

Abstract
Recent studies indicate that impairments in two cognitive domains characterize the cognitive abnormalities that appear earliest in the course of Alzheimer disease (AD). These cognitive domains pertain to memory and executive function ability; in particular, memory test scores reflecting the difference between immediate and delayed recall and tasks that assess cognitive flexibility (e.g., set-shifting). Preliminary data indicate that tasks of this nature, along with specific genetic information (i.e., APOE-4 status), are important in identifying which individuals with recent cognitive changes (considered to have “questionable” disease) will progress to the point where they meet criteria for AD over time. When this cognitive and genetic information is combined with neuroimaging measures targeted at the brain regions demonstrating pathology early in AD, it may serve as specific and accurate prognostic markers of AD.
Memory changes are generally the earliest cognitive change seen in Alzheimer disease (AD). Changes in executive function ability (which include the abilities responsible for concurrent manipulation of information, concept formation, and goal-directed behavior) also show significant declines early in the course of AD. A variety of neurobiologic measures can be found that reflect these alterations in cognitive function. This paper will review the early cognitive changes associated with AD, their neurobiologic correlates, and present preliminary data concerning predictors of the development of AD in individuals who have changes in memory that suggest they may be developing AD.
Memory Changes in Early AD
Difficulty with the acquisition of new information is generally the first and most salient symptom to emerge in patients with AD. When clinical neuropsychological tests are used to evaluate memory in AD patients, it is clear that recall and recognition performance are impaired in both the verbal and nonverbal domain (1, 2).
Experimental studies have examined AD patients to determine whether the manner in which information is lost over brief delays is unique in any way to this patient group. The results of these studies suggest that a comparison of immediate and delayed recall performance may be a useful diagnostic measure for identifying patients with AD.
The first such study was conducted by Moss et al. (3). They compared patients with AD to a group of amnesic patients who had alcoholic Korsakoff syndrome (KS), a group of dementing patients with Huntington disease (HD), and a group of normal controls. All of the subjects were administered the delayed recognition span test (DRST). This task uses disks on which are placed a variety of stimuli (words, colors, faces, patterns, etc.). During the recognition portion of the task, the disks are placed on a board one at a time (there are 16 disks in all). As each disk is added, the board is hidden from view. The subject is then asked to point to the disk that was added during the delay interval. To do this, the subject must keep track of an increasingly long series of disks. The disks are added one at a time, until the subject makes an error. This yields a delayed recognition span for each of the stimuli sets. As shown in Fig. 1, all of the patient groups are impaired in their recognition performance with respect to controls, but there is overlap among the patient groups. There was no significant difference among the three patient groups in their ability to recognize new spatial, color, pattern, or facial stimuli; patients with HD performed significantly better than the other two groups when verbal stimuli were used.
Figure 1.
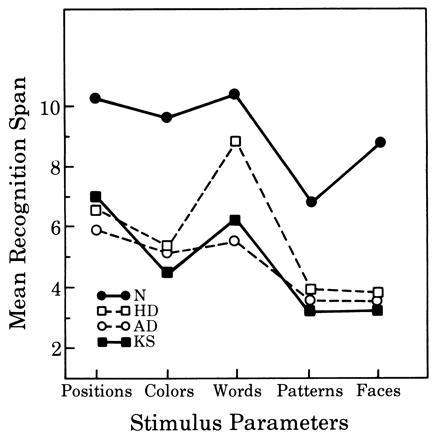
Performance on the recognition portion of the DRST. The groups compared are normal controls (N), patients with AD (AD), patients with HD (HD), and patients with alcoholic KS (KS).
In the verbal condition of the DRST task (with words on the disks), a recall paradigm was added. At both 15 sec and 2 min after completion of the last verbal recognition trial, the subject was asked to recall the words that had been on the disks. In this condition, the AD patients differ considerably from the other patients. They recall significantly fewer words over this brief delay interval (2 min) than either HD or KS patients. Although all three patient groups were equally impaired relative to normal controls at the 15-sec interval, patients with AD recalled significantly fewer words than either the HD or KS groups at the 2-min interval; in fact, only the AD group performed significantly worse at the longer interval, as compared with the shorter interval (Fig. 2). It is notable that by the end of the 2-min interval, 11 of the 12 patients in the AD group could recall fewer than 3 of the 16 words presented repeatedly during recognition testing. Of these 11 patients, 7 were unable to recall any of the 16 words at the longer interval. Whereas the KS, HD, and normal control subjects lost an average of 10% to 15% of the verbal information between the 15-sec and 2-min delay intervals, patients with AD lost an average of 75% of the material. This pattern of recall performance demonstrated for the first time that patients with AD lose more information over a brief delay than other patients with amnesic or dementing disorders.
