

Abstract
It is well recognized that poor dissolution rate and solubility of drug candidates are key limiting factors for oral bioavailability. While numerous technologies have been developed to enhance solubility of the drug candidates, poor water solubility continuously remains a challenge for drug delivery. Among those technologies, amorphous solid dispersions (SD) have been successfully employed to enhance both dissolution rate and solubility of poorly water-soluble drugs. This research reports a high-throughput screening technology developed by utilizing a 96-well plate system to identify optimal drug load and polymer using a solvent casting approach. A minimal amount of drug was required to evaluate optimal drug load in three different polymers with respect to solubility improvement and solid-state stability of the amorphous drug–polymer system. Validation of this method was demonstrated with three marketed drugs as well as with one internal compound. Scale up of the internal compound SD by spray drying further confirmed the validity of this method, and its quality was comparable to a larger scale process. Here, we demonstrate that our system is highly efficient, cost-effective, and robust to evaluate the feasibility of spray drying technology to produce amorphous solid dispersions.
Key words: 96-well, amorphous, bioavailability, HTPs, in vivo, SD, SDD, solid dispersions, spray dry
INTRODUCTION
Oral bioavailability is limited by factors such as the permeability, solubility, dissolution rate, chemical stability, and metabolism of the drug. Among those factors, solubility and dissolution rate of poorly water-soluble drugs are considered the most critical factors. In the past decade, there has been an increasing challenge for the pharmaceutical industry to achieve reasonable bioavailability after oral delivery of poorly water-soluble drug candidates (1–8). Thus, there is a continuous emphasis in industry to improve drug candidate solubility by encouraging chemists to utilize more soluble core molecules when building a drug candidate. Despite the effort, the difficulty of incorporating solubility into a drug candidate while retaining potency and selectivity remains a challenge to all medicinal chemists (1,9).
It is well acknowledged in the pharmaceutical industry today that an increasing number of lipophilic drug candidates are providing scientists with the growing challenge of reaching desired exposures in vivo. Approaches to improve solubility, enhance dissolution rate, and improve oral bioavailability of poorly soluble molecules have been developed for both clinical and preclinical studies. Methods such as inclusion complex, nanoparticles, pro-drugs, co-solvents, micelles/emulsions, salts, co-crystals, and amorphous solids are widely used. Investigations on utilizing amorphous solids of small molecule drugs to improve oral bioavailability of poorly soluble drugs have been reported (1,10–13). The advantage of amorphous solids versus crystalline solids on solubility is well studied (10–13). An amorphous solid can be defined as its molecular arrangement lacks long-range order, which is the unique feature of crystals. Therefore, the entropy and free energy of an amorphous solid are higher than those of its crystalline counterpart. This energy difference leads to significantly higher solubility and faster dissolution for the amorphous form than the crystalline form. Solubility (S) of a solid solute can be expressed by considering the three basic measurements in the following equation (14).
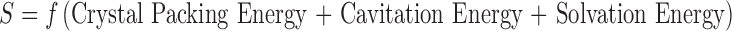 |
It is well understood that the first step to the solubilization of a solid solute is the disruption of its crystal packing where the crystal packing energy is accounted for. The cavitation energy is the energy required to disrupt water for creation of a cavity in which the solute is to be hosted, and solvation energy is the sum of favorable interactions between solute and solvent. A crystalline solid has a higher relative crystal packing energy as compared with an amorphous solid, which leads to the amorphous solid often exhibiting higher solubility. Therefore, when the solubility or dissolution rate of the drug in the gastrointestinal tract is the limiting factor for absorption, dosing an amorphous solid can result in improved oral bioavailability (15–17). Despite the promising features of amorphous solids, unfortunately, they are not problem-free. Stabilizing the amorphous solid to prevent recrystallization would be required. However, kinetic stabilization of the amorphous state of a drug below the glass transition temperatures poses great challenges (10).
The current strategy is to employ appropriate polymeric matrices to inhibit crystallization of the amorphous drug. The effect of different polymers on inhibition of crystallization of amorphous drugs has been well studied with a variety of pharmaceutically acceptable polymers including povidone, crospovidone, poloxamer, hydroxypropyl methylcellulose acetate succinate (HPMCAS), hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose (HPMC), hydroxypropyl-beta-cyclodextrin, polymethacrylates, and so forth. Recently, the application of the spray drying process to make amorphous solid dispersion drug polymer systems has drawn a lot of interest. Spray drying has been widely used in the chemical and food industries to dry aqueous solutions, organic solutions, and emulsions. For instance, dry milk powder, detergents, and dyes are a few spray-dried products currently on the market. Today, spray drying is widely utilized in the pharmaceutical industry to generate amorphous solids due to its fast drying capability. Spray-dried solid dispersion (SDD) technology, where the drug is dispersed in its amorphous form at the molecular or nanoparticle level within a solid matrix, is a proven technique for successfully improving drug solubility (1,7,10,12). In broad terms, SDDs are thermodynamically stable solid dispersions of the amorphous active pharmaceutical ingredient (API) dispersed in a polymeric matrix. Due to their morphology and thermodynamic properties described above, SDDs are capable of reducing drug crystallinity and stabilizing the system during storage and in vivo. In consequence, the oral bioavailability of a SDD drug is often found to be orders of magnitude higher than that of the purely crystalline drug form. In the past decade, SDDs have become more widely accepted by the pharmaceutical industry for drug delivery than other similar technologies due to its performance and cost efficiency. For example, SDDs are 30–50 times less expensive than freeze-drying (18).
