. Molecular docking score of ibuprofen, meloxicam and felodipine was found to be −10.0126, −16.8289, and −14.5670, respectively.
Fig. 6.

Possible interactions of drugs and MC in two dimensional view a, c, and e and in Pose view b, d, and f
Interaction Intensity
A quantitative evaluation investigated that upon the addition of MC the absorption intensity increased without significant shift on the λmax and it has been attributed to the presence of interactions between coagulant and drugs. In this case, these interactions correspond to the presence of the hydrogen bond. In order to evaluate existing interactions in the present study a representative factor (F) for the interaction intensity has been calculated. The interaction intensity of SDIM, SDMM, and SDFM at 1:6 proportion was found to be 0.038, 0.043, and 0.025, respectively (Fig. 7), and from that it concludes that strong interactions taking place in SDMM than SDIM and SDFM due to various proton acceptor and donor functionalities present in drugs as stated earlier.
Fig. 7.

Effect of moringa coagulant on interaction intensity (F)
CONCLUSION
The present work is a successful attempt to stabilize amorphous state solids using MC. The investigations have revealed that miscibility and decreased molecular mobility of drug–coagulant solid dispersion contribute towards amorphous state stabilization. The mechanism behind stabilization seems to be dilution effect of the moringa coagulant; and/or interactions occurring at molecular level (hydrogen bonding and weak electrostatic interactions confirmed by ATR-FTIR, molecular docking and interaction intensity). Amongst solid dispersions studied, SDMM was found to be most stable due to maximum hydrogen acceptor (5) and donor (2) functionalities of meloxicam, least devitrification and maximum dissolution. Since arginine is the most active amino acid present in moringa coagulant, its predominate role in dissolution enhancement and stabilization can be confirmed.
ACKNOWLEDGMENTS
One of the authors (Santosh Bhende) is thankful to All India Council of Technical Education (AICTE, New Delhi, India.) for the financial support in terms of Junior Research Fellowship. We would like to thank Veg India Pvt. Ltd., for kind gift of moringa seed powder.
REFERENCES
- 1.Ahlneck C, Zografi G. The molecular basis of moisture on the physical and chemical stability of drugs in the solid state. Int J Pharm. 1990;62:87–95. doi: 10.1016/0378-5173(90)90221-O. [DOI] [Google Scholar]
- 2.Hancock BC, Christensen K, Shamblin SL. Estimation the critical molecular mobility temperature (Tk) of amorphous pharmaceuticals. Pharm Res. 1998;15:1649–1651. doi: 10.1023/A:1011983923386. [DOI] [PubMed] [Google Scholar]
- 3.Yoshioka M, Hancock BC, Zografi G. Crystallization of indomethacin from amorphous state below and above its glass transition temperature. J Pharm Sci. 1994;83:1700–1705. doi: 10.1002/jps.2600831211. [DOI] [PubMed] [Google Scholar]
- 4.Hancock BC, Shamblin SL, Zografi G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperature. Pharm Res. 1995;12:709–806. doi: 10.1023/A:1016292416526. [DOI] [PubMed] [Google Scholar]
- 5.Pokharkar V, Mandpe L, Padamwar M, Ambike A, Mahadik K, Paradkar A. Development, characterization and stabilization of amorphous form of a low Tg drug. Powder Technol. 2006;167:20–25. doi: 10.1016/j.powtec.2006.05.012. [DOI] [Google Scholar]
- 6.Matsumoto T, Zografi G. Physical properties of solid molecular (vinylpyrrolidone-co vinylacetate) in relation to indomethacin crystallization. Pharm Res. 1999;16:1722–1728. doi: 10.1023/A:1018906132279. [DOI] [PubMed] [Google Scholar]
- 7.Pignatello R, Spadaro D, Vandelli MA, Forni F, Puglisi G. Characterization of the mechanism of interaction in ibuprofen- Eudragit RL100 coevaporates. Drug Dev Ind Pharm. 2004;30:277–288. doi: 10.1081/DDC-120030421. [DOI] [PubMed] [Google Scholar]