Figure 2.
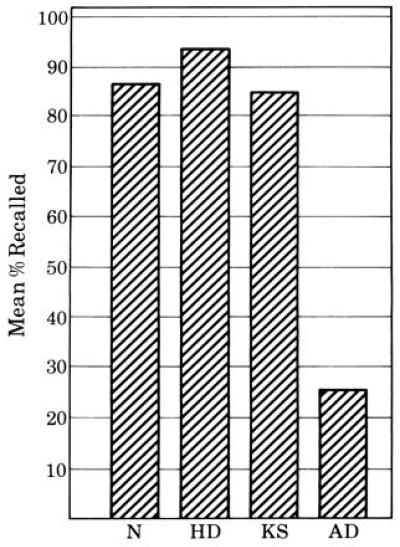
The difference between immediate and delayed recall on the verbal recall portion of the DRST. The groups compared are normal controls (N), patients with AD (AD), patients with HD (HD), and patients with alcoholic KS (KS).
A similar pattern of results has since been reported by numerous other investigators (e.g., refs. 4, 5, 6). The findings of Hart et al. (5) are particularly notable. They administered a continuous recognition task to AD patients and controls, and equated both groups of subjects for retention 90 sec after the task was completed. They then retested the subjects at 10 min, 2 hr, and 48 hr after completion of the task. The AD patients showed a greater loss of information than the controls between the 90-sec interval and the 10-min interval, but not between the 10-min and 2-hr or 48-hr intervals, suggesting that intervals of 10-min or less may be optimal for differentiating AD patients from other patient groups and from controls.
Since these findings were first reported, additional patient groups have been compared with AD patients on tasks of this nature. These groups, likewise, appear to recall more information after a delay than patients with AD. Milberg and Albert (7) compared the performance of AD patients with that of progressive supranuclear palsy patients. The two groups were equated for overall level of impairment on the basis of the Mattis dementia rating scale (8) and were equivalent in years of education. There was no difference between the patient groups on most of the tasks administered (e.g., vocabulary, digit span forward, similarities, block design); there was, however, a striking difference between the groups on both of the memory tasks (see Fig. 3). The AD patients were significantly impaired in comparison to the progressive supranuclear palsy patients on tests of both verbal and nonverbal memory.
Figure 3.
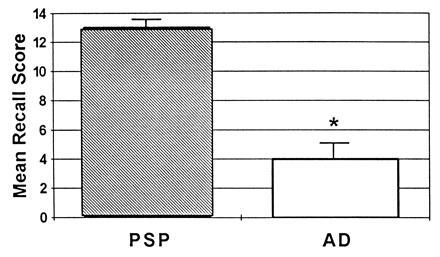
Verbal and nonverbal memory performance on the Wechsler memory scale (i.e., the logical memory and visual reproduction subtests). The groups compared are patients with AD (AD) and patients with progressive supranuclear palsy (PSP).
A comparison of patients with AD and patients with frontotemporal dementia (9) also demonstrates the severe recall deficits of the AD patients. Here again, patients with AD and patients with frontotemporal dementia, equated for overall level of cognitive impairment, were administered the DRST, which was described earlier. As in the earlier study, the difference in total recall between the 15-sec and the 2-min delay interval (i.e., the savings score) differentiated the groups (Fig. 4). The retention of the frontotemporal dementia patients over this delay interval approaches normality, whereas the AD patients lose a substantial amount of information.
Figure 4.
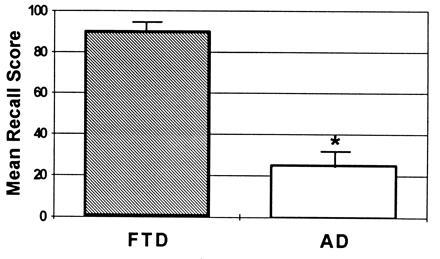
The difference between immediate and delayed recall on the verbal recall portion of the DRST. The groups compared are patients with AD (AD) and patients with frontotemporal dementia (FTD).