A robust formulation with optimal drug load and excipients is one of the key factors of successfully developing a SDD system. However, due to the large scale of conventional spray dryers, the amount of bulk drug needed for feasibility testing is high, and the process is very time-consuming. Moreover, each batch prepared can only evaluate a single formulation composition. It quickly becomes expensive and resource-limiting when multiple compounds need to be tested in discovery stage. This issue has limited the initial consideration of using SDDs as an oral delivery option. Such a limitation is particularly critical in the pharmaceutical discovery setting, since large amounts of candidates are made in small quantities and both in vitro and in vivo resources are often limited and costly. Previously, several articles have reported screening methods for making amorphous solids (19,20). Despite their success, the suitability, validity, and theory of utilizing the reported SDD screening method were not evaluated. Most importantly, the long-term product stability and detailed solid-state characterization, which are the most important factors for predicting long-term success, were not reported in those articles.
The scope of this work was to develop a fully automated high-throughput system for complete SDD feasibility screening. A 96-well plate vacuum dry system was applied for sample preparation, and several marketed drugs along with an internal drug candidate were used to validate the system. Powder X-ray (PXRD) pattern, solubility, and stability of the resulting SD in each well were evaluated to determine the best composition. This system allows batch processing (96-well) and offers comparable drying efficiency to that of a conventional spray dryer through a high vacuum centrifuge thermal evaporation system. Therefore, it enables us to evaluate both polymer type and drug load simultaneously with minimal API consumption and short processing time, while providing a full picture of stability and product shelf-life estimation.
It is well expected that, in order to be useful, a high-throughput screening (HTS) technique must be fully tested and validated. The system should be designed to provide flexibility, acceptable accuracy, low compound consumption, and most importantly, the ability to predict scalability. Our 96-well system was designed to accommodate all of the above qualities. In order to test our system, a total of four model drugs were used: acetaminophen, indomethacin, celebrex, and griseofulvin. For choice of polymers, HPMC-AS, polyvinylpyrrolidone (PVP) K90, and HPMC K100 were selected. These drug and polymer systems were selected based on the abundance of literature in amorphous solid efforts and in-house data (21,26–31). Based on the design of 96-well plates, any combination of drug(s), polymer(s), or surfactant(s) can be easily implemented. Furthermore, it is well known that, in order to fully leverage the advantage of amorphous materials for long-term drug delivery, stability of the material is the key. Only systems with suitable stability can be developed for long-term usage.
MATERIALS AND METHODS
Materials and Instrumentation System
High-performance liquid chromatography (HPLC)-grade acetonitrile was obtained from Burdick and Jackson (Muskegon, MI). The HPLC system used was an Agilent HP 1100 HPLC equipped with a diode array and a variable wavelength UV detectors and quaternary solvent delivery system (Palo Alto, CA). Several analytical columns were tested, and an Alltech Alltima C8 (5 μm, 4.6 × 150 mm) was selected and used for analysis. The water purification system used was a Millipore Milli-Q system. All chemicals used for system validation were either synthesized internally or obtained from Aldrich (St. Louis, MO) and were used without further purification. Compound A was manufactured by Genentech Inc. HPMCAS grade M was purchased from Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan), lot # 6113225; PVP, K90, and HPMC K100 were purchased from Aldrich (St. Louis, MO). An EZ-2 Plus centrifuge vacuum dry system from SP scientific (Stone Ridge, NY) was used for drying with maximum temperature set at 80°C. A typical vacuum of 6–8 mbar or lower is often achieved during the drying. The S141937 Crystallizer Block from with S120464 glass substrate from Freeslate was used for sample preparation. Bruker D8 Discover with GADDS HTS powder X-ray analysis and operating in reflectance mode at 40 kV and 40 mA. Scans were taken from 0 to 40° (2θ) within a time window of 8 min. The mDSC experiments were carried out on a Thermal Analysis (New castle, DE) DSC Q1000 system with two modulated scans from 0 to 180°C at a ramp rate of 2°C/min and modulation of ±0.5°C every 60 s.