- 8.Lin SY, Perng RI. Solid-state interaction studies of drugs/polymers. I: Indomethacin/Eudragit E, RL or S resins. STP Pharm Sci. 1993;3:465–471. [Google Scholar]
- 9.Boldyrev VV, Shakhtshneider TP, Burleva LP, Severtsev VA. Preparation of the disperse systems of sulphathiazole-poly(vinylpyrrolidone) by mechanical activation. Drug Dev Ind Pharm. 1994;20:1103–1114. doi: 10.3109/03639049409038355. [DOI] [Google Scholar]
- 10.Shakhtshneider TP, Danède F, Capet F, Willart JF, Descamps M, Paccou L, et al. Grinding of drugs with pharmaceutical excipients at cryogenic temperatures. Part I. Cryogenic grinding of piroxicam–polyvinylpyrrolidone mixtures. J Therm Anal Chem. 2007;89:699–707. doi: 10.1007/s10973-006-7958-7. [DOI] [Google Scholar]
- 11.Shakhtshneider TP, Danède F, Capet F, Willart JF, Descamps M, Paccou L, et al. Grinding of drugs with pharmaceutical excipients at cryogenic temperatures. Part II. Cryogenic grinding of indomethacin–polyvinylpyrrolidone mixtures. J Therm Anal Chem. 2007;89:709–715. doi: 10.1007/s10973-006-7959-6. [DOI] [Google Scholar]
- 12.Van den G, Wuyts M, Blaton N, Busson R, Grobet P, Augustijns P, et al. Physical stabilisation of amorphous ketoconazole in solid dispersions with polyvinylpyrrolidone K25. Eur J Pharm Sci. 2001;12:261–269. doi: 10.1016/S0928-0987(00)00173-1. [DOI] [PubMed] [Google Scholar]
- 13.Ambike AA, Mahadik KR, Paradkar AR. Stability study of amorphous valdecoxib. Int J Pharm. 2004;282:151–162. doi: 10.1016/j.ijpharm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 14.Shimpi SL, Mahadik KR, Paradkar AR. Study on mechanism for amorphous drug stabilization using Gelucire 50/13. Chem Pharm Bull. 2009;57:937–942. doi: 10.1248/cpb.57.937. [DOI] [PubMed] [Google Scholar]
- 15.Alja G, Uro M, Marjan B, Odon P, Stane S, Miran G, et al. Vitrification from solution in restricted space: formation and stabilization of amorphous nifedipine in a nanoporous silica xerogel carrier. Int J Pharm. 2007;343:131–140. doi: 10.1016/j.ijpharm.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Subrata M, Satyanarayan P, Kalpana S, Pintu K, Arindam S, Gaurisankar G, et al. Formation of physically stable amorphous phase of ibuprofen by solid state milling with kaolin. Eur J Pharm Biopharm. 2008;68:346–351. doi: 10.1016/j.ejpb.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Ambrogi V, Perioli L, Marmottini F, Rossi C. Use of Mg–Al–hydrocalcite to enhance the stability of celecoxib in amorphous form. Eur J Pharm Sci. 2007;66:253–259. doi: 10.1016/j.ejpb.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Folkard GK, Sutherland JP, Grant WD. Natural coagulant for appropriate water treatment—a novel approach. Waterlines. 1990;8(4):30–32. doi: 10.3362/0262-8104.1990.020. [DOI] [Google Scholar]
- 19.Ndabigengesere A, Narasiah KS, Talbot BG. Active agents and mechanism of coagulation of turbid waters using Moringa oleifera. Water Res. 1995;29:703–710. doi: 10.1016/0043-1354(94)00161-Y. [DOI] [Google Scholar]
- 20.Gassenschmidt U, Jany K, Tauscher B, Niebergall H. Isolation and characterization of a flocculating protein from Moringa oleifera Lam. Biochim Biophys Acta. 1995;1243:477–481. doi: 10.1016/0304-4165(94)00176-X. [DOI] [PubMed] [Google Scholar]
- 21.Maikokera R, Kwaambwa HM. Interfacial properties and fluorescence of a coagulating protein extracted from moringa oleifera seeds and its interaction with sodium dodecyl sulphate. Colloids Surf B Biointerfaces. 2007;55:173–178. doi: 10.1016/j.colsurfb.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Jadhav NR, Bhawale BK, Paradkar AR, Kadam SS. inventors. Novel pharmaceutical composition for solubility enhancement. Indian patent. 1883. June.28, 2010.
- 23.Jadhav NR, Bhende SA, More HN. Novel stabilized composition of amorphous state solids. Indian patent. 2046. July.19, 2011.