In general, these findings suggest that the nature and severity of the AD patients memory disturbance, in relation to delays spanning the first 10 min after encoding, are likely to be the result of a unique pattern of neuropathological and/or neurochemical dysfunction.
It is important to note that the alterations in memory associated with early AD are substantially different from those associated with age-related changes in memory. Although there are significant declines in delayed recall performance as individuals get older, much accumulated data indicates that these differences pertain to the fact that it takes older individuals longer to learn new information, but once learned, it is retained well over numerous delay intervals (e.g., refs. 10 and 11). For example, if one compares the difference between recall scores on immediate and delayed recall, there are no statistically significant age-related differences (12). Thus, if one allows healthy older subjects to learn material well (i.e., to the point where few errors are made), they do not forget what they have learned more rapidly than the young. However, if healthy older subjects are not given the ability to learn material to the same level of proficiency as younger individuals, after a delay, less information on average will be retained by the older person.
Executive Function Changes in Early AD
Recent neuropsychological data also indicate that deficits in executive function may occur early in the course of AD (13). Most of the tasks that reveal a significant deficit are those that require concurrent manipulation of information, i.e., tasks that require set-shifting, self-monitoring, or sequencing [e.g., the controlled word association task (14), the trail making test (part B) (15), and the self-ordering test (16)]. By contrast, performance on tasks that assess cue-directed attention and verbal concept formation do not appear to be significantly impaired in very mild patients (i.e., patients with mini-mental state exam scores ≥ 22).
It therefore seems most likely that the underlying disability of the mild AD patients on the executive function tasks is the result of a primary difficulty with the concurrent manipulation of information. For example, AD patients perform well on part A of the trail making test, a test that requires sustained attention and simple sequencing, but their performance is impaired on part B, which requires tracking two overlearned sequences simultaneously (i.e., numbers and letters) and switching rapidly from one sequence to the other.
This is consistent with the findings of Sahakian et al. (17) in mildly impaired patients, using a task that requires shifting between stimulus dimensions, and Baddeley et al. (18) who observed that mild-to-moderately impaired AD patients had difficulty coordinating two concurrent tasks. These results are also consistent with the findings of investigators reporting executive function deficits in AD patients with a broader range of severity (19, 20, 21, 22). Deficits in the central executive system (20) attributed to difficulties with “working memory” are also consistent with these findings.
Changes in Brain Structure in Early AD
The most likely explanation for the abnormalities in memory that characterize the early stage of AD pertains to the damage to the hippocampal formation seen in these patients (23, 24). In the hippocampal formation, neuronal loss and abnormal formations within the cells (e.g., neurofibrillary tangles and neuritic plaques) are seen primarily in the entorhinal cortex and subiculum, the primary pathways that convey information into and out of the hippocampus. It has been suggested that abnormalities in these regions produce a functional isolation of the hippocampus (24, 25). These findings suggest that neuropathological damage to medial temporal lobe structures may be responsible for the marked memory impairment evident in the early stages of AD.
These results were first observed in patients with end stage disease; however, they have recently been extended to patients with very mild disease (26). Most striking is the fact that the entorhinal cortex has neuronal loss of approximately 60% and 40% in layers 2 and 4 of the entorhinal cortex, respectively. Since this region is known to be critically important for the acquisition and retention of new information (27), abnormalities here are likely to be responsible for the severe anterograde memory loss evident early in the course of AD.
MRI studies focusing on mildly impaired AD patients are entirely consistent with the neuropathological data cited above. They have uniformly reported significant and striking differences between MRI measures of the medial temporal lobe in AD patients and controls. Measures of the hippocampal formation, the parahippocampal gyrus, the amygdalo–hippocampal complex, and the temporal horn of the lateral ventricles have demonstrated significant differences between mildly impaired AD patients and controls across a wide range of studies, using a variety of techniques (28, 29, 30, 31, 32, 33, 34, 35). These studies are consistent with computerized tomography (CT) studies showing suprasellar cistern/temporal horn abnormalities in AD (e.g., refs. 36, 37, 38).