96-Well Screening Sample Preparation
Screening samples were prepared using the solvent casting approach. For each compound, 10 mg/mL stock was made by dissolving compound into the most suitable organic solvent (acetaminophen in ethanol, and indomethacin, celebrex, and griseofulvin in acetone). For each polymer, various concentrations of stocks (2.1 to 45 mg/mL) were prepared in organic solvent (HPMC in 50/50 methylene chloride/ethanol, HPMCAS in acetone, and PVP in ethanol). Each individual compound stock was dispensed into the Freeslate (Santa Clara, CA) 96-well plate (40 uL) and then followed by 80 uL of each polymer solution. After dispensing, the plates were briefly vortexed to thoroughly mix the stock solutions. The solvent was quickly evaporated using the vacuum dry system and operating conditions described above. Duplicate plates were made for each set. One plate was used for PXRD, and the other plate was used for solubility measurement. Any combination of drugs, polymers, and surfactants can be evaluated using the same device. An example of a plate map is illustrated in Fig. 1. After drying, plates were allowed to equilibrate in a vacuum oven with house vacuum and ambient temperature overnight. Post-equilibration, one plate was transferred to a stability oven (controlled at 50°C and 75% RH) for a period of 2 weeks for stability evaluation. The other plate was disassembled to obtain the glass plate for HTPS PXRD. Following PXRD scan, the plate was placed in the same 50°C and 75% RH oven for physical stability evaluation and re-scanned on day 7 and day 14. In order to obtain key thermal data (Tg by DSC) to compare the quality of the material generated by 96-well plate against material generated by the SDD process (21), small batches of compound A were scaled-up by using the same methodology and glass vials in order to accommodate large scale. The amount of sample and polymer was increased by 20-fold (1 mL of 10 mg/mL compound stock and 2 mL of polymer stock).
Fig. 1.

Example of plate map used for screening of polymer and drug load. Each well contains 0.4 mg of compound
Spray-Dried Dispersion of Compound A
SDD preparation followed the procedure reported previously (21). Generally, solid molecular dispersions are reported as a percent drug load (by weight) in HPMCAS-M. For example, a 25% drug load consists of one part (by weight) compound and three parts (by weight) HPMCAS-M. Solutions were spray-dried on a Buchi B290 (Flawil, Switzerland) spray dryer using a high-performance cyclone and small-volume sample collector. After spray drying, samples were dried under ambient (23°C) conditions to remove the solvent. Compound A (1.49 g) and HPMCAS (4.48 g) were dissolved as a 5 wt.% solution in MeOH, for a total solution weight of 114 g. Spray drying yielded 5.43 g product (91%) as a powder which was stable upon standing and used in the preparation of suspensions for testing.
X-ray Powder Diffraction (PXRD)
The PXRD patterns were recorded at room temperature with a Bruker-AXS D8 Discover X-ray Powder Diffractometer equipped with a GADDS 2D detector. The detector was placed 15 cm from the sample. Radiation of Cu Kα at 40 KV × 40 mA was used. The collection range was 5–40° 2θ. A flat glass 96-well plate with samples on top was positioned on a plate , and data collection time was approximately 6 min for each powder sample plus video image.
Characterization: DSC Analysis and Glass Transition (Tg) Determination
Differential scanning calorimetry was performed on a TA Instruments Q1000 modulated DSC. A modulated differential scanning calorimeter (TA Instruments Q1000) was used to measure melting point, glass transition temperature, and heat capacity of both crystalline and amorphous solid dispersion of compound A. Samples were initially cooled to 0°C for 5 min and were heated to 200°C at 2°C/min with modulation of ±0.5°C every 60 s. High-purity indium was used to calibrate for the heat flow and heat capacity of the instrument.
Solubility Study
On day 14, the second 96-well plate was removed from the stability oven. The solubility test was performed by adding 0.5 mL of 50 mM pH 6.5 sodium phosphate buffer with 0.1% Tween 80 (preheated to 37°C) into each well. The plate was placed on a shaker heated to 37°C for an hour. The shaking speed was set at 500 rpm. The mixtures of each well were then transferred to a 96-well 0.2 μm PVDF membrane filter plate (Corning, New York, USA). The samples were pulled through the filtration plate using the Whatman 96-well vacuum filtration unit, and the filtrate was collected in V10636 1 mL 96-well autosampler vials from Freeslate . One hundred microliters of the filtrate was then transferred to a 96-well HPLC plate (Agilent) prefilled with 100 μL of 50/50 IPA/dimethyl sulfoxide (DMSO) and thoroughly mixed to prevent any further precipitation of the compound before analysis. The drug concentration was analyzed by HPLC against an external standard. The HPLC multiple solvent pump system was used for the gradient elution. A total of two mobile phases were used to prepare the gradient. Solvent line A contained acetonitrile with 0.1% trifluoroacetate (TFA) (v/v); solvent line B contained Milli-Q water with 0.1% TFA (v/v). Flow rate was set at 1.5 ml/min for fast elution. For the method in general, at T = 0 min the mobile phases (95% A, 5% B) were mixed by the HPLC pump and held for 0.5 min (isocratic elution). From T = 0.51 to T = 4.0 min, a linear gradient from 5% B to 100% B was applied and allowed to hold at 100% for 1 min (from 4.01 to 5.0 min). At T = 5.01 min, the system was set back to the initial condition, and the flow rate was set to 2.0 mL/min and allowed to equilibrate for 1 min to prepare for the next injection. The gradient program was occasionally changed in order to achieve the best elution condition. A total of five wavelengths (220, 240, 254, 280, and 330 nm) were used for data collection for best sensitivity. For each well containing SDs (with polymer), the solubility ratio against a control well (100% drug) was calculated by the following equation to estimate the degree of solubility improvement based on the polymer and drug load.