- 24.Mura P, Maestrelli F, Cirri M. Ternary systems of naproxen with hydroxypropyl-b-cyclodextrin and aminoacids. Int J Pharm. 2003;260:293–302. doi: 10.1016/S0378-5173(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 25.Mura P, Bettinetti GP, Cirri M, Maestrelli F, Sorrenti M, Catenacci L. Solid state characterization and dissolution properties of naproxen–arginine–hydroxypropyl beta-cyclodextrin ternary system. Eur J Pharm Biopharm. 2005;59:99–106. doi: 10.1016/j.ejpb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Arakawa T, Kita Y, Hajime K. Solubility enhancement of gluten and organic compounds by arginine. Int J Pharm. 2008;355:220–223. doi: 10.1016/j.ijpharm.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 27.El-Maradny HA, Mortada SA, Kamel OA, Hikal AH. Characterization of ternary complexes of meloxicam-HP_CD and PVP or l-arginine prepared by the spray-drying technique. Acta Pharm. 2008;58:455–466. doi: 10.2478/v10007-008-0029-9. [DOI] [PubMed] [Google Scholar]
- 28.Ariki R, Hirano A, Arakawa T, Shiraki K. Arginine increases the solubility of alkyl gallates through interaction with the aromatic ring. J Biochem. 2011;149:389–394. doi: 10.1093/jb/mvr004. [DOI] [PubMed] [Google Scholar]
- 29.Hirano A, Kameda T, Arakawa T, Shiraki K. Arginine-assisted solubilization system for drug substances: solubility experiment and simulation. J Phys Chem B. 2010;114:13455–13462. doi: 10.1021/jp101909a. [DOI] [PubMed] [Google Scholar]
- 30.Kawano M, Hatta T, Hwang J. Enhancement of dissolution rates of amorphous silica by interaction with amino acids in solution at pH 4. Clays Clay Miner. 2009;57:161–167. doi: 10.1346/CCMN.2009.0570203. [DOI] [Google Scholar]
- 31.Xiaodan Q, Jing Z, Wei W, Deying C. Solubility and stability of indomethacin in arginine-assisted solubilization system. Pharm Dev Technol. 2011. doi:10.3109/10837450.2011.595797. [DOI] [PubMed]
- 32.Bramhane D, Saindane N, Vavia P. Inclusion complexation of weakly acidic NSAID with β-cyclodextrin: selection of arginine, an amino acid, as a novel ternary component. Inclusion Phenom Macrocyclic Chem. 2011;69:453–460. doi: 10.1007/s10847-010-9783-7. [DOI] [Google Scholar]
- 33.Gutzow I, Schmelzer J. Crystal research technology: the vitreous state—thermodynamics, structure, rheology, and crystallization. Berlin: Springer; 1996. p. 178. [Google Scholar]
- 34.Nascimento MLF, Souza LA, Ferreira EB, Zanotto ED. Can glass stability parameters infer glass forming ability? Journal of Non-Crystalline Solids. 2005;351:3296–3308. doi: 10.1016/j.jnoncrysol.2005.08.013. [DOI] [Google Scholar]
- 35.Turnbull D. Under what conditions can a glass be formed? Contemp Phys. 1969;10:473–488. doi: 10.1080/00107516908204405. [DOI] [Google Scholar]
- 36.Chawla G, Bansal AK. Molecular mobility and physical stability of amorphous irbesartan. Sci Pharm. 2009;77:695–709. doi: 10.3797/scipharm.0806-09. [DOI] [Google Scholar]
- 37.Hernandez EA, Posada B, Irizarry R, Castro ME. Role of hydrogen bonding interactions in directing one-dimensional thiol-assisted growth of silver-based nanofibers. J Phys Chem B. 2005;109:7251–7257. doi: 10.1021/jp044242n. [DOI] [PubMed] [Google Scholar]
- 38.Evangelos K, Georgios K, Aristotelis X, Emmanouel G. Effect of hydrogen bonding interactions on the release mechanism of felodipine from nanodispersions with polyvinylpyrrolidone. Eur J Pharm Biopharm. 2006;63:103–114. doi: 10.1016/j.ejpb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Ambike AA, Mahadik KT, Paradkar A. Spray-dried amorphous solid dispersions of simvastatin, a low Tg drug: in vitro and in vivo evaluations. Pharm Res. 2005;22:990–998. doi: 10.1007/s11095-005-4594-z. [DOI] [PubMed] [Google Scholar]
- 40.Gordon M, Taylor JS. Ideal copolymers and the second-order transitions of synthetic rubbers. I. Non-crystalline copolymers. J Appl Chem. 1952;2:493–500. doi: 10.1002/jctb.5010020901. [DOI] [Google Scholar]
- 41.Ali W, Williams AC, Rawlinson CF. Stochiometrically governed molecular interactions in drug: poloxamer solid dispersions. Int J Pharm. 2010;391:162–168. doi: 10.1016/j.ijpharm.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Sharma S, Sher P, Badve S, Pawar AP. Adsorption of meloxicam on porous calcium silicate: characterization and tablet formulation. AAPS PharmSciTech. 2005;6(4):E618–E625. doi: 10.1208/pt060476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Evangelos K, Emmanouel G, Dimitrios B. Felodipine nanodispersions as active core for predictable pulsatile chronotherapeutics using PVP/HPMC blends as coating layer. Int J Pharm. 2006;313:189–197. doi: 10.1016/j.ijpharm.2006.01.015. [DOI] [PubMed] [Google Scholar]