A recent study comparing regional MRI measures in mildly impaired AD and HD patients indicates that, while several medial temporal lobe regions are sensitive to the presence of AD, the volume of the temporal horn appears to be a measure that is not only sensitive but may be specific in early AD (R. Killiany, M. Moss, M.S.A., T. Nicholson, D. Sax, T. Sandor, and F. Jolesz, unpublished work). The specificity of the temporal horn measure may be related to the fact that the fibers of passage from the entorhinal cortex run adjacent to it. Thus, the temporal horn may be an indirect reflection of the entorhinal cortex damage that appears to typify AD early in its course.
It is important to note that the changes in the brain seen in early AD differ considerably from what is seen with respect to age-related change. Data in monkeys and humans indicate that age-related neuronal loss is highly selective within the hippocampal formation. For example, the subiculum shows a significant age-related loss in humans and a similar trend in monkeys; however, the CA1, CA2, and CA3 fields show no evidence of an age-related neuronal loss (40, 41, 42), nor does the entorhinal cortex (26). These data suggest that a comparison of activation patterns in the subfields of the hippocampal formation (e.g., with newer functional imaging MR techniques) may differentiate patients in the very earliest stages of AD from those with age-related changes in memory.
The development of cognitive symptoms, in addition to memory, in AD patients (e.g., cognitive inflexibility and language dysfunction) is likely due to the progression of neuropathological change to cortical regions known to underly these functions or to dysfunction of subcortical regions that project to cortical areas. With respect to the executive function changes seen in the early stages of AD, two explanations appear likely. The first pertains to pathological changes in subcortical structures, such as the basal forebrain, that modulate cortical function (43). This is consistent with findings that the basal forebrain receives afferent projections from numerous subcortical structures and projects to numerous cortical and subcortical regions (44, 45); it can therefore serve as a source of integrated information to the cortex.
The second likely source for the executive function deficits seen in early AD pertains to the loss of neocortical synapses (46, 47, 48, 49) and long cortico–cortical projection systems (50, 51, 52) seen in AD. The partial degeneration of an intracortical projection system early in the course of disease could produce difficulties in tasks that require the rapid and simultaneous integration of multiple types of information. It is therefore of interest to note that recent reports indicate that decreased synaptic density correlates more highly with measures of cognitive function in AD patients than other neuropathological markers, such as plaques and tangles (46, 49). This is also consistent with the hypothesis that abnormalities in executive function can result from damage that is not anatomically based in the frontal lobes (53).
It appears unlikely that the executive function abnormalities in early AD can be attributed to dysfunction of the frontal lobes per se. There is little neuropathological evidence to suggest that the frontal lobes have extensive pathology early in the course of disease (54). For example, positron-emission tomography scan data generally demonstrate that frontal declines in glucose metabolism are a late phenomenon in AD (e.g., refs. 55, 56, 57).
Predictors of the Development of AD
Based on the foregoing findings, it seems likely that, among a group of older subjects with recent changes in cognitive function (so-called “questionable AD”), tests of delayed recall and executive function and measurements of medial temporal lobe regions on MRI (such as the temporal horn) would be significant predictors of which subjects would develop AD over time. To assess this possibility, a group of 165 subjects have been recruited and are being followed by me and my colleagues. Of these 165 individuals, 124 have recent changes in cognitive function, consistent with the clinical dementia rating (CDR) of questionable AD (i.e., CDR = 0.5).
The subjects were administered a lengthy neuropsychological battery and neuroimaging studies during the first year of the study, and are being followed annually to determine which subjects experience progression of their cognitive difficulties. Preliminary findings indicate that tests assessing immediate and delayed recall at baseline are significant predictors of who will be rated as CDR = 1 (i.e., probable AD) over time. When all of the scores from the neuropsychological battery were entered into a regression equation, the significant predictors of progression were: the first delayed recall trial from the California verbal learning test, immediate recall of the figures from the Wechsler memory scale, and the time to completion on trails B of the trail making test. When these baseline scores were adjusted by the ratings in each of the six areas used to generate the overall CDR rating (sometimes known as the sum of boxes), the predictive power of the memory tests was reduced, suggesting that the ratings that generate the sum of boxes reflect difficulties with delayed recall in a manner similar to that of the California verbal learning test. These results also confirm earlier reports that tests of executive function, particularly those related to cognitive flexibility, are altered early in the course of AD (13). MRI data are incomplete at this writing.