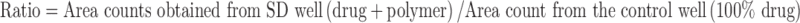 |
RESULTS AND DISCUSSION
It is understood that, in order to resemble the spray drying system, a fast solvent evaporation is essential. When the solvent evaporation rate becomes too slow, crystallization may occur. For example, it has been reported the acetaminophen SDD can be made only by the spray drying system and not by the evaporation method (22). In our system, both high vacuum and heat were used to ensure fast solvent removal. The solvent evaporation rate was calculated by the Hickman equation and the Clausius–Clapeyron equation.
Hickman Equation
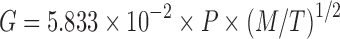 |
Where
- G
Drying rate
- P
Vapor pressure
- M
Molecular weight
- T
Temperature °K
Clausius–Clapeyron Equation
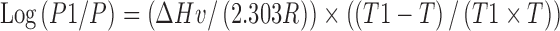 |
Where
- ΔHv
Heat of vaporization
- P1
Vapor pressure (at T1)
- P
Vapor pressure (at T)
- T1
Temperature (80°C/353°K in this case)
- R
Gas constant
Based on the equations, high drying temperature (highest setting of the instrument) was applied in our method to ensure short drying time. For example, the drying time of ethanol (120 uL) under the experimental conditions is calculated to be about 1 s, and DMSO (120 uL) is about 15 s. This fast and complete drying should avoid crystallization that may cause false-negatives.
In our system, the stability of post-processed materials was challenged by storing plates in a high temperature and high humidity chamber (50°C/75% RH). PXRD was run on the same plate on days 1, 7, and 14 to assess the physical stability of the product. For example, post-process, most samples were found to be amorphous on day 1, with the exception of the highest drug loads. On day 7, however, crystalline signals were found in all drugs with lower polymer content, although different polymers were found to have different cut-offs. On day 14, the same plate was scanned by PXRD again, and data were used to compare with results obtained on both day 1 and day 7. In general, stronger crystalline signals were found in wells showing crystallinity on day 7; meanwhile new crystalline signals were found in wells that showed no crystalline signal on day 7. Due to the need for drug product stability, it is believed that the data generated from the 14the day is more relevant to reflect the long-term stability of the solid dispersion. The physical stability difference of different polymer SD systems was not a surprise since the interaction between different polymers and drugs are different, and similar findings were reported by other researchers (23–32). Our 96-well plate results have good agreement with literature results that were made at much larger scales. For instance, stable SDDs of both acetaminophen and indomethacin were reported by researchers (26–29) and characterized where PVP was used as a polymer. In our 96-well system, when comparing drug load and stability (crystalline signal) post-storage, PVP worked well for acetaminophen, indomethacin, and celebrex (30) but not griseofulvin (31). At higher drug loads, the SD materials of acetaminophen, indomethacin, and celebrex made with PVP remained amorphous post-storage (50°C/75% RH), while griseofulvin did not. This finding was supported by the solubility data. For example, the solubility ratios of griseofulvin PVP SDs are much lower when compared with other polymers. An example of griseofulvin SD stability in different polymers on day 14 by PXRD is illustrated in Fig. 2, and a crystallinity evaluation summary for all 4 model compounds is tabulated in Table I. In general, the trend of solubility ratio vs. drug load can be correlated with the results of PXRD data where solubility ratio decreases with increasing drug load and corresponds to the increasing crystalline signal in the PXRD pattern. In several cases, higher than unity solubility ratios were found in sample wells that showed crystalline signals. This phenomenon is highly likely to be attributed to the partial crystallinity of the samples which may still result in higher solubility ratio.
Fig. 2.

PXRD data of griseofulvin SD on day 14 at 50°C/75% RH (drug load from top to bottom 100%, 10%, 20%,30%, 40%, 50%, 60%, and 70%)
Table I.
PXRD Results of Model Compounds
| Compound | Acetaminophen crystallinity (day 1/day 7/day 14) | Indomethacin crystallinity (day 1/day 7/day 14) | Celebrex crystallinity (day 1/day 7/day 14) | Griseofulvin crystallinity (day 1/day 7/day 14) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| % drug | ||||||||||||
| 100% | Y/Y/Y | Y/Y/Y | Y/Y/Y | N/Y/Y | N/Y/Y | N/Y/Y | Y/Y/Y | Y/Y/Y | Y/Y/Y | N/Y/Y | N/Y/Y | N/Y/Y |
| 10% | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/Y/Y |
| 20% | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/Y/Y |
| 30% | N/Y/Y | N/Y/Y | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/Y/Y | N/Y/Y | N/Y/Y |
| 40% | N/Y/Y | N/Y/Y | N/N/N | N/Y/Y | N/N/N | N/N/N | N/N/N | N/N/N | N/N/N | N/Y/Y | N/Y/Y | N/Y/Y |
| 50% | N/Y/Y | N/Y/Y | N/N/N | N/Y/Y | N/Y/Y | N/N/N | N/N/Y | N/N/N | N/N/N | N/Y/Y | N/Y/Y | N/Y/Y |
| 60% | N/Y/Y | Y/Y/Y | N/Y/Y | N/Y/Y | N/Y/Y | N/N/N | N/Y/Y | N/N/N | N/N/N | Y/Y/Y | Y/Y/Y | Y/Y/Y |
| 70% | Y/Y/Y | Y/Y/Y | N/N/Y | N/N/Y | N/N/Y | N/N/N | N/Y/Y | N/N/Y | N/N/N | Y/Y/Y | Y/Y/Y | Y/Y/Y |
PXRD powder X-ray, HPMC hydroxypropyl methylcellulose (polymer 1), HPMCAS hydroxypropyl methylcellulose acetate succinate (polymer 2), PVP polyvinylpyrrolidone (polymer 3), N no crystalline signal, Y crystalline signal
Overall, this solubility data further helped to narrow down the polymer selection and drug loads where a higher ratio was desired. It is worth noticing that, for acetaminophen, the solubility ratios of all SD materials are close to unity. It is not a surprise since acetaminophen is considered a BCS class I/III compound where solubility is considered high (32,33). The use of acetaminophen as a model compound in making amorphous materials is due to its high degree of crystallinity. A summary table is illustrated in Table II. Based on this data, recommendations of polymer system and maximum drug load can be made. For example, among these three polymers, the best polymer for indomethacin is PVP (based on the drug load) and maximum drug load is at least 70%. This finding is in agreement with other researchers where indomethacin PVP SDD was made up to 80% drug load (34,35). It is believed that the high degree of drug load of indomethacin with PVP is an attribute of indomethacin’s interaction with PVP in solid dispersions through hydrogen bonds formed between the drug hydroxyl and polymer carbonyl group (35–37). In contrast, PVP is less likely to work with griseofulvin due to the lack of a hydrogen bond accepter. Furthermore, both HPMC and HMPCAS worked as well. Compared with HPMC, HPMCAS was found to work slightly better for both indomethacin and celebrex and equally for acetaminophen and griseofulvin (30). It is worth noticing that although, both HPMC and HPMCAS work with griseofulvin, the drug load remains very low (<30%). A different polymer system may be needed to obtain a higher drug load. This finding is not a surprise since most of the recent griseofulvin SDD research has been focused on ternary systems (38,39).
Table II.
Summary of Model Compounds Solubility Ratio of SD Materials, Displayed as Fold Improvement Over 100% API
| Compound | Acetaminophen | Indomethacin | Celebrex | Griseofulvin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer | HPMC | HPMCAS | PVP | HPMC | HPMCAS | PVP | HPMC | HPMCAS | PVP | HPMC | HPMCAS | PVP |
| % drug | ||||||||||||
| 100% | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 10% | 1 | 1 | 2 | 6 | 3 | 3 | 4 | 6 | 7 | 12 | 22 | 2 |
| 20% | 1 | 1 | 2 | 4 | 2 | 3 | 3 | 3 | 4 | 7 | 18 | 2 |
| 30% | 1 | 1 | 1 | 4 | 3 | 3 | 2 | 3 | 3 | 5 | 15 | 2 |
| 40% | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 6 | 8 | 2 |
| 50% | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 5 | 4 | 1 |
| 60% | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 3 | 3 | 1 |
| 70% | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 3 | 2 | 1 |
SD solid dispersions, API active pharmaceutical ingredient, HPMC hydroxypropyl methylcellulose, PVP polyvinylpyrrolidone
These findings are very critical, since drug load, correct polymer(s), and thermal stability of the amorphous material are essential for the success of utilizing SDs as a drug delivery option. The selection of polymer will ultimately influence the quality and stability of SD materials, hence, the in vivo performance. For example, if PVP was selected to make griseofulvin SD with high drug loading, the risk of crystallization during storage would be higher. If during storage, amorphous material converts back to a crystalline or partially crystalline form, there may be an impact on dissolution rate and solubility. This impact may translate to reduced exposure and large variability in vivo. Both cases are not acceptable for long-term usage. Furthermore, the best polymer systems will allow maximum drug load and reduce the pill burden. These findings allow the formulator to narrow down polymer(s) selection and drug load early on. These results combined with solubility data enable one to quickly rule out the undesired drug load against a certain polymer.