In addition to the factors mentioned earlier, genetic factors also appear to add to the ability to predict who will develop AD over time. To date, four genes have been associated with the development of AD. Three of these genes [on chromosomes 21, 14, and 1 (58, 59, 60)] lead to the development of early-onset AD (onset before age 60 or 65). One gene, the apolipoprotein E gene (on chromosome 19), is associated primarily with late-onset AD (onset after age 60 to 65) (61, 62). The apolipoprotein E gene has three alleles, designated 2, 3, and 4. The 4 allele (APOE-4) has been shown in numerous studies to be associated with AD (for review, see ref. 39). There is a general consensus that APOE-4 is acting as a risk factor for AD, rather than as an etiologic gene.
To examine the effect of apolipoprotein E gene status on the progression of cognitive problems, the apolipoprotein E gene status of each of the subjects in the study of “questionable” AD is being evaluated. Although, to date data have only been collected on 101 of the 165 subjects, the results indicate that when a variable representing APOE-4 carrier status was added to the regression equation in which the neuropsychological tests scores that predict progression had been entered, the APOE-4 variable was significant, indicating that knowledge of genetic status improves predictive accuracy.
Conclusion
Taken together, these findings suggest several major conclusions. They indicate that tests of immediate and delayed recall, administered over brief delays, can be used to differentiate AD patients from controls, and from patients with a wide variety of dementing disorders. Preliminary data suggest that these measures, along with tests that assess cognitive flexibility and APOE-4 status, may also be used to identify which persons with recent cognitive changes will develop AD over time.
These findings also suggest that selected, and differing, alterations in the brain are responsible for the cognitive changes with age and for those related to the development of AD. Understanding the nature of these cognitive changes and the brain alterations associated with them, is the first step in developing methods for changing them.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: AD, Alzheimer disease; KS, Korsakoff syndrome; HD, Huntington disease; DRST, delayed recognition span test; CDR, clinical dementia rating.
References
- 1.Wilson R, Bacon L, Fox P, Kaszniak A. J Clin Neuropsychol. 1983;5:337–344. doi: 10.1080/01688638308401181. [DOI] [PubMed] [Google Scholar]
- 2.Storandt M, Hill R D. Arch Neurol. 1989;46:383–386. doi: 10.1001/archneur.1989.00520400037017. [DOI] [PubMed] [Google Scholar]
- 3.Moss M, Albert M, Butters N, Payne M. Arch Neurol. 1986;43:239–246. doi: 10.1001/archneur.1986.00520030031008. [DOI] [PubMed] [Google Scholar]
- 4.Butters N, Salmon D, Heindel W, Granholm E. In: Aging and the Brain. Terry R D, editor. New York: Raven; 1988. pp. 63–87. [Google Scholar]
- 5.Hart R, Kwentus J, Harkins S, Taylor J. Brain Cognit. 1988;7:31–38. doi: 10.1016/0278-2626(88)90019-x. [DOI] [PubMed] [Google Scholar]
- 6.Welsh K, Butters N, Hughes J, Mohs R, Heyman A. Arch Neurol. 1992;49:448–452. doi: 10.1001/archneur.1992.00530290030008. [DOI] [PubMed] [Google Scholar]
- 7.Milberg W, Albert M. J Clin Exp Neuropsychol. 1989;11:605–614. doi: 10.1080/01688638908400919. [DOI] [PubMed] [Google Scholar]
- 8.Mattis S. In: Geriatric Psychiatry. Bellack L, Karasu T, editors. New York: Grune & Stratton; 1976. pp. 71–121. [Google Scholar]