In order to further test the system, compound A, a low-solubility b-Raf kinase inhibitor (21), was used as a benchmark compound for further comparisons. Compound A was made in-house and found to have solubility limited absorption in vivo (21). Several compositions of SDDs of compound A were made by a spray dry process in large scales to improve oral BA (21). The materials generated by the spray drying process were tested both in vitro and in vivo previously. A direct comparison of SD materials generated by our process against the spray dry process will further strengthen the validity of our method. The same 96-well procedure and conditions were used for compound A sample preparation. For the PXRD study, it was found that all samples were amorphous at day 1. At day 7, all HPMC, HPMCAS, and low drug load PVP samples still remained amorphous, but pure API and high drug load (70%) PVP samples showed crystalline signals. At day 14, all of the HPMC and HPMCAS samples were still amorphous, while pure API and several high drug load PVP samples (50%, 60%, and 70% drug load) showed crystalline signals. An example of PXRD data on PVP samples is illustrated in Fig. 3. This data generated by our 96-well plate method is in good agreement with the data generated by the larger-scale SDD process. It was reported that, upon co-spray drying with HPMCAS, compound A remains amorphous up to 80% drug load (unpublished data). Solubility data comparison is illustrated in Fig. 4. The degree of solubility increase was found to be drug load- and polymer-dependent. In general, material made with HPMCAS has the highest solubility followed by HPMC, then PVP. The solubility improvement versus drug load was non- linear. For example, the solubility of 20% drug load material made with HPMCAS had a 14-fold increase when compared with sprayed drug alone (control). The ratio dropped quickly from 14 to 9 when drug load increased to 30%. The ratio further dropped to sixfold with increasing drug load to 40%. This is not a surprise since the amount of polymer needed to stabilize amorphous materials in aqueous media is very sensitive from compound to compound. The solubility trend generated by our system is very similar to the materials made by large-scale SDD with HPMCAS (21). While they are all amorphous, when compared with the API, the solubility increase of compound A HPMCAS SDD with drug loads of 40%, 60%, and 80% were 30, 10, and 5-fold, respectively (21). The superiority of HPMCAS in increasing compound A solubility in the aqueous medium is likely due to two key properties. First, it is reported the polymer is partially ionized at pH above 5. This charge prevents nano-sized drug polymer aggregates (colloidal particles) from merging into larger aggregates that may not be capable of facilitating release of free drug. Secondly, as HPMCAS is amphiphilic; the hydrophobic regions on the polymer provide sites for drug association, while hydrophilic regions permit the formation of stable, hydrated, nano-colloidal structures in aqueous media (28).
Fig. 3.

PXRD scans of compound A amorphous materials (with PVP) stored at 50°C/75% RH. From left to right: day 0, day 7, and day14. From top to bottom—100%, 10%, 20%, 30%, 40%, 50%, 60%, and 70% drug load
Fig. 4.

Solubility comparison of compound A amorphous materials stored at 50°C/75% RH for 14 days
In order to further compare the SD materials generated by our process to those obtained from the large-scale spray dry process, a small scale-up was conducted by using the same procedure (“Materials and Methods” section). Since the large scale-up SDD was only made with HPMCAS due to its best in vitro performance (21), only HPMCAS was used for our small scale-up. Both 25% and 40% drug load SD materials were made and characterized by DSC and HPLC. The results were compared with SDDs obtained from the spray drying process. DSC data suggest that the SD materials made by our process were very close to those made with large-scale spray dry process. For 25% drug load, Tg of compound A in SDDs generated from large-scale spray drying process was 116°C (SDD) and 119°C (SD) from our process. For 40% drug load, Tg of the solid molecular dispersion of compound A from the large-scale batch was 123°C (SDD) and 124°C (SD) from our process. No evidence of compound crystallization was seen at temperatures near the melting point (Tm = 200°C). A small deviation was attributed to a slight difference of the final drug loads and purity of the API used. Detailed information is illustrated in Fig. 5.
Fig. 5.

DSC comparison of SD (small in-house scale-up) and SDD (large-scale process) materials of compound A
Based on the data, it is concluded that the SD materials made from our process are very similar (if not equal) to those made from large-scale SDD. We are fairly confident that our well-validated process can be used as SD formulation screening method to identify suitable polymer(s) and drug load. With our device, the same conclusion was drawn based on PXRD, stability, and solubility data with much less time and resources. It has been proven that this method is simple, cost-effective, accurate, capable of providing multiple outputs, and can be used in a high throughput mode. A flow chart of this HTS assay is illustrated in Fig. 6. One limitation of this method is that, due to the small well size, only one time point sample can be drawn for solubility testing. The current design forbids us from constructing a full solubility profile to potentially investigate dissolution rate, which may help to further narrow down the selections for scale up. Since amorphous material is in a high-energy metastable state, amorphous drug in SDDs will eventually convert to the crystalline form in the aqueous medium over time. Ideally, multiple solubility measurements would be made to get a clearer picture of solubility improvement and the effect on dissolution rate. For future development, a plate with larger well can be used to easily overcome this limit. Furthermore, a fully automated robotic system may be needed to enhance the speed and accuracy of our method.
Fig. 6.

Flow chart
CONCLUSIONS
It is well understood that one of the biggest challenges in the pharmaceutical industry nowadays is achieving reasonable exposure after oral delivery of poorly water-soluble drug candidates in both the preclinical and clinical settings. In order to overcome such an issue, researchers have developed many technologies/platforms to enable oral delivery of problematic compounds. Within those technologies, polymer-based amorphous systems such as hot melt extrusion and SDD have drawn a lot of interest in the past decade. Despite the high interest level, the implementation of these technologies early on was limited mainly due to high material and time demands. A 96-well SDD screening system has been developed and validated. This system combines sensitivity, selectivity, and convenience. The setup is very flexible: Well size, amount of materials, type of materials, and detection method can be changed easily to accommodate all needs. In addition, minimal compound requirement makes it an ideal assay in the drug discovery lab setting. Although other efforts have been made to answer the same question, none provided all the advantages such as accuracy, ease of use, cost-effectiveness, and scalability as our system did. One of the biggest challenges the pharmaceutical industry is facing today is the need to lower costs and to reduce time to market. Therefore, generating high-quality data is one of the top priorities for the industry. We believe that, by using the system described above, reliable data for decision making can be obtained early in the process without large investments, and such effort cannot be overemphasized.
Acknowledgment
Authors thank Array BioPharma (Boulder, CO) and Genentech Chemists which provided compound A.
References
- 1.Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JAS. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm. 2008;5(6):1003–1019. doi: 10.1021/mp8000793. [DOI] [PubMed] [Google Scholar]
- 2.Lipinski CA. Poor aqueous solubility—an industry wide problem in drug discovery. Am Pharm. 2002;5:82–85. [Google Scholar]
- 3.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2002;44:235–249. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 4.Lipinski CA. Physicochemical properties and the discovery of orally active drugs: technical and people issues. Molecular informatics: confronting complexity. Proceedings of the Beilstein-Institut Workshop. Germany: Frankfurt; 2003. [Google Scholar]
- 5.Gardner CR, Walsh CT, Almarsson O. Drugs as materials: valuing physical form in drug discovery. Nat Rev Drug Discov. 2004;3:926–934. doi: 10.1038/nrd1550. [DOI] [PubMed] [Google Scholar]
- 6.Schroter C. Prioritizing molecules based on physicochemical characteristics. Am Pharm. 2006;9:60–67. doi: 10.1002/jps.3080090114. [DOI] [Google Scholar]
- 7.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 8.Anon. November 2006. New drug development. GAO Report to Congress. GAO-07-49.
- 9.Ruben AJ, Kiso Y, Freire E. Overcoming roadblocks in lead optimization: a thermodynamic perspective. Chem Biol Drug Des. 2006;67:2–4. doi: 10.1111/j.1747-0285.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 10.Gao P. Amorphous pharmaceutical solids: characterization, stabilization, and development of marketable formulations of poorly soluble drugs with improved oral absorption. Mol Pharm. 2008;5(6):903–904. doi: 10.1021/mp800203k. [DOI] [PubMed] [Google Scholar]
- 11.Hageman MJ, Miyake PJ, Stefanski KJ, He X, Rohrs BR, Mackin LA, Kararli TT. Solid state form of celecoxib having enhanced bioavailability. 2001;WO0141536.
- 12.Gupta P, Kakumanu VK, Bansal AK. Stability and solubility of celecoxib-PVP amorphous dispersions: a molecular perspective. Pharm Res. 2004;21(10):1762–1769. doi: 10.1023/B:PHAM.0000045226.42859.b8. [DOI] [PubMed] [Google Scholar]
- 13.DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams RO., III Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm. 2008;5(6):968–980. doi: 10.1021/mp800042d. [DOI] [PubMed] [Google Scholar]
- 14.Lipinski C, Lombardo F, Dominy B, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 15.Hancock B, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17:397–404. doi: 10.1023/A:1007516718048. [DOI] [PubMed] [Google Scholar]
- 16.Imaizumi H. Stability and several physical properties of amorphous and crystalline forms of indomethacin. Chem Pharm Bull(Tokyo) 1980;29:983–987. doi: 10.1248/cpb.28.2565. [DOI] [PubMed] [Google Scholar]
- 17.Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design and performance. Crit Rev Ther Drug Carrier Syst. 2004;21:133–193. doi: 10.1615/CritRevTherDrugCarrierSyst.v21.i3.10. [DOI] [PubMed] [Google Scholar]
- 18.Patel RC, Masnoon S, Patel MM, Patel NM. Formulation strategies for improving drug solubility using solid dispersions. Pharm Rev. 2009;7(6):1918–5561. [Google Scholar]
- 19.Mansky P, Dai W, Li S, Pollock-Dove C, Daehne K, Dong L, Eichenbaum G. Screening method to identify preclinical liquid and semi-solid formulations for low solubility compounds: miniaturization and automation of solvent casting and dissolution testing. J Pharm Sci. 2007;96(6):1548–1563. doi: 10.1002/jps.20799. [DOI] [PubMed] [Google Scholar]
- 20.Shanbhag A, Rabel S, Nauka E, Casadevall G, Shivanand P, Eichenbaum G, Mansky P. Method for screening of solid dispersion formulations of low-solubility compounds—miniaturization and automation of solvent casting and dissolution testing. Int J Pharm. 2008;351:209–218. doi: 10.1016/j.ijpharm.2007.09.042. [DOI] [PubMed] [Google Scholar]
- 21.Wenglowsky S, Ren L, Ahrendt KA, Laird ER, Aliagas I, Alicke B, Buckmelter AJ, Choo EF, Dinkel V, Feng B, Gloor SL, Gould SE, Gross S, Gunzner-Toste J, Hansen JD, Hatzivassiliou G, Liu B, Malesky K, Mathieu S, Newhouse B, Raddatz NJ, Ran Y, Rana S, Randolph N, Risom T, Rudolph J, Savage S, Selby LT, Shrag M, Song K, Sturgis HL, Voegtli WC, Wen Z, Willis BS, Woessner RD, Wu W, Young WB, Grina J. Pyrazolopyridine inhibitors of B-RafV600E. Part 1: the development of selective, orally bioavailable, and efficacious inhibitors. ACS Med Chem. 2011;2:342–347. doi: 10.1021/ml200025q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuji Chemical/APIs & Pharmaceuticals Website: http://www.fujichemical.co.jp/english/medical/spray_dry/solution/detail.html