- 9.Moss M, Albert M. In: Geriatric Neuropsychology. Albert M, Moss M, editors. New York: Guilford; 1988. pp. 145–178. [Google Scholar]
- 10.Craik F. Q J Exp Psychol. 1971;23:316–323. [Google Scholar]
- 11.Wickelgren W. Dev Psychol. 1975;11:165–169. [Google Scholar]
- 12.Petersen R, Smith G, Kokmen E, Ivnik R, Tangalos E. Neurol. 1992;42:396–401. doi: 10.1212/wnl.42.2.396. [DOI] [PubMed] [Google Scholar]
- 13.Lafleche G, Albert M. Neuropsychology. 1995;9:313–320. [Google Scholar]
- 14.Benton A L, Hamsher K. Multilingual Aphasia Examination. Iowa City: Univ. Iowa Press; 1976. [Google Scholar]
- 15.Reitan R M. Perceptual Motor Skills. 1958;8:271–276. [Google Scholar]
- 16.Petrides M, Milner B. Neuropsychologia. 1982;20:249–262. doi: 10.1016/0028-3932(82)90100-2. [DOI] [PubMed] [Google Scholar]
- 17.Sahakian B J, Downes J J, Eagger S, Evenden J L, Levy R, Philpot M P, Roberts A C, Robbins T W. Neuropsychologia. 1990;28:1197–1213. doi: 10.1016/0028-3932(90)90055-s. [DOI] [PubMed] [Google Scholar]
- 18.Baddeley A D, Logie R H, Bressi S, Della Sala S, Spinnler H. Q J Exp Psychol. 1986;38a:603–618. doi: 10.1080/14640748608401616. [DOI] [PubMed] [Google Scholar]
- 19.Becker J T. J Clin Exp Neuropsychol. 1988;10:739–753. doi: 10.1080/01688638808402811. [DOI] [PubMed] [Google Scholar]
- 20.Morris R G, Baddeley A D. J Clin Exp Neuropsychol. 1988;10:279–276. doi: 10.1080/01688638808408242. [DOI] [PubMed] [Google Scholar]
- 21.Lafleche G C, Stuss D T, Nelson R F, Picton T W. Can J Aging. 1990;9:120–134. [Google Scholar]
- 22.Nestor P G, Parasuraman R, Haxby J V. Dev Neuropsychol. 1991;7:236–243. [Google Scholar]
- 23.Ball M. Acta Neuropathol. 1977;37:111–118. doi: 10.1007/BF00692056. [DOI] [PubMed] [Google Scholar]
- 24.Hyman B, Van Hoesen G, Kromer C, Damasio A. Science. 1985;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 25.Hyman B, Van Hoesen G, Kromer L, Damasio A. Ann Neurol. 1986;20:472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Isla T, Price J, McKeel D, Morris J, Growdon J, Hyman B. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zola-Morgan S, Squire L, Ramus S. Hippocampus. 1994;4:483–495. doi: 10.1002/hipo.450040410. [DOI] [PubMed] [Google Scholar]
- 28.Seab J, Jagust W, Wong S, Roos M, Reed B, Budinger T. Magn Reson Med. 1988;8:200–208. doi: 10.1002/mrm.1910080210. [DOI] [PubMed] [Google Scholar]
- 29.Kesslak J, Nalcioglu O, Cotman C. Neurology. 1991;41:51–54. doi: 10.1212/wnl.41.1.51. [DOI] [PubMed] [Google Scholar]
- 30.Jack C, Petersen R, O’Brien P, Tangalos E. Neurology. 1992;1992:183–188. doi: 10.1212/wnl.42.1.183. [DOI] [PubMed] [Google Scholar]
- 31.Killiany R, Moss M, Albert M, Sandor T, Tieman J, Jolesz F. Arch Neurol. 1993;50:949–954. doi: 10.1001/archneur.1993.00540090052010. [DOI] [PubMed] [Google Scholar]
- 32.Convit A, deLeon M, Golomb J, George A, Tarshish C, Bobinski M, Tsui W, De S, Wegiel J, Wisniewski H. Psychiatr Q. 1993;64:371–387. doi: 10.1007/BF01064929. [DOI] [PubMed] [Google Scholar]
- 33.Lehericy S, Baulac M, Chiras J, Pierot L, Martin N, Pillon B, Deweer B, Dubois B, Marsault C. Am J Neuroradiol. 1994;15:929–937. [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda M, Tanabe H, Nakagawa Y, Kazui H, Oi H, Yamazaki H, Harada K, Nishimura T. Neuroradiology. 1994;36:7–10. doi: 10.1007/BF00599184. [DOI] [PubMed] [Google Scholar]
- 35.Laasko M, Soininen H, Partanen K, Hakilainen M, Lehtovirta M, Hanninen T, Vainio P, Riekkinen P. Am J Neuroradiol. 1995;16:727–734. [PMC free article] [PubMed] [Google Scholar]
- 36.Sandor T, Albert M, Stafford J, Harpley S. Am J Neuroradiol. 1988;9:1181–1187. [PMC free article] [PubMed] [Google Scholar]