- 23.Bee T, Rahman M. Using polymer technology to enhance bioavailability. Pharm Technol. 2010;34(9):6. [Google Scholar]
- 24.Broman E, Khoo C, Taylor LS. A comparison of alternative polymer excipients and processing methods for making solid dispersions of a poorly water soluble drug. Int J Pharm. 2001;222(1):139–151. doi: 10.1016/S0378-5173(01)00709-8. [DOI] [PubMed] [Google Scholar]
- 25.Damian F, Blaton N, Kinget R, Van den Mooter G. Physical stability of solid dispersions of the antiviral agent UC-781 with PEG 6000, Gelucire 44/14 and PVP K30. Int J Pharm. 2002;244(1–2):87–98. doi: 10.1016/S0378-5173(02)00316-2. [DOI] [PubMed] [Google Scholar]
- 26.Thybo P, Hovgaard L, Lindelov JS, Brask A, Andersen SK. Scaling up the spray drying process from pilot to production scale using an atomized droplet size criterion. Pharm Res. 2008;25(7):1610–1620. doi: 10.1007/s11095-008-9565-8. [DOI] [PubMed] [Google Scholar]
- 27.De Villiers MM, Wuster DE, Van der Watt JG, Ketkar A. X-Ray powder diffraction determination of the relative amount of crystalline acetaminophen in solid dispersions with polyvinylpyrrolidone. Int J Pharm. 1998;163(1–2):219–224. doi: 10.1016/S0378-5173(97)00367-0. [DOI] [Google Scholar]
- 28.Gong HK, Viboonkiat R, Rehman IU, Buckton G, Darr JA. Formation and characterization of porous indomethacin-PVP coprecipitates prepared using solvent-free supercritical fluid processing. J Pharm Sci. 2005;94(12):2583–2590. doi: 10.1002/jps.20474. [DOI] [PubMed] [Google Scholar]
- 29.Corrigan OI, Holohan EM, Reilly MR. Physicochemical properties of indomethacin and related compounds co-spray dried with polyvinyl-pyrrolidone. Drug Dev Ind Pharm. 1985;11(2&3):677–695. doi: 10.3109/03639048509056895. [DOI] [Google Scholar]
- 30.Appel LE, Friesen DT, Herbig SM, Ketner RJ, Shamblin SL. Dosage Forms Comprising celecoxib providing both rapid and sustained pain relief. Patent application number: 20100233272. 2010.
- 31.Vasanthavada M, Tong W, Joshi Y, Kislalioglu MS. Phase behavior of amorphous molecular dispersions II: role of hydrogen bonding in solid solubility and phase separation kinetics. Pharm Res. 2005;22(3):440–448. doi: 10.1007/s11095-004-1882-y. [DOI] [PubMed] [Google Scholar]
- 32.Curatolo W, Nightingale JA, Herbig SM. Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm Res. 2009;26(6):1419–1431. doi: 10.1007/s11095-009-9852-z. [DOI] [PubMed] [Google Scholar]
- 33.Neirinckx E, Vervaet C, De Boever S, Remon JP, Gommeren K, Daminet S, De Backer P, Croubels S. Species comparison of oral bioavailability, first-pass metabolism and pharmacokinetics of acetaminophen. Res Vet Sci. 2010;89(1):113–119. doi: 10.1016/j.rvsc.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Kalantzi L, Reppas C, Dressman JB, Amidon GL, Junginger HE, Midha KK, Shah VP, Stavchansky SA, Barends DM. Biowaiver monographs for immediate release solid oral dosage forms: acetaminophen (paracetamol) J Pharm Sci. 2006;95(1):4–14. doi: 10.1002/jps.20477. [DOI] [PubMed] [Google Scholar]
- 35.Yoshioka M, Hancock BC, Zografi G. Inhibition of indomethacin crystallization in poly(vinylpyrrolidone) coprecipitates. J Pharm Sci. 1995;84(8):983–986. doi: 10.1002/jps.2600840814. [DOI] [PubMed] [Google Scholar]
- 36.Taylor LS, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res. 1997;14(12):1691–1698. doi: 10.1023/A:1012167410376. [DOI] [PubMed] [Google Scholar]
- 37.Rumondor AC, Marsac PJ, Stanford LA, Taylor LS. Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol Pharm. 2009;6(5):1492–1505. doi: 10.1021/mp900050c. [DOI] [PubMed] [Google Scholar]
- 38.Al-Obaidi H, Brocchini S, Buckton G. Anomalous properties of spray dried solid dispersions. J Pharm Sci. 2009;98(12):4724–4737. doi: 10.1002/jps.21782. [DOI] [PubMed] [Google Scholar]
- 39.Al-Obaidi H, Buckton G. Evaluation of griseofulvin binary and ternary solid dispersions with HPMCAS. AAPS PharmSciTech. 2009;10(4):1172–1177. doi: 10.1208/s12249-009-9319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]