- 37.George A, deLeon M, Stylopoulos L, Miller J, Kluger A, Smith G, Miller D. Am J Neuroradiol. 1990;11:101–107. [PMC free article] [PubMed] [Google Scholar]
- 38.Sandor T, Jolesz F, Tieman J, Kikinis R, Jones K, Albert M. Arch Neurol. 1992;49:381–384. doi: 10.1001/archneur.1992.00530280069024. [DOI] [PubMed] [Google Scholar]
- 39.Roses A, Pericak-Vance M. In: Principles and Practice of Medical Genetics. 3rd Ed. Emery E, Rimoin D, editors. Edinburgh: Churchill-Livingstone; 1995. [Google Scholar]
- 40.Amaral D. Neurobiol Aging. 1993;14:671–672. doi: 10.1016/0197-4580(93)90066-k. [DOI] [PubMed] [Google Scholar]
- 41.Rosene D. Neurobiol Aging. 1993;14:669–670. doi: 10.1016/0197-4580(93)90065-j. [DOI] [PubMed] [Google Scholar]
- 42.West M, Coleman P, Flood D, Troncoso J. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 43.Whitehouse P, Price D, Clark A, Coyle J, DeLong M. Ann Neurol. 1981;14:1–14. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 44.Arendt T, Bigl V, Tennsted A, Arendt A. Neuroscience. 1985;14:1–14. doi: 10.1016/0306-4522(85)90160-5. [DOI] [PubMed] [Google Scholar]
- 45.Mesulam M M, Mufson E J, Levey A I, Wainer B H. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 46.DeKosky S T, Scheff S W. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 47.Davies C A, Mann D M, Sumpster P Q. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 48.Masliah E, Mallory B S, Hanson L, DeTeresa R, Terry R D. Neurology. 1993;43:192–197. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 49.Terry R D, Masliah E, Salmon D P, Butters N, DeTeresa R, Hill R, Hansen L A, Katzman R. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 50.Lewis D A, Campbell M J, Terry R D, Morrison J H. J Neurosci. 1987;7:1799–1808. doi: 10.1523/JNEUROSCI.07-06-01799.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morrison J H, Scherr S, Lewis D A, Campbell M J, Bloom F E, Rogers J, Benoit R. In: The Biological Substrates of Alzheimer’s Disease. Scheibel A B, Weschler A F, editors. New York: Academic; 1986. pp. 115–131. [Google Scholar]
- 52.Scheff S W, Price D A. Ann Neurol. 1993;33:190–199. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- 53.Stuss D T, Gow C A. Neuropsychiatr Neuropsychol Behav Neurol. 1992;5:272–282. [Google Scholar]
- 54.Brun A, Gustafson L. Arch Psychiatr Nervenkrankheiten. 1976;223:15–33. doi: 10.1007/BF00367450. [DOI] [PubMed] [Google Scholar]
- 55.Chase T N, Foster N L, Fedio P, Brooks R, Mansi L, Di Chiro G. Ann Neurol. 1984;15:S170–S174. doi: 10.1002/ana.410150732. [DOI] [PubMed] [Google Scholar]
- 56.Cutler N R, Haxby J V, Duara R, Grady C L, Kay A D, Kessler R M, Sundaram M, Rapoport S. Ann Neurol. 1985;18:298–309. doi: 10.1002/ana.410180305. [DOI] [PubMed] [Google Scholar]
- 57.Haxby J V, Grady C L, Koss E, Horwitz B, Schapiro M, Friedland R P, Rapoport S I. Neurology. 1988;38:1853–1863. doi: 10.1212/wnl.38.12.1853. [DOI] [PubMed] [Google Scholar]
- 58.Goate A, Chartier-Harlin M, Mullan M, Brown J, Crawford F, et al. Nature (London) 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 59.Sherrington R, Rogaev E I, Liang Y, Rogaeva E, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 60.Levy-Lehad E, Wasco W, Poorkaj P, Romano D, Oshima J, et al. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 61.Strittmatter W, Saunders A, Schmechel D, Pericak-Vance M, Enghild J, et al. Proc Natl Acad Sci USA. 1993;190:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saunders A, Strittmatter W, Schmechel D, St George-Hyslop P, Pericak-Vance M, et al. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]